Abstract
Venous thromboembolism (VTE) refers to deep vein thrombosis (DVT), whose consequence may be a pulmonary embolism (PE). Thrombosis is associated with significant morbidity and mortality and is the third most common cardiovascular disease after myocardial infarction and stroke. DVT is associated with the formation of a blood clot in a deep vein in the body. Thrombosis promotes slowed blood flow, hypoxia, cell activation, and the associated release of many active substances involved in blood clot formation. All thrombi which adhere to endothelium consist of fibrin, platelets, and trapped red and white blood cells. In this review, we summarise the impact of various factors affecting haemostatic disorders leading to blood clot formation. The paper discusses the causes of thrombosis, the mechanism of blood clot formation, and factors such as hypoxia, the involvement of endothelial cells (ECs), and the activation of platelets and neutrophils along with the effects of bacteria and reactive oxygen species (ROS). Mechanisms related to the action of anticoagulants affecting coagulation factors including antiplatelet drugs have also been discussed. However, many aspects related to the pathogenesis of thrombosis still need to be clarified. A review of the drugs used to treat and prevent thrombosis and natural anticoagulants that occur in the plant world and are traditionally used in Far Eastern medicine has also been carried out.
1. Introduction
Thrombosis can occur in either veins or arteries. In the veins, it leads to DVT and a pulmonary embolism, referred to as VTE. In turn, in the arteries, it most often causes myocardial infarction, ischemic stroke, and acute limb ischemia [1]. The term DVT usually refers to a blood clot that forms in a deep vein, most often in the large veins that run through the calf muscles and eventually leads to pain and swelling in the leg. If the thrombosis is not treated, a pulmonary embolism may occur [2].
Venous thrombosis is becoming more common and is associated with our lifestyle. It can occur very quickly within an hour or over a long period with worsening symptoms. Long hours of travel, e.g., by plane or car, contribute to thrombosis. Moreover, hormonal therapy, including contraception, is also a contributing factor. Other factors conducive to the development of thrombosis can also include surgery, immobilization as a consequence of a procedure or caused by a long-term treatment, delivery, infectious diseases, pre-existing vein diseases, excessive coagulation, high haematocrit (increase blood viscosity), the use of drugs that irritate the endothelium, and varicose veins [3].
Thrombosis may also occur in pregnant and postpartum women. It affects not only people sitting motionless but may occur after intensive effort as well, and it can also occur after dehydration of the body. It also applies to patients undergoing an orthopaedic surgery. Diabetes, hypertension, but also smoking and being overweight, are additional risk factors for thrombosis [4].
Other factors favouring thrombosis are the age of the patients, a sedentary or standing lifestyle, lack of physical activity, obesity, as well as existing cardiovascular diseases such as hypercoagulability of the blood and the presence of varicose vein and recent surgery but also rheumatism, diabetes, smoking, and others [5,6]. Cases of thrombosis are also associated with intravenous drug users (IDUs) in whom 22% of DVT cases were reported. It is estimated that the number of drug users in the world ranges from 11 to 21 million including 4–47.6% of people with DVT [7].
DVT is associated with the formation of a blood clot in a deep vein in the body. However, blood clots can occur most often in the large veins of the calf and thigh muscles. Depending on the size, a blood clot that forms in a vein can narrow the vessel or close it completely. The consequence is the appearance of pain and swelling in the leg, as well as of serious complications such as a pulmonary embolism when a blood clot travels to the lungs leading to the inhibition of blood flow [5].
The clots can also break away and be transferred from the flowing blood to the cerebral vessels leading to an ischemic stroke. Therefore, it is very important to quickly diagnose DVT and immediately start its treatment [2]. Essential in the formation of thrombosis are endothelial cells (ECs), which provide many haemostatic regulatory molecules. The normal endothelium has anticoagulant and antithrombotic properties. Damage to the vessel causes adhesion of the platelets with the participation of the von Willebrand factor (vWF) secreted by the cells. ECs also produce tissue plasminogen activator (t-PA), its inhibitor, and an inhibitor of the tissue factor (TF) pathway. The endothelial surface is additionally the site of thrombin inactivation by antithrombin (AT) and conversion of prothrombotic thrombin by thrombomodulin into anticoagulant protein. In addition, thrombomodulin cooperates with protein C, and the thrombin-activated fibrinolysis inhibitor maintains the endothelial microenvironment in an anti-inflammatory and anticoagulant state [8,9]. Additionally, platelet activation performs a pivotal role in both infection-induced thrombosis and the immune response against pathogens.
Common symptoms of venous thrombosis include swelling, red and tense skin, sometimes a bluish tinge, and an increase in temperature in the extremity. Other symptoms are pain in the foot, calf, and lower knee, which disappears when the limb is lifted. However, one of the dangers of this serious disease is that in many cases it develops asymptomatically, and the patient is unaware of thrombosis. This is the case when the clot is attached to the wall of the vessel and does not completely close the vessel lumen, and some of the flowing blood is taken over by the surrounding smaller vessels. In addition, the blood clot may shrink, which results in an increased blood flow. In the best situation, the blood clot dissolves, and the vessel is restored. Unfortunately, endogenous protective and repair mechanisms are not always sufficient, and the formation of blood clots often leads to complications.
This review describes the causes of vein thrombosis and the basic mechanism of clot formation regarding the intrinsic and extrinsic pathways. Then, we discuss the influence of platelets, endothelial cells, and microorganisms on the development of thrombosis. In the next chapter, we show the role of hypoxic conditions in endothelial dysfunction (ED) by different factors including ROS. The paper reviews available drugs commonly used to fight venous thrombosis. We discuss various treatment strategies used in thrombosis including FX inhibitors, thrombin, antiplatelet therapy (GP IIb/IIIa, GP VI, GP Ib, and other inhibitors), and emerging drugs such as PAR-1 and PAR-4 antagonists and protein disulphide-isomerase (PDI) and phosphatidylinositol 3 kinase β (PI3Kβ) inhibitors. In addition, we describe other natural compounds that have been shown to potentially reduce thrombosis. We present natural anticoagulants from the plant world and traditionally used in Far Eastern medicine.
2. The Basic Mechanism of Blood Clotting
This section presents blood coagulation pathways and the participation of various coagulation factors in haemostasis. Discussing this topic is important as the rest of the work will present drug-inhibitors of pathways and coagulation factors in thrombosis. Blood clotting is a natural physiological process that prevents blood loss in the case of a damaged vessel. The blood clotting process is one of the body’s defences when the continuity of the vascular bed is interrupted. The basis of the blood coagulation process is the formation of a clot from fibrin with the participation of thrombin [10].
Thrombin, an important molecule in the clotting process, cleaves fibrinogen, transforming it into fibrin, which is the main component of the clot. In a further stage, fibrin is cross-linked with the active FXIII to a form resistant to the action of fibrin-degrading enzymes (Figure 1) [11].
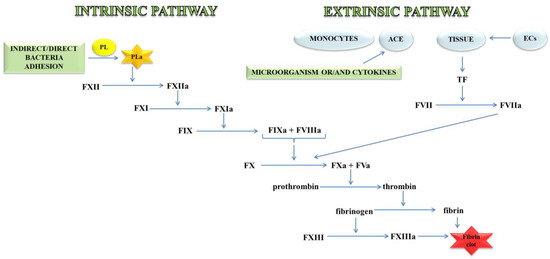
Figure 1.
A blood coagulation cascade including the internal and external paths. The former of these pathways is initiated by the contact of FXII with a negatively charged surface or indirectly through the involvement of polyP and blood platelets (PL). In contrast, the external pathway is initiated by FVII activation by tissue factor (TF) on the surface of monocytes, microparticles, activated endothelial cells (ECs), or cells in the damaged vessel wall, which exhibit TF. Both pathways lead to the activation of FX factor (prothrombinase), which converts inactive prothrombin into thrombin. The next stage is the transformation of fibrinogen to fibrin, which forms a fibrin clot; abbreviation: activated platelets (PL-a), angiotensin-converting enzyme (ACE).
The external mechanisms are associated with TF, initiated by tissues or monocytes. TF is an integral type I membrane protein (glycoprotein). A fragment of this glycoprotein is exposed on the membrane surface and binds with very high affinity to FVII. Complex TF-FVII is rapidly converted by limited proteolysis to the activated form of FVII [12]. The internal mechanism occurs due to the action of FXII with the surface having a negative charge. Both mechanisms lead to the activation of the enzymatic transformation cascade FX. The spatial network of a clot is stabilised by transglutaminase, FXIII. In addition, damage to the vessel leads to the discovery of the subendothelial layer, to which the platelets of blood immediately adhere that, as a result of activation, form a clot, inhibiting bleeding from a damaged arteriole, small vein or capillaries. The formation of the clot is caused by the presence of adhesive proteins that participate in the bonding of the tiles between them. The most important protein involved in this process is the platelet integrin αIIbβ3 also called the glycoprotein (GP) IIb/IIIa receptor. In the blood coagulation cascade, other factors such as FVII, FIX, FX, FXI, FXII and activated forms are also involved: FVa, FVIIa, FVIIIa, FIXa, Fxa, FXIa, FXIIa, FXIIIa, and PolyP [13]. Briefly, the process of blood clotting under conditions of haemostasis has been described, taking into consideration the internal and external pathways and coagulation factors.
3. Causes of Vein Thrombosis
Already more than 100 years ago, Rudolf Virchow stressed that the cause of thrombosis is the damage to the sinus, endothelial damage, coagulation disorders that lead to an increase in blood viscosity and slow its flow. Arterial thrombosis occurs under conditions of severe shear and is associated with platelets. In contrast, venous thrombosis occurs with low shear flow and mainly around the intact endothelial wall. Arterial thrombosis is associated with the formation of platelet-rich clots in the arteries as a result of atherosclerotic plaque rupture. The detachment of the thrombus can lead to a heart attack, stroke, transient ischaemic attack and limb and intestinal ischemia. On the other hand, thrombosis is related to the formation of clots in the veins of blood cells, low in platelets but rich in fibrin and plasma clotting factors. In thrombosis under hypoxic conditions, a thrombus forms, which causes increased blood clotting activity. Unlike arterial thrombosis, thrombosis causes ischemia at the site of origin. Thrombosis is promoted by the presence of cancer, especially lung and pancreatic cancers, which is associated with disturbances in homeostasis and the clotting process [14].
Blood flow in deep veins is very slow and, hence, the most common thrombosis occurs in these vessels, where the main mechanism forcing blood flow is the spasm of the shins. However, this mechanism stops working when the muscles do not shrink for a long time. Such a phenomenon occurs most often after surgery, in diseases leading to being bedridden, and in people with lower-limb paralysis. The frequency of complications is higher in patients undergoing an orthopaedic surgery than in others; this applies to, for example, a hip fracture. DVT in operated people is initiated by several of factors, such as the release of TF in the operative field, the release and absorption of thrombin from the surgical wound, and imbalance in the fibrinolytic system [15]. Besides, intravenous administration of general anaesthetics leads to a decrease in blood flow. In turn, medications from the curare or succinylcholine group that cause immediate muscle relaxation, reduce the effect of the muscle on the venous vessels and lead to a slowdown in blood flow to a minimum [16].
In most patients, sepsis also leads to the activation of the coagulation system and, consequently, thrombosis. Inflammation in the vein wall initiates the formation of a thrombus in a normal vessel. The first stage of clot formation is the activation of ECs, platelets, and leukocytes associated with the induction of inflammation and the release of microparticles that trigger the coagulation system by induction of the TF [17,18]. The thrombus is formed when platelets and leukocytes adhere to the vessel wall, and it is further promoted by inflammation and tissue factor as well as proinflammatory cytokines that activate coagulation. Leukocyte adhesion occurs when P-selectin glycoprotein ligand-1 (PSGL-1) present on the surface binds P-selectin expressed on activated endothelial cells. When P-selectin is also exposed to the surface of blood platelets, it binds to leukocytes leading to inflammation. Furthermore, exposure of P-selectin to the surface of platelets leads to rapid exposure of TF in the blood through monocytes contributing to intravascular coagulation [19].
