Unveiling the Hidden Treasury: CIITA-Driven MHC Class II Expression in Tumor Cells to Dig up the Relevant Repertoire of Tumor Antigens for Optimal Stimulation of Tumor Specific CD4+ T Helper Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
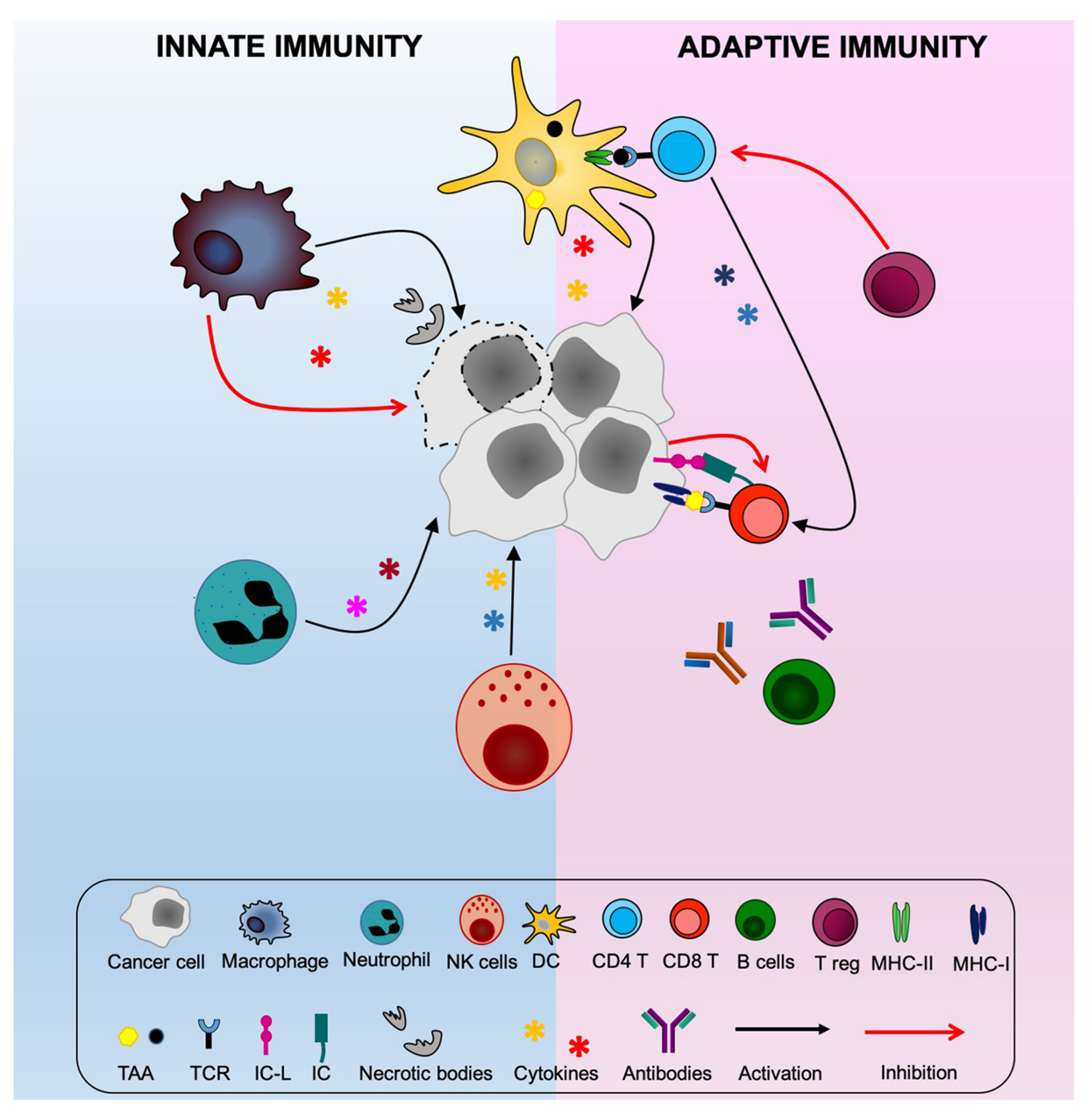
2. Exploring New Avenues for Optimal Stimulation of Anti-Tumor CD4+ Th Cells
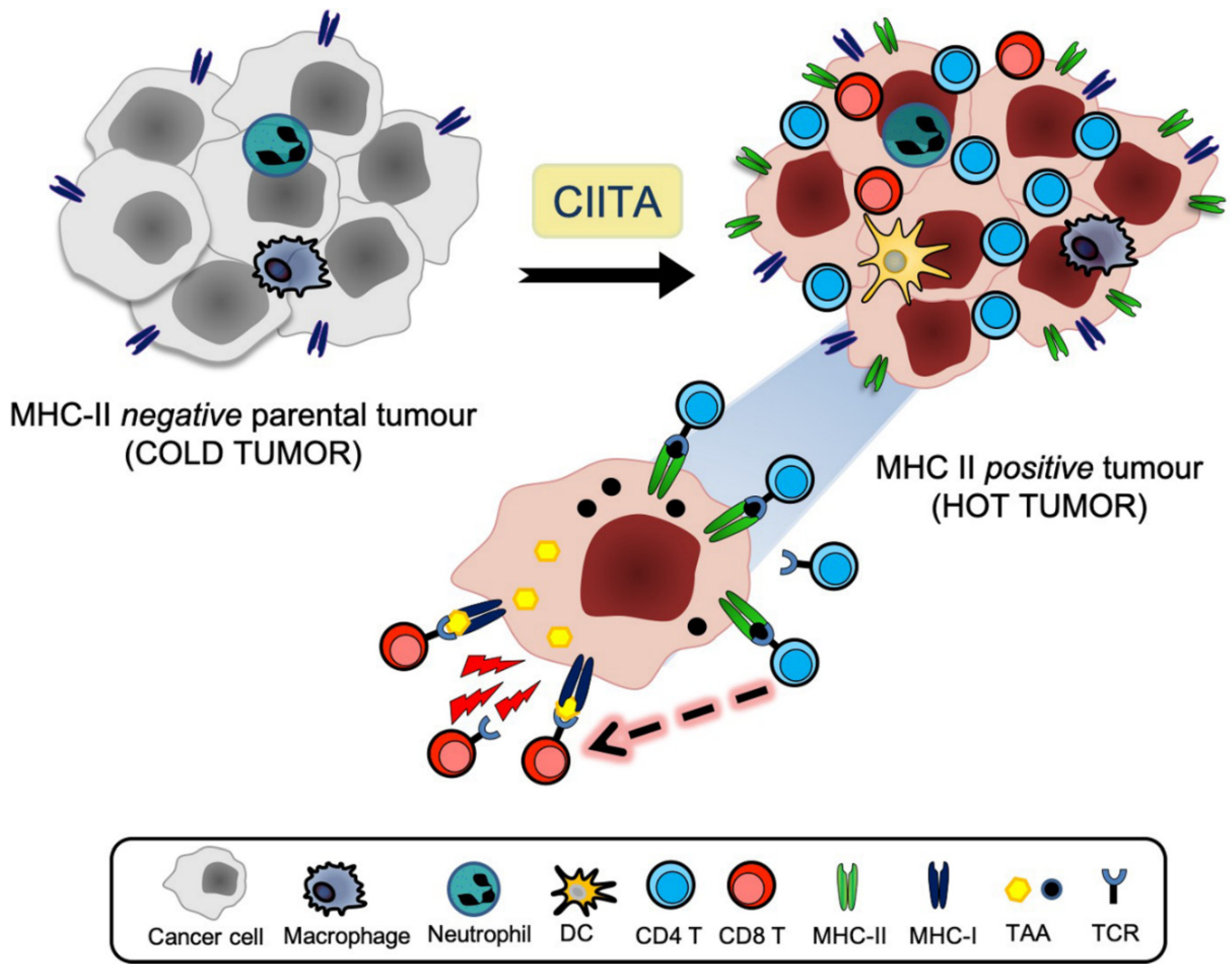
3. The CIITA Approach
4. Optimal Stimulation of Tumor-Specific CD4+ Th Cells by CIITA-Driven MHC Class II Expressing Cells
5. The Tumor Microenvironment and the APC Function of CIITA-Driven MHC-II-Expressing Tumor Cells
6. From Bench to Bedside: New Hope for the Future of Immmunotherapy?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Algarra, I.; Garcia-Lora, A.M.; García-Lora, Á.M. The escape of cancer from T lymphocytes: Immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol. Immunother. 2010, 59, 1601–1606. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Boon, T.; Cerottini, J.-C.; Van den Eynde, B.; van der Bruggen, P.; Van Pel, A. Tumor Antigens Recognized by T Lymphocytes. Annu. Rev. Immunol. 1994, 12, 337–365. [Google Scholar] [CrossRef]
- Hung, K.; Hayashi, R.J.; Lafond-Walker, A.; Lowenstein, C.J.; Pardoll, D.M.; Levitsky, H. The Central Role of CD4+ T Cells in the Antitumor Immune Response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M.; Topalian, S.L. The role of CD4+ T cell responses in anti-tumor immunity. Curr. Opin. Immunol. 1998, 10, 588–594. [Google Scholar] [CrossRef]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.-C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+T Cell Immunity Against a Tumor/Self-Antigen Is Augmented by CD4+T Helper Cells and Hindered by Naturally Occurring T Regulatory Cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Daar, A.S.; Fuggle, S.V.; Fabre, J.W.; Ting, A.; Morris, P.J. The detailed distribution of Mhc class II antigens in normal human organs. Transplantation 1984, 38, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Dendritic cells: Versatile controllers of the immune system. Nat. Med. 2007, 13, 1155–1159. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P.R.; Ploegh, H.L. How MHC class II molecules acquire peptide cargo: Biosynthesis and trafficking through the endocytic pathway. Annu. Rev. Cell Dev. Biol. 1995, 11, 267–306. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S.; Adorini, L.; Sartoris, S.; Sinigaglia, F.; Guardiola, J. MHC: Orchestrating the immune response. Immunol. Today 1995, 16, 8–11. [Google Scholar] [CrossRef]
