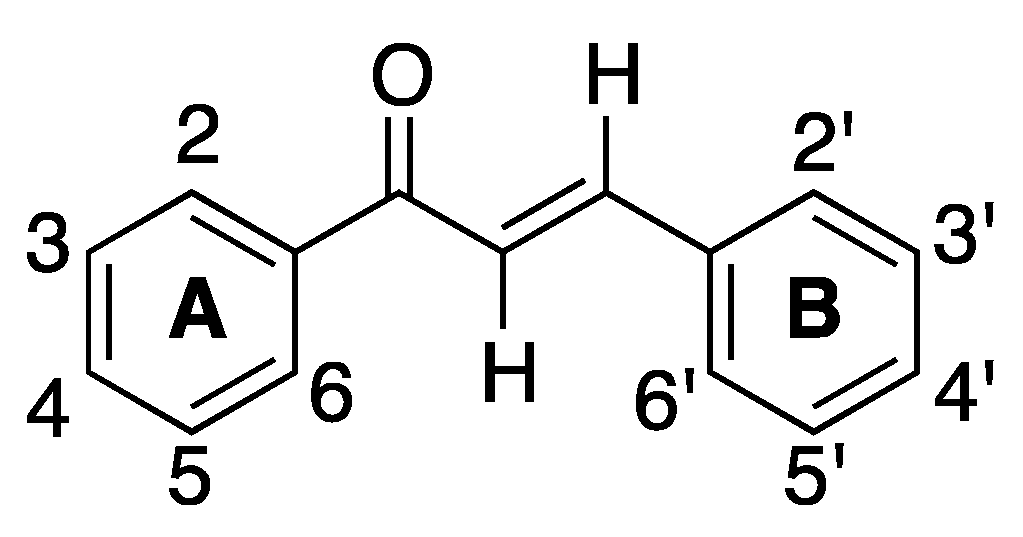
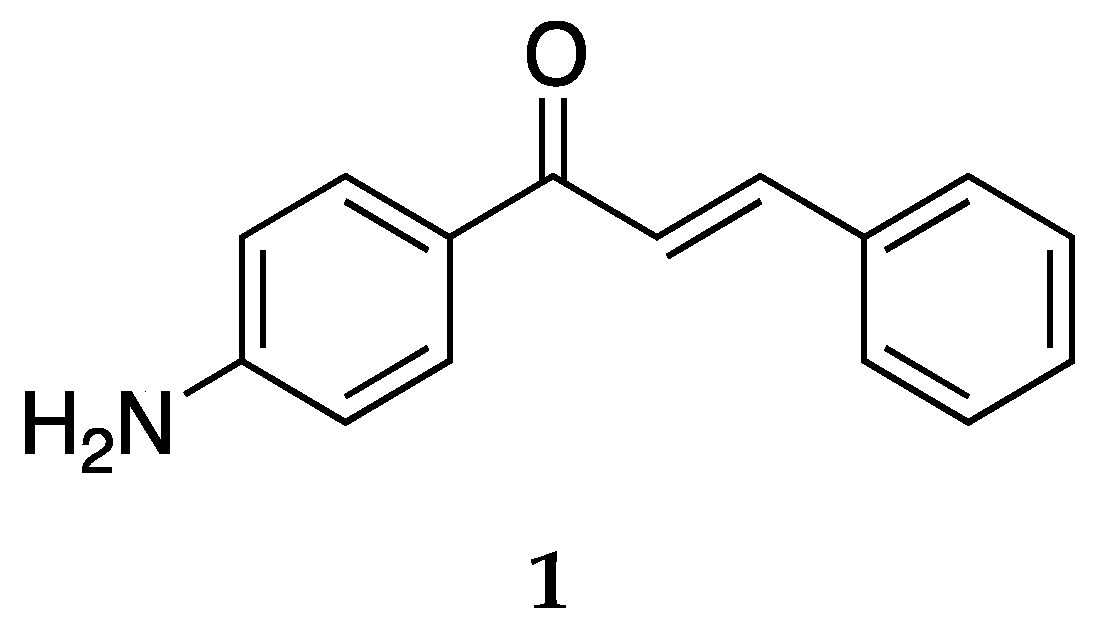
2.1.1. 4-Aminochalcone and Derivatives
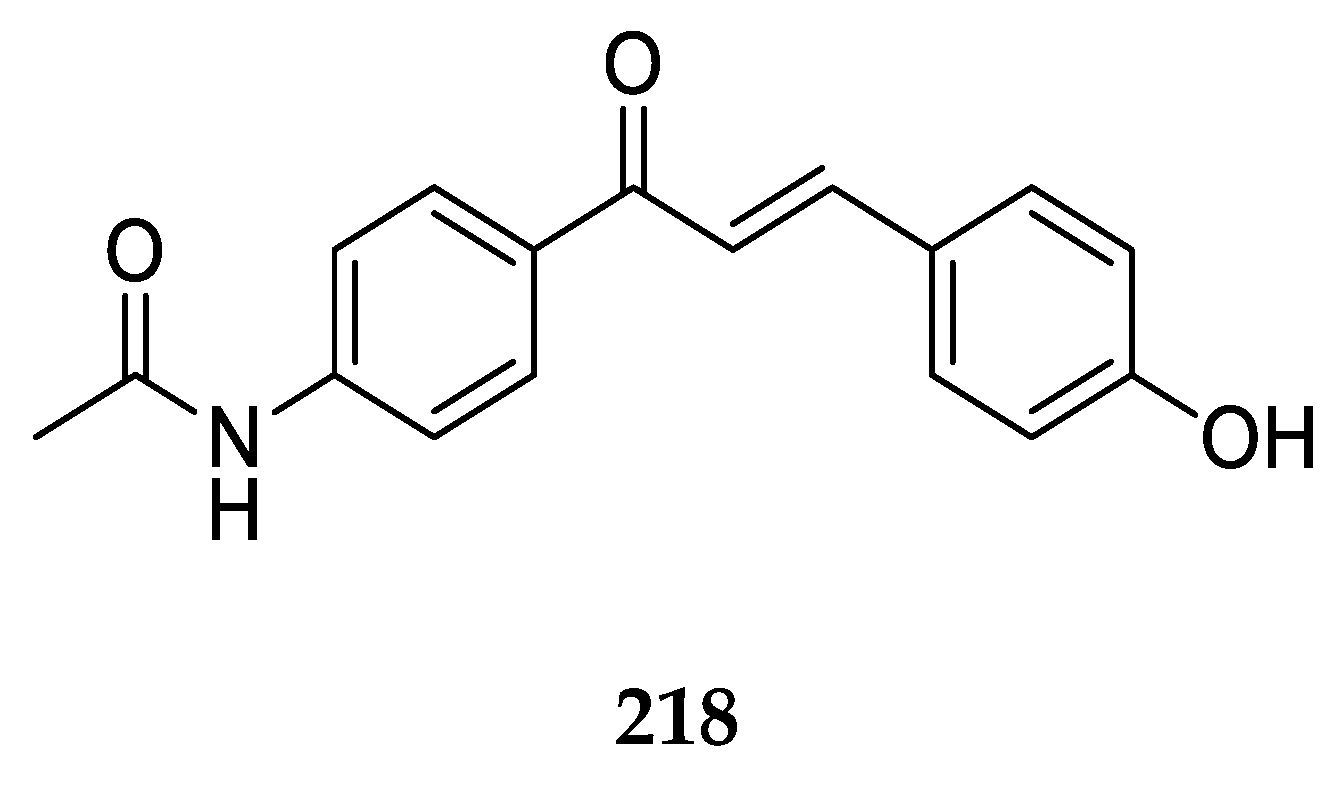
The most studied aminochalcone
1 contains a -NH
2 substitution at position 4 on ring A (
Figure 3). The amino group on ring A increases the magnitude of the positive charge on the olefinic carbon atom adjacent to ring B. This has been proposed to enhance the electrophilic attack of these compounds on cancerous cells [
36].
Compound
1 has been extensively studied for its anticancer activity on multiple cell lines along with antibacterial and antifungal activities. Dimmock et al. [
36] synthesized and evaluated the cytotoxic activities of several 4-aminochalcones, including
1 against the human Molt 4/C8, CEM T-lymphocytes, murine P388, and L1210 cancer cell lines. Compound
1 has also been prepared and tested by Romagnoli et al. [
37] on five different cancer cell lines. Moreover, other groups have also tested
1 on several other cancer cell lines. The IC
50 values are summarized in
Table 1.
The IC
50 values of a simple chalcone against the Molt 4/C8, CEM, P388, and L1210 cell lines are 12.7, 12.5, 9.63, and 41.4 µM, respectively [
36]. The IC
50 values against the same cell lines for
1 are lower than this, which shows that the presence of an amino group at position 4 leads to a more potent compound. Of all the compounds tested by Novilla et al. [
41], compound
1 showed the highest level of cytotoxicity against the K562 cell line with an IC
50 value of 5.87 ± 0.15 µg/mL. Compound
1 showed better activity against this cell lines when compared with Accutane (IC
50 = 39.47 ± 3.14 µg/mL) and comparable activity to imatinib (IC
50 = 2.67 ± 0.53 µg/mL). Compound
1 was also more potent than imatinib (IC
50 = 16.59 ± 0.77 µg/mL) for the HL-60 cell line. Overall, the IC
50 values against multiple cancer cell lines show the potency of compound
1 against cancer cells. Novilla et al. [
41] have also tested
1 on lymphocytes with an IC
50 value of 4.33 ± 0.97 µg/mL. Compound
1 was tested against the canine malignant histiocytic cell line DH82 [
43] and showed a mean percentage cell death of DH82 at 59.2 ± 4.2 in 15.0 µg/mL of the compound. This activity was higher than the simple chalcone (mean percent = 27.7 ± 7.3), which was attributed to the presence of the amino group in
1. The IC
50 values determined by Mai et al. [
38] against
1 showed that the compound had cytotoxic properties as it was potent against a few cell lines. The IC
50 value for
1 against the normal embryonic kidney HEK-293 cells was 95.34 ± 2.48 µM, and the selectivity ratio with HT-29 was <1. Thus, compound
1 could also be toxic to normal cells if used as a cancer therapy.
Along with its cytotoxic activity,
1 has also been tested for its activity against microbes. The antimicrobial activity of
1 against
Escherichia coli ATCC 25923,
Staphylococcus aureus ATCC 25922, and
Candida albicans ATCC 10231 was also reported by Suwito et al. [
40] The diameter of inhibition values for these microbes were reported to be 10.25 ± 0.13 mm, 9.59 ± 0.16 mm, and 9.69 ± 0.02 mm, respectively, tested at a concentration of 500 µg/mL. The inhibition activity of
1 against
Escherichia coli ATCC 25923 and
Candida albicans ATCC 10231 was as strong as the antibiotic sulfamerazine and as strong as sulfadiazine against
Staphylococcus aureus ATCC 25922. The antibacterial and antifungal activities against different strains were also tested by Kozłowska et al. [
42], as shown in
Table 2.
Compound
1 causes complete growth inhibition in the case of
S. aureus DSM799 and
A. niger DSM1957 microbial strains and showed significant prevention of growth for the other strains [
42]. The results of these investigations [
40,
42] show
1 as a promising antibacterial agent.
Apart from cytotoxic and antimicrobial activities, compound
1 has also been tested for its inhibitory activity against the chlorinating activity of the myeloperoxidase (MPO) enzyme [
44]. MPO has been explored as a target for anti-inflammatory therapy due to its ability to generate hypochlorous acid. Compound
1 was identified as a potent inhibitor of the chlorinating activity of MPO with an IC
50 value of 0.26 ± 0.04 µmol/L in a cell-free, purified MPO system. Interestingly, for a simple chalcone and aniline, both did not show any inhibition to the chlorinating activity of MPO, thus showing that the presence of both the amino and chalcone groups is important for any significant activity. The activity of
1 has also been compared with 4-hydroxychalcone, which also did not show any inhibitory activity. Thus, the presence of an amino group seems important compared to an electron-donating group. Moreover, the IC
50 value for
1 is comparable to 5-fluorotryptamine, which is considered a potent MPO inhibitor.
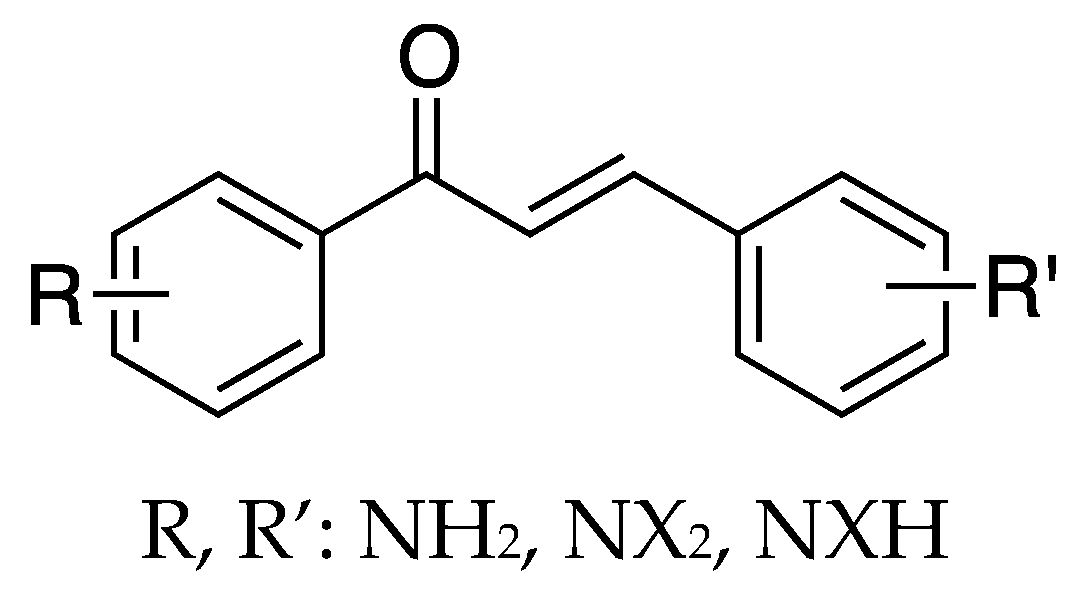
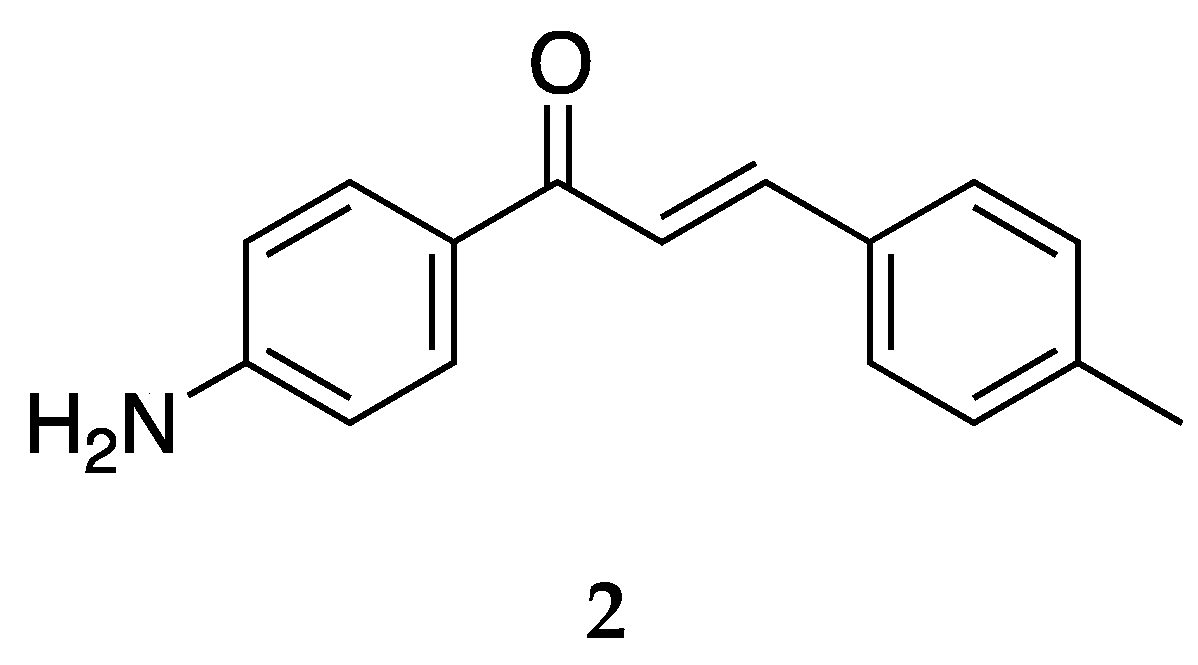
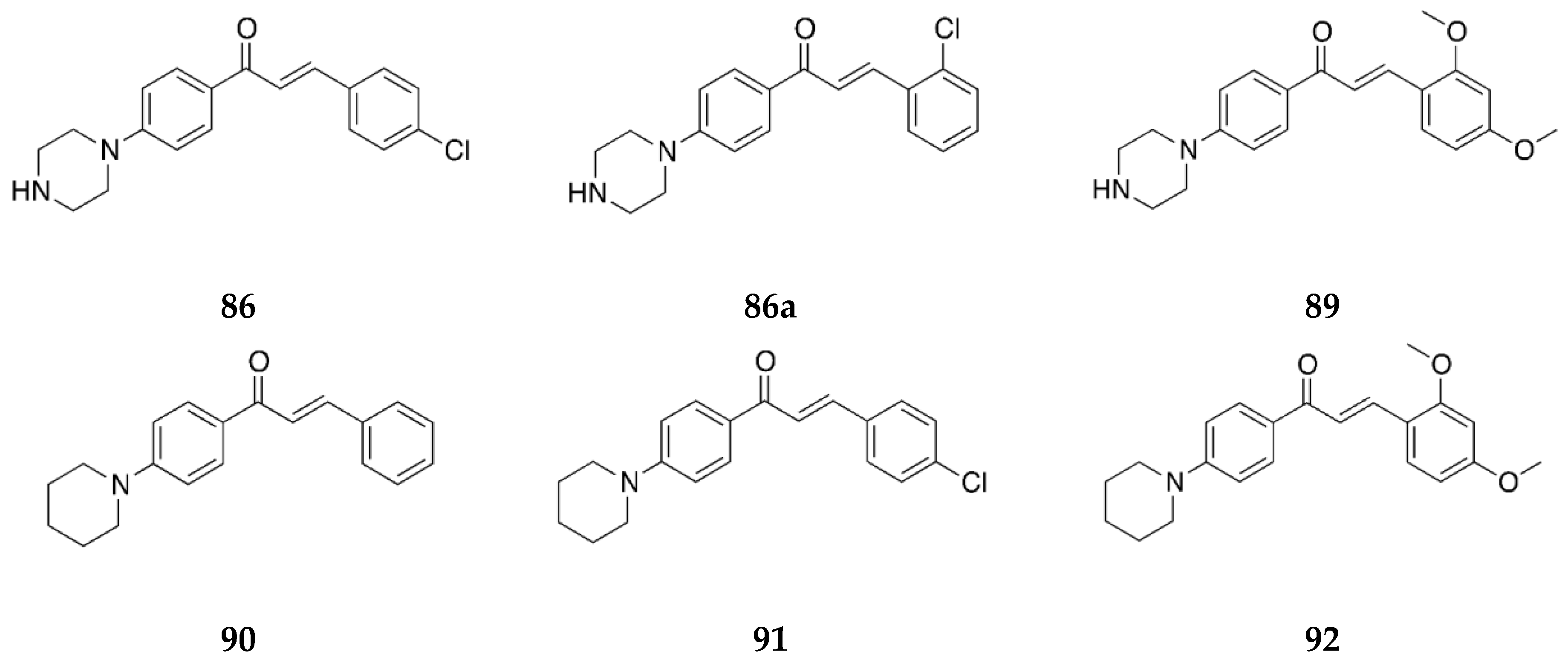
Several derivates of 4-aminochalcone have been synthesized by various groups over the years, with one of them being derivates with aliphatic alkylation on position 4 of ring B (
Figure 4 and
Figure 5).
Compound
2 was tested for its cytotoxic activities by Dimmock et al. [
36], Santos et al. [
43], and Santos et al. [
39], shown in
Table 3.