Thrombosis is not always associated with the occurrence of complications. Occasionally, the thrombus is partially or completely dissolved in the vessel. Sometimes this process may last longer before it is completely dissolved, displaying practically no symptoms. During this period, part of the function related to the blood flow will be taken over by the smaller vessels. DVT has typical signs and symptoms, including pain, swelling, heat, redness or discoloration, and distention of surface veins. However, about half of people with this disease have no symptoms [20]. The presence of a clot can be diagnosed with an X-ray or less invasive methods such as conventional contrast venography, ultrasound, computed tomography (CT), or magnetic resonance imaging (MRI) scans [5].
Thrombosis risk factors are summarized and the mechanism of thrombus formation and the symptoms accompanying this process are described.
3.1. Hypoxia and Thrombus Formation
It is believed that hypoxia is the main cause of the development of mechanisms that lead to thrombosis. Hypoxia regulates endothelial pro- and anticoagulant properties. Its function is also to regulate the expression of P-selectin on the endothelium. This process leads to the recruitment of leukocytes or leukocyte microparticles containing TF, which can be an impulse to initiate a thrombotic response [21]. It has recently been shown that hypoxia of the whole body causes venous thrombosis [22,23]. It was reported that 73% of mice developed a thrombus after one-hour stenosis of the inferior vena cava (IVC) [22].
As shown in the mouse models, the size of the thrombus reaches a maximum after 1–2 days, followed by the process of its fragmentation, which depends on the composition, the participation of leukocytes, cytokines, metalloproteinases, and T lymphocytes in it. Clot formation may be related to flow stagnation. The result of stagnation is hypoxia, which develops in the pockets of the venous valves [24]. Such a situation may be caused by a drop in blood pressure and insufficient efficiency of the muscle pump of the calf. In about 50% of patients in whom venous thrombosis occurs, it is impossible to determine the underlying causes. Such cases of VTE are defined as spontaneous or idiopathic. The cause of stagnation in the flow may also be a sitting position during long-haul flights or mountain climbing. In addition, blood stasis is positively correlated with age [25]. Reduced blood flow leads to local hypoxia of the veins. It is known that low oxygen pressure may be harmful to tissues [26].
Hypoxia has been shown to initiate exocytosis and the release of vWF and P-selectin, supporting neutrophil binding [27]. Low oxygen pressure leads to the expression of intercellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule (VCAM-1), but also the expression of angiotensin-converting enzyme (ACE); metalloproteinases; and the activation of leukocytes, mast cells, and ECs in the venous wall, as well as the release of inflammatory mediators (Figure 2) [28]. The increase in the expression of adhesion molecules induced by proinflammatory cytokines such as tumour necrosis factor alfa (TNF-α), interleukin-1 (IL-1), and IL-6, interferon gamma (IFN-γ) leads to the proliferation of smooth muscle cells, which accumulate in the place of the damaged vessel causing an increase in the thickness of its wall. In this condition, recruit leukocytes and platelets lead to thrombosis. Moreover, Gupta et al. [23] showed on the model of venous thrombosis that hypoxia initiates the formation of thromboembolic systems. The same group of researchers reported that a marked, increased expression of inflammasome is a component of the innate immune system (NLRP3), caspase-1, and IL-1β in patients with clinically established venous thrombosis. It has been proved that the activation of this complex is an early response to low oxygen pressure and is regulated by hypoxia-inducible factor-1 alfa (HIF-1α). It was shown that activated macrophages lead to endothelial dysfunction (ED) in hypoxia. Under hypoxic conditions, macrophages release HIF-1α, HIF-2α, which leads to an increase in induced nitric oxide synthase (iNOS), endothelin converting enzyme (ECE-1), and cyclooxygenase 2 (COX-2) and a simultaneous decrease in prostacyclin synthase (SBSI) (Figure 2) [29].
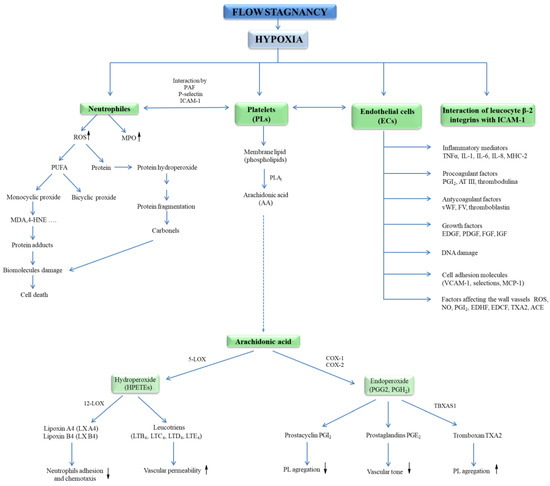
Figure 2.
Neutrophils, platelets, and endothelial cells interactions in hypoxia lead to blood cell response. Activation of neutrophils causes a respiratory burst and an increase in myeloperoxidase (MPO) activity. Release of ROS initiates the oxidation of proteins, lipids, and DNA. In contrast, activation of ECs leads to the secretion of procoagulant factors (PGI, AT III, thrombomodulin), growth factors (EDGF, FGF, IGF, PDGF), inflammatory mediators (IL-1, IL-6, IL-8, TNF-α, MHCII), factors regulating vascular wall tension (ROS, NO•, PGI2, EDHF, ACE, TXA2, EDCF) as well as release of anticoagulant agents (vWF, FV, thromboplastin), endothelial cell-adhesion molecules (VCAM-1, selectins, MCP-1). In turn, activation of blood platelets leads to the release of PGG2, PGH2, TXA2. Abbreviation: angiotensin-converting enzyme (ACE), antithrombin III – (AT III), deoxyribonucleic acid (DNA), endothelial cells (ECs), endothelium-derived contracting factor (EDCF), endothelium-derived growth factor (EDGF), endothelium-derived hyperpolarizing factor (EDHF), fibroblast growth factor (FGF), insulin-like growth factor (IGF), interleukin (IL), monocyte chemoattractant protein 1 (MCP-1), major histocompatibility complex II (MHCII), myeloperoxidase (MPO), nitric oxide (NO•), platelet-derived growth factor (PDGF), prostaglandin G2 (PGG2), prostaglandin H2 (PGH2), prostacycline (PGI), reactive oxygen species (ROS), tumour necrosis factor α (TNF-α), tromboxan A2 (TXA2), vascular cell adhesion molecule (VCAM), von Willebrand factor (vWF).
Additionally, hypoxia promotes inflammation. The accumulation of inflammatory cells in many organs, vascular leakage, an increase in the level of serum cytokines, and leukocyte activation have been demonstrated in mice exposed to low oxygen pressure [30,31,32]. The authors also provide evidence that HIF-1 may be involved in bacterial infections. It was shown that bacterial lipopolysaccharide (LPS) initiated the increase in HIF-1α transcription [33]. This problem is of particular importance because ischemia of organ transplants increases the risk of inflammation and failure of the transplanted organs or rejection of the transplant. Hypoxia leads to the migration of HIF-1α and the HIF-1β subunit into the cell nucleus where, after binding to the hormone response element (HRE), they induce transcription of many genes, including nuclear factor kappa B (NF-κB) and toll-like receptor (TLR), and deoxyribonucleic acid (DNA) damage. It was noticed that TLR4 contributes to the inflammation in the transplanted organ and its damage. Affecting the TLR4 signalling may be important in the prevention or treatment of acute renal transplant injury [34,35]. In turn, NF-κB and its family regulate inflammation and affect immune responses and control tissue homeostasis [36].
Hypoxia stimulates the production of ROS, which may initiate the activation of the ECs, leading to the expression of adhesion receptors and the recruitment and extravasation of leukocytes [34,37]. Moreover, the action of ROS leads to the formation of an immune complex, complement and neutrophil activation, and secondary production of oxygen free radicals and other reactive compounds. Activation of the complement system causes its deposition on the wall of the vessel, initiating the inflammation progression [38,39]. Hypoxia leads to oxidative stress in the cells, which is the cause of inhibition of the iron-sulphur (Rieske) protein in complex III of electron transport chain (ETC) by damaged cytochrome. Under this condition, ubiquinol is oxidised to ubisemiquinone, which enhances the oxidative stress [40]. In physiological conditions (normoxia), the cytochrome oxidase present in complex IV of ETC reduces oxygen to water but under hypoxia conditions, it produces nitrous oxide from nitrite [41].
Cytochrome c oxidase of the mitochondrial respiratory chain is one way of inactivating nitric oxide (NO•) in cells under hypoxic conditions [42]. Additionally, in hypoxia and inflammation, superoxide (O2•−) and hydrogen peroxide (H2O2) can be generated with xanthine oxidase [43]. In hypoxic conditions, NO• synthesis can be catalysed in the blood by haemoglobin (Hb), while in the tissues by xanthine oxidase, neuroglobin, myoglobin, and cytoglobin [44]. At low oxygen pressure, a strong peroxynitrite (ONOO−) oxidant can be produced instead of superoxide in the reaction of NO• with O2•− [40]. The influence of hypoxia on the release of various active substances leading to the formation of a vascular clot is shown.
3.2. Role of Patogens and Oxidtive Stress
ROS production is the response of the host’s innate immunity to microbial invasion. Oxygen free radicals are an effective weapon against pathogens that prevent tissue colonisation by microorganisms [45]. Generation of ROS can be stimulated by “danger signal molecules” such as extracellular adenosine triphosphate (eATP) released by cells infected or near death. Production of ROS in host cells via the eATP-P2X7 receptor has been shown to be an important modulator for controlling chronic infection and inflammation. This mechanism can explain the persistence of microbes through regulation of ROS signalling under physiological conditions [46].
Bacteria are involved in vascular thrombosis and their strategy is related to the release of various toxins, enzymes and other substances. For example, superantigens (type I toxins) produced by Staphylococcus aureus and Streptococcus pyogenes damage host cells. Haemolysins and phospholipases (type II toxins) destroy cell membranes. In contrast, type III toxins, also known as A/B toxins, interfere with host cell defence systems to allow dissemination in organs [47]. Streptococcal haemolysins, streptolysins, lead to complete lysis of red blood cells (RBCs). There are two streptolysins: streptolysin O (SLO) and streptolysin S (SLS). SLO exotoxin secreted by group A streptococci and Streptococcus dysgalactiae is sensitive to oxygen. Group A streptococci and Streptococcus dysgalactiae interact with membrane of red and white blood cells but also macrophages, and platelets. SLS, in turn, is also released by group A streptococci, but is resistant to oxygen. SLS also affects immune cells, including polymorphonuclear leukocytes and lymphocytes. In addition, SLS is thought to prevent the regeneration of the host’s immune system [48]. Shigatoxigenic Escherichia coli (STEC) produces Shiga toxin, but the pathogenic mechanism of this bacterium is also associated with the release of enterohaemolysin. This toxin causes cell lysis by a mechanism similar to that of a detergent action [49,50].
In turn, bacterium Staphylococcus aureus produces a cytolytic toxin that leads to the release of oxyhaemoglobin (oxyHb). The breakdown of oxyHb leads to the secretion of haem, which bacteria use as a source of iron for their growth. In the host organism both haemoglobin molecule and released haem exhibit bactericidal activity by catalysing the reaction with the production of ROS. Additionally, oxyhaemoglobin as a result of autoxidation turns into methaemoglobin (metHb) with superoxide generation. In turn, MetHb exhibits pseudoperoxidase activity capable of catalysing the production of O2•-, which is a precursor to other ROS [51,52,53].
On the other hand, release of ROS is bactericidal, but it is also associated with damage to the host’s tissues. However, the presence of glycoproteins such as haptoglobin (Hp), hemopexin (Hx) and apoplipoprotein A-I (apoAI) binding oxyHb inhibits oxidative stress [53]. Haemolysis, as a consequence of bacterial infection, releases oxyHb, which promotes proinflammatory and thrombotic reactions. The presence of free oxyHb in plasma leads to a disruption of homeostasis. In response to free oxyHb, TF, the initiator of blood coagulation by macrophages, is expressed. However, the binding of oxyHb to TF leads to the weakening of oxyHb’s pro-oxidative properties [54]. Bacteria release many toxins, which can lead to vein thrombosis.