- Rudensky, A.Y.; Preston-Hurlburt, P.; Hong, S.-C.; Barlow, A.K.; Janeway, C.A. Sequence analysis of peptides bound to MHC class II molecules. Nat. Cell Biol. 1991, 353, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-Loading Compartments for Major Histocompatibility Complex Class II Molecules Continuously Receive Input from Autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, A.N.; Van Der Meijden, E.D.; Honders, M.W.; Goeman, J.J.; Wiertz, E.J.H.J.; Falkenburg, J.H.F.; Griffioen, M. Endogenous HLA class II epitopes that are immunogenic in vivo show distinct behavior toward HLA-DM and its natural inhibitor HLA-DO. Blood 2012, 120, 3246–3255. [Google Scholar] [CrossRef]
- Chicz, R.M.; Urban, R.G.; Gorga, J.C.; Vignali, D.A.; Lane, W.S.; Strominger, J.L. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J. Exp. Med. 1993, 178, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Jaraquemada, D.; Marti, M.; Long, E. An endogenous processing pathway in vaccinia virus-infected cells for presentation of cytoplasmic antigens to class II-restricted T cells. J. Exp. Med. 1990, 172, 947–954. [Google Scholar] [CrossRef]
- Melief, C.J.M.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Eggermont, A.M. Immunotherapy: Vaccine trials in melanoma--time for reflection. Nat. Rev. Clin. Oncol. 2009, 6, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S.; Tosi, G. Optimal MHC-II-restricted tumor antigen presentation to CD4+ T helper cells: The key issue for development of anti-tumor vaccines. J. Transl. Med. 2012, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accolla, R.S.; Lombardo, L.; Abdallah, R.; Raval, G.; Forlani, G.; Tosi, G. Boosting the MHC Class II-Restricted Tumor Antigen Presentation to CD4+ T Helper Cells: A Critical Issue for Triggering Protective Immunity and Re-Orienting the Tumor Microenvironment Toward an Anti-Tumor State. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nat. Cell Biol. 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Accolla, R.S.; Tosi, G. Adequate Antigen Availability: A Key Issue for Novel Approaches to Tumor Vaccination and Tumor Immunotherapy. J. Neuroimmune Pharmacol. 2012, 8, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Bou Nasser Eddine, F.; Ramia, E.; Tosi, G.; Forlani, G.; Accolla, R.S. Tumor Immunology meets…Immunology: Modified cancer cells as professional APC for priming naïve tumor-specific CD4+ T cells. OncoImmunology 2017, 6, e1356149. [Google Scholar] [CrossRef]
- Accolla, R.S.; Ramia, E.; Tedeschi, A.; Forlani, G. CIITA-Driven MHC Class II Expressing Tumor Cells as Antigen Presenting Cell Performers: Toward the Construction of an Optimal Anti-tumor Vaccine. Front. Immunol. 2019, 10, 1806. [Google Scholar] [CrossRef]