Aminochalcone
2 has IC
50 values similar to
1; thus, the anticancer activity of 4-aminochalcone does not increase or significantly decrease with the addition of a methyl group on ring B. Santos et al. [
43] also reported the activity against canine malignant histiocytic cell line DH82 for
2, which was 74.7 ± 1.7%. This value is higher than
1, showing that the addition of a methyl group on ring B increased the potency of the compound.
In addition to the cytotoxic activity, the antioxidant activity of
2 has also been tested by Prasad et al. [
45]. Many degenerative diseases such as atherosclerosis, cancer, inflammatory joint disease, asthma, diabetes, senile dementia, and degenerative eye disease originate from deleterious free radical reactions. Most free radical damage is caused by reactive oxidant species (ROS) which can damage genetic material, cause lipid peroxidation in cell membranes, and inactivate membrane-bound enzymes [
46]. While the human body has mechanisms to protect itself from these ROS, often times, exogenous antioxidants are needed to overcome the effects. Prasad et al. [
45] tested the antioxidant activity of a series of aminochalcones. Unfortunately, compound
2 did not show any significant activity with IC
50 values of 86.02 µg/mL, 192.15 µg/mL, and 64.85 µg/mL against superoxide radical, hydroxyl radical, and ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) cation radical, respectively. Compound
2 at 50 µg showed inhibition of 17.33% against lipid peroxidation, which is lower than the percentage inhibition of ascorbic acid 20.46% at the same concentration.
Just like compound
1, compound
2 was also tested for its inhibitory activity against the chlorinating activity of the myeloperoxidase (MPO) enzyme by Zeraik et al. [
44]. Compound
2 showed IC
50 values similar to
1 and was 0.25 ± 0.01 µmol/L for a cell-free, purified MPO system. Thus, the methylation on ring B does not change the potency of aminochalones towards MPO.
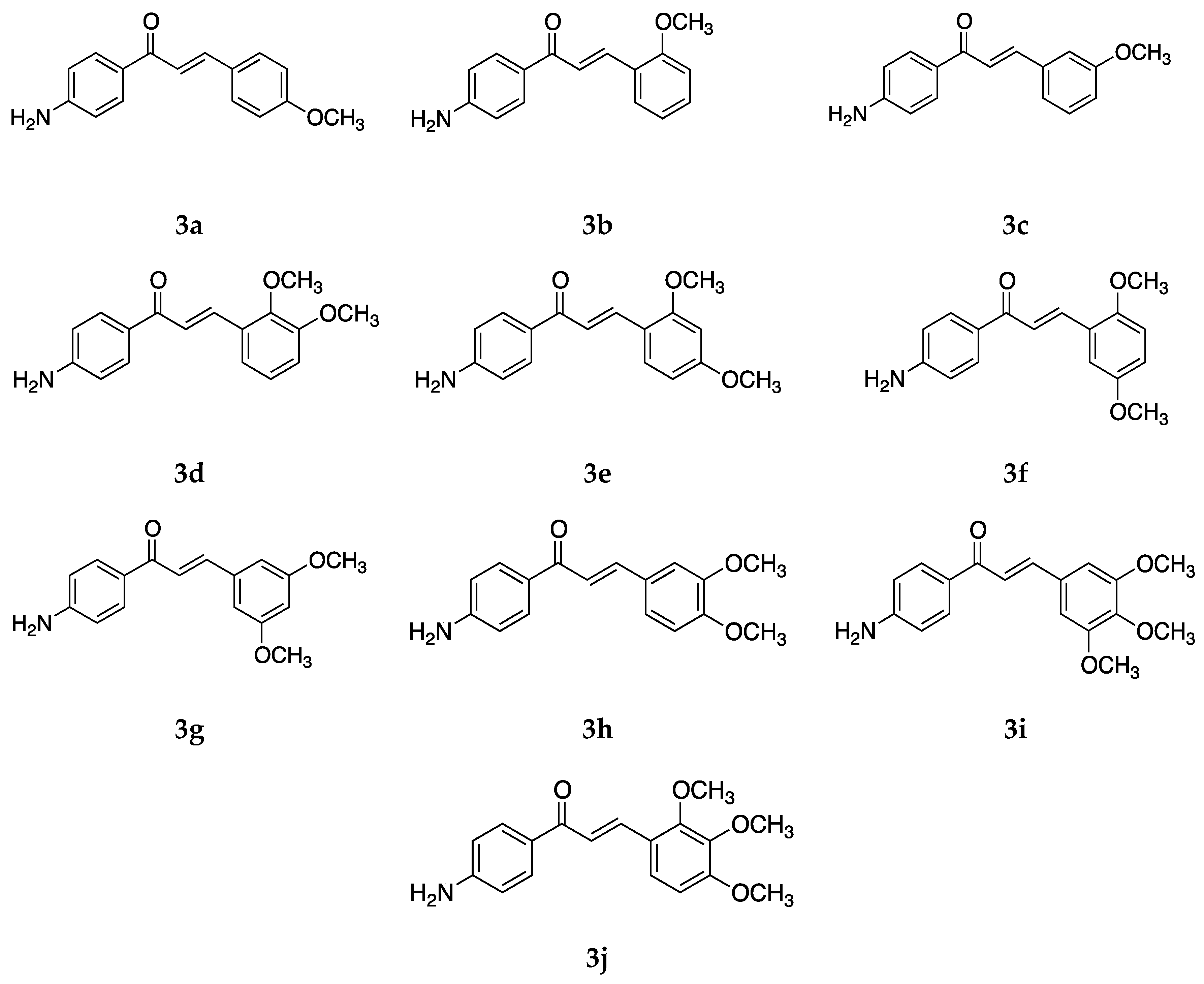
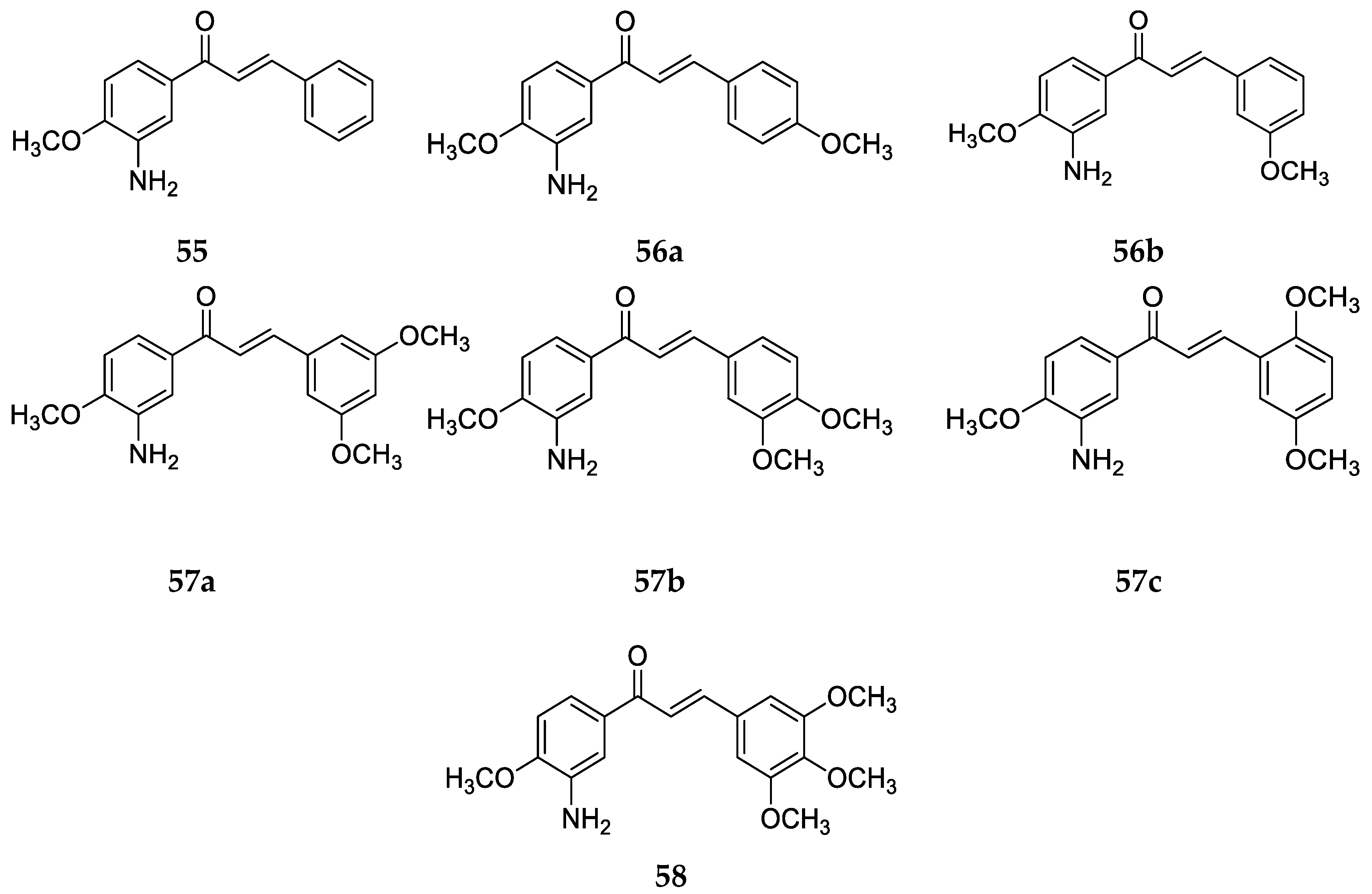
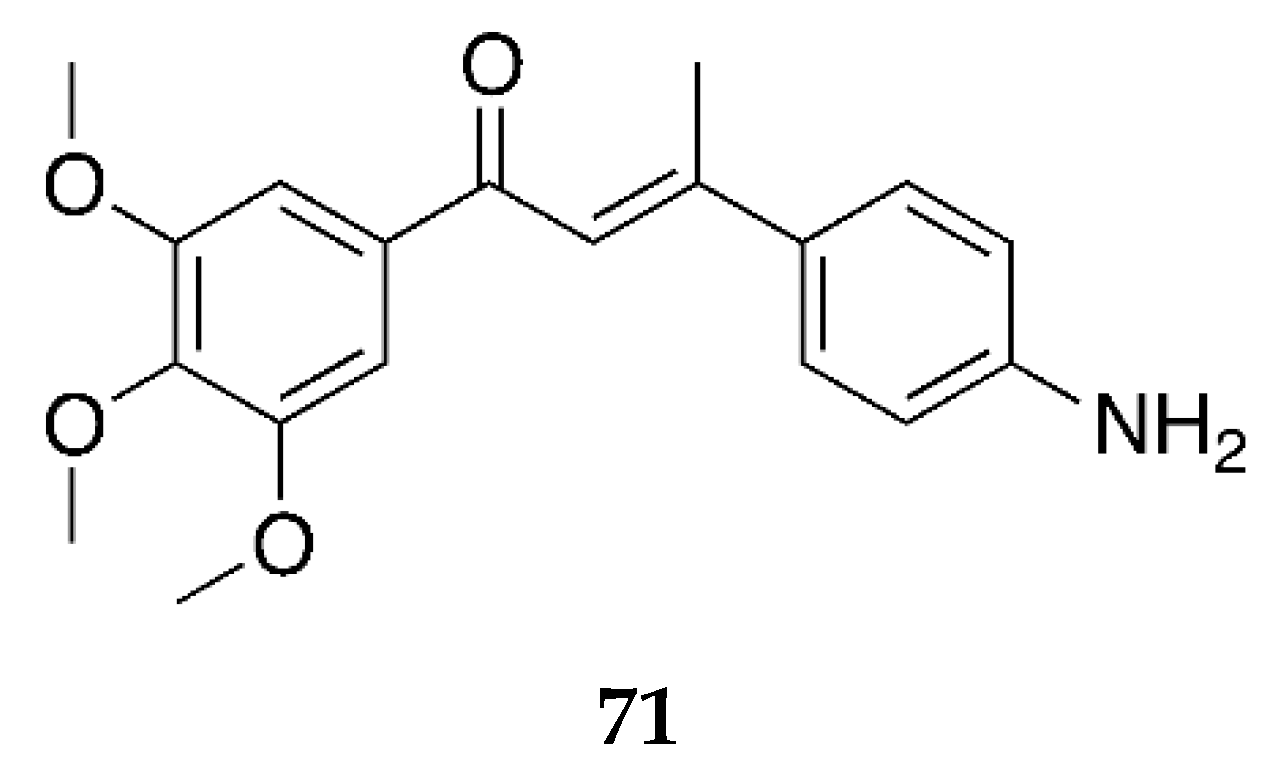
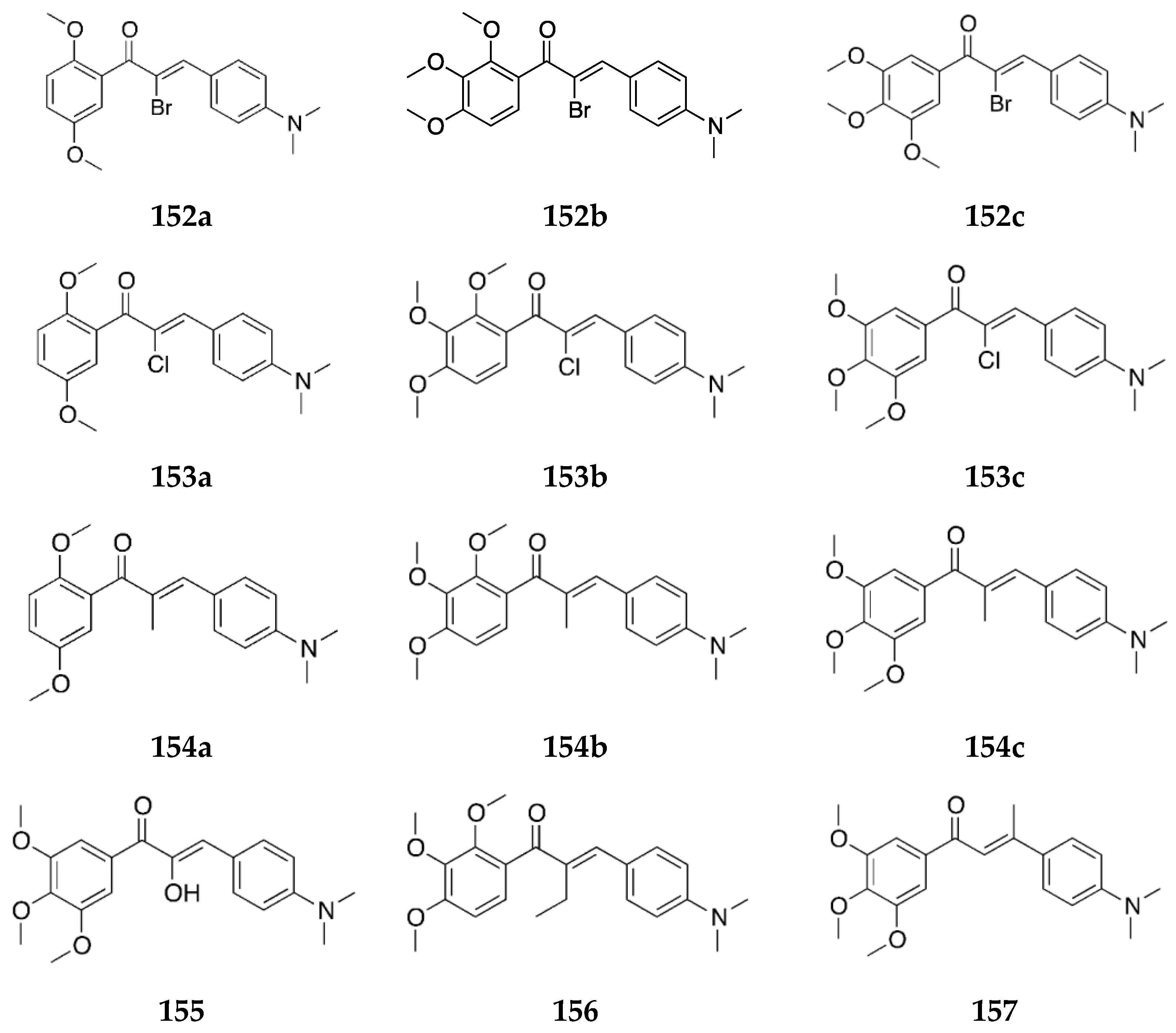
Several 4-aminochalcones have been reported with methoxy side chains on ring B (
Figure 5). These compounds have been tested for some biological activities.
The cytotoxic activity for these compounds on different cell lines has been reported by various research groups, which are shown in
Table 4.
Compounds
1 and
3a differ by a methoxy group on ring B. Though the cytotoxic activity between the two does not greatly differ in some cell lines, in others such as CEM and L1210, the IC
50 value for
1 is lower than that of
3a. Between the mono methoxylated compounds
3a–
c, the values for
3c are higher than the other two for the Molt 4/C8, CEM, L1210, HeLa, FM3A, and HL-60 cell lines. The cytotoxic activity seems to decrease when the methoxy group is placed on position three on ring B. The most active compound for Molt 4/C8, CEM, L1210, HeLa, and FM3A is
3i, which is a tri-methoxylated aminochalcone. For the K562 cell line, compound
3d was the most potent compound, with activity comparable to imatinib and greater than Accutane. Imatinib and Accutane are commercial drugs used to treat different types of cancers. Compounds
3b and
3f showed activity higher than both imatinib and Accutane. Suwito et al. [
40] showed that the methoxy-4-aminochalcones show better anticancer activity compared with methoxy chalcones and methoxy chalcones with 4-bromo; thus, the presence of 4-amino on ring A is essential for significant antiproliferative activity. The number and position of the methoxy group play a part in the anticancer activity of the compounds. Among known structures that bind to tubulin effectively are combretastatin A-4-5 and colchicine, which seem to have some structural overlap with some of these methoxy-substituted chalcones [
41].
Apart from these cancer cell lines, the cytotoxic activity of
3a was also tested against a canine malignant histiocytic cell line (DH82) by Santos et al. [
43], with the mean percentage of DH82 cell death equal to 46.6 ± 1.0%, which is quite less when compared with
1 (59.2 ± 4.2%) and
2 (74.7 ± 1.7%), thus showing that the substitution of a strong electron-donating group on ring B decreased cytotoxicity. A similar conclusion was made by Santos et al. [
39] when they tested
3a against estrogen-receptor-positive breast cancer cells MCF-7 and the triple-negative breast cancer (TNBC) cells MDA-MB-23. Compound
3a did not show any significant activity against the two cell lines.
The antioxidant activities of
3a,
3h, and
3i were tested by Prasad et al. [
45]. Compound
3i had an IC
50 value of 78.32 ± 1.59 µg/mL against the superoxide radical which was the best among all of the tested chalcones. Compounds
3a and
3h had IC
50 values of 80.09 ± 1.21 and 80.65 ± 1.58 µg/mL, respectively, against the superoxide radical. Compounds
3a,
3h, and
3i all showed a dose-dependent inhibition activity against the hydroxyl radical, with IC
50 values of 179.01 ± 1.58, 168.63 ± 1.55, and 158.18 ± 1.63 µg/mL, respectively. Against the ABTS radical cation, all three
3a,
3h, and
3i showed remarkable activities which were comparable to the standard drug, ascorbic acid. The IC
50 values were 56.91 ± 0.68, 52.89 ± 1.31, and 50.06 ± 1.41 µg/mL, respectively, amongst which
3i showed maximum activity from the tested compounds. For the inhibition of lipid peroxide,
3i showed the most potency as well (IC
50 value = 547.18 ± 2.01 µg/mL). The results of this study show that chalcones with electron-releasing groups such as methoxy have greater activity against oxidative species. Between
3a,
3h, and
3i, the scavenging activity increased as the number of methoxy substituents increased on ring B.
Apart from the cytotoxic activities of methoxy-substituted 4-aminochalcomes, the antimicrobial activity of
3a–
f has also been tested by Suwito et al. [
40] against
Escherichia coli ATCC 25923,
Staphylococcus aureus ATCC 259, and
Candida albicans ATCC 10231 using different concentrations of the compounds, similar to
1. All of the compounds showed promising antibacterial activity, especially
3d, which showed the strongest inhibition activity against
Escherichia coli ATCC 25923 (diameter of inhibition = 10.68 ± 0.16 mm at 500 µg/mL),
Staphylococcus aureus ATCC 259 (diameter of inhibition = 10.33 ± 0.01 mm at 500 µg/mL), and
Candida albicans ATCC 10231 (diameter of inhibition = 11.30 ± 0.15 mm at 500 µg/mL). The activity of compound
3d was comparable to the positive controls sulfamerazine and sulfadiazine.