In addition to bacteria, also viruses are involved in thrombosis. For example, the influenza A virus (IAV) that infects the respiratory tract of humans leads to uncontrolled thrombosis, disseminated intravascular coagulation, and formation of pulmonary microembolisms with consequent haemorrhage and pulmonary oedema [55]. However, over-activation of the clotting system can cause lung damages and cardiovascular disease. For example, IAV infections lead to an increased risk of deep vein thrombosis and pulmonary embolism, acute coronary syndromes, and acute myocardial infarction [56]. In turn, influenza A virus subtype H1N1 increases the expression of genes promoting haemostasis, increasing platelet aggregation and platelet signature genes related to acute myocardial infarction (AMI) [57]. Dramatically increased PE associated with abnormal coagulation profile has been observed in patients with COVID-19. These patients had a significantly higher inflammatory syndrome, a more distal thrombosis process involving the deep vein system of the shin, and a higher disease severity score on computed tomography SARSCoV-2. The authors hypothesized that COVID-19 patients may have local pulmonary thrombosis rather than an embolism [58].
4. Regulation of Cells Activation in Development of VTE
4.1. Platelets Activation
A blood platelet is sensitive to factors from the surrounding environment via a contact with receptors present on its surface and takes part in the interactions of the platelets between each other and with other cells. Activation of platelets by inflammatory signals or infection results in P-selectin exposure and expression of cluster of differentiation-154 (CD-154) and CD-40 and leads to interactions with blood cells such as neutrophils, eosinophils, T-cells, B-cells, and ECs. Interactions with ECs lead to the expression of E-selectin and release of ICAM-1 and VCAM-1 adhesion molecules and to leukocyte recruitment [59]. Moreover, activation of platelet leads to exposure of glycoprotein (GP) IIb/IIIa, which is a receptor for fibrinogen. In turn, the GP Ib-IX-V complex is involved in the binding of platelet to the damaged epithelium via vWF. In contrast, GPs from the protease-activated receptor (PAR) family participate in platelet stimulation by thrombin. Other receptors associated with platelet activation include the thromboxane A2 (TXA2) receptor and platelet activating factor (PAF) receptor and the platelet C-type lectin-like receptor (CLEC-2). In homeostasis, GP Ia/IIa and GP VI also have a crucial role, being involved in the platelet response to collagen [60].
Activated platelets (PLa) release a number of active substances from the α and β granules, which additionally intensify their activation. The platelets also liberate serotonin, which causes narrowing of the blood vessels in the wound [13]. In addition, stimulated platelets release other adhesive GPs from granules such as fibrinogen; thrombospondin; vWF; coagulation factors: FV, FXI, FXIII, protein S; mitogenic factors: platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), epidermal growth factor (EGF); angiogenic factors: vascular endothelial growth factor (VEGF), PF4 inhibitor; chemokines: chemokine CXC ligand (CXCL7), CXCL4, CHCL1, CXCL5, CCL5 (RANTES), CCL3 and cytokines: IL-1β, TGF-β, fibrinolysis inhibitors, plasminogen activator inhibitor-1 (PAI-I) [61].
Numerous experimental and clinical studies confirm the key role of ROS in the mechanism of platelet stimulation. There are many activation pathways, one of which is an increased isoprostane formation. The main source of superoxide and other ROS is NADPH oxidase 2 (NOX2) [62]. One source is the arachidonic acid (AA) metabolism and associated lipoxygenase (LOX) expression, the other is NADPH oxidase (NOX) [63]. ROS form within PLa and regulate their responses to collagen-mediated thrombus formation [64].
It was shown that a platelet can be strongly stimulated by advanced glycation end products (AGEs) that result from the non-enzymatic reaction of amino acids with sugars in pathological conditions such as diabetes and/or uraemia, but also during aging, which lead to expression of CD62 and CD63. In addition, AGEs induce the expression of blood TF. It was shown that high TF expression on monocytes and vascular endothelium regulates the inflammatory response. The adhesion of PLa to leukocytes has a key role in thrombus formation [65].
Additionally, inflammation initiated by pathogens also leads to the activation of blood platelets, if accompanied by damage to the endothelium, then fibrin builds up and a clot forms [66]. Bacteria can also release substances that activate platelets. One of them is gingipain, an enzyme released by Porphyromonas gingivalis, which activates the thrombin receptor on platelets [67]. In turn, STEC strains Escherichia coli releases and the Shiga toxin activates platelets through platelet glycosphingolipid [68].
In veins and larger arteries, the flow is fast enough that the blood behaves like a Newtonian fluid, and the shear stress generated is proportional to the shear rate [69]. In the formation of thrombosis, an important role is held by shear forces induced by blood flow, because they contribute to the excessive activation of platelets, their aggregation leading to the formation of a clot. Studies have shown that in clogged blood vessels, shear forces can be up to two orders of magnitude higher than in healthy vessels [70]. Additionally, shear stress leads to activation and alterations in vWF structure. High shear stress and associated platelet activation is responsible for venous and arterial thrombosis. At low shear stress, the main role is held by fibrinogen, while at high shear stress, the adhesion and aggregation of platelets is determined by vWF, but not fibrinogen [71]. Higher level of vWF is a risk factor in acute coronary syndromes, and patients with vWF deficiency show resistance to thrombosis [72,73]. In in vivo studies, a significant increase in vWF levels occurred below the site of coronary occlusion and endothelial damage, which led to increased platelet reactivity to high shear stress. In vitro tests have revealed markedly increased platelet activity by doubling the vWF concentration, which indicates a link between vWF concentration and platelet reactivity [74].
Platelet hyperactivity promotes the pathogenesis of cardiovascular disease and may be affected by elevated levels of circulating growth factors, which include insulin-like growth factor-1 (IGF-1). Although IGF-1 cannot initiate platelet activation, in combination with physiological stimuli, it can intensify this process through a phosphoinositide 3 dependent mechanism [75]. During sluggish blood flow, the levels of coagulation factors and thrombin are locally very high, which promotes platelet adhesion and aggregation and, consequently, leads to the formation of a blood clot [76,77].
Both infection and inflammation can directly or indirectly activate platelets through different receptors, triggering aggregation and thrombi formation within the vasculature. However, many reviews based on current literature summarize the impact of potential risk factors related to the involvement of platelets in the pathogenesis of VTE, as well as in predicting the risk of VTE events [78]. Several platelet protein polymorphisms have been found to be associated with VTE and are considered hereditary risk factors. These include polymorphisms of glycoproteins GPIa/IIa, GPIIb/IIIa and GPVI as well as the Janus kinase 2 (JAK-2) mutation. Additional risk factors may be expression markers such as tissue factor containing microparticles and P-selectin, which is a platelet activation marker, enhances microparticle production, tissue factor expression, and platelet aggregation. Additional factors of thrombosis and recurrent VTE in antiphospholipid syndrome (APS) are the increased mean platelets volume (MPV) and their increased number [79].
Various mechanisms of platelet activation, release of ROS by these cells as well as the involvement of platelets and vWF in clot formation have been described.
4.2. Endothelial Cells Activation
Endothelial cells have a key role in homeostasis by having a direct contact between the blood and the vessel wall. They are involved in various functions such as regulation of blood flow, vascular permeability, recruitment of inflammatory cells, thrombosis, and angiogenesis. These cells secrete molecules that act on smooth muscle cells associated with vascular tone regulation and leukocytes that regulate inflammation state. ECs release prostacycline (PGI) and NO•, both substances that inhibit platelet adhesion and vasodilators (Figure 3) [8,80].
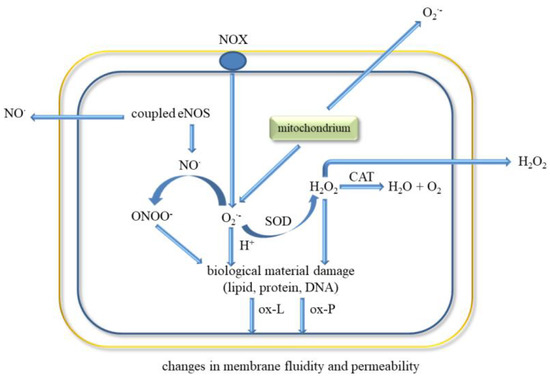
Figure 3.
ROS release and oxidative biological material damage in endothelial cell. Abbreviations: catalase (CAT), water (H2O), hydrogen peroxide (H2O2), endothelial nitric oxide synthase (eNOS), nitric oxide (NO•), NADPH oxidase (NOX), oxygen (O2), superoxide anion radical (O2•−), peroxynitrite (ONOO−), oxidised lipids (ox-L), oxidised proteins (ox-P), superoxide dismutase (SOD).
The classic factors leading to endothelium dysfunction include dyslipidaemia, smoking, hypertension diabetes mellitus, and aging. Other factors are infection inflammation, physical inactivity, high level of homocysteine, post prandial state, and obesity [80]. Excessive generation of ROS in the vascular wall is a common feature of many cardiovascular diseases (Figure 3). It was shown that EC-derived ROS have a key role in aortic wall susceptibility to dissection [81].
Endothelial dysfunction occurs in both varicose veins and DVT [82]. It has been shown that ECs can be damaged by the endotoxin part of outer membrane of Gram-negative bacteria in bacterial pathogenesis and endotoxic shock (Figure 4) [83]. Activation of ECs is associated with increased expression of cellular adhesion molecules VCAM-1 and ICAM-1, which causes ED and promotes monocyte adhesion [84]. This process can be triggered by ischemia, catecholamines, angiotensin II, cytokines (IL-1, IL-6, TNF-α, TGF-β), and endotoxins (Figure 4). The presence of adhesive molecules leads to increased interaction between inflammatory cells and the endothelium and their migration into the endothelial space [37,85,86].

Figure 4.
Factors leading to the development of endothelium dysfunction. These include ROS, proinflammatory cytokines (IL-1, IL-6, IF-8, TNF-α), TGF-β, Hb and oxyHb, Th-1 lymphocytes, sepsis, macrophages, and neutrophils.
Stimulation of ECs leads to the release of microparticles of endothelial cells (EMP). Their presence in plasma has been found in many immuno-inflammatory diseases such as atherosclerosis, sepsis, and others [87]. Other agents released by endothelium include PAF, vWF, thrombomodulin, TF, t-PA, ectonucleotidases, and type I PAF [8].
Inflammatory mediators promote the adherence of neutrophils to the endothelium and their activation. Activated neutrophils, being inflammatory cells, as a result of respiratory burst release large amounts of ROS such as superoxide, H2O2, hydroxyl radical (HO•), nitric oxide, and hypochlorous acid (HClO) (Figure 5). ROS produced during inflammation lead to oxidative stress but are also used to remove pathogens [88]. In addition, ED leads to increased production of O2•- and H2O2 catalysed by xanthine oxidase, which is accompanied by an increase in the synthesis of peroxynitrite, the factor responsible for damaging the biological material [43]. Activation of endothelial cells releases many active substances that may be involved in clot formation.
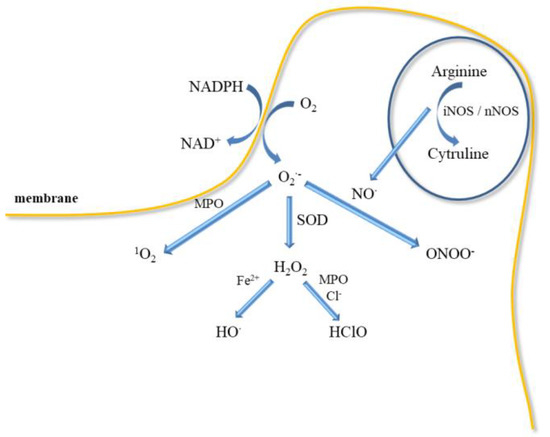
Figure 5.
