
Circulating Ligands of the Receptor for Advanced Glycation End Products and the Soluble Form of the Receptor Modulate Cardiovascular Cell Apoptosis in Diabetes


 ,
,
Abstract
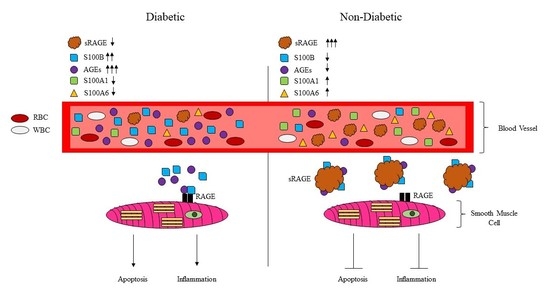
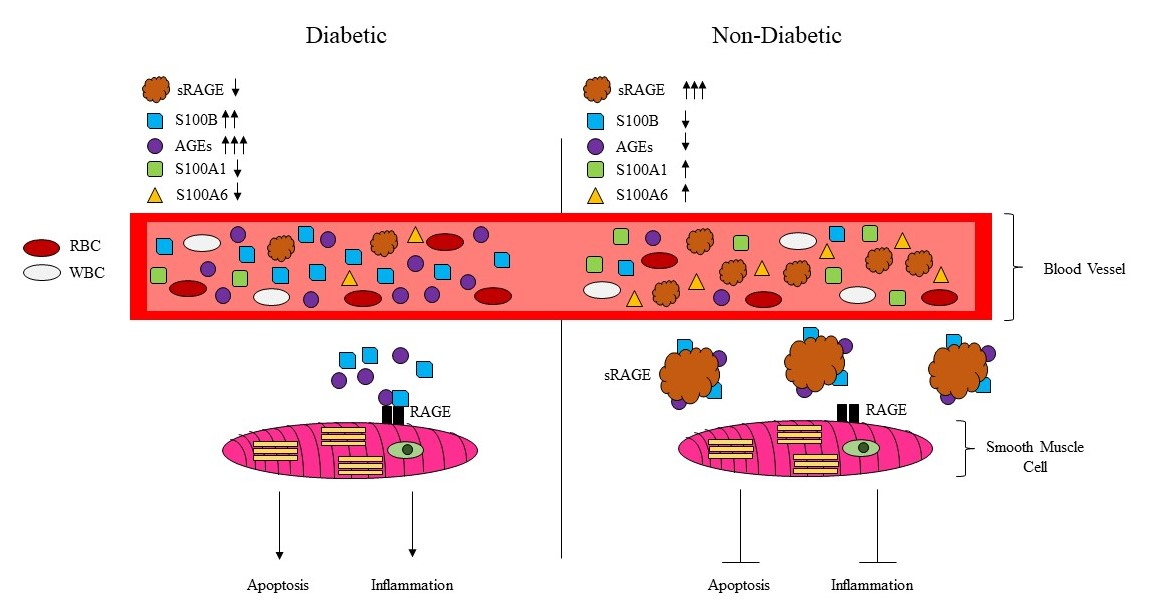
:
1. Introduction
2. Results
2.1. Anthropometric and Metabolic Characteristics
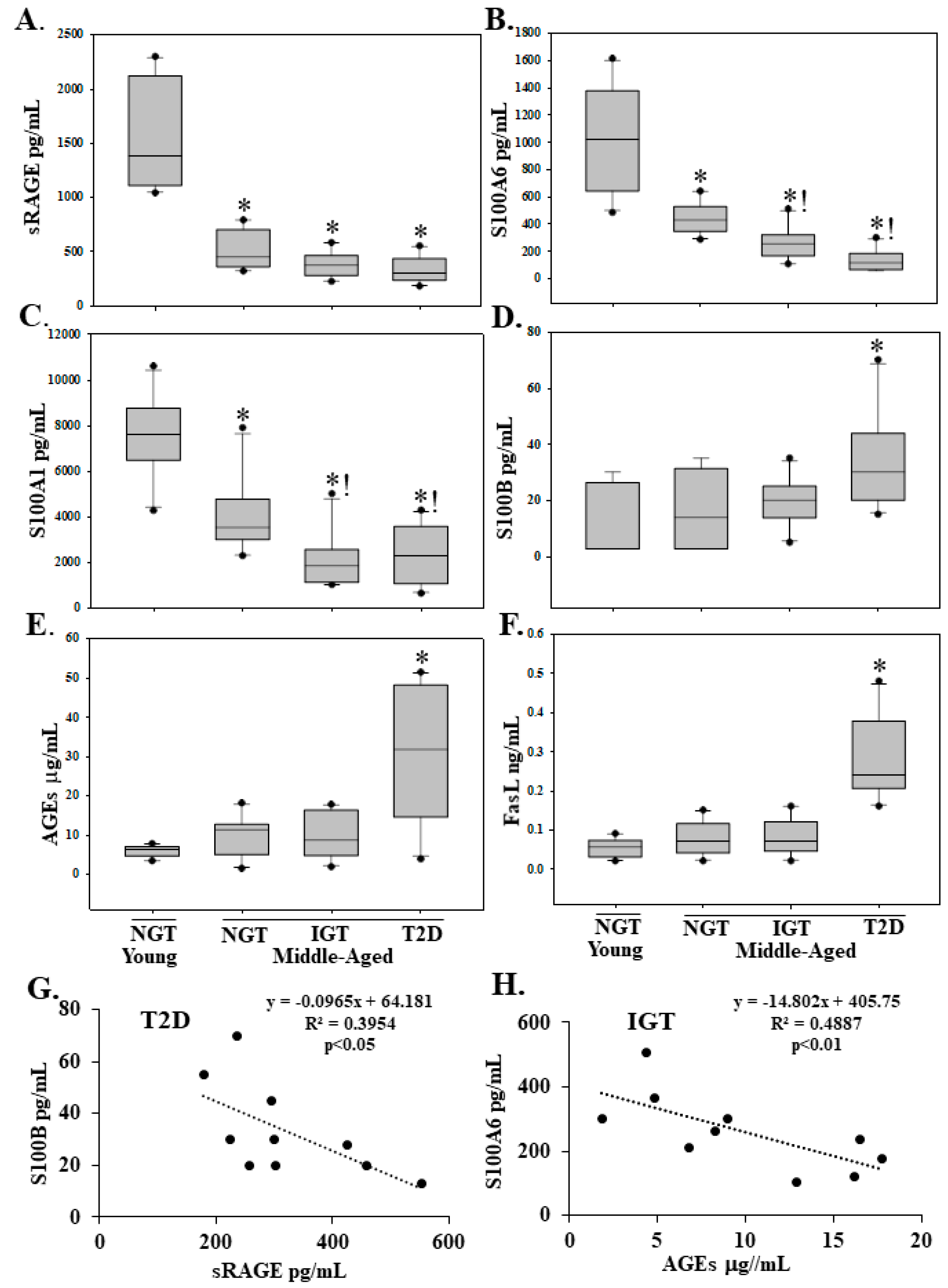
2.2. Serum sRAGE, S100A1, S100A6, AGEs, S100B and FasL Levels Are Differentially Regulated with Age and Diabetes
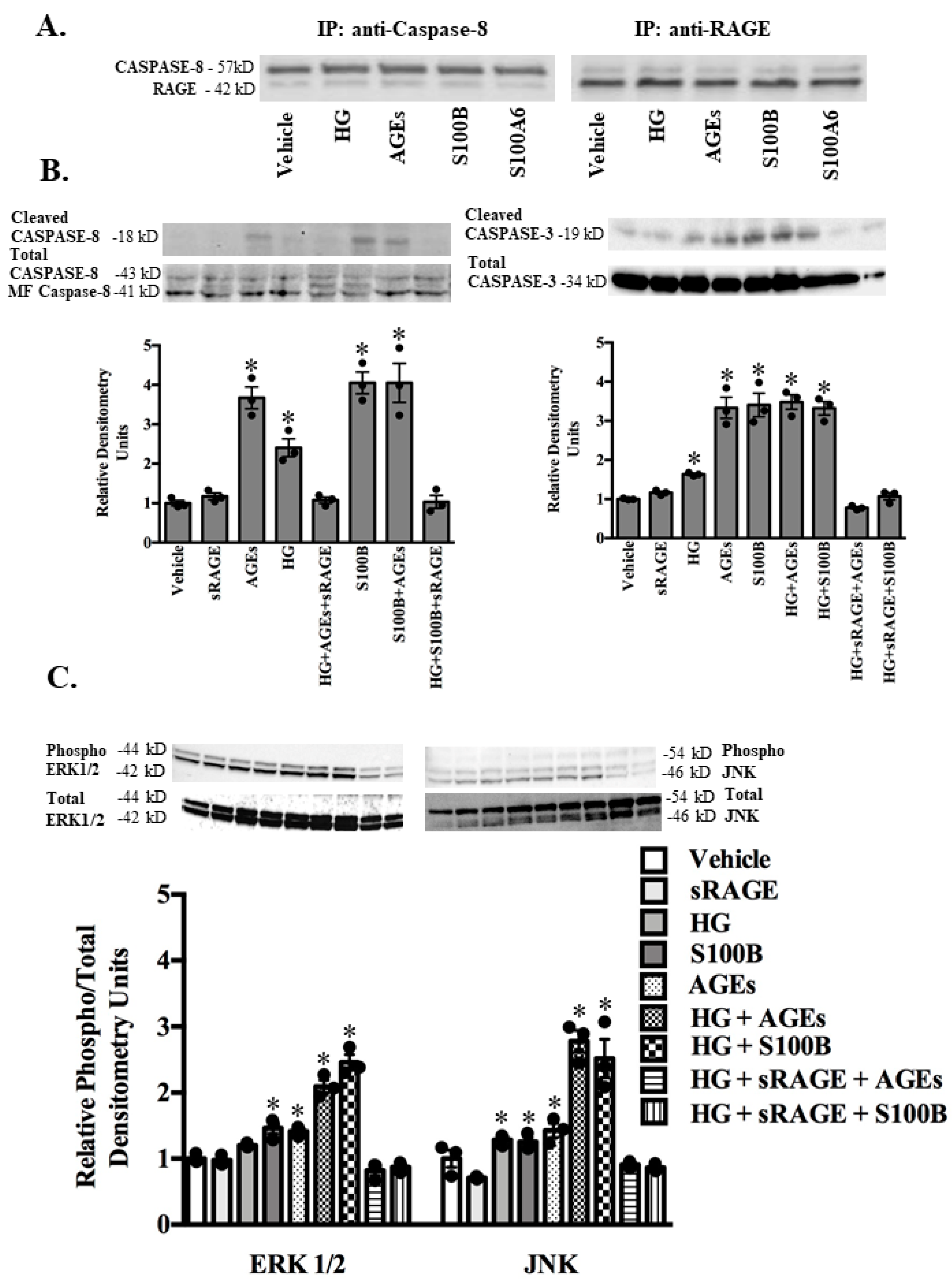
2.3. RAGE Ligands (AGES, S100B, S100A1, S100A6) and sRAGE Differentially Regulate Parameters of Apoptosis (BAX/BCL2 Ratio-Mitochondrial Pathway; Fas Ligand, Fas Receptor-Death Receptor Pathway) and Markers of Inflammation (TNF-α) in ASMC
2.4. RAGE-Ligand Binding Induces the MAPK Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Study Populations
4.2. Ethics Statement
4.3. Laboratory Analyses
4.4. Blood Sampling and Laboratory Methods
4.5. Cell Culture
4.6. Immunoprecipitation and Western Blot Analysis
4.7. Real-Time Quantitative RT-PCR
4.8. Measurements of Apoptosis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Herman, W.H.; Zimmet, P. Type 2 diabetes: An epidemic requiring global attention and urgent action. Diabetes Care 2012, 35, 943–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, E.T.; Prosser, L.A.; Hoerger, T.J.; Lieu, T.; Ludwig, D.S.; Laffel, L.M. Estimated morbidity and mortality in adolescents and young adults diagnosed with type 2 diabetes mellitus. Diabet. Med. 2012, 29, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Yap, F.Y.T.; Tong, D.C.K.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.I.; Kay, T.W.; Slattery, R.M.; et al. Advanced glycation end products are direct modulators of beta-cell function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.; Imaizumi, T. Diabetic vascular complications: Pathophysiology, biochemical basis and potential therapeutic strategy. Curr. Pharm. Des. 2005, 11, 2279–2299. [Google Scholar] [CrossRef]