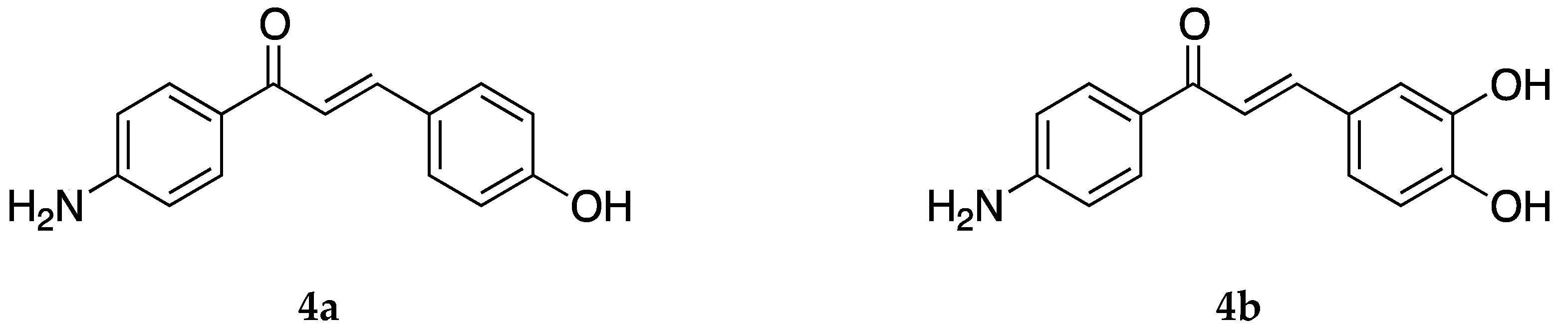
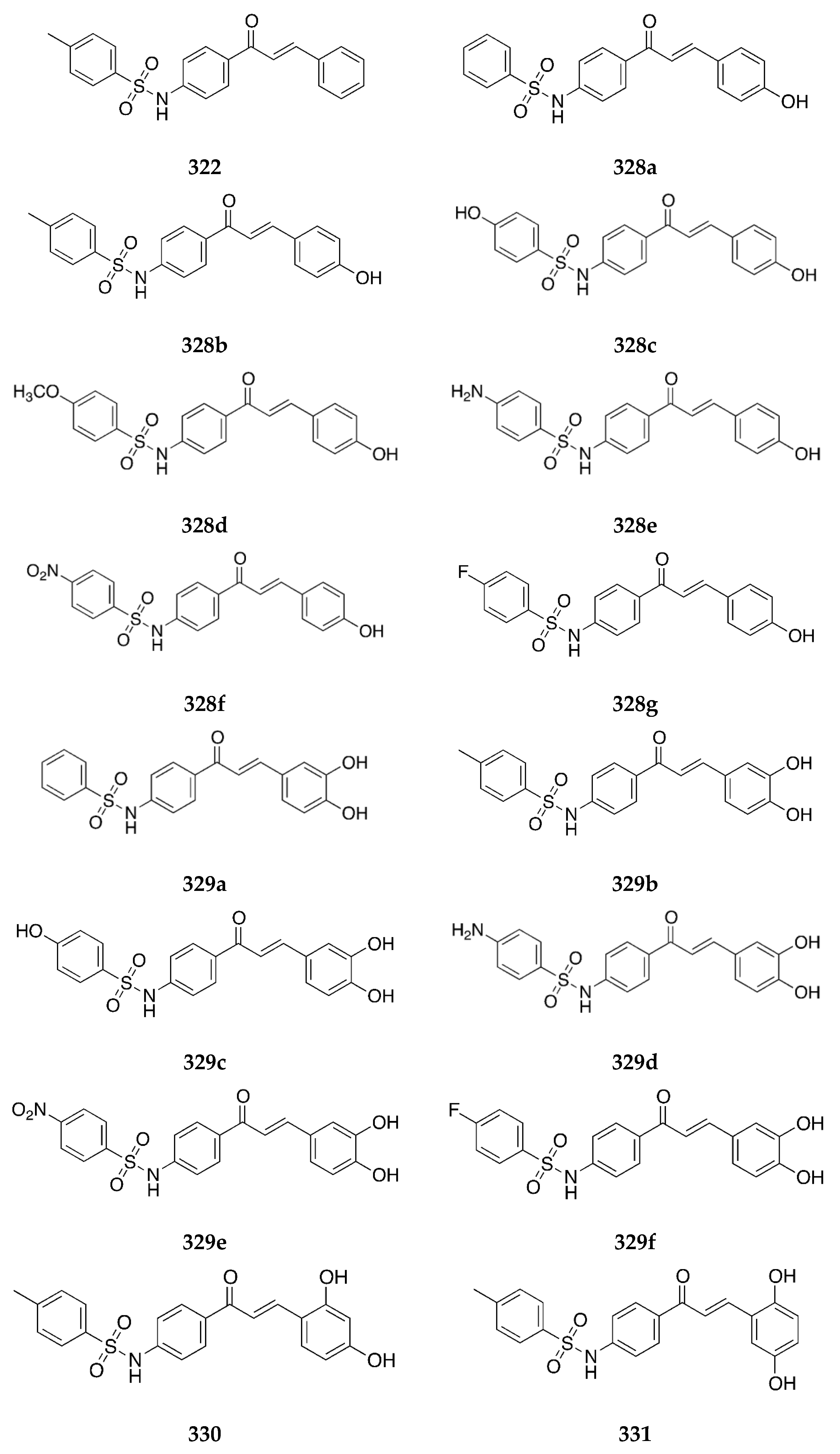
4-Aminochalcones with hydroxy groups on ring B (
Figure 6) have also been synthesized and tested for their biological activities.
Both
4a and
4b were tested as α-glucosidase inhibitors, along with several sulfonamide chalcones [
47]. α-Glucosidase inhibitors can be used in the treatment of cancer, diabetes, and viral diseases. As glycosidase enzymes are responsible for the processing and synthesis of complex carbohydrates, inhibitors of these molecules can be important tools in glycobiology and can help modulate cellular functions along with biological recognition processes. α-Glucosidases catalyze the release of α-D-glucopyranose from the non-reducing ends of various substrates, inhibitors of which can control the uptake of dietary carbohydrates and, thus, can decrease postprandial hyperglycemia, which may be useful in treating diabetes or obesity. Compounds
4a and
4b showed strong inhibitory activities against the three glycosidases that it was tested on. The IC
50 values for
4a were 41.0 and 268.9 µM respectively against α-glucosidase from baker’s yeast and α-amylase from
Bacillus licheniformis. The values for
4b were 62.1 and 126.8 µM against α-glucosidase from the baker’s yeast and ß-amylase from barley. While this activity did not exceed that of the sulfonamides, it was still considered significant, especially when compared with the respective 3-aminochalcones.
Kang et al. [
48] tested the inhibitory activities of
4a and
4b against ß-secretase (BACE1), which is strongly associated with the onset of Alzheimer’s disease (AD) and had IC
50 values of 48.2 ± 1.2 and 17.7 ± 0.8 µM, respectively. The 3,4-dihydroxy derivate was more potent than the one bearing a 4-hydroxy substituent. While they exhibited inhibitory activity, the respective sulfonamide chalcones were manyfold more potent than these two compounds (and will be discussed in their section).
Similar to compounds
1 and
2,
4a was tested for its inhibitory activity against the chlorinating activity of the myeloperoxidase (MPO) [
44]. While both
1 and
2 showed potency towards MPO, the IC
50 value for
4a was reported to be greater than 50.00 µM. This indicated that the presence of an easily oxidizable group cancels the inhibitory activity of the aminochalcone.
Seo et al. [
49] also reported the anti-pigmentary effect of
4a. It had an IC
50 value of 4.8 µM for tyrosinase enzyme and 48.7 µM against the melanin formation cell. The compound was more potent than kojic acid (IC
50 = 16.3 µM), a known inhibitor of tyrosinase.
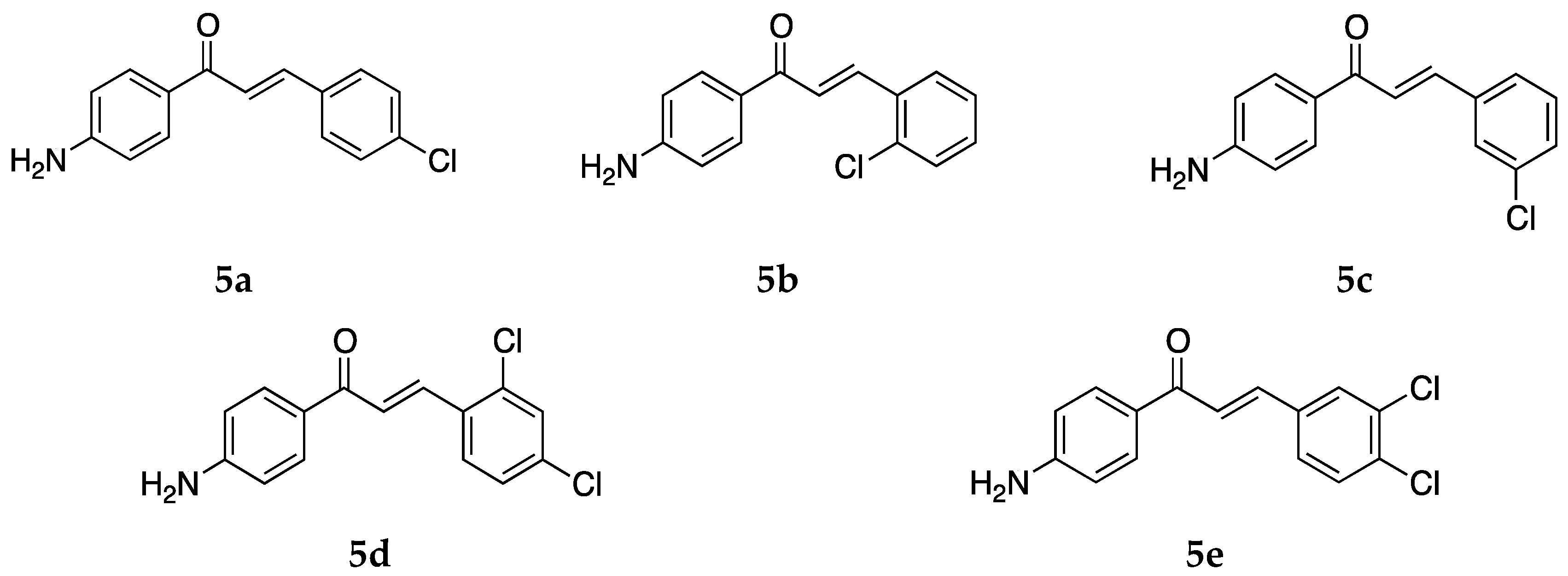
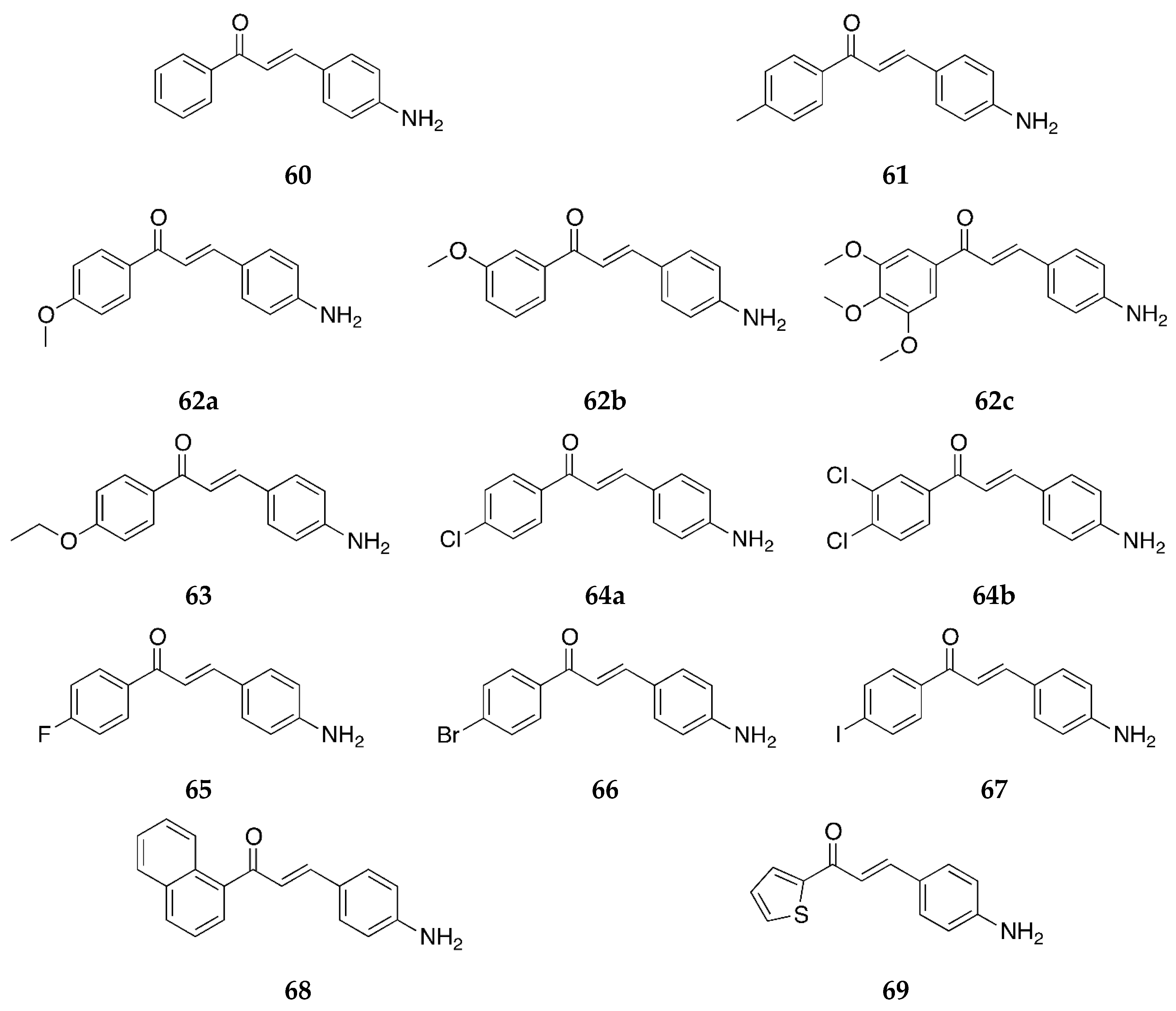
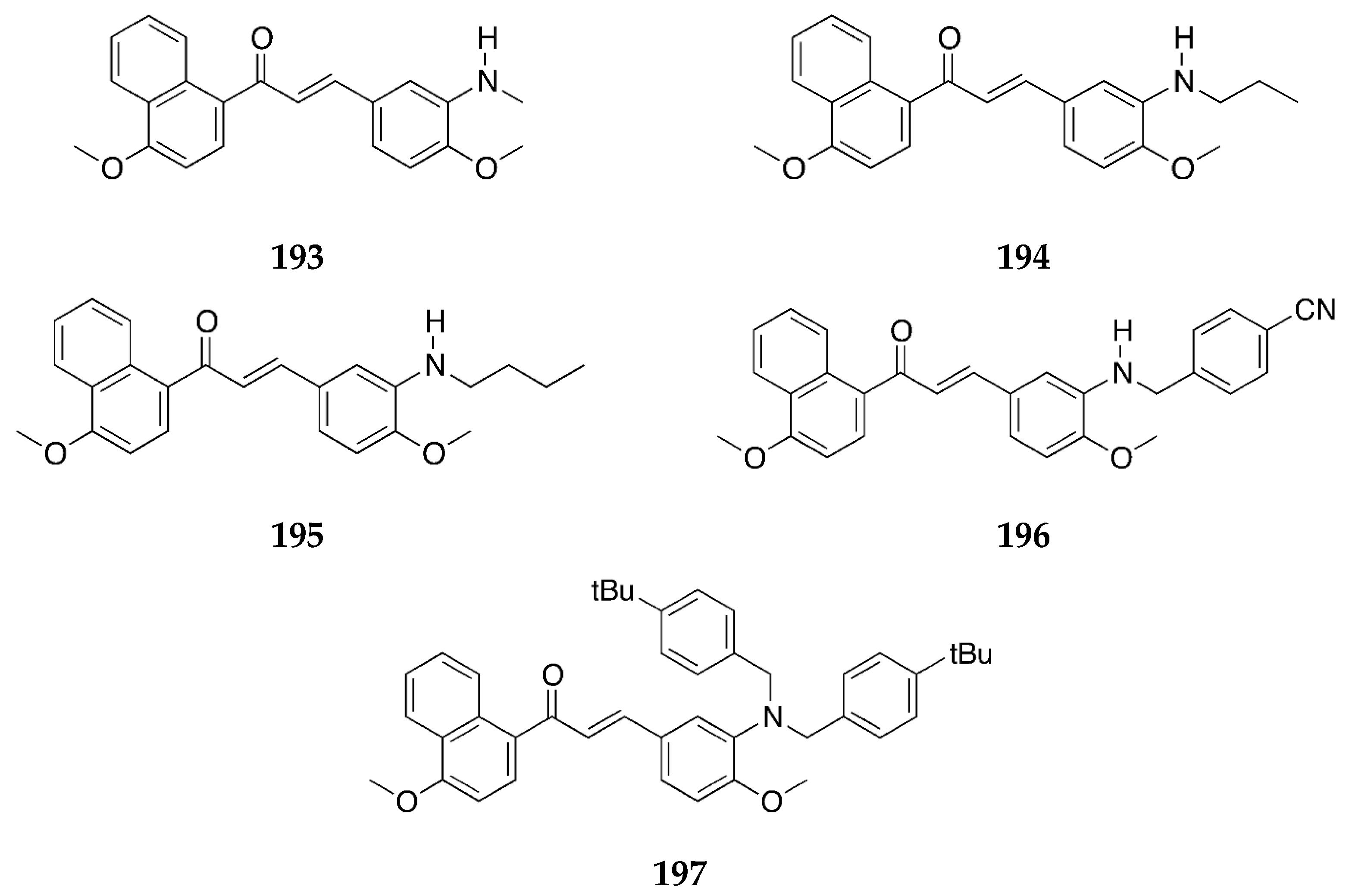
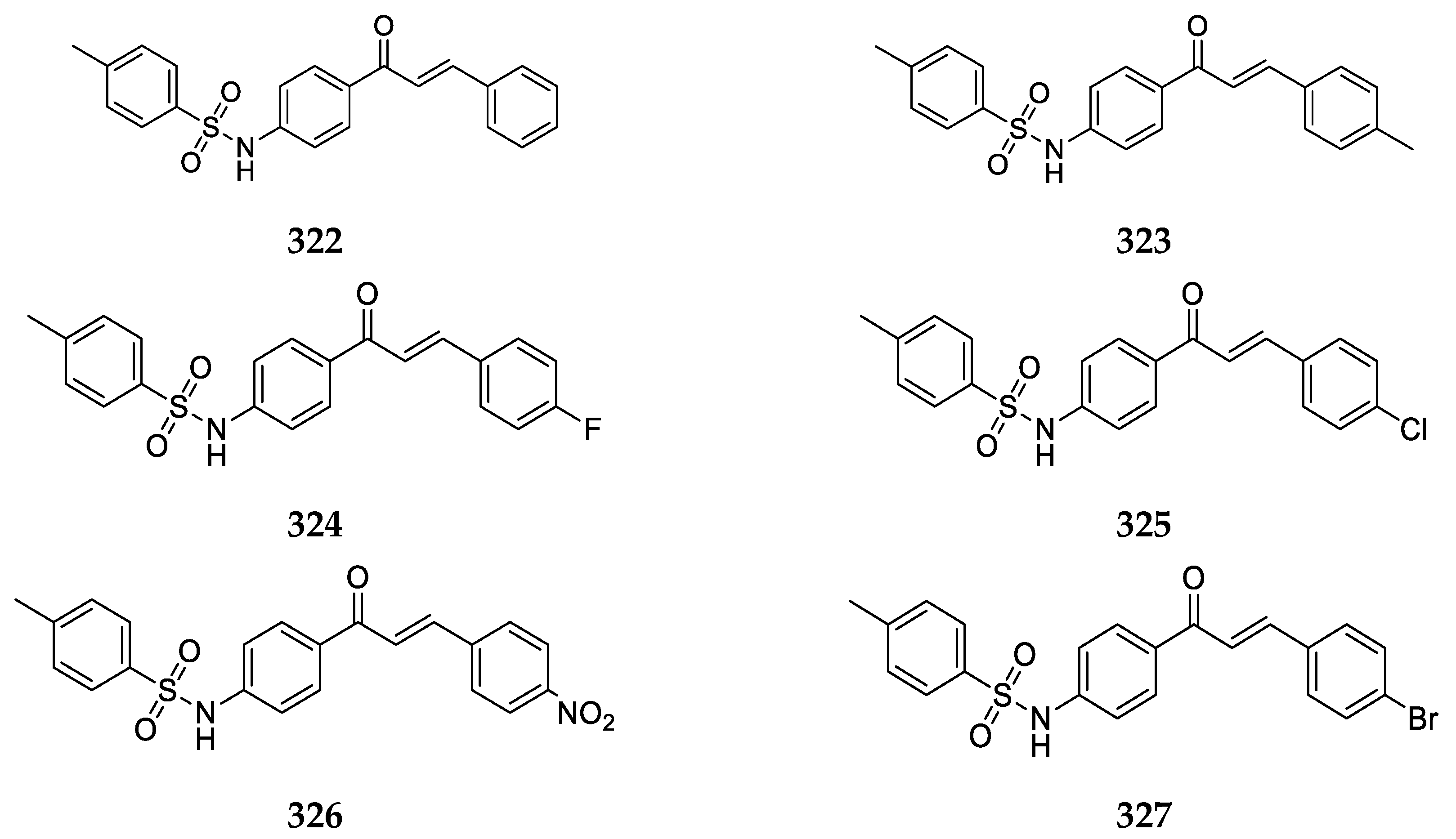
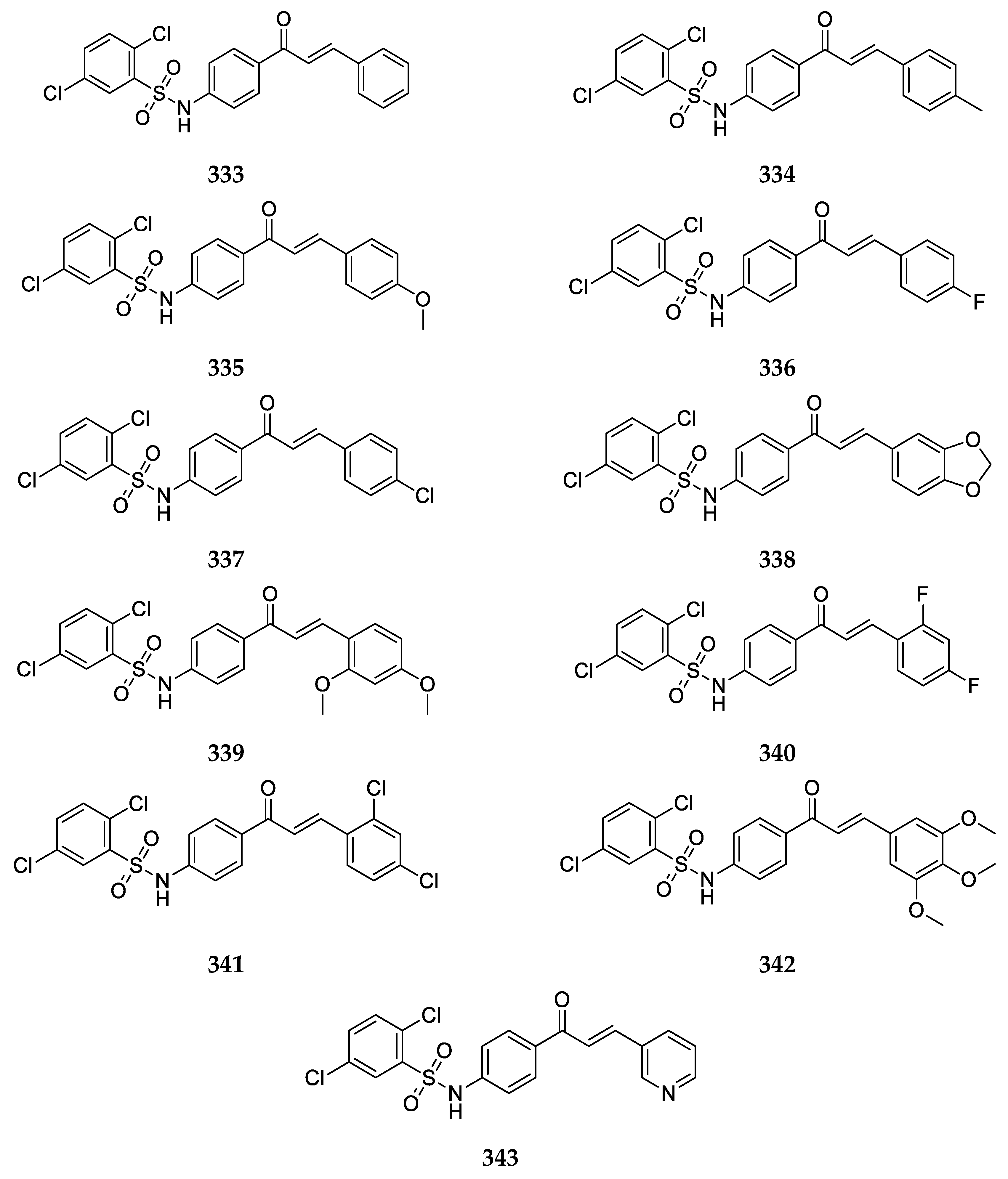
Several 4-aminochalcones with halogen substitutions, particularly chloro-, fluoro-, and bromo- on ring B have also been reported by various research groups over the years. The 4-aminochalcones with various chlorine substitutions have been reported by four research groups and are shown in
Figure 7. The anticancer activity reported for
5a–
e by various research groups is shown in
Table 5.
The cytotoxicity for Molt 4/C8, CEM, P388, and L1210 was retained when chlorine substitutions were introduced on ring B when compared with
1. The IC
50 values for both
5a and
5e were found comparable to those of melphalan against some cell lines (IC
50 values of melphalan: 3.24 ± 0.79, 2.47 ± 0.30, 0.22 ± 0.01, and 2.13 ± 0.03 µM against Molt 4/C8, CEM, P388, and L1210, respectively). Both
5a and
5e were more potent to the cell lines as compared to the acylated versions of these aminochalcones [
36]. Mai et al. [
38] also determined the antiproliferative activities of
5a and
5d, and the study revealed that
5a was more potent than
5d against most of the cancer cell lines, with its best activity against the CEM-SS cell line. The activities of
5d were poor against all the cell lines investigated in this study [
38]. The 3-chlorophenyl derivative,
5c, was the most potent compound against the MCF-7 cell line amongst the tested chloro-4-aminochalcones; on the other hand,
5a showed better activity against the MDA-MB-231 cells. These compounds were more potent than other derivatives with cyano, nitro, and bromo groups [
39]. Compound
5a was the more potent compound amongst all tested by Santos et al. against the canine malignant histiocytic cell line DH82 [
43]. The mean percentage cell death of DH82 was 91.0 ± 1.3 at 15.0 µg/mL of the compound. Compound
5a was subsequently selected for cytotoxic assay against DH82 and MDCK (non-tumorigenic Madin–Darby canine kidney) cells. The IC
50 values were 31.4 and 57.2 µM, respectively. The selectivity ratio was 1.8. The activity of
5a against DH82 was greater than etoposide (IC
50 = 95.5 µM), the reference drug used.
Compounds
5a,
5b, and
5d were also tested for their antioxidant activity by Prasad et al. [
45] (
Table 6). Amongst the three chloro-4-aminochalcones,
5a, the 4-chloro-4-aminochalcone was found to be the more potent and showed dose-dependent inhibition against the superoxide radical, the hydroxyl radical, and lipid peroxidation. The activity of
5a was comparable to the reference ascorbic acid (IC
50 = 45.42 ± 1.24 µg/mL) for the ABTS cation radical.
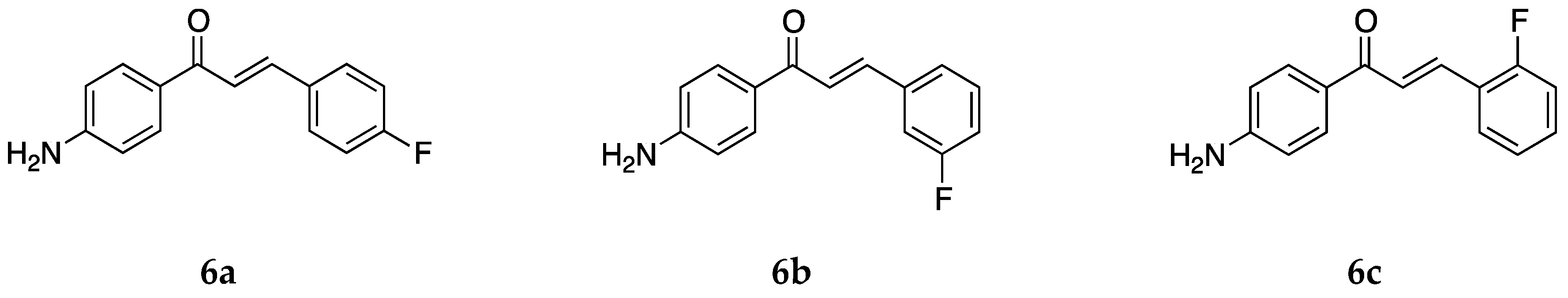
Continuing with halogens, research groups also reported the synthesis and activity of fluoro-4-aminochalcones (
Figure 8).
The anticancer activity of the three mono fluoro derivatives was reported by Santos et al. [
39] against estrogen-receptor-positive breast cancer cells MCF-7 and the triple-negative breast cancer (TNBC) cells MDA-MB-231. Compound
6a did not show any significant activity, and the IC