ROS can destroy phagocytized pathogens. Respiratory burst of neutrophil, which leads to the activation of NADPH-oxidase producing O2•−, precursor of other ROS, such as, H2O2, which can oxidize chloride to HClO in the presence of MPO, HO• release in Fenton reaction.
5. ROS and Oxidative Stress
The release of lower ROS levels by host cells is associated with their physiological function in regulating processes such as haemostasis, apoptosis, immune response, and microbial colonisation [46]. ROS are produced of numerous enzymatic reactions in various cell compartments, including the cytoplasm, cell membrane, endoplasmic reticulum, mitochondria, and reactions peroxisome as intra- and intercellular signalling molecules. The precursor of ROS is the superoxide anion radical (O2•−), which is formed as a result of a one-electron reduction of molecular oxygen. Superoxide anion radical is formed by the enzymatic process, autooxidation reaction, and by nonenzymatic electron transfer reactions in which an electron is transferred to molecular oxygen [89]. Superoxide is mostly produced within the mitochondria in electron transport chain during electron leakage. There are 11 sites in the respiratory chain in mammalian mitochondria where superoxide and/or hydrogen peroxide is formed [90]. Other enzymes that can produce superoxide are lipoxygenase, cyclooxygenase [91]. Superoxide can exist in two forms such as O2•− or hydroperoxyl radical (HO2•) at low pH [92]. Its reactivity with the biomolecules is low while the reactivity of hydroperoxyl radical is much higher. Hydroperoxyl radical can penetrate the phospholipid bilayer easier than the charged form (O2•−). Polymorphonuclear leukocytes (PMNs), monocytes, and macrophages perform an important role in the innate immune response to pathogens. They can produce and release ROS in significant amounts [93]. One of the main sources of peroxide in phagocytic cells is membrane NAD(P)H oxidase [94]. Spontaneous or enzymatic dismutation of superoxide leads to hydrogen peroxide. Hydrogen peroxide can diffuse through the mitochondrial membranes into the cytoplasm [95]. The superoxide produced during phagocytosis is transformed into other ROS, which are directed to the absorbed material in the phagolysosome. Depending on the way used to activate the NAD(P)H oxidase, the production of superoxide can also target the extracellular environment [96]. ROS perform a physiological role as signalling molecules; however, their increased production and/or failure of antioxidative systems may lead to oxidative stress, which is associated with damage/modification of life-important molecules such as DNA, proteins, and lipids. It has recently been shown that certain molecules produced during the blood clotting process can stimulate PMNs to produce ROS. Released ROS by PMNs can damage the endothelium and affect the coagulation process [97].
The release of high ROS levels due to acute infection or inflammation may lead to oxidative damage to the host’s biological material. ROS have a crucial role in vascular function. Redox balance is important in cell signalling and haemostasis. However, perturbation of redox state leads to ROS interaction with important vital macromolecules such as proteins, enzymes, nucleic acids that can lead to vascular pathology [98,99]. ROS are associated with the pathogenesis of many diseases, including cardiovascular diseases such as hypertension, ischemic heart disease, ischemia-reperfusion injury, and other vascular diseases including thrombosis. ROS may also lead to other pathological conditions, such as pulmonary fibrosis and vascular retinopathy [37,100].
In pathological conditions, there is a continuous recruitment of inflammatory cells that release ROS [101]. This process is observed, for example, in atherosclerosis and a long-term venous disease. Excessive ROS formation and/or a decrease of antioxidant potential leads to changes in the vessel wall [102]. Eukaryotic cells have numerous organelles in which ROS can be produced. The largest amounts of ROS are released in the ETC in the mitochondria [103].
In the vascular system, ROS are generated mainly by NOX but also uncoupled NOS, xanthine oxidase, COX, and myeloperoxidase (MPO). NOX occurs in the isoforms NOX1, NOX2, NOX4, and NOX5. It was shown that the expression of isoforms 1, 2, 4 is the cause of cardiovascular diseases [100,104,105]. NOX4 participates in the production of oxygen active compounds through internal mitochondrial channels, including ATP-dependent potassium channels and mitochondrial permeability transition pore (mPTP) through which ROS can flow from the mitochondria to the cytosol [106,107]. NOX4 induced by TGF-β lead to ROS production, inhibition of mitogen-activated protein kinases (MAPK), MKP-1, and activation of RhoA [108]. NOX4 expression leads to DNA damage by H2O2, which was also shown diffused through nuclei membrane [109]. NOX can be enhanced by exogenous factors such as ultraviolet (UV) radiation, smoking, high calorie diets, and diabetes [100]. All these enzymes are complexes present in the membrane and are expressed in vascular tissue only to produce ROS. For example, NOX2 and NOX4 produce ROS in cardiomyocytes and fibroblasts, while NOX1, NOX4, and NOX5 do so in vascular smooth muscle cells (VSMCs) [108]. Other sources of ROS include peroxisomes, endoplasmic reticulum, and lysosomes, where they are produced by NOX, xanthine oxidases, cytochrome P450 (CYP), etc. [110].
However, large amounts of ROS are released by neutrophils during respiratory burst in response to infection because they have a crucial role in host defence and inflammation (Figure 5). Neutrophils participate in the immune responses associated with bacterial invasion, and they also produce nitric oxide, which is synthesised by neutrophilic inducible nitric oxide synthase. In addition to various physiological functions, their participation is associated with pathology [111]. Neutrophils may participate in numerous pathologies such as glomerulonephritis, diabetes, stroke, septic shock, whooping cough, sepsis, encephalitis, and ulcerative colitis. NO• has been shown to mediate neutrophil extracellular traps (NETs) formation at the site of inflammation/infection by increasing ROS production. Activated neutrophils can form NETs, which include proteins, histones, chromatin, and DNA [90]. In turn, NETs increase coagulation by stimulating platelet aggregation, erythrocyte mobilization, and fibrin accumulation as well as endothelial activation and damage [112,113]. NO• is also a mediator of neutrophil response in a neurodegenerative disease (Parkinson’s disease, schizophrenia, depression) and cardiovascular diseases, thrombosis, and hypertension [111,114]. Higher iNOS activity in neutrophils was observed in patients with congestive heart failure that positively correlated with elevated levels of IL-6 and noradrenaline in circulation [115].
The mechanism of bacteria removal by ROS is associated with an attack on membranes, but also with their penetration into the interior of bacteria, where they can damage nucleic acids, proteins, and enzymes [116]. However, NOX function is crucial for the formation of NETs [117]. NETs formation is also induced by proteases, which can be activated by ROS. Neutrophils can generate more ROS by responding to AGEs through the NOX complex [118,119]. Additionally, superoxide may be also released in mitochondria by NAD(P)H-oxidase, cytochrome c peroxidase or xanthine oxidase (Figure 5). Myeloperoxidase located in neutrophil granules can, with the participation of H2O2, oxidise chlorides to HClO, which is a strong oxidising agent and bacteria killing agent. MPO can also directly convert superoxide to singlet oxygen (1O2). Singlet oxygen can also be produced in a reaction of hypochlorite with hydrogen peroxide. However, the released H2O2 can be reduced in the presence of iron II to the HO• (Fenton reaction). In addition, both ROS stimulate the production of proinflammatory cytokines as TNF-α and macrophage inflammatory protein 2 (MIP-2) [116,117,120].
Another important source of ROS in platelets are NOX occurring in various cell types, including ECs and other vascular cells. NOX expression can be triggered by hypoxia and endogenous factors, such as pro-inflammatory molecules, growth factors, PDGF, and hormones: angiotensin II and insulin [121,122]. ROS are also generated in the cells of VSMCs. This mechanism involves extracellular cyclophilin A (iCypA), which increases ROS production by angiotensin II [123].
Oxidative stress initiates the NF-κB transcription factor, which can cause the expression of AGEs and TNF-α [124]. Additionally, AGEs may promote oxidative stress which leads to the formation of oxidative protein products [125]. TNF-α is a pro-inflammatory cytokine that is released by active monocytes and macrophages and is associated with the inflammatory process in the host’s response to infection [126].
Proper functioning of the endothelium is also associated with the release of NO• by endothelial nitric oxide synthase (eNOS) in which calcium ions participate [124]. Nitric oxide characterised by a low reactivity, free of electric charge, easily penetrates through the membranes of ECs and other cells. However, it can react with oxygen, yielding a strong oxidant and a nitrating agent, nitric dioxide [127]. In turn, the reaction of NO• and the O2•− leads to formation of ONOO-, which is characterised by similar properties as nitrogen dioxide, both inflammatory agents [128,129]. The constant rate of reaction of NO• with O2•− is about three times higher than dismutation of superoxide by superoxide dismutase (SOD) [130,131,132]. This supports the thesis that higher levels of superoxide and nitric oxide are harmful and in pathology lead to a damage to biological material [133]. In turn, oxyHb released in plasma can react with NO• and produce ONOO− and metHb [134,135]. It was shown that treatment of RBCs with nitric oxide led to metHb formation and oxidative damage of lipids and proteins in these cells [136]. It was reported that the lipid peroxidation product, 4-hydroxy-2-nonenal (HNE), causes changes in cryptic TF on the surface of blood macrophages and monocytic cell line, THP-1, converting it to TF procoagulant [137].
ROS affecting the processes of coagulation, fibrinolysis, proteolysis and complement, as well as the activation of platelets, endothelial cells, erythrocytes, neutrophils, monocytes may lead to the formation of a venous thrombus [138]. Furthermore, ROS can initiate coagulation inducing increased expression of tissue factor (TF) in endothelial cells, monocytes, and vascular smooth muscle cells, but NOX enzymes are necessary for ROS production [139,140,141]. It has recently been shown that certain molecules produced during the blood clotting process can stimulate PMNs to produce ROS. Released ROS by PMNs can damage the endothelium and affect the coagulation process [97].
This section describes the various sources of ROS and their effect on cell activation and thrombus formation as well as on damage to biological material.
6. Advances in Treatment of Thromboembolism
Conservative therapy is based on the administration of anticoagulants that reduce the risk of a pulmonary embolism. Anticoagulants have become the basis of DVT therapy and are used to prevent PE progression and recurrence of thrombosis. It was reported than the 30-day mortality risk for VTE patients was 3% for DVT not treated with anticoagulants. However, mortality risk increases to 31% in patients who have developed PE. In addition, anticoagulants inhibit the growth of existing thrombi and prevent the emergence of new ones. These medicines include low molecular weight heparin (LMWH), acenocoumarol, aspirin, and other antiplatelet inhibitors [142]. In addition, natural phlebotropic drugs such as rutin, aescin, pycnogenol, Ruscus or Centella extracts, Ginkgo biloba, and others strengthen and protect the walls of venous vessels [143].
The therapy uses oral anticoagulants for 3–6 months. Discontinuation of treatment in patients after the first episode of idiopathic VTE causes recurrent thrombosis with a frequency of approximately 5–10% in the first 2 years and in the following 1–3%. On the other hand, the long-term use of anticoagulants is associated with haemorrhagic complications. It is very important to clarify in which patients there is an increased risk of VTE recurrence. It is connected with the decision to extend the therapy and the isolation of the group of patients with low risk of VTE relapse, in whom anticoagulant therapy can be safely discontinued [144].
The variety of anticoagulants is associated with their use for the treatment and prevention different disease such as atrial fibrillation, deep vein thrombosis, pulmonary embolism, Ischemic stroke, myocardial infarction (heart attack), hip or knee replacement surgery, and others. Some drugs do not dissolve thrombi but prevent the growth of those already formed and the formation of new ones. In contrast, another group of drugs called thrombolytics (“clot busters”) can dissolve a clot during a period of a few days.
6.1. Endovascular Treatment of DVT
Various methods are used to treat intravascular venous thrombosis, such as catheter-directed thrombolysis (CDT), catheter-directed pharmacomechanical thrombolysis (PCDT), percutaneous aspiration thrombectomy (PAT), vena cava filter protection, venous balloon dilatation, and venous stent implantation [145]. In addition, an intravascular PAT complementary technique is used, such as introducing a balloon to break up the thrombus, stent implantation and placement of a venous filter [145,146].