- Sourris, K.C.; Forbes, J.M. Interactions between advanced glycation end-products (AGE) and their receptors in the development and progression of diabetic nephropathy–are these receptors valid therapeutic targets. Curr. Drug Targets 2009, 10, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Daffu, G.; del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int. J. Mol. Sci. 2013, 14, 19891–19910. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Yamagishi, S.; Okuda, S. Role of AGEs-RAGE system in cardiovascular disease. Curr. Pharm. Des. 2014, 20, 2395–23402. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE axis: A fundamental axis signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. The diverse ligand repertoire of the receptor for advanced glycation endproducts & pathways to the complications of diabetes. Vascul. Pharmacol. 2012, 57, 160–167. [Google Scholar] [PubMed] [Green Version]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Bierhaus, A.; Hofmann, M.A.; Ziegler, R.; Nawroth, P.P. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc. Res. 1998, 37, 586–600. [Google Scholar] [CrossRef] [Green Version]
- The Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [PubMed]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Kislinger, T.K.; Fu, C.B.; Huber, B.; Qu, W.; Taguchi, A.; Yan, S.D.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. Nε-(carboxymethyl)lysine adducts of proteins are ligands for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [Green Version]
- Beisswenger, P.J.; Makita, Z.; Curphey, T.J.; Moore, L.L.; Jean, S.; Brinck-Johnsen, T.; Bucala, R.; Vlassara, H. Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes 1995, 44, 824–829. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Makita, Z.; Shiroshita, K.; Ueda, T.; Fusegawa, T.; Kuwajima, S.; Takeuchi, M.; Koike, T. Specific fluorescence assay for advanced glycation end products in blood and urine of diabetic patients. Metabolism 1998, 47, 1348–1353. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Rouhiainen, A.; Kuja-Panula, J.; Tumova, S.; Rauvala, H. RAGE-mediated cell signaling. Methods Mol. Biol. 2013, 963, 239–263. [Google Scholar] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehrenbach, H.; Kasper, M.; Tschernig, T.; Shearman, M.S.; Schuh, D.; Müller, M. Receptor for advanced glycation end products (RAGE) exhibits highly differential cellular and subcellular localization in rat and human lung. Cell. Mol. Biol. 1998, 44, 1147–1157. [Google Scholar]
- Vazzana, N.; Santilli, F.; Cuccurullo, C.; Davì, G. Soluble forms of RAGE in internal medicine. Intern. Emerg. Med. 2009, 4, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Soluble form of a receptor for advanced glycation end products (sRAGE) as a biomarker. Front. Biosci. 2010, 2, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Hori, O.; Stern, D.; Schmidt, A.M. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J. Clin. Investig. 1996, 97, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef]
- Wendt, T.; Harja, E.; Bucciarelli, L.; Qu, W.; Lu, Y.; Rong, L.L.; Jenkins, D.G.; Stein, G.; Schmidt, A.M.; Yan, S.F. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis 2006, 185, 70–77. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. Low levels of serum soluble receptors for advanced glycation end products, biomarkers for disease state: Myth or reality. Int. J. Angiol. 2014, 23, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, C.; Emanuele, E.; D’Angelo, A.; Buzzi, M.P.; Belvito, C.; Cuccia, M.; Geroldi, D. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1032–1037. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, J.B.; de Lemos, J.A.; Cipollone, F.; Cipollone, F.; Ayers, C.R.; Rohatgi, A.; Morrow, D.A.; Khera, A.; McGuire, D.K. Association between circulating soluble receptor for advanced glycation end products and atherosclerosis: Observations from the dallas heart study. Diabetes Care 2009, 32, 1218–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.I.; Moon, Y.P.; Kalea, A.Z.; Khatri, M.; Marquez, C.; Schmidt, A.M.; Paik, M.C.; Yoshita, M.; Sacco, R.L.; De Carli, C.; et al. Association of