50 values were not determined. The IC
50 values for
6b were >100 and 74.1 ± 1.2 µM for MCF-7 and MDA-MB-231 cancer cells, respectively. Compound
6c was the more potent against these cells among all of the compounds tested with IC
50 values 13.2 ± 3.5 and 34.7 ± 5.2 µM, respectively. Compound
6c induced apoptosis rather than necrosis in both cells. The 3-chloro-4-aminochalcone compound
5c was also very active against the MCF-7 cell line. A comparison among values for the different compounds tested showed that more electronegative halogens on ring B showed a higher degree of antiproliferative activity against the MCF-7 and MDA-MB-231 cancer cell lines. Electron-donating groups, on the other hand, had a negative effect on bioactivity, as can be seen from the values of compounds
2 and
3a.
The cytotoxic activity for
6a was also tested against the canine malignant histiocytic cell line DH82 and non-tumorigenic Madin–Darby canine kidney MDCK cells [
43] (IC
50 = 34.4 and 70.1 µM, respectively). The selectivity ratio was 2.0. Thus,
6a had a greater selectivity against the DH82 cancer cell line. The three most active compounds in this paper were
2,
5a, and
6a. Compound
2, having a methyl group, is electron-donating, and the halogen-substituted compounds are electron-withdrawing atoms. Thus, it was concluded that the electronic effect of the substituent did not matter much for the antiproliferative activity against these particular cell lines. The halogen chalcones
5a and
6a along with the methyl chalcone
2 are lipophilic and more active than the other compounds tested. Thus, compounds with hydrophobic substitutions on ring B were more potent.
Apart from the anticancer activity, the antioxidant activity for
6a was also tested by Prasad et al. [
45] with IC
50 values of 79.73 ± 1.58, 175.71 ± 1.61, 61.06 ± 1.78, and 552.18 ± 2.98 µg/mL against the superoxide radical, hydroxyl radical, ABTS cation radical, and lipid peroxidation, respectively. While it was not the most potent compound among all those tested,
6a showed dose-dependent activity against all of the radicals tested. Its activity was comparable to the standard ascorbic acid for the ABTS cation and superoxide radical. Compound
6a was also tested for its inhibitory activity against the chlorinating activity of the myeloperoxidase (MPO) enzyme by Zeraik et al. [
44] and had an IC
50 value of 0.25 ± 0.08 µmol/L in a cell-free, purified MPO system. This value is similar to those of
1 and
2. Similar values for compounds substituted with methyl and fluorine on ring B showed that the main property contributing to MPO inhibition is the presence of both amino and chalcone groups in the compound.
4-Aminochalcone compounds with bromine substitutions on ring B (
Figure 9) have also been reported.
Both
7a and
7b were also tested against the estrogen-receptor-positive breast cancer cells MCF-7 and the triple-negative breast cancer (TNBC) cells MDA-MB-231 [
39]. However, these derivatives were not very potent towards the cell lines, especially when compared with compounds substituted with fluorine and chlorine.
The antioxidant activity [
45] for
7b was tested, and the IC
50 values were reported to be 85.65 ± 1.30, 204.33 ± 1.53, 74.45 ± 0.96, and 600.85 ± 2.38 µg/mL against the superoxide radical, hydroxyl radical, ABTS cation radical, and lipid peroxidation, respectively. While this activity was not terrible, it was not comparable to that of ascorbic acid or other derivatives of 4-aminochalcones.
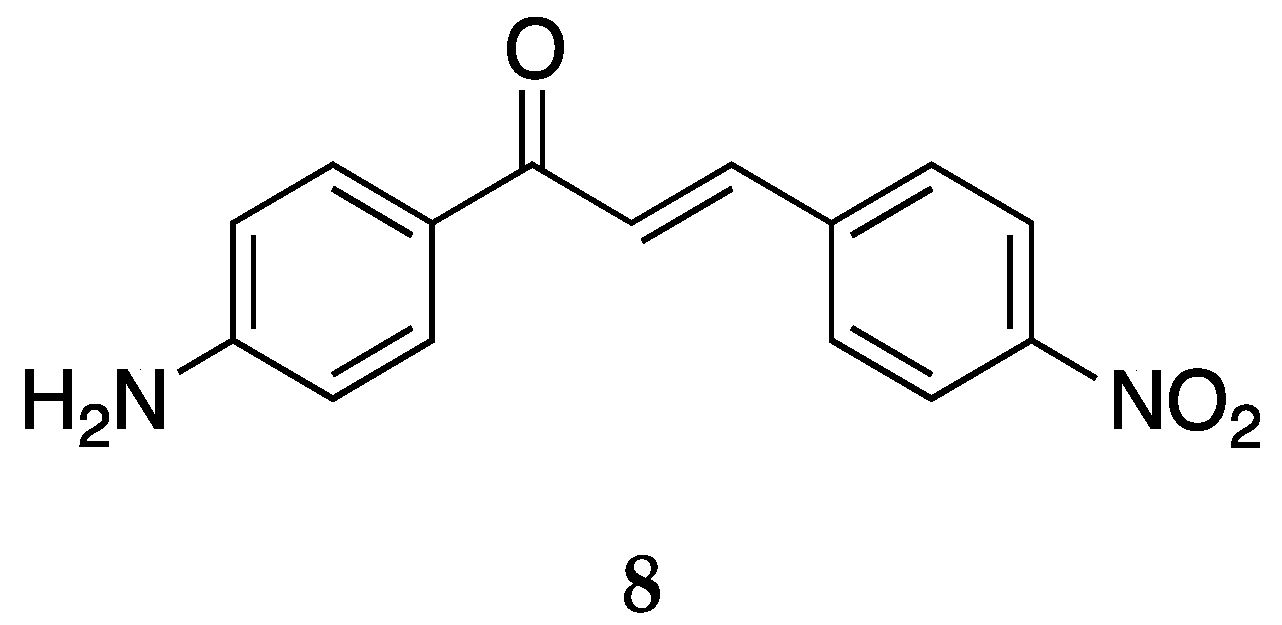
The 4-nitro-4-aminochalcone compound (
Figure 10) has also been reported in many articles of some groups.
The cytotoxic activity for
8 was tested against many different cancer cell lines (
Table 7).
The IC
50 values [
36] for the Molt 4/C8, CEM, P388, and L1210 cell lines against compound
8 were better when compared with the simple 4-aminochalcone
1. The activity for
8 was better than all derivatives, including chloro and methyl, tested for the Molt 4/C8, P388, and L1210 cells. The cell lines tested by Kozlowska et al. [
42] showed
8 to be decently potent. Among these cell lines, compound
8 exhibited the highest cytotoxic activity against the HT-29 cell line. Compound
8 exhibited activity better than cisplatin against the HT-29, LoVo, and LoVo/DX cell lines (IC
50 values of cisplatin: 16.73 ± 0.58, 1.49 ± 0.13, 2.09 ± 0.12, and 2.03 ± 0.17 µg/mL against HT-29, LS180, LoVo, and LoVo/DX, respectively). However, it was also toxic to the normal green monkey fibroblast line COS7 (IC
50 = 3.10 ± 0.50 µg/mL) used to determine the toxicity of the compounds. Compound
8 was also tested for its anticancer activity against MCF-7 and MDA-MB-231 [
39]; however, no significant activity was observed. The mean percentage of DH82 cell death [
43] was 52.4 ± 6.7% for
8.
Kozlowska et al. [
42] tested the antimicrobial activity of
8 against two strains of bacteria,
E. coli ATCC10536 and
S. aureus DSM799; one strain of yeast,
C. albicans DSM1386; and three strains of fungi,
A. alternata CBS1526,
F. linii KB-F1, and
A. niger DSM1957. Compound
8 prevented the growth of
E. coli with minimum inhibitory concentration (MIC) value equal to 0.25 mg/mL, two times stronger than reference oxytetracycline (MIC = 0.5 mg/mL). Compound
8 also prevented the growth of
S. aureus and
F. linii (MIC = 0.25 and 1.0 mg/mL).
Prasad et al. [
45] also tested the antioxidant activity and scavenging effects for
8. It had IC
50 values of 85.31 ± 1.30, 194.22 ± 1.76, 62.32 ± 1.33, and 612.46 ± 2.99 µg/mL, respectively.
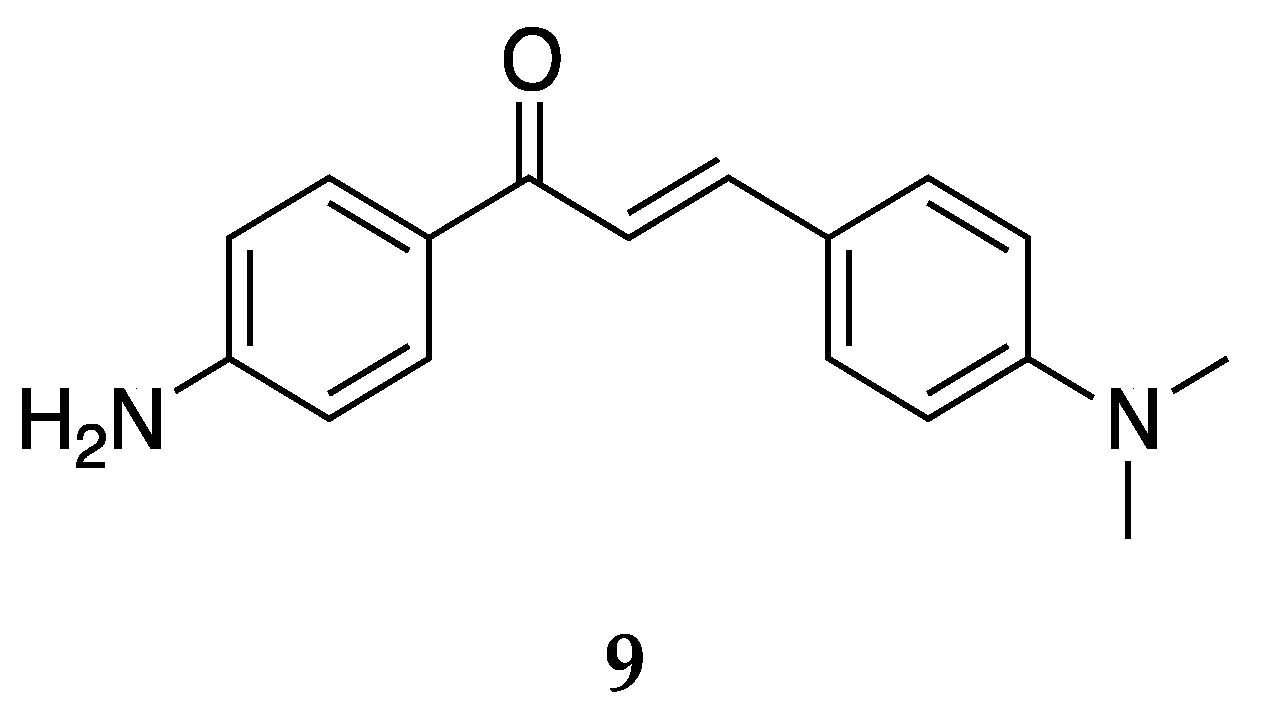
The antioxidant and anticancer activities were also tested for another very interesting compound with a dialkylated amino group on ring B (
Figure 11).