6.2. Anticoagulants
Drugs used to treat or prevent thrombosis can be divided into three groups: (1) anticoagulants that prevent the coagulation system and interfere with further plaque expansion, (2) antiplatelet agents that reduce platelet aggregation and inhibit blood clot formation, (3) fibrinolytic drugs that directly dissolve the resulting clot [147].
Under conditions of haemostasis, a balance is maintained through the interaction between platelets and vascular endothelium, which is also controlled by the cascade of roots and fibrinolysis. Continued cascade is associated with the transformation of prothrombin into thrombin, followed by the conversion of fibrinogen to fibrin, which forms a vascular clot with blood platelets (Figure 1). Additionally, thrombin activates other coagulation cascade factors like FV and FVIII, activates and releases FVIII from binding to vWF [148]. The standard pharmacological method of treating thrombosis is the use of anticoagulants, which reduce the formation of clots and prevent the further development of a thrombotic disease [149].
6.2.1. Inhibitors of FX
In thromboembolic syndromes, coagulation FXa has a key role in the blood coagulation cascade. Factor Xa participates in both the external and internal coagulation pathways and is important in conversion of prothrombin to thrombin. The advantage of FXa inhibitors is the predictable anticoagulant effect and no need for routine monitoring. The introduction of selective inhibitors for FXa may reduce thrombin production causing an anticoagulant effect leading to smaller disturbances of primary haemostasis [150,151].
A number of FX inhibitory drugs have been synthesised that have different effects. Their variety allows choosing the drug with the highest effectiveness and the smallest side effect depending on the type of disease and the patient’s condition [152]. Direct FXa inhibitors (Figure 6) may include: apixaban (1), betrixaban (2), darexaban (3), edoxaban (4), nokxaban (5), rivaroxaban (6) (BAY59-7939), and indirect FXa inhibitors: fondaparinux (7) and idraparinux (8) (antithrombin-binding pentasaccharide), dabigatran and LMWH. These drugs are used to reduce the risk of stroke and embolism in people with nonvalvular atrial fibrillation. They are also used to prevent deep vein thrombosis (DVT), which can lead to a pulmonary embolism (PE) in patients after a knee or hip surgery, as well as to treat DVT and PE, and to reduce the risk of both diseases coming back. Apixaban is metabolised by CYP mainly by CYP3A4, rivaroxaban by enzymes CYPA415, CYP2J2, in turn, nokxaban (GCC-4401C) by CYP2D6 and CYP2A4 [153,154,155].
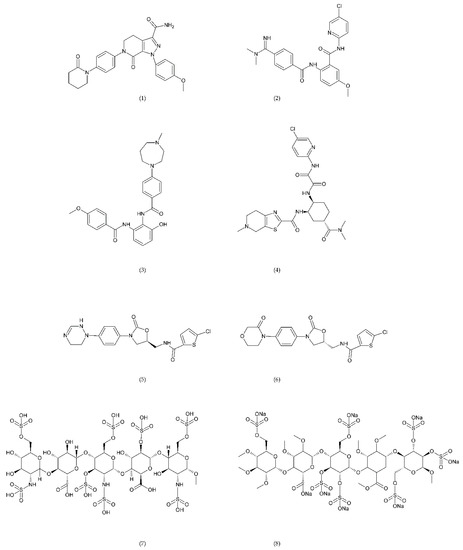
Figure 6.
Direct factor Xa inhibitors: apixaban (1), betrixaban (2), darexaban (3), edoxaban (4), nokxaban (5), rivaroxaban (6), fondaparinux (7), and idraparinux (8).
Apixaban and other anticoagulants such as dabigatran, edoxaban, and rivaroxaban are effective, similar to warfarin in preventing haemorrhagic stroke in people with atrial fibrillation, and carry a lower risk of intracranial bleeding [156]. Apixaban is an anticoagulant medicine used to treat and prevent thrombosis, and it is also used to prevent stroke in patients with non-valvular atrial fibrillation. Apixaban is a drug that is more effective than aspirin or warfarin in reducing the risk of stroke and bleeding, reducing the mortality rate in this patient group. Apixaban, but also dabigatran or rivaroxaban, may be an effective alternative to warfarin in the prevention of stroke and thromboembolism, especially in women with paroxysmal or persistent atrial fibrillation [157]. However, patients should not have prosthetic heart valves, haemodynamic valvular disease, severe kidney failure, and advanced liver disease (impaired baseline clotting function). Factors such as individual risk of stroke and bleeding should be taken into consideration when choosing the appropriate anticoagulant, along with comorbidities, availability of agents that reverse the anticoagulant effect in the event of bleeding complications, international normalized ratio (INR) control possibility—a way of standardising prothrombin time (PT) measurement across labs [158]. Apixaban is also used in prevention of postoperative DVT in patients after total hip or knee arthroplasty [159].
Betrixaban, like apixaban, is used in the prevention of embolism following knee surgery and in the prevention of stroke following non-valvular atrial fibrillation and extended prophylaxis in high-VTE-risk [160].
Darexaban is an anticoagulant drug that is used in venous thromboembolism, after a major orthopaedic surgery, stroke in patients with atrial fibrillation, and in ischemic incidents in acute coronary syndrome [161,162]. Darexaban and its glucuronide or maleate specifically and competitively inhibit FXa. By inhibiting prothrombin at the sites where a blood clot (thrombus) forms, it leads to a reduction in the formation of blood clots in a dose-dependent manner [161]. Increasing the risk of myocardial infarction, unstable angina, venous thrombosis, and ischemic stroke [163].
Dabigatran (Dabigatran etexilate), an oral anticoagulant, is used in Europe and Canada for the prevention of stroke and secondary VTE following atrial fibrillation and joint replacement surgery [164].
Edoxaban is a medicine used to treat deep vein thrombosis and pulmonary embolism and to prevent the disease from coming back. Edoxaban is also used to prevent blood clots in patients with non-valvular atrial fibrillation who have at least one risk factor such as a history of stroke, high blood pressure, diabetes, heart failure, and age [165].
Rivaroxaban is a highly selective drug with a rapid onset of action that inhibits FXa by disrupting the internal and external pathways of the blood clotting cascade. In addition, it inhibits the formation of thrombin and the development of blood clots. This drug is used to prevent stroke and blood clots in patients with nonvalvular atrial fibrillation [156]. It is used together with aspirin to reduce the risk of heart attack and stroke in patients with coronary artery disease. This drug is also used to prevent VTE in some hospitalised patients who are at risk of blood clots due to reduced mobility and who do not have a high risk of bleeding. Rivaroxaban is associated with a lower frequency of severe and fatal bleeding than warfarin. However, rivaroxaban shows a higher rate of gastrointestinal bleeding [166].
Nokxaban is a new drug used to prevent VTE in patients who have undergone a hip or knee replacement surgery. An additional application of Nokxaban is the treatment of acute coronary syndromes for stroke prevention in patients with atrial fibrillation [167].
The risk of VTE with many anticoagulants has increased from low to high in the following order: rivaroxaban, apixaban, edoxaban, enoxaparin, darexaban, and betrixaban. In turn, the classification of clinically significant bleeding from low to high for the same drugs was as follows: betrixaban, enoxaparin, darexaban, edoxaban, apixaban, and rivaroxaban [168].
Another anticoagulant is c2 (NAPc2), an 85 amino acid peptide isolated from the nematode Ancylostoma caninum. NAPc2 binds to FX and FXa at a non-catalytic site and inhibits FVIIa. It has a half-life of almost 50 h, so it can be administered once every two days [169].
6.2.2. Thrombin Inhibitors
Thrombin inhibitors are another group of drugs that are involved in thrombus formation. Thrombin activates FV, FVIII, and FXI, which participate in the release of more thrombin and activate FXIII, a protein involved in fibrin cross-linking and clot stabilisation. Thrombin also participates in the transformation of soluble fibrinogen into insoluble fibrin. Thrombin is a strong platelet agonist, and activated platelets allow the formation of clotting factor complexes that increase thrombin production by more than 1000-fold [170].
Inhibitors that inhibit free thrombin are commonly used anticoagulants, heparins such as unfractionated heparin (UFH) and LMWH (9). Molar mass of natural heparin chains ranges from 5000–40,000. In contrast, LMWH obtained in the 1980s contains chains with an average molecular weight of approx. 8000. UFH, or briefly heparin (10), is a naturally occurring glycosaminoglycan that binds to AT III, changes its conformation, and leads to its activation.
The activated form of AT III causes strong inactivation of thrombin FIIa and FXa and to a lesser extent of IXa, XIa, and XIIa, preventing the formation of clots. In turn, LMWHs also show affinity for AT III and have a greater effect on FXa. Ultimately, both heparins inhibit thrombin activation. Heparin not only blocks the formation of fibrin but also inhibits the thrombin-induced activation of FV and FVIII [154,155].
Direct thrombin inhibitors can be divided into bivalent, monovalent, and allosteric depending on their interaction with the thrombin molecule. Bivalent inhibitors such as bivalirudin (11) and hirudin (12) form a bond with the active centre and the external site of thrombin molecule (Figure 7). In contrast, monovalent inhibitors dabigatran (13), argatroban (14), and ximelagatran (15) bind only to the active site (Figure 8). In turn, allosteric inhibitors represent a third group that has been developed recently.
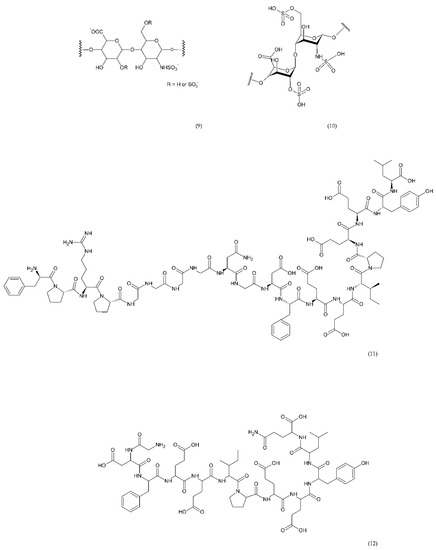
Figure 7.
Direct bivalent thrombin inhibitors: low molecular weight heparin (LMWH) monomer structure (9), heparin (10), bivalirudin (11), and hirudin (12).
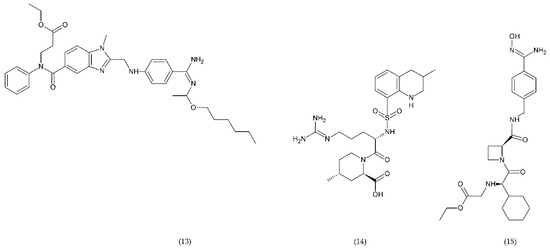
Figure 8.
Direct monovalent thrombin inhibitors: dabigatran (13), argatroban (14), ximelagatran (15).
Hirudin is a natural protein isolated from leech’s saliva which has a significant anticoagulant effect and has been used in many countries as a natural anticoagulant [171,172]. The use of hirudin in combination with LMWH for perioperative treatment of senile intertrochanteric fractures for the prevention of DVT was more effective than LMWH-calcium alone. In addition, the drug combination did not affect the platelet count during treatment [173]. Direct thrombin inhibitors like bivalirudin are used instead of unfractionated heparin in in the treatment of thromboembolic events [174]. Bivalirudin is a synthetic congener of the naturally occurring drug hirudin. Bivalirudin is a dual inhibitor that inhibits collagen-dependent thrombin and platelet aggregation [175]. However, it has also been shown that bivalirudin blocks the endothelial bioavailability of NO• and leads to vascular immobilization of MPO, a mediator of vascular function.
Dabigatran inhibits prothrombin but also inhibits the conversion of fibrinogen to fibrin. Table 1 presents the drugs used as anticoagulants in the treatment of thrombosis disease, the doses used and the mechanism of action. It has been shown that long-term use of Ximelagatran led to idiosyncratic liver toxicity and death, and it was withdrawn from the market [164].

Table 1.