serum soluble receptor for advanced glycation end-products with subclinical cerebrovascular disease: The Northern Manhattan Study (NOMAS). Atherosclerosis 2011, 216, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvin, E.M.; Halushka, M.K.; Rawlings, A.M.; Selvin, E.; Halushka, M.K.; Rawlings, A.M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. sRAGE and risk of diabetes, cardiovascular disease, and death. Diabetes 2013, 62, 2116–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.; Hitman, G.; Neil, A.; Livingstone, S.; Menys, V.C.; Bao, W.; Demicco, D.A.; Preston, G.M.; et al. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: An analysis from the CARDS trial. Diabetes 2011, 60, 2379–2385. [Google Scholar] [CrossRef] [Green Version]
- Farhan, S.S.; Hussain, S.A. Advanced glycation end products (AGEs) and their soluble receptors (sRAGE) as early predictors of reno-vascular complications in patients with uncontrolled type 2 diabetes mellitus. Diabetes Metab. Syndr. 2019, 4, 2457–2461. [Google Scholar] [CrossRef]
- Fujisawa, K.; Katakami, N.; Kaneto, H.; Naka, T.; Takahara, M.; Sakamoto, F.; Irie, Y.; Miyashita, M.; Kubo, F.; Yasuda, T.; et al. Circulating soluble RAGE as a predictive biomarker of cardiovascular event risk in patients with type 2 diabetes. Atherosclerosis 2013, 227, 425–428. [Google Scholar] [CrossRef]
- Tan, K.C.B.; Shiu, S.W.M.; Chow, W.S.; Leng, L.; Bucala, R.; Betteridge, D.J. Association between serum levels of soluble receptor for advanced glycation end products and circulating advanced glycation end products in type 2 diabetes. Diabetologia 2006, 49, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Challier, M.; Jacqueminet, S.; Benabdesselam, O.; Grimaldi, A.; Beaudeux, J.L. Increased serum concentrations of soluble receptor for advanced glycation endproducts in patients with type 1 diabetes. Clin. Chem. 2005, 51, 1749–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D.A. Higher plasma soluble Receptor for Advanced Glycation End Products (sRAGE) levels are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: A 12-year follow-up study. Diabetes 2010, 59, 2027–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadzadeh, F.; Tsoporis, J.N.; Izhar, S.; Desjardins, J.F.; Parker, T.G. Deficiency of S100B confers resistance to experimental diabetes in mice. Exp. Cell Res. 2018, 365, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Edwin, R.; Miranda, E.R.; Somal, V.S.; Blackburn, B.K.; Wang, E.; Farabi, S.; Karstoft, K.; Fealy, C.E.; Kashyap, S.; Kirwan, J.P.; et al. Circulating soluble RAGE isoforms are attenuated in obese, impaired-glucose-tolerant individuals and are associated with the development of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E631–E640. [Google Scholar]
- Gupta, S.; Lee, C.M.; Wang, J.F.; Parodo, J.; Jia, S.H.; Hu, J.; Marshall, J.C. Heat-shock protein-90 prolongs septic neutrophil survival by protecting c-Src kinase and caspase-8 from proteasomal degradation. J. Leukoc. Biol. 2018, 103, 933–944. [Google Scholar] [CrossRef]
- Maelfait, J.; Vercammen, E.; Janssens, S.; Schotte, P.; Haegman, M.; Magez, S.; Beyaert, R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med. 2008, 205, 1967–1973. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Tsoporis, J.N.; Overgaard, C.B.; Izhar, S.; Parker, T.G. S100B modulates the hemodynamic response to norepinephrine stimulation. Am. J. Hypertens. 2009, 22, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Mohammadzadeh, F.; Desjardins, J.F.; Tsoporis, J.N.; Proteau, G.; Leong-Poi, H.; Parker, T.G. S100B: Role in cardiac remodeling and function following myocardial infarction in diabetes. Life Sci. 2013, 92, 639–647. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | NGT-A | NGT-B | IGT | T2D |
|---|---|---|---|---|
| Young | Middle-Aged | Middle-Aged | Middle-Aged | |
| n(%men) | 10 (50) | 10 (50) | 10 (50) | 10 (70) |
| Age, years | 32.1 ± 1.5 | 59.6 ± 1.2 # | 59.8 ± 1.6 # | 59.8 ± 1.6 # |
| Weight, kg | 73.9 ± 4.7 | 75.1 ± 2.9 | 80.5 ± 4.3 | 91.5 ± 3.1 # |
| BMI, kg/m2 | 24.0 ± 0.9 | 27.9 ± 0.7 # | 28.1 ± 0.66 # | 31.8 ± 0.9 *# |