Romagnoli et al. [
37] tested
9 against the L1210, FM3A, Molt4, CEM, and HeLa cell lines with IC
50 values equaling 67 ± 33, 96 ± 32, 9.9 ± 1.0, 20 ± 3, and 28 ± 10 µM. Compound
9 was concluded to show weak antiproliferative activity towards these cell lines.
The potency of
9 against oxidative species, on the other hand, was quite good. The IC
50 values [
45] against the superoxide radical, hydroxyl radical, ABTS cation radical, and lipid peroxidation were 78.87 ± 1.32, 151.48 ± 1.09, 50.36 ± 1.22, and 423.08 ± 2.26 µg/mL, respectively. Compound
9 was one of the most potent compounds tested in this analysis and showed dose-dependent inhibition activities against all assays. It showed the maximum activity, comparable to the standard ascorbic acid against the hydroxyl radical, ABTS radical cation, and inhibition of lipid peroxidation. Compound
8, having an electron-withdrawing group on ring B, was not very potent against these assays. It was, thus, concluded that electron-releasing substitutions like methoxy and dimethylamino on ring B contribute to the antioxidant activity.
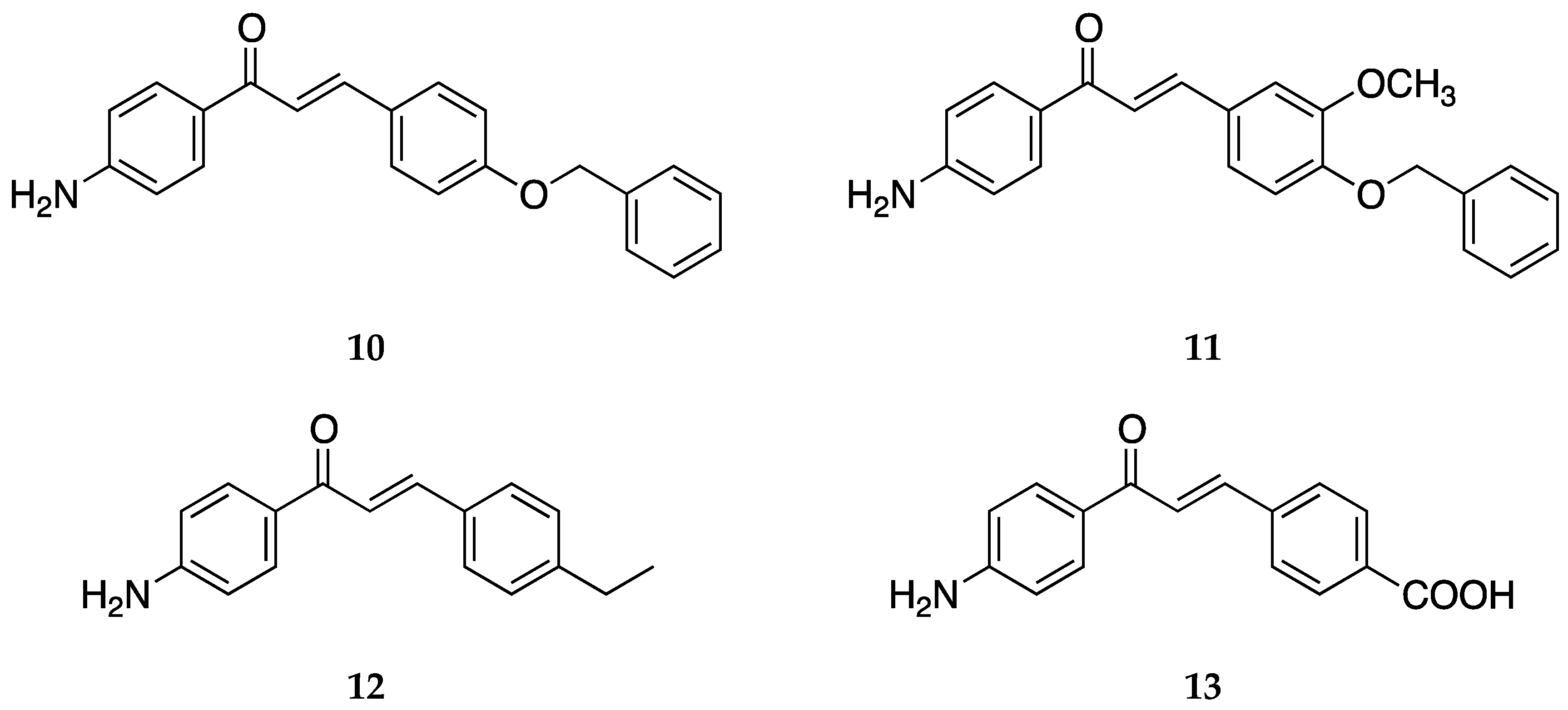
4-Aminochalcone derivatives with some other substitutions on ring B (
Figure 12) were also synthesized and tested by Kozłowska et al. [
42].
The anticancer activities of
10–
13 are shown in
Table 8.
The 4-benzyloxy moieties, 10, 11, and 4-amino-4-ethylchalcone 12 exhibited inhibition stronger than the reference drug cisplatin (IC50 = 16.73 ± 0.58 µg/mL) against the HT-29 cell line. Compound 13 showed the poorest inhibition among all the reported aminochalcones.
The antimicrobial activity for
10,
11, and
13 were reported [
42] against
E. coli ATCC10536. The minimal inhibitory concentrations were 0.125, 0.5, and 0.125 mg/mL respectively. Both
10 and
13 showed better inhibition when compared to reference compound oxytetracycline (MIC = 0.5 mg/mL against
E. coli ATCC10536).
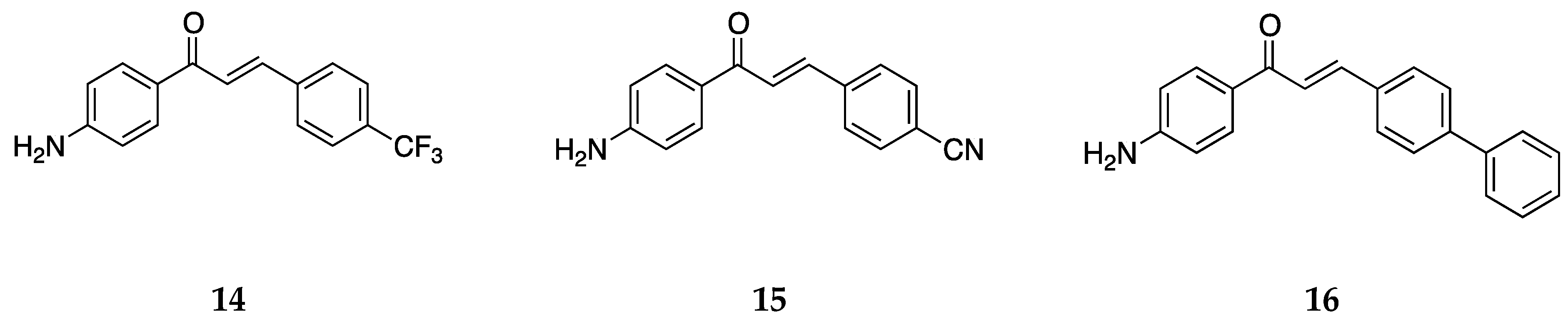
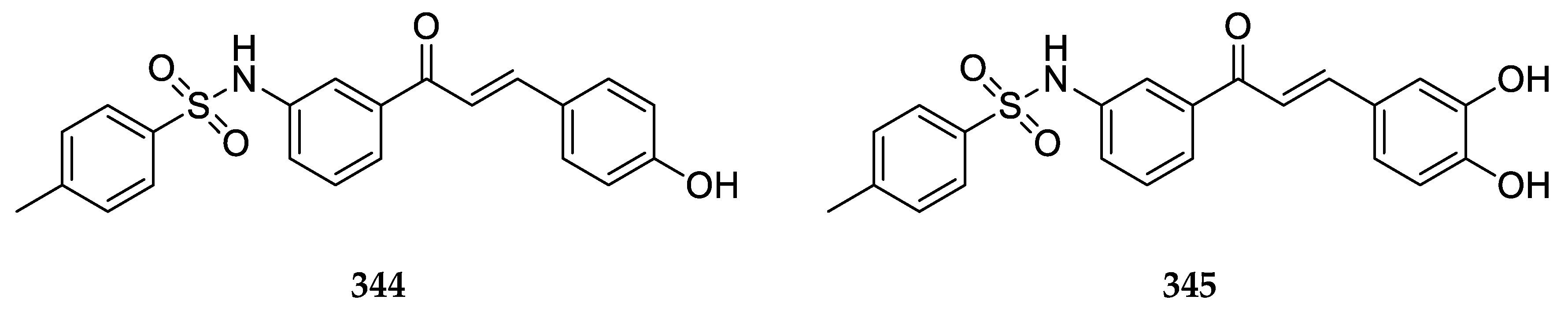
The 4-aminochalcones synthesized by Santos et al. [
39] included those shown in
Figure 13.
These compounds were tested against the estrogen-receptor-positive breast cancer cells MCF-7 and the triple-negative breast cancer (TNBC) cells MDA-MB-231. Compounds 14 and 15 did not show very significant antiproliferative activity as their percentage of metabolically viable cells (%MVC) was greater than 98% against both cell lines. The IC50 values for 16 were 22.7 ± 6.0 and >100 µM against MCF-7 and MDA-MB-231, respectively. Compound 16 had a %MVC value of 44.2 ± 3.3 against MCF-7.
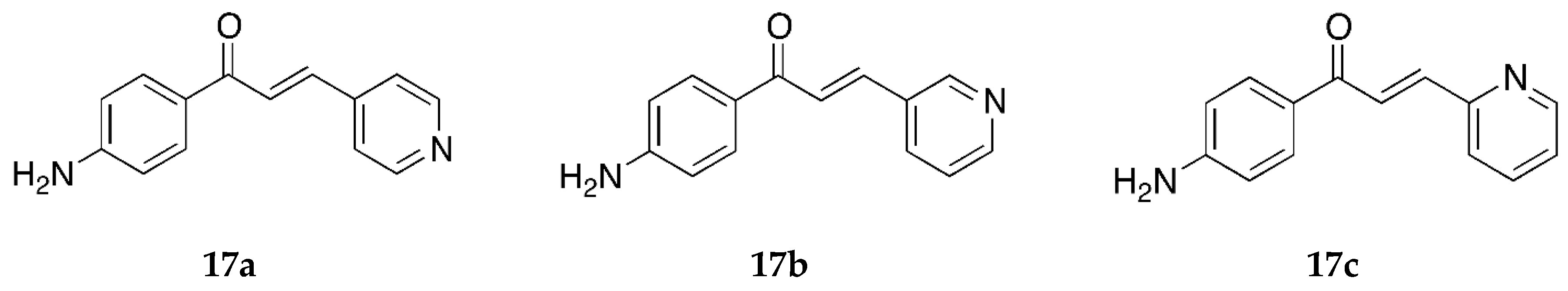
Some derivatives containing pyridine rings have also been reported (
Figure 14).
Compounds
17a and
17b were synthesized by Santos et al. [
39] using the Claisen-Schmidt aldol condensation method, and their cytotoxic activity was tested against the MCF-7 and MDA-MB-231 cancer cell lines as well. Compound
17b showed significant antiproliferative activity against the MCF-7 cell line with an IC
50 value of 15.7 ± 5.9 µM. Compound
17b was also potent against the MDA-MB-231 cell line (IC
50 = 33.9 ± 7.1 µM). Apart from the 4-fluorophenyl derivative
6c,
17b was the only compound among those reported in the manuscript which showed significant potency against both cell lines. Compound
17b induced apoptosis rather than necrosis in both cells and upregulated the p53 expression in the MCF-7 cell line. Compound
17a was active against the MCF-7 and showed a percentage of metabolically viable cells (%MVC) value of 61.3 ± 3.4. Its IC
50 value was not determined.
The antioxidant activity for
17a,
17b, and
17c was tested by Prasad et al. [
45] Many 4-aminochalcones reported in this manuscript have already been discussed earlier, with some of them showing significant antioxidant activity comparable to the reference ascorbic acid. Compounds
17a,
17b, and
17c, however, did not show very significant activity against the superoxide, hydroxyl, ABTS cation radicals, and lipid peroxidation (
Table 9). The IC
50 values for these compounds were not comparable to ascorbic acid or the more potent compounds tested by the group.
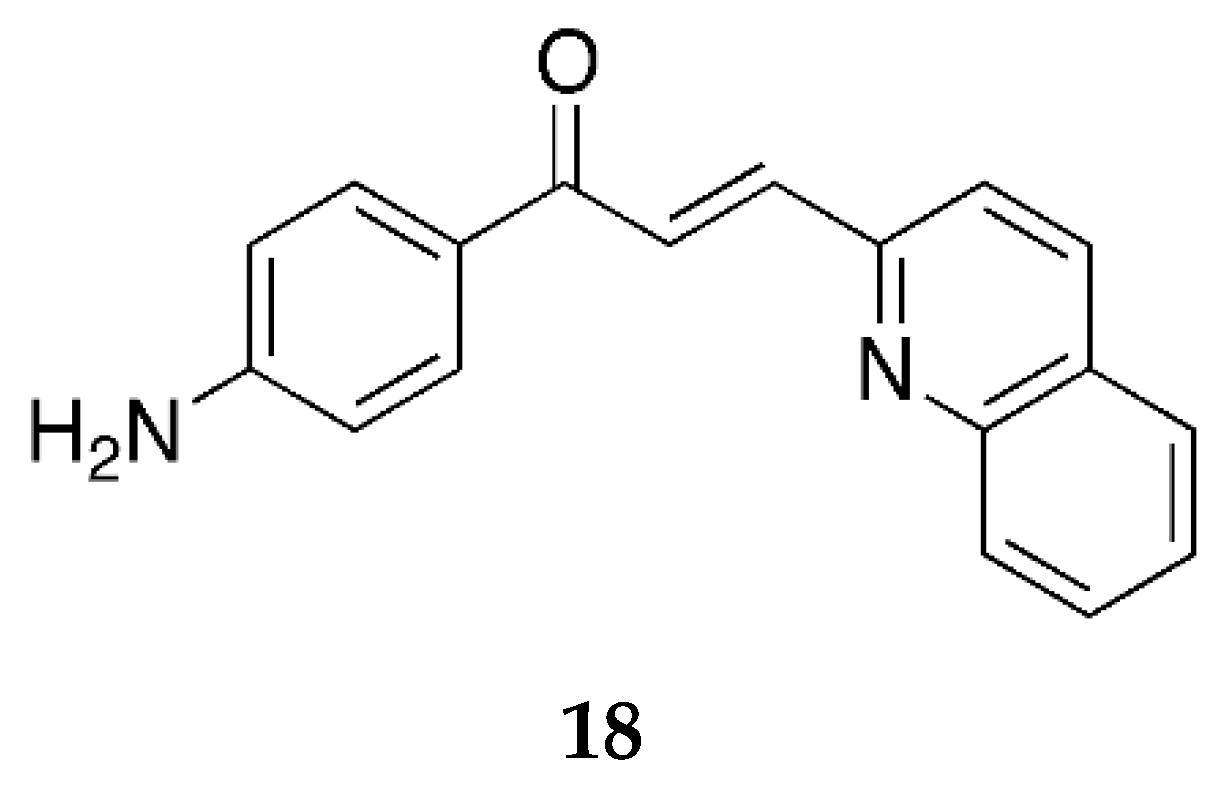
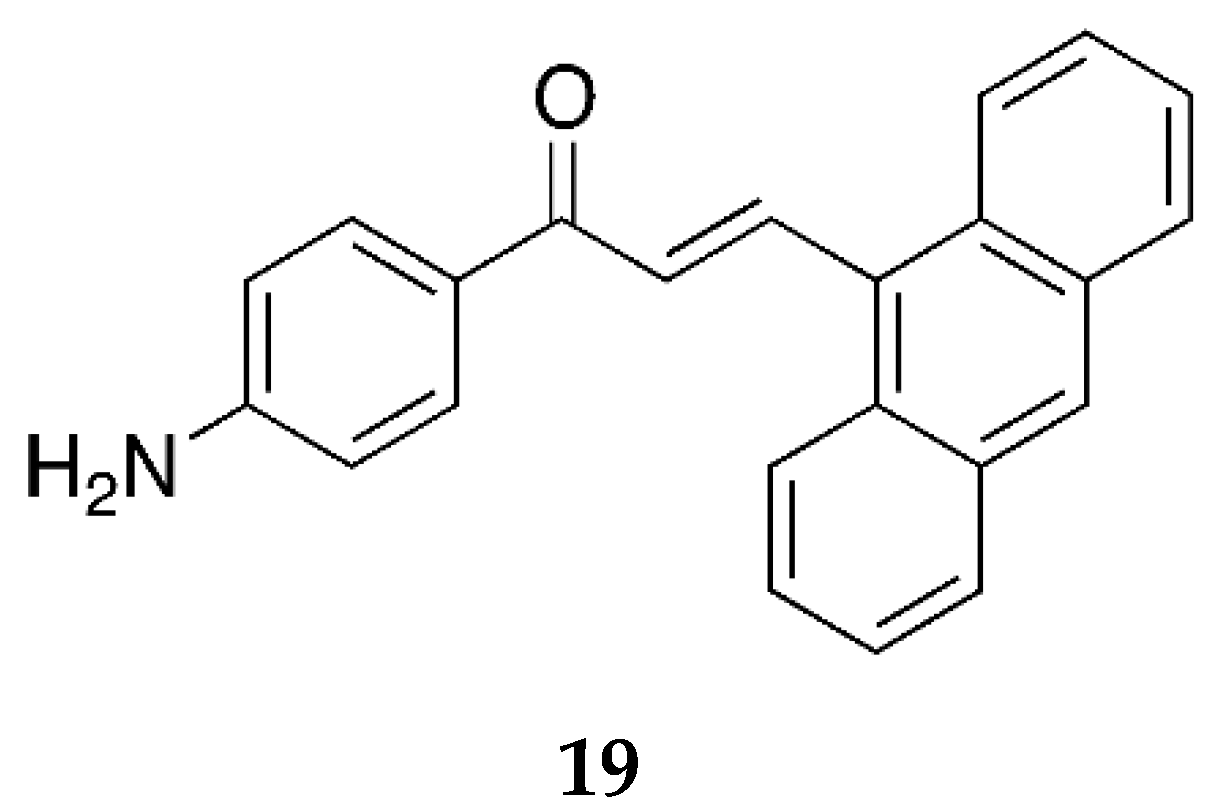
Prasad et al. [
45] have also reported the 2-quinolinyl 4-aminochalcone derivative (
Figure 15) and aminochalcone containing anthracene ring (
Figure 16).
Compound 18 showed values similar to the pyridinyl derivatives (17a, 17b, and 17c). Its IC50 values against the superoxide radical, hydroxyl radical, ABTS cation radical, and lipid peroxidation were 88.87 ± 1.78, 197.87 ± 1.48, 77.89 ± 2.52, and 656.54 ± 1.75 µg/mL, respectively.