Effect of drug anticoagulants for atherothrombotic diseases.
6.2.3. Other Compounds
Another drug commonly used to treat blood clots such as deep vein thrombosis and a pulmonary embolism is warfarin (16) (Figure 9), a coumarin derivative, which is an antagonist of vitamin K. Warfarin is an inhibitor of the vitamin K epoxide reductase complex (VKORC1), which performs a key role in activating vitamin K in the body. Warfarin lowers vitamin K reserves, leading to a reduction in the synthesis of the active clotting factors such as II, VII, IX, and X. Warfarin is used in medicine as an oral anticoagulant preventing stroke in people who have atrial fibrillation. It is also used in valvular heart disease or artificial heart valves and to prevent stroke in people who have atrial fibrillation, valvular heart disease or artificial heart valves. Warfarin is used for long-term anticoagulation or after a thrombotic event. It is also used to prevent thrombosis in high-risk patients, as well as in postoperative conditions. Warfarin combined with other anticoagulants is an effective medicine for the prevention of DVT in patients who have undergone DVT. It is also an effective drug in the prevention of DVT in cancer patients, especially those treated with chemotherapy [176].
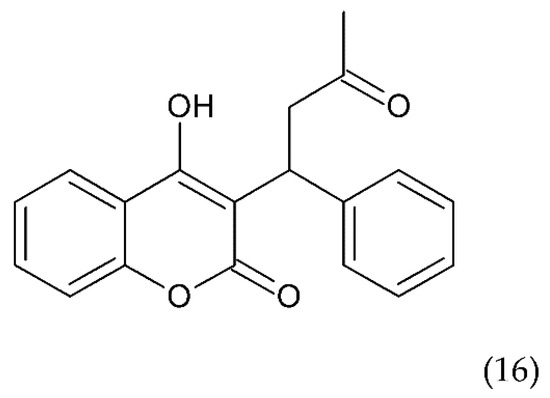
Figure 9.
Chemical structure of warfarin (16).
Recently, a fully human antibody MAA868 was prepared. It binds to the catalytic domain of both forms of the enzyme FXI (zymogen) and the FXIa with high binding affinity. The safe and well-tolerated effects of single subcutaneous doses of MAA868 have been demonstrated in healthy subjects. MAA868 resulted in dose-dependent and time-dependent sustained prolongation of partial thromboplastin time after activation and FXI suppression to 4 weeks or more. Clinical trials have shown the possibility of a potential subcutaneous anticoagulant therapy once a month [177].
6.3. Antiplatelet Therapy
In thrombosis, pathological activation of platelets occurs, which in turn leads to uncontrolled clot growth, embolism, or blockage of the blood vessel. The corollary of this is organ ischemia. In the treatment of thrombosis, antiplatelet drugs (Table 2) are used, which include: COX-1 inhibitors, adenosine diphosphate (ADP) P2Y12 receptor antagonists, GP IIb/IIIa inhibitors, GP VI, GP IB/IX/V, prostaglandin E (PGE) synthase inhibitors, serotonin receptor 2A (5HT2A), PAR, and TXA2 inhibitors [191,192].
Table 2.
Antiplatelets drugs in the treatment of atherothrombosis, the doses used, and the receptor inhibitors/antagonists.
The most commonly used antiplatelet agents (Figure 10) include acetylsalicylic acid (ASA) and clopidogrel as well as dipyridamole and cilostazol as phosphodiesterase inhibitors [3,193,194]. Dipyridamole (27), antithrombotic drug, enhances the potential of endogenous anticoagulants and the patency of small vessels and capillaries, thereby preventing further tissue damage. In addition, it has a protective effect against oxidative stress [195]. Dipyridamole inhibits the release of proinflammatory cytokines and monocyte chemoattractant protein (MCP-1), matrix metalloproteinase (MMP-9), and leads to a decrease in high-sensitivity C-reactive protein (hsCRP) in patients [196].
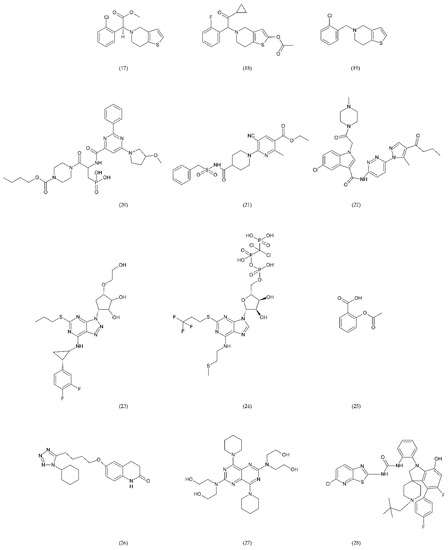
Figure 10.
Antiplatelet drugs: clopidogrel (17), prasugrel (18), ticlopidine (19) as well as novel compounds ACT246475 (selatogrel) (20), AZD1283 (21), and SAR216471 (22) are active antiplatelet drugs irreversible, competitive, thienopyridine P2Y12 receptor antagonists, while ticagrelor (23) and cangrelor (24) are reversible P2Y12 receptor antagonists. ASA (25) inactivates cyclooxygenase (COX) by irreversible acetylation in the metabolism of arachidonic acid (AA). Cilostazol (26) inhibits the platelet aggregation induced by collagen, ADP, epinephrine, and AA. Dipyridamole (27) the potential of endogenous anticoagulants and the patency of small vessels and capillaries. BMS-884775 (28) and P2Y1 antagonist demonstrated similar anticoagulant properties such as prasugrel.
Cilostazol (26) belongs to phosphodiesterase type 3 (PDE3) inhibitors, and its use causes an increase in cyclic adenosine monophosphate (cAMP). In turn, the increase in cAMP leads to an increase in protein kinase A (PKA), which directly affects the inhibition of platelet aggregation. In addition, PKA inhibits the activation of the myosin light chain kinase, which has an important role in the contraction of smooth muscle cells, which ultimately leads to vasodilatation [197]. It was shown that cilostazol was more effective in long-term use than aspirin and clopidogrel alone in patients with a previous ischemic stroke and had a significantly lower risk of bleeding. Cilostazol can be used in patients without heart failure to treat intermittent claudication [198].
ASA (25) inhibits platelet aggregation by inactivating COX through irreversible acetylation, which is involved in the metabolism of AA. Additionally, ASA inhibits the synthesis of MCP-1, inhibits the increased expression of matrix metalloproteinase-9, and attenuates the nuclear translocation of NF-κB [199].
Clopidogrel (17), prasugrel (18), and ticlopidine (19) are active antiplatelet drugs, irreversible, competitive, thienopyridine P2Y12 receptor antagonists. In contrast, ticagrelor (23), cangrelor (24), and ACT246475 (selatogrel) belong to the competitive reversible P2Y12 receptor blockade group. The P2Y12 receptor is G-protein coupled that binds ADP. ADP receptor binding results in inhibition of adenyl cyclase and cAMP secretion which leads to activation of the intracellular signal and alteration of glycoprotein conformation IIb/IIIa, enhancing fibrinogen affinity [200,201].
In addition to the P2Y12 inhibitors that initiate ADP-induced platelet aggregation in the study phase, there are other very potent inhibitors such as ACT246475 (20), AZD1283 (21), and SAR216471 (22) [202,203]. For example, ACT246475 undergoing phase II testing was characterised by less bleeding, higher selectivity, and ticagrelor-like anticoagulant activity. One of the methods used in anticoagulation therapy is blocking the P2Y12 receptor. In turn, P2Y1 antagonists show similar anticoagulant efficacy as P2Y12 inhibitors. BMS-884775 (28) is an antiplatelet drug, reversible and potent receptor antagonist of human P2Y1 receptors, it has good anticoagulant efficacy with less bleeding compared to prasugrel (P2Y12 antagonist) [202,204,205]. SAR216471 is a potent highly selective and reversible inhibitor, which has higher therapeutic index than clopidogrel, prasugrel, and ticagrelor [206]. AZD1283 inhibitor of P2Y12 has high potential almost 2-fold higher than ticagrelor and 4-fold higher than clopidogrel [206,207].
Glycoprotein IIb/IIIa has an important role in the adhesion and aggregation of platelets and occurs quite abundantly on the surface of platelets being fibrinogen binding site. Eptifibatide (Figure 11) is a cyclic natural heptapeptide that inhibits platelet aggregation found in the venom of the rattlesnake, Sistrurus miliarus barbouri. Eptifibatide is characterised by its antithrombotic activity, which selectively and reversibly binds to the platelet GP IIb/IIIa receptors by blocking the binding of fibrinogen, vWF and other adhesive molecules [208].
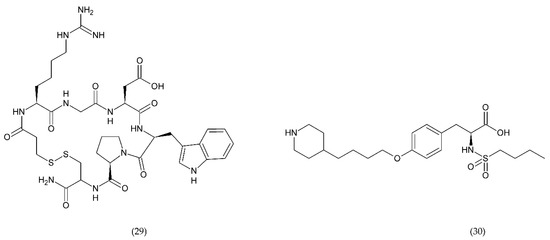
Figure 11.
Antiplatelet drugs which selectively and reversibly bind to the platelet IIb/IIIa receptor glycoprotein: eptifibatide (29) and tirofiban (30).
Tirofiban (Figure 11) belongs to L-tyrosine derivatives in which the hydroxyl group in the ring is involved in the binding of piperidine residue, and in the amino group of the amino acid there is a butylsulfonyl residue. Tirofiban is an antiplatelet drug that inhibits aggregation of platelets and belongs to the reversible antagonists of GP IIb/IIIa receptors. The drug blocks the binding of fibrinogen molecules to GP IIb/IIIa receptors [192,208].
6.4. Emerging Drugs
6.4.1. RUC-1, RUC-2, and RUC-4
In turn, the new drugs under study are RUC-1 (31), RUC-2 (32), and RUC-4 (33) (Figure 12), which are inhibitors of fibrinogen binding, specific for the α(IIb)β subunit of the GP IIb/IIIa receptor. These drugs, interacting with the receptor, prevent fibrinogen from binding. RUC-1 is an α(IIb)β inhibitor that binds selectively to the α(IIb) subunit. In contrast, RUC-2, which is a derivative of RUC-1, has approximately ~ 100-fold higher affinity for binding to the same subunit α(IIb) as RUC-1 [220]. RUC-2 additionally forms a bond with the β residue of metal ion-dependent adhesion site (MIDAS) of glutamic acid 220. In turn, RUC-4 showed approximately 20% greater inhibition of ADP-induced platelet aggregation than RUC-2. The reaction of RUC-4 with the receptor does not depend on whether it is in an active state or not, which is why it affects the entire platelet population. Both RUC-2 and RUC-4 have a unique mechanism of action consisting in blocking the receptor in its inactive conformation. In general, other drugs can lead to a conformational change and can contribute to thrombocytopenia in a small percentage of treated patients who have developed antibodies to the altered conformation [221,222].
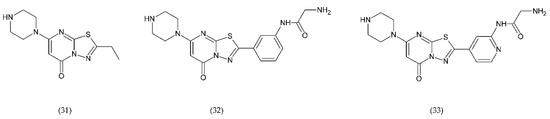
Figure 12.
Chemical structures of RUC-1 (31), RUC-2 (32), RUC-4 (33).
6.4.2. PAR-1 and PAR-4 Antagonist
Protease-activated receptors (PAR) have a special role in the development of chronic inflammatory diseases. The PAR family is conjugated to the G protein through the G domain. Activated receptors are involved in the transmission of signals by the recruitment of G proteins [223]. Thrombin, which is one of the key mediators of blood coagulation, activates a specific G-protein coupled receptor, called PAR-1. Vorapaxar (34) (himbacine analogue from Galbulimima baccata) is a drug that inhibits thrombin-mediated platelet activation (Figure 13). Its mechanism of action is related to the inhibition of protease-activated receptor-1. It is used in patients at high risk of ischemia, with an ischemic heart disease, and with a history of myocardial infarction. Moreover, it is characterized by a low risk of bleeding [224].