- Accolla, R.S.; Carra, G.; Guardiola, J. Reactivation by a trans-acting factor of human major histocompatibility complex Ia gene expression in interspecies hybrids between an Ia-negative human B-cell variant and an Ia-positive mouse B-cell lymphoma. Proc. Natl. Acad. Sci. USA 1985, 82, 5145–5149. [Google Scholar] [CrossRef] [Green Version]
- Accolla, R.S.; Scarpellino, L.; Carra, G.; Guardiola, J. Trans-acting element(s) operating across species barriers positively regulate expression of major histocompatibility complex class II genes. J. Exp. Med. 1985, 162, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S.; Jotterand-Bellomo, M.; Scarpellino, L.; Maffei, A.; Carra, G.; Guardiola, J. aIr-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. J. Exp. Med. 1986, 164, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, J.; Scarpellino, L.; Carra, G.; Accolla, R.S. Stable integration of mouse DNA into Ia-negative human B-lymphoma cells causes reexpression of the human Ia-positive phenotype. Proc. Natl. Acad. Sci. USA 1986, 83, 7415–7418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steimle, V.; Otten, L.; Zufferey, M.; Mach, B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 1993, 75, 135–146. [Google Scholar] [CrossRef]
- Accolla, R.S. Human B cell variants immunoselected against a single Ia antigen subset have lost expression of several Ia antigen subsets. J. Exp. Med. 1983, 157, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Mach, B.; Steimle, V.; Martinez-Soria, E.; Reith, W. REGULATION OF MHC CLASS II GENES: Lessons from a Disease. Annu. Rev. Immunol. 1996, 14, 301–331. [Google Scholar] [CrossRef]
- Accolla, R.S.; Carra, G.; Buchegger, F.; Carrel, S.; Mach, J.P. The human Ia-associated invariant chain is synthesized in Ia-negative B cell variants and is not expressed on the cell surface of both Ia-negative and Ia-positive parental cells. J. Immunol. 1985, 134, 3265–3271. [Google Scholar]
- Accolla, R.S.; Barbaro, A.D.L.; Mazza, S.; Casoli, C.; De Maria, A.; Tosi, G. The MHC class II transactivator: Prey and hunter in infectious diseases. Trends Immunol. 2001, 22, 560–563. [Google Scholar] [CrossRef]
- Sartoris, S.; Valle, M.T.; Barbaro, A.D.L.; Tosi, G.; Cestari, T.; D’Agostino, A. HLA Class II Expression in Uninducible Hepatocarcinoma Cells After Transfection of AIR-1 Gene Product CIITA: Acquisition of Antigen Processing and Presentation Capacity. J. Immunol. 1998, 161, 814–820. [Google Scholar]