Compound 19 also did not show very significant antioxidant activity with IC50 values 89.25 ± 1.99, 198.22 ± 1.69, 76.32 ± 2.66, and 652.25 ± 1.44 µg/mL against the superoxide radical, hydroxyl radical, ABTS cation radical, and lipid peroxidation, respectively. Lack of significant activity might be due to the absence of any strong electron-releasing group such as methoxy or dimethylamino on ring B, as derivatives containing these substitutions were more potent.
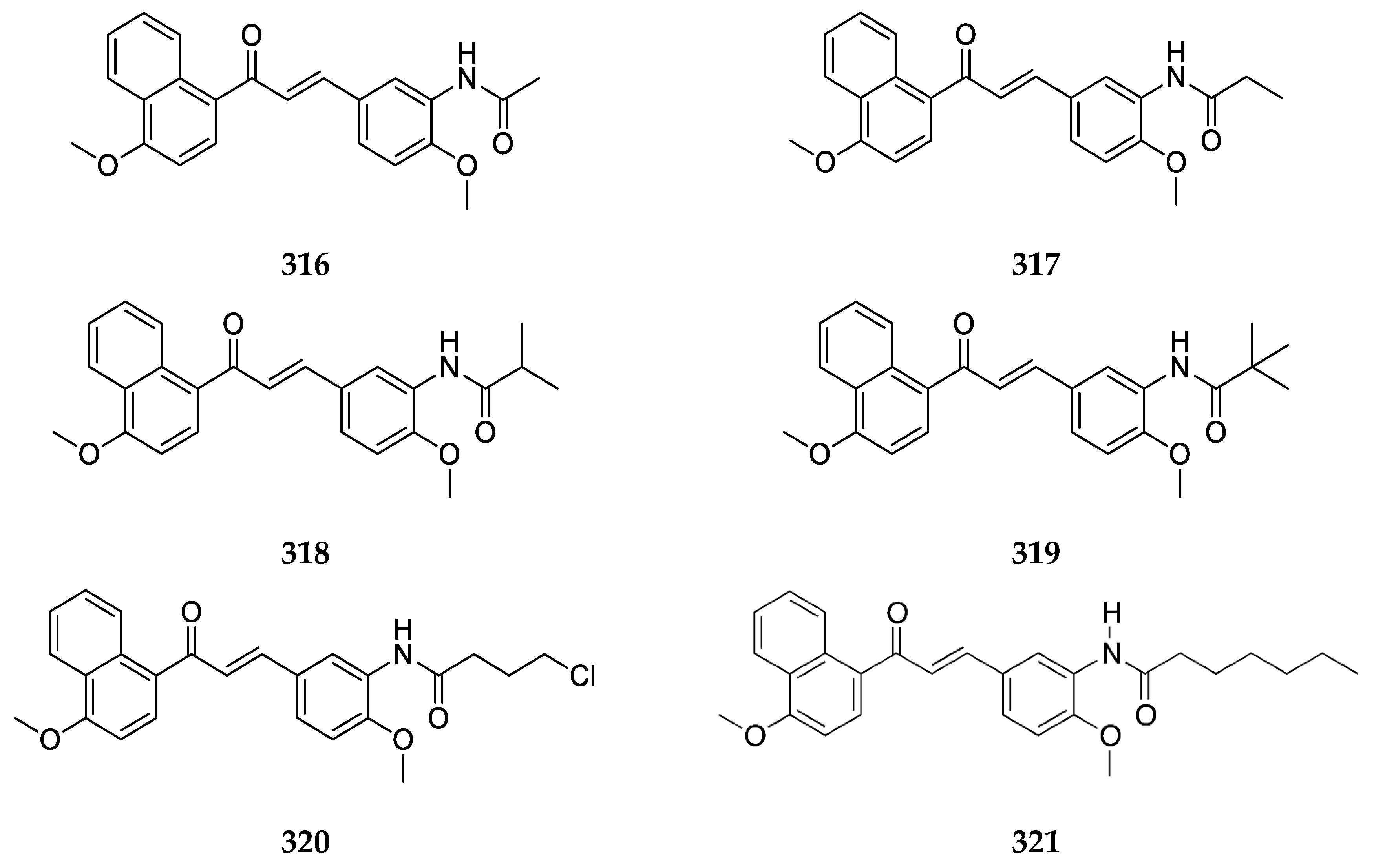
4-amino-1-naphthylchalcone
20 and its methylated derivative
21 (
Figure 17) were synthesized by Seba et al. [
50], and their activity against osteosarcoma cells was reported.
Osteosarcoma is the most recurrent malignant bone tumor. This tumor shows a high propensity for local invasion and distant metastasis. Due to this, the long-term survival rate of osteosarcoma patients with metastasis is decreased by 20–30% [
51]. Seba et al. [
50] studied the inhibitory effects of
20 and
21 against osteosarcoma migration. They found that, while these compounds neither induce apoptosis in osteosarcoma cells can suppress tumor cell lines U2OS and SOAS, they showed inhibitory effect against migration and invasion of osteosarcoma cells. This effect was more potent in p53-expressing cells, namely, U2OS. They increased p53 expression and reduced the activity and expression of two matrix metalloproteinases: MMP-2 and MMP-9. These compounds also downregulated the epithelial–mesenchymal transition (EMT). The migration and invasion of tumor cells towards the vascular/lymphatic system involves MMPs, which ultimately triggers metastasis. Tumor cells undergo various changes during their EMT, which relate to amplified migration, invasion, and metastasis of these cells. Thus, the targets of compounds
20 and
21 are important in the process of migration and invasion of osteosarcoma cells.
Compound
20 was also reported by Santos et al. [
39]. It showed significant antiproliferative activity against the estrogen-receptor-positive breast cancer cells MCF-7 (IC
50 = 14.3 ± 2.9 µM). It was inactive against the triple-negative breast cancer (TNBC) cells MDA-MB-231 with an IC
50 value of >100 µM.
Santos et al. [
43] reported the cytotoxic activity of
20 against the canine malignant histiocytic cell line DH82 as the mean percentage of DH82 cell death, which was 40.3 ± 0.6.
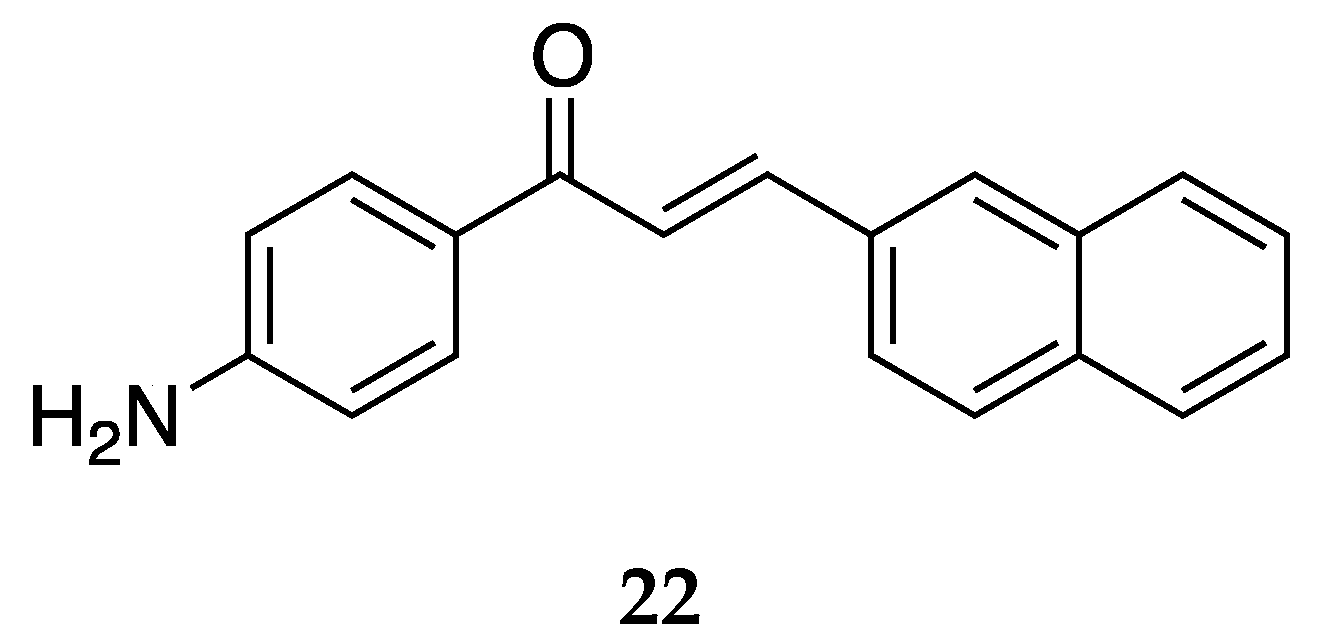
A 2-napthyl derivative of 4-aminochalcone,
22, was also synthesized and tested by Santos et al. [
43] (
Figure 18). It showed a mean percentage of DH82 cell death as 40.4 ± 3.7.
Both chalcones with naphthyl rings showed poor activity against the DH82 cell line.
4-aminochalcones with heterocycles instead of the phenyl ring B have also been synthesized (
Figure 19).
The antiproliferative activity of compounds
23 and
24 was reported by Santos et al. [
43] against the canine malignant histiocytic cell line DH82. The mean percentages of DH82 cell death values were 18.8 ± 4.0 and 43.6 ± 3.7, respectively. Thus, neither compound showed significant activity against the cell line and
23 had the poorest activity amongst all of the compounds reported in the manuscript. In addition to this,
23 was also tested by Santos et al. [
39] against the estrogen-receptor-positive breast cancer cells MCF-7 and the triple-negative breast cancer (TNBC) cells MDA-MB-231. It showed %MVC values of 104.5 ± 3.6 and 100.1 ± 7.1 against the cell lines, respectively. Thus, it can be concluded that these compounds do not show significant cytotoxic activity.
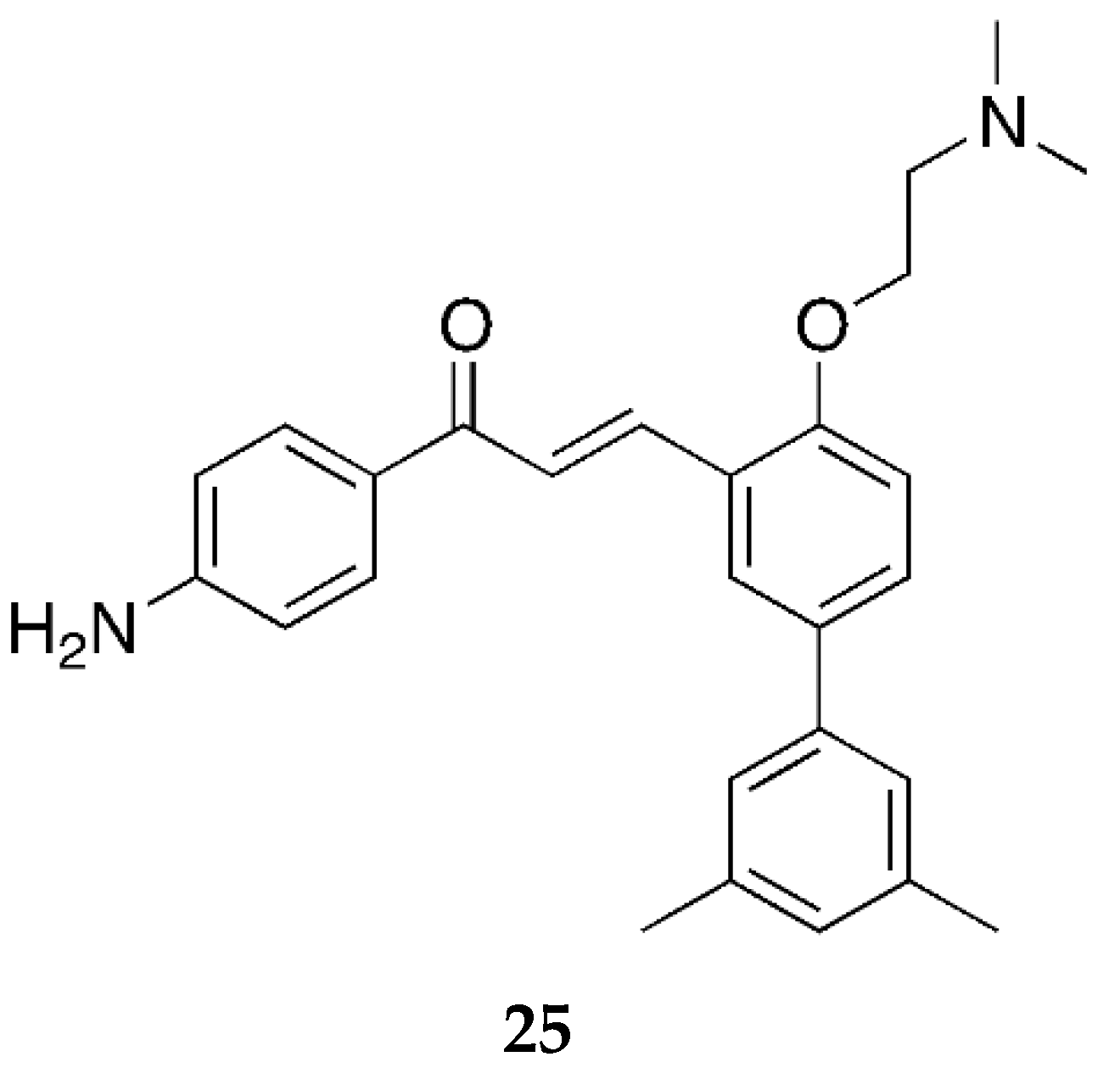
Moving towards the end of our discussion for 4-aminochalcones, compound
25 (
Figure 20) was synthesized by Nielsen et al. [
52] and tested for its antibiotic activity along with many other chalcones.
Compound 25 was tested against the S. aureus ATCC33591 (resistant to methicillin), E. faecium 17501 (vancomycin-resistant clinical isolate), and E. faecium ATCC29212 bacterial cell lines and showed MIC values of 10, 10, and 20 µM, respectively. As the values indicate, 25 had no significant effect on the activity. However, it showed some influence on the bacterial membrane. This is likely due to the protonation of the dimethylamino group on ring B.
2.1.2. 2-Aminochalcone and Derivatives
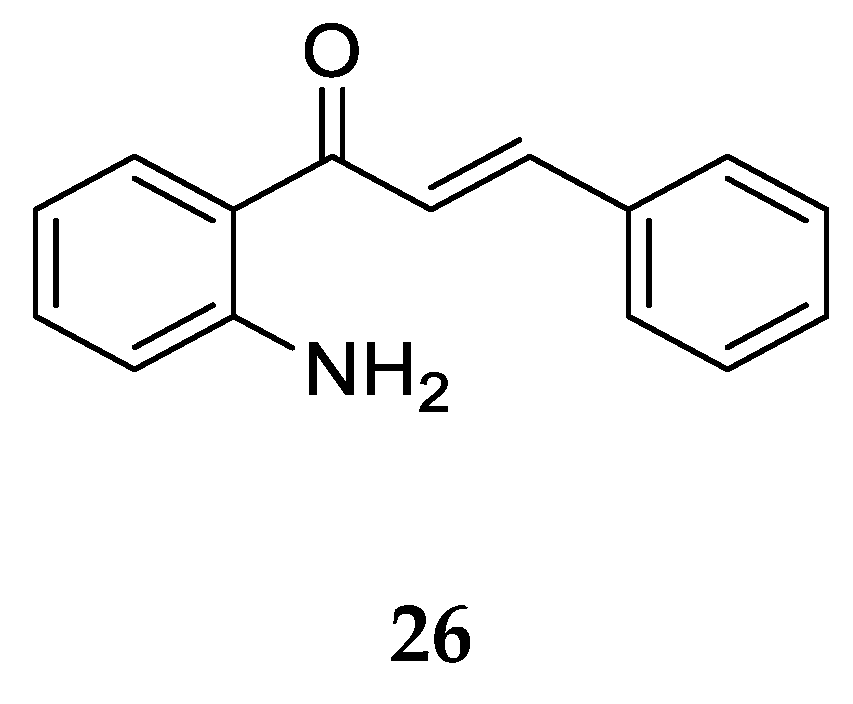
Aminochalcone
26 (
Figure 21) with a -NH2 substitution at position 2 on ring A is the simplest among 2-aminochalcones. Kozlowska et al. [
42] obtained it through a modified protocol for a base-catalyzed Claisen-Schmidt condensation described by Amir et al. [
53] using 2-aminoacetophenone and benzaldehyde.
Following synthesis, its anticancer activity was tested against various cancer cell lines using green monkey fibroblast COS7, selected as the normal cell line to determine toxicity (
Table 10) [
42]. Besides, Mai et al. [
38] also synthesized and tested the antiproliferative effect of
26 using the methyl thiazolyl tetrazolium (MTT) assay on ten different cancer cell lines, namely, human estrogen receptor-positive breast cancer cells (MCF7), human estrogen receptor-negative breast cancer cells (MDA-MB-231), human cervical cancer cells (HeLa), human ovarian cancer cells (Caov-3), human lung cancer cells (A549), human liver cancer cells, (HepG2), human colorectal cancer cells (HT-29), human nasopharyngeal cancer cells (CNE-1), human T-lymphoblastoid leukaemia cells (CEM-SS), human erythromyeloblastoid leukaemia cells (K562), and normal human embryonic kidney (HEK-293) cells.
Aminochalcone
26 was the strongest inhibitor of HT-29 cell line proliferation, as indicated by the values in
Table 10. It exhibits activity almost 12 folds higher than that of reference compound cisplatin (IC
50 = 16.73 ± 0.58 µg/mL) and 4 folds lower than that of doxorubicin (IC
50 = 0.33 ± 0.02 µg/mL). Its cytotoxicity against the COS7 normal cell line indicated its poor selectivity against cancer cells [
42]. Additionally, in comparison to the 4-aminochalcone discussed earlier, the IC
50 values of
26 are lower for the cell lines in
Table 1. This increase in potency is caused just by the change in position of the amine group from 4 to 2 on ring A.
Mai et al. [
38] reported that the selectivity ratio (IC
50 HEK-293/IC
50 HT-29) of
26 (ratio > 22.78) fulfilled the threshold limit (ratio ≥ 5); it was one of the promising anticancer agents. They also observed that, among all the chalcones tested,
26 showed the most potent antiproliferative effect against HT-29 cells, indicating that the -NH2 group on ring A plays an important role. The anti-proliferative effects exhibited by the aminochalcone
26 were due to apoptosis rather than necrosis and were dose-dependent.
Kozlowska et al. [
42] investigated the antimicrobial activity of
26 against bacterial strain
Staphylococcus aureus DSM799 and strains of fungi,
Alternaria alternata CBS1526 and
Aspergillus niger DSM1957. The minimum inhibitory concentration (MIC) values were evaluated and compared with reference substances, such as oxytetracycline (against bacteria) and nystatin (against fungi). The MIC value of
26 was 1.0 mg/mL for
A. alternata. Complete growth inhibition was observed in cases of
S. aureus (MIC = 0.25 mg/mL) and particularly
A. niger (MIC = 0.125 mg/mL), which was two times more effective than nystatin.
Furthermore, the antimicrobial activity of
26 against several bacteria was tested and reported by Ruanwas et al. [
54] Against all the gram-positive bacteria—
Bacillus subtilis,
Staphylococcus aureus,
Enterococcus faecalis TISTR 459, methicillin-resistant
Staphylococcus aureus (MRSA) ATCC 43300, and vancomycin-resistant
Enterococcus faecalis (VRE) ATCC 51299—and gram-negative bacteria—
Salmonella typhi,
Shigella sonei, and
Pseudomonas aeruginosa—compound
26 showed MIC values >300 µg/mL, which were significantly greater than the vancomycin standard (MIC < 2.34 µg/mL). These results are similar to the antimicrobial activity of 4-aminochalcone, which differs only by the position of the -NH2 group on ring A. This suggests that a change in position from 4 to 2 does not cause a big difference in antimicrobial activity. The same group also tested the antioxidant activity of compound
26 via a DPPH (2,2-diphenyl-1-picrylhydrazyl radical) antioxidant assay, which was later compared to the standard ascorbic acid. Aminochalcone
26 showed a percentage inhibition of 18.7%, which was the highest among the aminochalcones tested for free radical scavenging. However, it was notably less than the ascorbic acid standard, which exhibits 92.1% inhibition [
54].