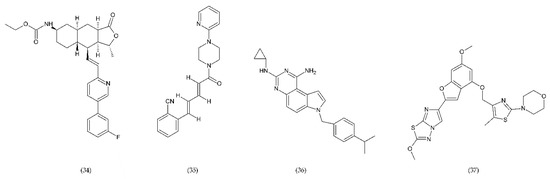
Figure 13.
PAR-1 and PAR-4 antagonists: vorapaxar (34), F16618 (35), SCH79797 (36), BMS986120 (37).
BMS986120 (37), which belongs to PAR4 antagonists, is a novel, selective and reversible drug that reduces the platelet aggregation during high shear stress [202,225].
A strong antithrombotic activity by F16618 (35), antagonist of PAR1, was demonstrated both in in vivo and ex vivo studies [226]. In addition, F16618 after oral or intravenous treatments reduced by 30 and 50%, respectively, the expression of the inflammatory cytokine TNF-α, 24 h after angioplasty [227].
In turn, SCH79797 (36), the PAR1 antagonist, was effective in reducing thrombi of fibrin-rich platelets at high shear rates (Figure 11). SCH79797 studies on experimental animals have shown a high potential for PAR1 antagonism in the treatment of diseases such as vascular thrombosis [228]. The antithrombotic effect of SCH79797 was significantly increased in combination with aspirin [229].
6.4.3. Inhibitors of PDI and PI3Kβ
In the process of haemostasis and thrombosis, the PI3Kβ/protein kinase B (Akt) signalling pathway associated with platelet activation and aggregation is important. The family of phosphoinositide 3-kinases (PI3Ks), which catalyse the phosphorylation of the inositol ring of phosphatidylinositol, acts as regulator of cellular function supporting platelet activation and thrombus formation. PI3K enzymes are also involved in cardiovascular diseases including angiogenesis, hypertension, and heart failure, and are the target of promising treatment for the prevention of thrombosis [219].
The use of drugs that inhibit this pathway can be a method of treating thrombosis in patients with implanted blood contacting medical devices [230]. A promising anticoagulant is isoquercetin (38), inhibitor of PDI found on the surface of PLa. Preliminary studies using this flavonoid have shown a reduction in platelet aggregation and limitation of thrombus formation. Isoquercetin reduces platelet-dependent thrombin production by blocking the production of platelet factor (PF) Va (Figure 14) [202].
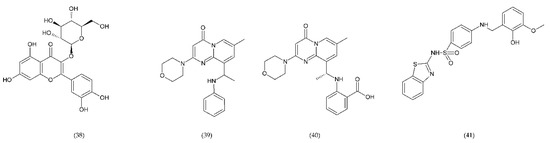
Figure 14.
Inhibitors of PDI and PI3Kβ: Isoquercetin (38), TGX221 (39), and AZD6482 (40); inhibitor of 12-lipoxygenase ML355 (41).
TGX221 (39) related pyrido[1, 2-a]pyrimidin-4-one belongs to a new target for antithrombotic therapy, and it is a PI3Kβ inhibitor. In platelets, PI3Kβ is an effector of various cell surface receptors including GP Ib, GP VI, and P2Y12. TGX221 is a potent and selective inhibitor [231]. The TGX-221 p110β inhibitor blocked IGF-1 initiated phosphorylation of Akt in platelets deficient in p110α, indicating that both p110α and p110β were involved in IGF-1 mediated phosphorylation of Akt [75]. Additionally, TGX221 has a particular role in shear stress conditions, becoming a new anticoagulant drug aimed at PI3Kβ [232]. In vivo studies have shown that inhibition of PI3Kβ provides protection against arterial thrombosis, with a limited effect on normal haemostasis [233].
AZD6482 (40), is an improved structural analogue of TGX-221 and is also PI3Kβ inhibitor that underwent preclinical and early clinical examination (phase I). AZD6482 exhibited antithrombotic effects without any increase in bleeding time [234,235]. AZD6482 inhibited agonist-induced platelet aggregation and shearing, showing concentration-dependent anticoagulation in vivo in dogs, with no detectable increase in bleeding time or blood loss [219]. Research conducted on healthy volunteers showed that the properties of the drug in inhibiting platelet activation placed it at levels between aspirin and clopidogrel. However, the maximum antiplatelet effect was evaluated clinically in combination of AZD6482 with ASA and clopidogrel [202].
ML355 (41) is a drug that inhibits platelet aggregation in humans. ML355 is a selective inhibitor of platelet 12-LOX, an enzyme in the AA pathway that leads to the formation of 12-HPET (Figure 14). It does not affect the activity of COX-1 and COX-2. ML355 inhibited dose-dependent aggregation of human platelets. ML355 treatment impaired thrombus growth in an arteriole in the mouse model [236,237].
6.4.4. Other Compounds
During trauma and/or high shear stress, the GP Ib/IX/V receptor, which binds to vWF, has a key role, resulting in platelet adhesion to the endothelium. The drug that binds the A1 domain of vWF is aptamer ARC1779, 40-mer modified DNA/RNA oligonucleotide. Additionally, ARC1779 is the ligand for GP Ib receptor on platelets [238,239]. In people with acute myocardial infarction, the level of vWF was 2 times higher than in healthy people. ARC1779 blocks binding of the vWF A1 domain to GP Ib receptors. Aptamer inhibited vWF activity and shear-dependent platelet function. Unlike GP IIb/IIIa antagonists, ARC1779 did not inhibit platelet aggregation induced by ADP, collagen, or AA [240]. Compared with abciximab, ARC1779 showed comparable anticoagulant efficacy, however, with less prolonged bleeding time.
In a partial phase I/II clinical trial, ARC1779 used in patients with congenital thrombocytopenic purpura (TTP) inhibited vWF-induced platelet aggregation and stabilized platelet counts without any associated bleeding [241].
The next anticoagulant was the AJvW-2n monoclonal antibody directed against human vWF A1 domain, which inhibited platelet aggregation induced by high shear stress in vitro. Antibody also counteracted clot formation in vivo in photochemically induced thrombosis in the carotid artery of guinea pigs without affecting bleeding time [242].
Another aptamer of the second generation inhibiting vWF activity, as a result of shearing forces, and as a consequence suppressing plate aggregation, was ARC15015. This compound inhibited platelet adhesion stimulated in whole blood by ristocetin at 90% in the aortic segment under strong shear conditions [243].
A promising drug seems to be the humanised, bivalent Caplacizumab nanobody (ALX-0081) that binds to GP Iba on vWF. ALX-0081 has been approved by FDA in the treatment of adult patients with TTP [244,245]. The bivalency of ALX-0081 results in higher affinity for vWF. In vitro studies in the perfusion chamber using blood of healthy individuals and patients after percutaneous coronary intervention (PCI) receiving a standard therapy (aspirin, clopidogrel, and unfractionated heparin) showed inhibition of platelet adhesion to type III collagen by Caplacizumab under arterial shear conditions (high shear stress) without side effects under low shear conditions. Caplacizumab has recently been approved and is a new drug for the treatment of TTP. This drug specifically binds to the A1 domain of vWF, which blocks vWF from binding to platelets. Caplacizumab is the first drug in its class to show significant efficacy in the treatment of TTP and its complications in clinical trials [244].
Another vWF-binding drug, which is also a GP Iba antagonist, is the snake venom anfibatide of Agkistrodon acutus, which inhibits the interaction between vWF and PLa and consequently blocks platelet adhesion and aggregation [246]. In turn, the tripeptide of the isolated venom Agkistrodon acutus successfully inhibited the binding of fibrinogen to GP IIb/IIIa [247]. In addition, anfibatide had also a protective effect on cerebral ischaemia and reperfusion injury and its use could be beneficial for the treatment of ischaemic stroke [248,249]. Anfibatide is currently undergoing a clinical trial in acquired TTP which ends in 2021.
Recently, a new therapeutic antibody against platelet GP VI, ACT017, has been shown to have a strong and selective antiplatelet activity. GP VI present on the surface of the platelets is a physiological collagen receptor. The interaction of collagen with the GP VI receptor leads to the release of ADP and TXA2, which in turn causes the activation and adhesion of platelets and the activation of the GP IIb/IIIa receptor. Studies in healthy volunteers have shown that antibody ACT017 is well tolerated and safe to use [250]. Despite the wide variety of drugs used in thrombosis, they are characterised by bleeding risk, cardiovascular complications, and mortality. The introduction of new stronger antiplatelet agents is associated with an increased risk of bleeding. In vivo research related to haemostasis and thrombosis has revealed significant differences between these phenomena that have been helpful in developing new antiplatelet drugs that would not lead to increased bleeding. Preclinical therapies have been introduced in preclinical studies that inhibit thrombosis but do not affect the haemostasis process. This group of drugs includes PI3Kβ inhibitors, protein disulphide isomerase, activated GP IIb/IIIa, receptors activated by protease, and platelet-mediated adhesion pathways [192].
An interesting method to reduce the possibility of vascular thrombus formation is the use of CX3C chemokine receptor 1 (CX3CR1) and fractalkine (CX3CL1) receptor inhibitors. Monocyte-endothelial cells interactions are partly mediated by expression of monocyte CX3CR1 protein and endothelial cell CX3CL1. CX3CL1 is a membrane protein synthesized in ECs. ECs produce CX3CL1 after stimulation with TNF-α, IL-1, IFN-γ, and LPS. It was shown than the weakening of interaction between the pair CX3CL1-CX3CR1 receptor can reduce monocyte binding to the endothelium [251,252,253].
A new strategy in the treatment of thrombosis is to use FXI and FXII inhibitors. Both factors are important in thrombosis but not in haemostasis. Factor FXI has a much more important role in thrombosis than FXII. In addition, the anti-FXI antibody inhibits the activation of platelets and fibrin deposition more than in the case of FXII [254]. However, FXII may be a possible, therapeutic, and safe target to reduce platelet adhesion and fibrin formation during medical interventions [255]. One of the methods of action is to inhibit FXI by antisense oligonucleotides (ASO), which inhibit FXI synthesis in liver [256]. In turn, monoclonal antibodies may block FXI activation or FXIa activity [177,257], whereas aptamers binding to FXI or FXIa and small molecules block the active site of FXIa or/and induce allosteric alterations [258,259].
This section summarises the various anticoagulants such as FX inhibitors, thrombin inhibitors, antiplatelet drugs, emerging drugs, PAR antagonists, and PDI and PI3K inhibitors as well as other active compounds.
7. Natural Compounds
There is an abundance of drugs used in thrombosis; however, they lead to non-desirable side effects, mainly bleeding. The increase in cardiovascular incidents requires the introduction of new drugs that would be effective in the treatment and prevention of thrombosis and at the same time be free of side effects. Many plant compounds such as saponins, flavonoids, and others have an important role in preventing various diseases. Oxidative stress promotes thrombotic complications. Therefore, various substances of natural origin reduce the level of oxidative stress and may be important in the treatment of thrombotic complications [260]. Inactivation of ROS and alleviation of oxidative stress can reduce the risk of platelet hyperactivation, which leads to cardiovascular diseases including thrombosis [261]. Numerous works have shown that compounds of natural origin can reduce the risk of cardiovascular diseases including thrombosis. In Far Eastern medicine, medicines of vegetable origin have been used that have good healing properties and no side effects. These substances affect the mechanisms of thrombosis in various ways, affecting the coagulation system and platelet activation and aggregation and changing blood rheological conditions.
Drugs used in anticoagulation therapy with anticoagulation, inhibiting platelet aggregation and associated with fibrinolysis, have been presented.
7.1. Flavonoids
Natural products such as dietary supplements are important in ensuring health but have an important role in the prevention of cardiovascular and other diseases. Flavonoids as antioxidants reduce ROS levels and thus inhibit platelet aggregation/function or prothrombotic effects [262]. Flavonoids can alleviate ED, which is one of the factors leading to thrombosis, by inhibiting the excessive availability of TF in the endothelium [261]. Flavonoids can inhibit platelet activity by inhibiting the AA pathway [263] or may bind to the TXA2 platelet agonist and this way inhibit their aggregation.