| Serum Lipids | ||||
| Triglycerides, mg/dL | 114 ± 16.1 | 140 ± 20.2 * | 140 ± 21.8 * | |
| Total Cholesterol, mg/dL | 229 ± 11.3 | 215 ± 13.1 | 217 ± 13.6 | |
| HDL Cholesterol, mg/dL | 59.7 ± 4.5 | 51.8 ± 4.7 | 44.2 ± 3.9 * | |
| LDL, Cholesterol, mg/dL | 138 ± 8.9 | 129 ± 11.9 | 140 ± 9.7 | |
| Liver Enzymes | ||||
| AST, U/L | 32 (17, 62) | 30 (19, 47) | 28 (12, 54) | |
| ALT, U/L | 54 (15, 124) | 43 (27, 80) * | 44 (26, 65) * | |
| g-GT, U/L | 60 (12, 138) | 57 (12, 174) | 63 (6, 165) | |
| Measures of Glycemia | ||||
| Fasting glucose, mg/dL | 86.1 ± 3.3 | 82.5 ± 3.9 | 108.2 ± 9.3 * | |
| 2-h glucose, mg/dL | 113 ± 4.3 | 154.8 ± 4.7 * | 251.9 ± 15.5 *! | |
| Fasting insulin, μU/mL | 10.8 ± 1.4 | 11.3 ± 1.3 | 15.9 ± 2.8 * | |
| 2-h insulin μU/mL | 94.3 ± 16.9 | 162.2 ± 37.4 * | 150.9 ± 22.1 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoporis, J.N.; Hatziagelaki, E.; Gupta, S.; Izhar, S.; Salpeas, V.; Tsiavou, A.; Rigopoulos, A.G.; Triantafyllis, A.S.; Marshall, J.C.; Parker, T.G.; et al. Circulating Ligands of the Receptor for Advanced Glycation End Products and the Soluble Form of the Receptor Modulate Cardiovascular Cell Apoptosis in Diabetes. Molecules 2020, 25, 5235. https://doi.org/10.3390/molecules25225235
Tsoporis JN, Hatziagelaki E, Gupta S, Izhar S, Salpeas V, Tsiavou A, Rigopoulos AG, Triantafyllis AS, Marshall JC, Parker TG, et al. Circulating Ligands of the Receptor for Advanced Glycation End Products and the Soluble Form of the Receptor Modulate Cardiovascular Cell Apoptosis in Diabetes. Molecules. 2020; 25(22):5235. https://doi.org/10.3390/molecules25225235
Chicago/Turabian StyleTsoporis, James N., Erifili Hatziagelaki, Sahil Gupta, Shehla Izhar, Vasileos Salpeas, Anastasia Tsiavou, Angelos G. Rigopoulos, Andreas S. Triantafyllis, John C. Marshall, Thomas G. Parker, and et al. 2020. "Circulating Ligands of the Receptor for Advanced Glycation End Products and the Soluble Form of the Receptor Modulate Cardiovascular Cell Apoptosis in Diabetes" Molecules 25, no. 22: 5235. https://doi.org/10.3390/molecules25225235