A variety of 2-aminochalcone derivatives with substitutions on ring B have been synthesized and reported for biological activity by Santos et al. [
43].
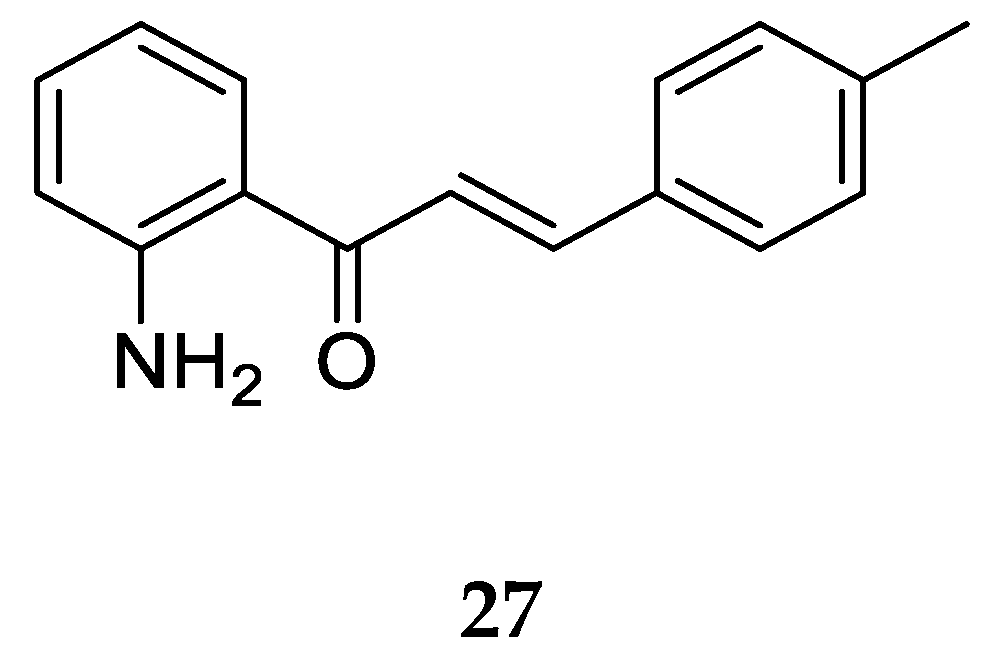
Aminochalcone
27 (
Figure 22) is an analog of the methylated 4-aminochalcone discussed earlier. It was synthesized and evaluated for anticancer activity against the canine malignant histiocytic cell line (DH82). The mean percentage of cell death was found to be 88.9 ± 1.5 at 15.0 µg/mL of the compound. Compound
27 was one of the three most active compounds tested due to its selective toxicity and apoptosis induction in DH82 cells compared to a nontumorigenic canine cell line (Madin-Darby canine kidney cells, MDCK). Compound
27 also showed a slight increase in cytotoxicity in comparison to its 4-aminochalcone analog. The compound largely owes its activity to the presence of a hydrophobic group on ring B. Similarly, against DH82 and MDCK cells, a nontumorigenic cell line, the IC
50 values of
27 were 38.2 µM and 23.3 µM, reflecting approximately three times more potency against DH82 than the reference antitumor drug etoposide (IC
50 = 95.5 µM). It was also observed that
27 induces cell death through apoptosis rather than necrosis in canine cells. Therefore, it can be deduced that compound
27 possesses a remarkable potential for anticancer activity against the tested cell lines.
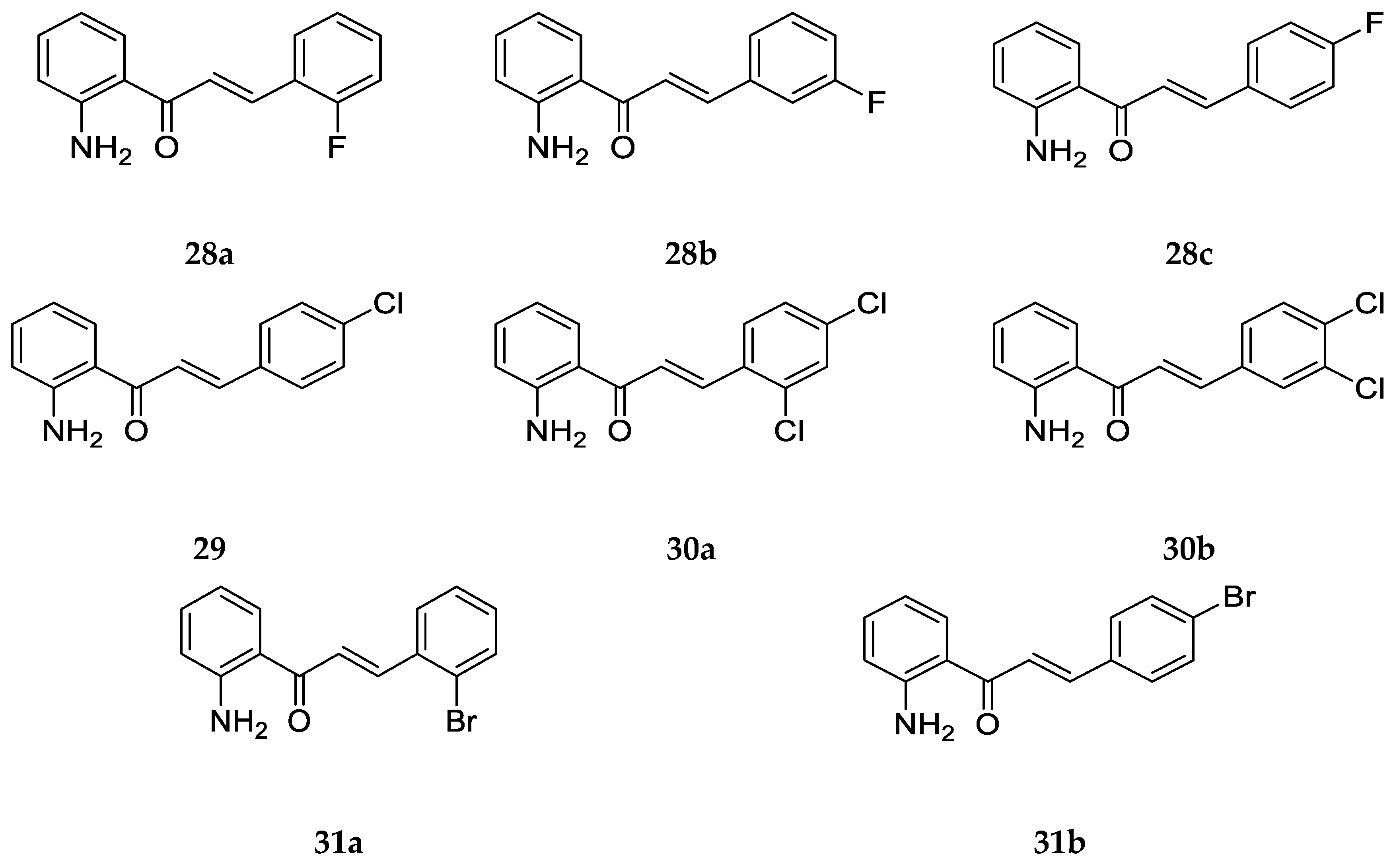
Santos et al. [
43] and Zhang et al. [
55] also prepared and investigated the biological activity of a variety of 2-aminochalcones derivatives with halogens substituted on ring B (
Figure 23).
The anticancer activity of
28c,
29, and
31b against canine malignant histiocytic cell line (DH82) was determined as the mean percentage of DH82 cell death ± SEM at 15.0 mg/mL of the compound. The activities of
28c,
29, and
31b were 76.6 ± 0.1, 83.2 ± 5.7, and 87.0 ± 7.5, respectively. These halogenated 2-aminochalcones exhibited cytotoxic activity similar to that of their halogenated 4-aminochalcone analogs [
43].
Like
26, compound
29 was also prepared and tested against ten cancer cell lines for antiproliferative activity by Mai et al. [
38]. The IC
50 values of
29 are summarized in
Table 11.
From the values in
Table 11, we can infer that the activity of
29 is the best against the HT-29 cell line. It is seen from the comparison of the activity of
29 with
26 that compound
26 is more potent against most cell lines including A549, HEPG2, CNE-1, K562, and CEM-SS whereas
29 has a higher antiproliferative activity against the Caov-3 cell line and HT-29 shows similar values for both
26 and
29. Therefore, the presence of -NH2 on ring A position 2 has a more significant contribution to the anticancer activity as opposed to the substitutions on ring B.
Additionally, Zhang et al. [
55] tested the antifungal activity of compounds
28a–
c,
30a–
b, and
31a against a single strain
Trichophyton rubrum. Their MIC80 values were found to be 1 µg/mL, 0.5 µg/mL, 1 µg/mL, >64 µg/mL, >64 µg/mL, and 1 µg/mL, respectively. Compound
28b was the most potent and
30a–b was the least potent among the compounds, and most of the MIC80 values recorded were greater than that of the reference drug Fluconazole (MIC80 = 0.25 µg/mL).
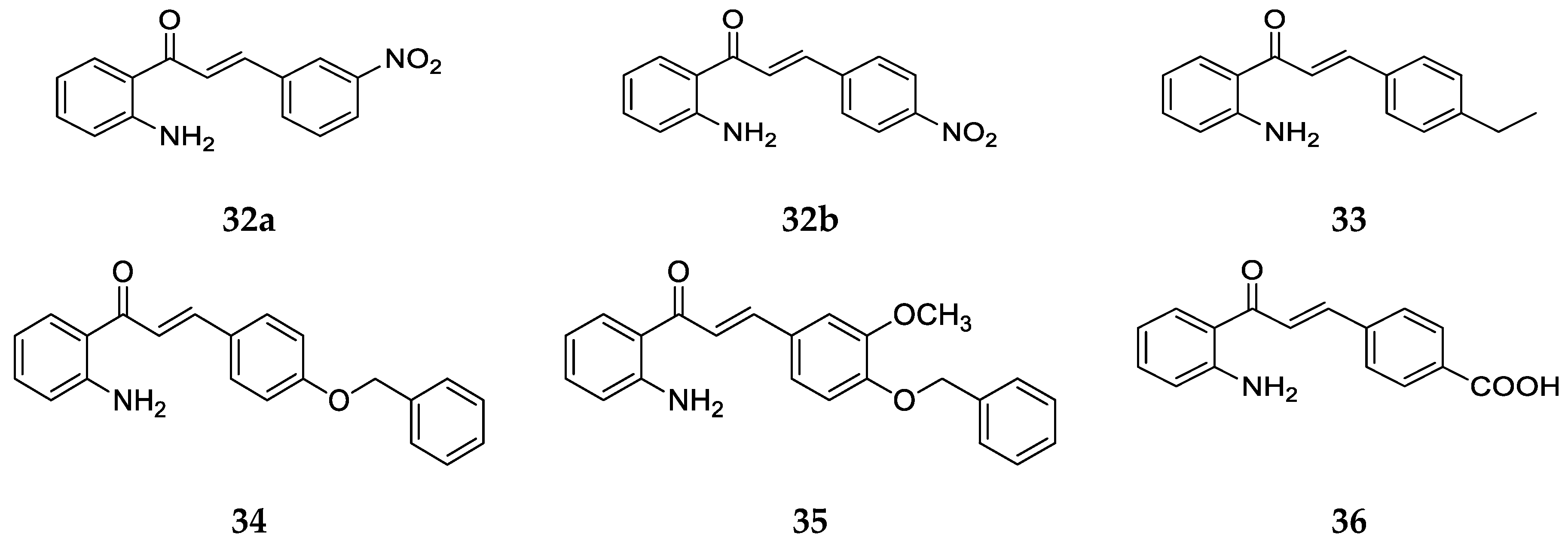
More derivatives of 2-aminochalcones, some of which were novel with different substituents on ring B (
Figure 24), were synthesized by Kozlowska et al. [
42], Mai et al. [
38], and Zhang at al. [
55].
The biological activity of the compounds was investigated against cancer cell lines and different microbes.
Table 12 shows the IC
50 values of compounds
32b,
33,
35, and
36 against cancer cell lines as investigated by Kozlowska et al. [
42] and Mai et al. [
38].
Compound
33 showed IC
50 values lower than
36 but higher than
32b for the cancer cell lines tested. Similar to the anticancer effect of compound
27 with the methyl group on ring B, the higher potency of
33 compared to
36 is also due to the hydrophobicity of the ethyl group. Also, the cytotoxicity of
33 against the COS7 normal cell line is lower than that of
26, the simple 2-aminochalcone which indicates the positive effect of ethyl substituent on ring B. Besides, the anticancer activity of
36 is observed to be the weakest and it was not tested against some cancer cell lines. The activity of
32b is impressive in comparison to the others tested. This compound was made and investigated for bioactivity by both groups [
38,
42] as shown in
Table 12 as IC
50 values. Kozlowska and colleagues [
42] found compound
32b to be slightly more potent against the common cell line HT-29 but still less active than compound
26. When compared with
33 and
36,
32b was more potent against all four cancer cell lines while also exhibiting toxicity against the COS7 normal cell line. Compound
35 showed moderate anticancer activity, with lower toxicity against the COS7 normal cell line, but there was no notable feature observed in the overall activity of
35 [
42]. Mai et al. [
38] reported that, since
32b exhibited good anticancer activity against most of the cell lines tested and the selectivity ratio (IC
50 HEK-293/IC
50 HT-29) of
32b (ratio = 6.77) was greater than the threshold value (ratio ≥ 5), it was one of the promising anticancer agents like
26.
In addition to anticancer activity, Kozlowska et al. [
42] also determined the antimicrobial activity in terms of minimum inhibitory concentrations (MIC) of compounds
33,
34,
36, and
32b against bacterial and fungal strains. Compound
32b was tested against
Escherichia coli ATCC10536 with (MIC = 0.25 mg/mL),
Candida albicans DSM1386, and
F. linii KB-F1, with MIC values = 0.5 mg/mL each. These values are almost equivalent to the standard drugs against which they were compared, and
32b showed MIC values lower than its 4-aminochalcone analog, thus revealing its antimicrobial potential and the significant impact of the position of the NH2 group (ring A) on the biological activity of aminochalcones. Compound
33 showed MIC value = 0.5 mg/mL when tested against
Escherichia coli ATCC10536, which was equal to the standard oxytetracycline. Compound
34 was only tested for antimicrobial activity and displayed MIC values of 0.25 mg/mL, 0.25 mg/mL, and 0.5 mg/mL against
Escherichia coli ATCC10536,
Staphylococcus aureus DSM799, and
Candida albicans DSM1386, respectively, and a better activity than the standard oxytetracycline (MIC = 0.5 mg/mL) for
E. coli. The MIC values of
36 were 0.125 mg/mL against
Escherichia coli ATCC10536,
Staphylococcus aureus DSM799,
Candida albicans DSM1386, and
Aspergillus niger DSM1957 and 0.5 mg/mL against
Alternaria alternata CBS1526. Compound
36 was found to be the best inhibitor of
C. albicans among the compounds tested, with four times higher activity than the standard cycloheximide. For the fungal strains,
36 showed a lower MIC value and thus higher activity than the standard drug nystatin, overall exhibiting a remarkable antimicrobial activity. Finally, the minimal inhibitory concentration of compound
35 was determined for
Escherichia coli ATCC10536,
Staphylococcus aureus DSM799, and
F. linii KB-F1 and was found to be 0.5 mg/mL for all three strains. Overall, it displayed an equal or poorer performance in comparison to the respective standard drugs and in comparison to other chalcones tested. The MIC values for
35 were higher than its structural analog
34, meaning that the addition of the methoxy group on ring B along with the benzyloxy group does not improve bioactivity but decreases it.
Lastly, values of the minimum inhibitory concentration required to inhibit 80% of cells (MIC80) for compounds
32a–
b were determined by Zhang et al. [
55]. The compounds were found to be potent against
Trichophyton rubrum, with MIC80 values of 0.5 µg/mL each. This value was greater than the standard reference, but
32a–
b was among the three most active antifungal agents compared to other chalcones tested.
Sakata et al. [
56] synthesized a series of 2-aminochalcones (
Figure 25) and tested their activity against the neurodegenerative Alzheimer’s disease markers, namely, acetylcholinesterase (AChE), β-secretase (BACE-1), and the amyloid-β (Aβ). As discussed earlier, some of these compounds have been also reported by other groups.
The activities of these compounds against AChE and BACE-1 are shown in
Table 13.
Tacrine was used a positive control for AChE inhibition, and quercetine was used for BACE-1. Against AChE, all compounds tested showed some inhibition activity, with compounds
27,
30c,
36a,
36b, and
36c showing IC
50 values lower than 10 µM. The 2′,3′-dichloro substituted aminochalcone
30c exhibited the highest potency, with IC
50 equal to 0.08 µM. These compounds were docked into the active site of AChE, and it was concluded that the high potency for some of these compounds was due to the hydrophobic features of the analogues as the aromatic ring helped in the adoption of a proper conformation at the ligand cleft. The IC
50 values for BACE-1 inhibition were obtained only for compounds that showed decent percentage inhibition values. Against BACE-1, compound
30c again showed the highest potency (IC
50 = 2.71 µM). Some chalcones without the amino group having structural similarities with compounds in
Figure 25 were tested against BACE-1. These chalcones did not inhibit the enzyme, thus showing the importance of the amino group on ring A. The molecular docking studies performed showed that the amino group on ring A interacts with the aspartic acid residues of the catalytic diade of BACE-1. The inhibitory effect on the Aβ amyloid aggregation was determined using thioflavin-T (ThT), which binds to the β-amyloid fibrils. This interaction increases the emission of ThT fluorescence; thus, the presence of an inhibitor will decrease the fluorescence. Aminochalcones
29a,
29,
30c, and
36b showed a good effect against Aβ fibrils formation and reduced the ThT fluorescence emission by 37, 52, 33, and 29%, respectively. The most potent compound was
29, as it was comparable to the reference compound curcumin (inhibition of fibril formation = 56%). Moreover, the cytotoxicity for these 2-aminochalcones was tested against the human glioblastoma M059J, mouse fibroblast Vero, and kidney epithelial 3 T3 cell lines. While 3 T3 was more sensitive to the chalcones, the overall IC
50 values obtained were quite high, showing that they were not potentially cytotoxic. Furthermore, it was determined that these compounds have low water solubility, which might be due to the presence of crystal packing. In conclusion, these compounds are promising leads against AD especially if the hydrosolubility is improved.