- Mortara, L.; Castellani, P.; Meazza, R.; Tosi, G.; Barbaro, A.D.L.; Procopio, F.A.; Comes, A.; Zardi, L.; Ferrini, S.; Accolla, R.S. CIITA-Induced MHC Class II Expression in Mammary Adenocarcinoma Leads to a Th1 Polarization of the Tumor Microenvironment, Tumor Rejection, and Specific Antitumor Memory. Clin. Cancer Res. 2006, 12, 3435–3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meazza, R.; Comes, A.; Orengo, A.M.; Ferrini, S.; Accolla, R.S. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur. J. Immunol. 2003, 33, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Frangione, V.; Mortara, L.; Castellani, P.; Barbaro, A.D.L.; Accolla, R.S. CIITA-driven MHC-II positive tumor cells: Preventive vaccines and superior generators of antitumor CD4+ T lymphocytes for immunotherapy. Int. J. Cancer 2010, 127, 1614–1624. [Google Scholar] [CrossRef]
- Armstrong, T.D.; Clements, V.K.; Martin, B.K.; Ting, J.P.-Y.; Ostrand-Rosenberg, S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc. Natl. Acad. Sci. USA 1997, 94, 6886–6891. [Google Scholar] [CrossRef] [Green Version]
- Dolan, B.P.; Gibbs, K.D.; Ostrand-Rosenberg, S. Tumor-Specific CD4+T Cells Are Activated by “Cross-Dressed” Dendritic Cells Presenting Peptide-MHC Class II Complexes Acquired from Cell-Based Cancer Vaccines. J. Immunol. 2006, 176, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Bikoff, E.K.; Huang, L.Y.; Episkopou, V.; Van Meerwijk, J.; Germain, R.N.; Robertson, E.J. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+ T cell selection in mice lacking invariant chain expression. J. Exp. Med. 1993, 177, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Viville, S.; Neefjes, J.; Lotteau, V.; Dierich, A.; LeMeur, M.; Ploegh, H.; Benoist, C.; Mathis, D. Mice lacking the MHC class II-associated invariant chain. Cell 1993, 72, 635–648. [Google Scholar] [CrossRef]
- Mortara, L.; Frangione, V.; Castellani, P.; Barbaro, A.D.L.; Accolla, R.S. Irradiated CIITA-positive mammary adenocarcinoma cells act as a potent anti-tumor-preventive vaccine by inducing tumor-specific CD4+ T cell priming and CD8+ T cell effector functions. Int. Immunol. 2009, 21, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheroutre, H.; Husain, M.M. CD4 CTL: Living up to the challenge. Semin. Immunol. 2013, 25, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.-C.S.; Rancan, C.; et al. Intratumoral CD4+ T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e13. [Google Scholar] [CrossRef] [PubMed]
- Sacher, A.G.; Paul, M.S.; Paige, C.J.; Ohashi, P.S. Cytotoxic CD4+ T Cells in Bladder Cancer—A New License to Kill. Cancer Cell 2020, 38, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Bevan, M.J. Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nat. Cell Biol. 2011, 471, 629–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhland, M.K.; Roberts, E.W.; Cai, E.; Mujal, A.M.; Marchuk, K.; Beppler, C.; Nam, D.; Serwas, N.K.; Binnewies, M.; Krummel, M.F. Visualizing Synaptic Transfer of Tumor Antigens among Dendritic Cells. Cancer Cell 2020, 37, 786–799.e5. [Google Scholar] [CrossRef]