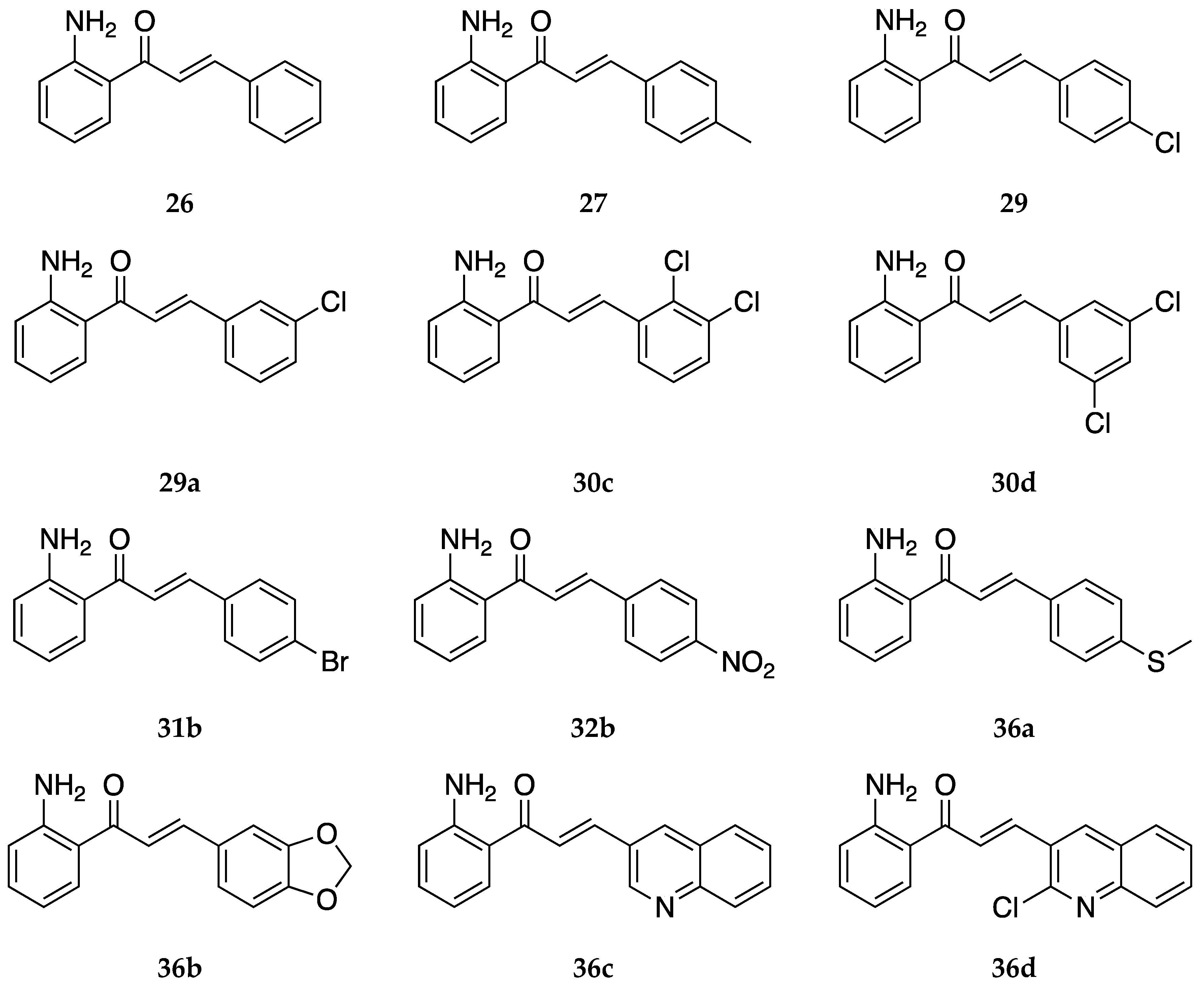
Moreover, 2-aminochalcone derivatives with methoxy and other substitutions on ring B (
Figure 26) were synthesized by Zhang et al. [
55], who tested the antifungal activity, and Ruanwas et al. [
54], who screened them for antioxidant and antibacterial activity.
Aminochalcone
37 was tested for antifungal activity against
Trichophyton rubrum, and its minimum inhibitory concentration needed to inhibit 80% of cells (MIC
80) was determined to be greater than 64 µg/mL, which was higher than most of the chalcones tested and overall demonstrated that
37 was not a good antifungal agent [
55].
2-Aminochalcones
38,
39, and
40 also showed poor antibacterial activity against the gram-positive bacteria—
Bacillus subtilis,
Staphylococcus aureus,
Enterococcus faecalis TISTR 459, methicillin-resistant
Staphylococcus aureus (MRSA) ATCC 43300, and vancomycin-resistant
Enterococcus faecalis (VRE) ATCC 51299—and the gram-negative bacteria—
Salmonella typhi,
Shigella sonei, and
Pseudomonas aeruginosa [
54]. The MIC values of these compounds for all bacterial strains were ≥300 μg/mL, like compound
26. The low MIC value of the vancomycin standard (<2.34 μg/mL) demonstrates the inadequate activity of the compounds against the strains tested. The antioxidant activities of compounds
38–
40 were tested through a DPPH radical scavenging method and were compared to ascorbic acid as a standard reference. The range of the percentage inhibition of free radical scavenging activity of
38–
40 was 0.41% to 3.27%, with
38 showing the highest and
39b showing the lowest activity among these compounds. These activities are lower than that of
26 (18.71%) found during the same investigation and significantly weaker than the ascorbic acid reference (92.06% inhibition). The results suggest that the general antioxidant activity by the α,β-unsaturated carbonyl group was reduced due to the substituents on ring B in
38–
40 [
54].
Adding diversity to the plethora of 2-aminochalcones, we will discuss Xia et al.’s [
57] prepared derivatives of 2-aminochalcone with substituents on both rings ranging from simple methyl to methylenedioxy substitution (
Figure 27).
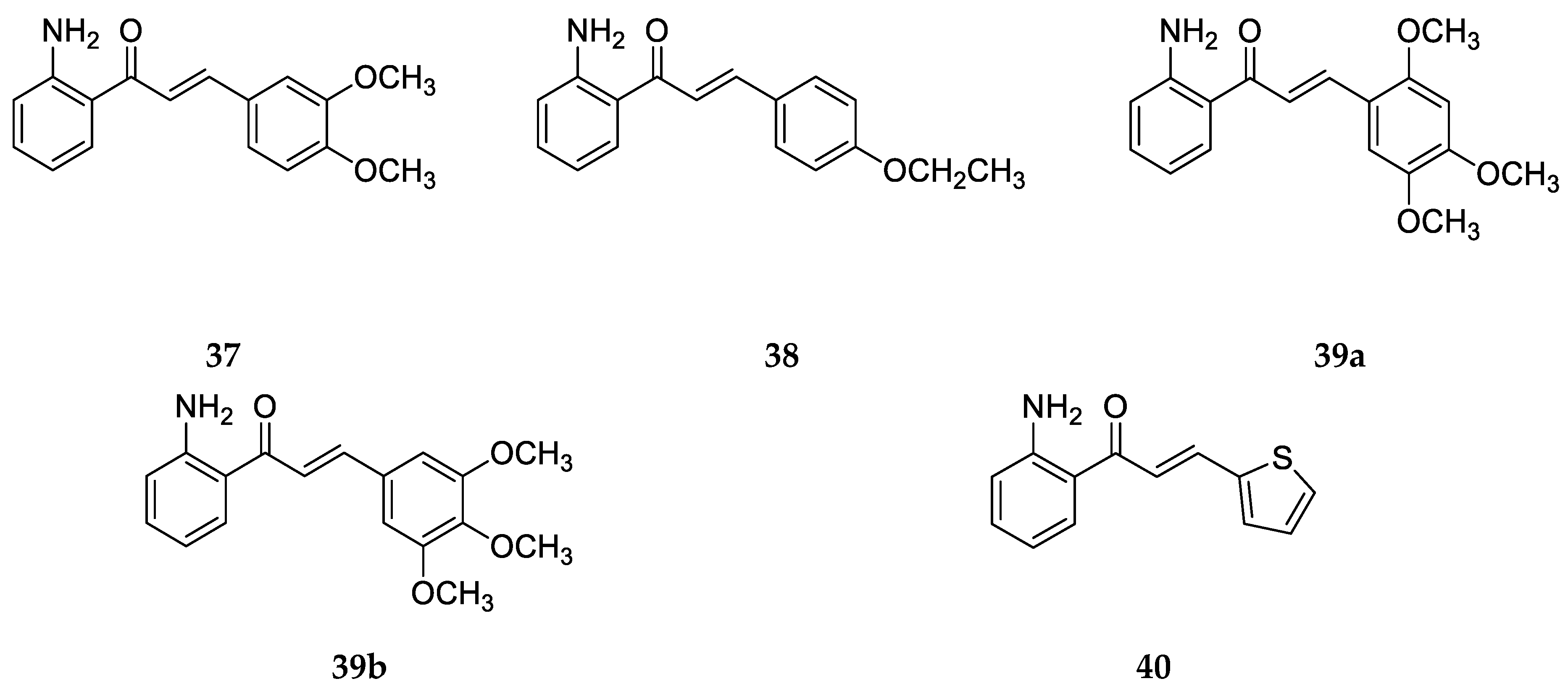
These compounds (
41–
43, 44a, and
44b) were synthesized by a base-catalyzed condensation of 2-amino acetophenones and aldehydes with appropriate substitutions. Then, they were assayed for cytotoxicity in vitro against nine human tumor cell lines [
57], namely, epidermoid carcinoma of the nasopharynx (KB), P-gp-expressing epidermoid carcinoma of the masopharynx (KB-VIN), osteosarcoma (Hos), melanoma (SKMEL-2), ileocecal carcinoma (HCT-8), breast cancer (MCF-7), lung carcinoma (A-549), glioblastoma (U87-MG), and ovarian cancer (1A9) cell lines. The ED
50 values of the compounds are listed in
Table 14.
Compounds
41–
44b showed significant cytotoxic activity (ED
50 ≤ 4.0 mg/mL) and pronounced selectivity against the KB, KBVIN, and 1A9 cell lines. Compound
42 was more potent than
41 against these cancer cell lines, with notably lower values for the compound concentration, affording a 50% reduction in cell number. However, compound
43, which is a structural analog of
42 and contains a methylenedioxy moiety on ring A (positions 4 and 5), was the most active in this study, with the lowest ED
50 values, making it a potential lead for antitumor agents. Among
44a and
44b, only differing in the position of fluorine on ring B, a relatively better activity was shown by
44a and the highest ED
50 values; thus, the least potency was displayed by
44b for most of the cancer cell lines. Also, when compared to a similar chalcone only without a –NH
2 group at position 2 on ring A, compound
44a was about 40-fold more active, demonstrating the importance of the amino group for biological activity. It was concluded that the introduction of a methylenedioxy moiety on ring A led to enhanced cytotoxic activity compared with the A-ring-unsubstituted compound
42 and that the presence of a methoxy group at position 3 on ring B was beneficial for increased cytotoxicity [
57].
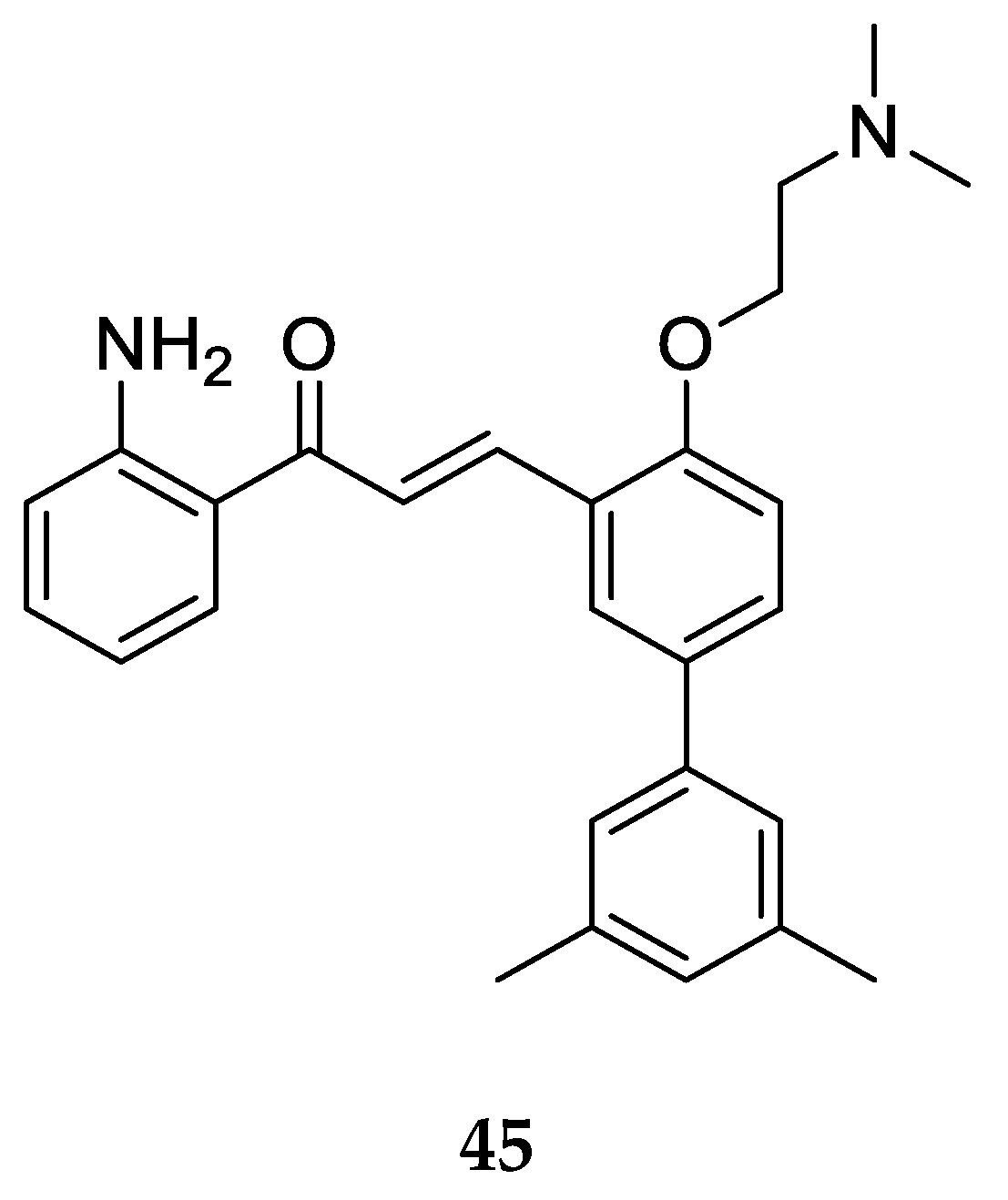
The 2-aminochalcone derivative with larger substitutions on ring B was prepared and tested for antibacterial activity by Nielsen et al. [
52] (
Figure 28).
Compound 45 was tested against bacterial strains Staphylococcus aureus ATCC33591 (resistant to methicillin), Enterococcus faecium 17501 (vancomycin-resistant clinical isolate), and Enterococcus faecium ATCC29212, with MIC values 5 µM, 5 µM, and 10 µM, respectively. Compound 45 and similar 2-aminochalcones with ring B substitutions were found to be more selective for the bacterial membrane. The MIC values also illustrated better antibacterial activity of 45 as compared to its 3-aminochalcone analog (MIC = 10 µM each) and 4-aminochalcone analog 25 (MIC = 10 µM and 20 µM), indicating that the position of the -NH2 group on ring A matters for antibacterial activity.
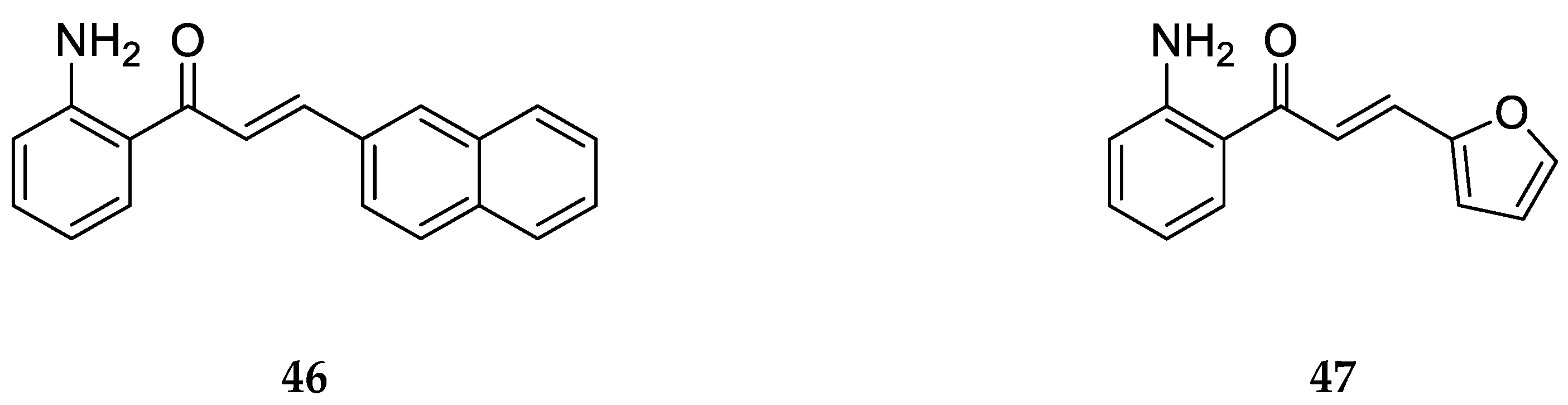
Similarly, 2-aminochalcones
46 and
47 are derivatives with substitutions on ring B synthesized and screened for antifungal activity by Zhang et al. [
55] (
Figure 29).
Compounds
46 and
47 displayed no significant antifungal activity against
Trichophyton rubrum with MIC
80 values >64 µg/mL. This shows that the substitutions on ring B of these compounds are not suitable to inhibit fungal activity, and this might be due to the bulk of the groups as the 2-aminochalcones with smaller substituents showed potent antifungal activity [
55].


























































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
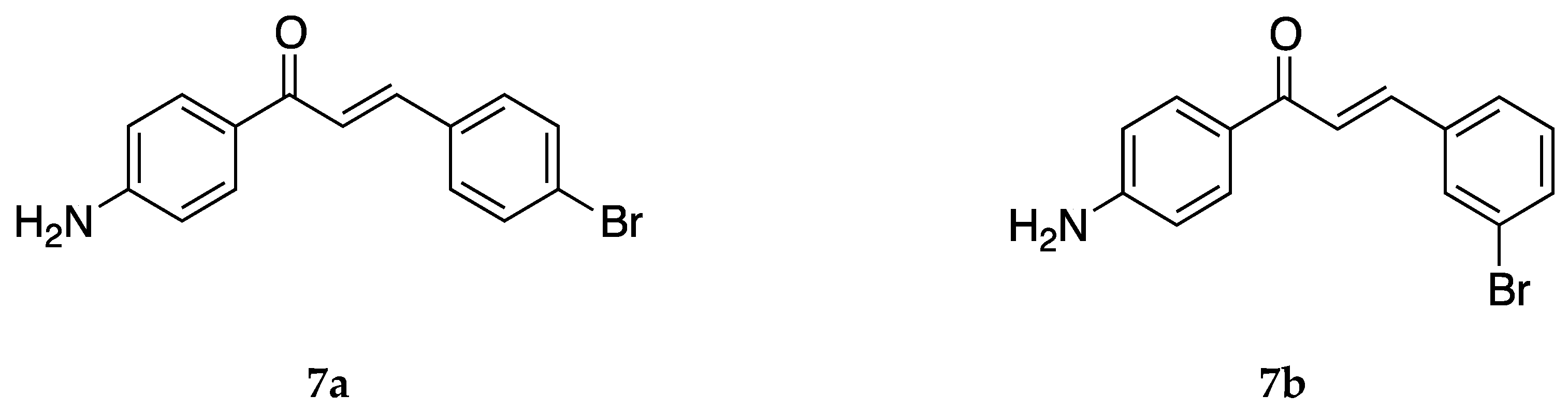{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
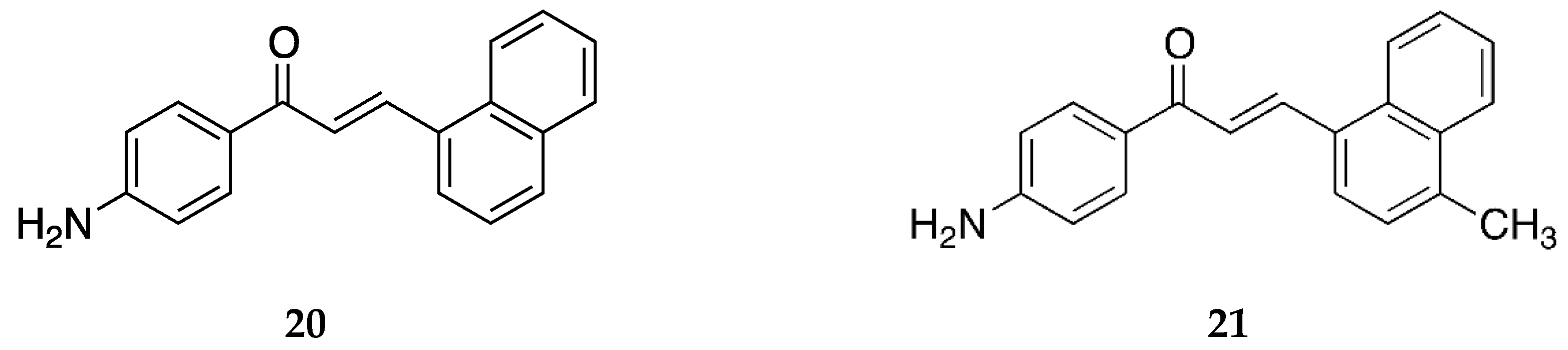{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}