- Hochweller, K.; Striegler, J.; Hämmerling, G.J.; Garbi, N. A novel CD11c.DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur. J. Immunol. 2008, 38, 2776–2783. [Google Scholar] [CrossRef]
- Bou Nasser Eddine, F.; Forlani, G.; Lombardo, L.; Tedeschi, A.; Tosi, G.; Accolla, R.S. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4+ TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. OncoImmunology 2016, 6, e1261777. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nat. Cell Biol. 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Bell, J. Amplifying with α-GalCer. Nat. Rev. Immunol. 2002, 2, 224. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Irvine, D.J.; Stachowiak, A.N.; Hori, Y. Lymphoid tissue engineering: Invoking lymphoid tissue neogenesis in immunotherapy and models of immunity. Semin. Immunol. 2008, 20, 137–146. [Google Scholar] [CrossRef]
- Singh-Jasuja, H.; Emmerich, N.P.N.; Rammensee, H.-G. The Tübingen approach: Identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol. Immunother. 2004, 53, 187–195. [Google Scholar] [CrossRef]
- Ramia, E.; Chiaravalli, A.M.; Eddine, F.B.N.; Tedeschi, A.; Sessa, F.; Accolla, R.S.; Forlani, G. CIITA-related block of HLA class II expression, upregulation of HLA class I, and heterogeneous expression of immune checkpoints in hepatocarcinomas: Implications for new therapeutic approaches. OncoImmunology 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Correnti, C.E.; Laszlo, G.S.; De Van Der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forlani, G.; Shallak, M.; Celesti, F.; Accolla, R.S. Unveiling the Hidden Treasury: CIITA-Driven MHC Class II Expression in Tumor Cells to Dig up the Relevant Repertoire of Tumor Antigens for Optimal Stimulation of Tumor Specific CD4+ T Helper Cells. Cancers 2020, 12, 3181. https://doi.org/10.3390/cancers12113181
Forlani G, Shallak M, Celesti F, Accolla RS. Unveiling the Hidden Treasury: CIITA-Driven MHC Class II Expression in Tumor Cells to Dig up the Relevant Repertoire of Tumor Antigens for Optimal Stimulation of Tumor Specific CD4+ T Helper Cells. Cancers. 2020; 12(11):3181. https://doi.org/10.3390/cancers12113181
Chicago/Turabian StyleForlani, Greta, Mariam Shallak, Fabrizio Celesti, and Roberto S. Accolla. 2020. "Unveiling the Hidden Treasury: CIITA-Driven MHC Class II Expression in Tumor Cells to Dig up the Relevant Repertoire of Tumor Antigens for Optimal Stimulation of Tumor Specific CD4+ T Helper Cells" Cancers 12, no. 11: 3181. https://doi.org/10.3390/cancers12113181