Natural dietary supplements including flavonoids are widely distributed in the plant world, especially in a variety of fruits, coffee, tea, cocoa, legumes, and other plant foods. The most popular flavonoids, including quercetin and its derivatives, and tea flavonoids, such as catechin, epictechin, epigallocatechin, and epigallocatechin gallate, have antioxidant, anti-inflammatory, anti-cancer, and anticoagulant properties, and are found in green tea [264]. But other less popular flavonoids have such properties as well like wogonine (42) from Scutellaria baicalensis Georgi and flavonoid wogonoside (43), which is a metabolite of wogonine isolated from Scutellaria baicalensis (Figure 15) [265].

Figure 15.
Chemical structures of flavonoids: wogonine (42) wogonoside (43), galangin (44), pinocembrin (45), nobiletin (46), and morin (47).
7.1.1. Antithrombotic Properties of Flavonoids
Anticoagulant activity associated with inhibition of platelet aggregation initiated by AA, ADP, and collagen as well as elongation of activated partial thromboplastin time (aPTT), PT, was shown using the extract of persimmon (Diospyros kaki) leaves, whose main ingredients are flavonoids such as catechins, epicatechin, and epicatechin gallate [266,267]. Similar results, associated with inhibition of platelet aggregation, were obtained using propolis, whose main ingredients are caffeic acid and its phenethyl ester, ferrulic acid; galangin (44); apigenin; quercetin; kaempferol; rutin; chrysin; pinostrobin; and pinocembrin (45) (Figure 15). The process of inhibiting platelet aggregation was induced by ADP, thrombin receptor activator peptide (TRAP), and collagen and was dose-dependent, which led to inhibition of thrombus formation [268].
Another flavonoid (Figure 15) with anti-inflammatory, antioxidant, anticancer, and anti-platelet inhibiting properties was nobiletin (46), a polymethoxylated flavone isolated from citrus fruit. Its mechanism of action is associated with inhibition of phosphorylation of phospholipase Cγ2 (PLCγ2), protein kinase C (PKC), Akt in mitogen stimulated MAPK protein kinase in collagen activated human platelets. In addition, nobiletin significantly reduced intracellular calcium mobilisation and hydroxyl radical generation, ultimately leading to inhibiting platelet activation [269].
The use of wogonins and wogonoside led to inhibition of thrombin and FXa activity and inhibited thrombin production in human umbilical vein endothelial cells (HUVEC), as well as resulted in prolonged aPTT and PT. Furthermore, both flavonoids inhibited thrombin-catalysed fibrin polymerisation and platelet aggregation and showed anticoagulant activity in vivo in mice. Wogonin and wogonoside reduced the ratio of PAI-1 to t-PA. These properties testify to the good anticoagulant effect of both flavonoids [270].
Flavonoids and their derivatives inhibited thrombus formation in vivo. For example, pentamethyl quercetin (PMQ), a methylated quercetin derivative, inhibited thrombus formation in mice with acute pulmonary thrombosis, leading to 90% of the mice surviving. It has also been shown that PMQ significantly improved blood flow in mice with damaged carotid artery showing an inhibitory effect on thrombus formation [271].
Diosmin (diosmetin 7-O-rutinoside), a disaccharide derivative that consists of diosmetin is a natural flavonoid found in the Rutaceae family or the Citrus family. Diosmin has the properties of an antioxidant, antihyperglycemic, anti-inflammatory, antimutagenic, and antiulcer agent and is commonly used to treat varicose vein [143]. Diosmin isolated from Galium verum also showed anticoagulant properties, and its mechanism of action was associated with regulation of centrosome-associated protein 350 (CEP 350), which is related to spindle assembly. Its use led to the improvement of venous thrombosis [272].
Another bioactive flavonoid with anticoagulant properties is morin. Morin (47) belongs to the flavonol group and is found in old fustic (Chlorophora tinctoria), osage orange (Maclura pomifera), almond, mill (Prunus dulcis), fig (Chlorophora tinctoria), onion, and apple. This flavonoid is a compound that prevents inflammation and apoptosis of cells and also has antioxidant activity (inhibits xanthine oxidase activity). Morin significantly inhibited human platelet aggregation stimulated by collagen, but not stimulated by other agonists. In collagen PLa, morin was shown to inhibit ATP release, intracellular Ca2+ ions mobilisation, P-selectin expression, PLCγ2 phosphorylation, PKC, and Akt [273].
The anticoagulant properties of the extract from Chaenomeles sinensis have been shown. Thirteen flavonoids were isolated from fruit of Chaenomeles sinensis, and five of them, including hovertrichozide (48), luteolin-7-O-β-D-glucuronide, hyperin (quercetin-3-O-galactoside), avicularin (49) (quercetin 3-alpha-L-arabinofuranoside) (Figure 16), and quercitrin, were active in inhibiting TF expression induced by calcium ions in rat plasma in vitro. The activity of the hovertrichozide benzofuran derivative was higher than that of classic flavonoid derivatives of chroman, such as luteolin-7-O-β-D-glucuronide, hyperin, avicularin, and quercitrin [274].
Figure 16.
Chemical structure of hovertrichozide (48) and avicularin (49).
7.1.2. Isoflavonoids
In addition to flavonoids, isoflavonoids (Figure 17) also show antiplatelet activity. Isoflavonoids include genistein (50) and daidzein (51), which are COX-1 inhibitors and functional TXA2 receptor antagonists, affecting the platelet aggregation cascade. COX-1 inhibition by these isoflavonoids is similar to that of acetylsalicylic acid. The use of both isoflavonoids may have a positive inhibitory effect on the aggregation of human platelets in cardiovascular diseases in which they are associated with increased platelet activity [275].
Figure 17.
Chemical structure of isoflavonoids: genistein (50), daidzein (51), silybin A (52), silybin B (53), and silychristine (54) flavonoid.
Silymarin is the mixture of substances derived from Silybum marianum seed that exhibits antioxidant, anticancer, anti-inflammatory, and antiviral properties [276]. Silymarin inhibits cell proliferation and induces apoptosis, while also having anti-angiogenic properties. Silymarin consists mainly of silybin A (52) and silybin B (53), and other compounds. Both isomers of flavonoids belong to the group of flavonolignans. Silybin has been shown to be a signal transducer and activator of transcription 3 (STAT3) protein inhibitor whose function is to control the expression of genes that are involved in cell survival, proliferation, chemo-resistance, and angiogenesis. Silybin is also an inhibitor of expression of the programmed death-ligand 1 (PD-L1) checkpoint immune regulator as well as epithelial-mesenchymal transition (EMT) regulators, and it can be a promising adjuvant in non-small-cell lung carcinoma (NSCLC) [277].
Another flavonolignan, silychristine (54), is a 1-benzofuran derivative isolated from Silybum marianum or from silymarin. Silychristine also has antioxidant properties and is an inhibitor of lipid peroxidation in vivo and in vitro, LOX, and prostaglandin synthetase [276]. Silybin and silychristine inhibited collagen-initiated platelet activation. Significant decreases in platelet aggregation and microparticle aggregation were observed, as well as reduced expression of P-selectin and activation of αIIbβ3 integrin but also reduced adhesion and secretion of PF-4. Both can be used to prevent primary and secondary thrombotic events [278]. Strong inhibitors effects of silybin and silychristine on platelet activation induced by ADP have been shown [279].
This chapter discusses the anticoagulant mechanisms of action of popular flavonoids and isoflavonoids.
7.1.3. Traditional Korean and Chinese Medicine
Lindera obtusiloba (LLE) is used in traditional Korean and Chinese medicine for the treatment of blood stasis and inflammation. Additionally, the leaf extract showed various physiological effects. The main flavonoids in the extract are afzelin (55) (Figure 18) and quercetrin. Leaf extract was shown to significantly inhibit agonist-induced platelet aggregation in vitro and ex vivo and to inhibit collagen-induced TXA2 production in blood platelets. In addition, in vivo, the extract showed a protective effect on clot formation induced by intravenous injection of a collagen and adrenaline mixture. Further studies showed that the extract did not significantly affect PT and aPTT [280].
Figure 18.
Chemical structure of afzelin (55), biochanine A (56), sophorabioside (57), tatectoridin (58), apigenin (59), isorhamnetin glucosides (60), and rutin (61).
Buds and fruits of Sophora japonica (Leguminosae) plants widely distributed in Korea, China, and Japan have been used as haemostatic agents in traditional Korean medicine. Sophora japonica extract has antiplatelet activity. Four flavonoids and six flavonoid-glycosides were isolated from the extract, such as: biochanine A (56), eriodictyol, genistein, taxifoline, sophorabioside (57), genistin, tatectoridin (58), apigenin (59), quercetin, kaempferol, and its 3-O-rutinoside and 8-metoxykaempferol, isorhamnetin glucosides (60), rutin (61), and six derivatives of quercetin (Figure 18) [281].
7.2. Saponins
New natural substances with anticoagulant properties are currently being sought that would be free of side effects such as bleeding that may occur when using classic anticoagulants. Natural anticoagulants occur in the world of plants, which are traditionally used in Chinese medicine, but they also may be substances of animal origin [282]. The group of natural compounds with anticoagulant properties includes saponins, which occur in the world of plants and are produced by some marine organisms, including star fish and sea cucumbers. Saponins belong to amphipathic glycosides, which have one or more hydrophilic glycosides linked to a lipophilic triterpene or a steroid derivative [283]. Molecular weight depending on the basic backbone of the saponins and the number of sugar residues. We have corrected the molecular weight of saponins. Saponins have a high molecular weight ranging from 741 to 1808 g/mol [284].
7.2.1. Plant to the Genus Panax
Ginseng is a plant belonging to the genus Panax, which has long been known for its medicinal properties that help alleviate disease symptoms and prevent potential diseases. Ginseng contains many bioactive components, mainly saponins, most of which are glycosides of triterpenoid aglycons, also known as ginsenosides [285].
Saponins Panax notoginseng (PNS) are part of a traditional herb used in Chinese medicine to treat blood clotting, swelling, and pain relief. The main components of Panax notoginseng are ginsenoside Rg1 (64), ginsenoside Rb1 (62), and notoginsenoside R1 (67) (Figure 19). Panax saponins inhibit platelet aggregation, preventing thrombosis, but also regulate many signalling pathways by acting on cardiovascular protection. In addition, they show anti-inflammatory effects mediated by peroxisome proliferator-activated receptor γ (PPAR-γ) in human endothelial cells. In vitro inhibited platelet aggregation induced by thrombin overexpression of PPAR-γ increased expression of PI3Kβ/Akt/eNOS pathway in platelets. In vivo studied saponin Panax notoginseng reversed thrombin-induced hypercoagulable state saponins Panax induced PPAR-γ activation and also had a key role in the PI3Kβ/Akt/eNOS pathway [286]. Other PNS saponins have anti-inflammatory, antioxidant action, they inhibit platelet aggregation, regulate glucose and blood pressure, inhibit neuronal apoptosis, and provide neuronal protection [287].
Figure 19.
Chemical structure of saponins: ginsenoside rb1 (62), ginsenoside rb3 (63), ginsenoside rg1 (64), ginsenoside rg3 (65), ginsenoside Ro (66), notoginsenoside R1 (67), Panax bipinnatifidus, araloside A methyl ester (68), and Panax bipinnatifidus stipuleanoside R2 (69).
The platelet aggregation inhibiting properties have been demonstrated by the next two saponins (Figure 19) isolated from Panax bipinnatifidus, a plant used in traditional Vietnamese medicine. These include araloside A methyl ester (68) and stipuleanoside R2 (69), which are the main components isolated from this plant [288]. However, their mechanism of action has not yet been described. In turn, ginsenoside Ro (66), Panax ginseng oleanane-type saponin, inhibiting IIb/IIIa mediated fibrinogen binding by preventing platelet aggregation may be an effective anticoagulant [285].
7.2.2. Liriope muscari
Anticoagulant properties were demonstrated by natural saponin D39 (70) (Figure 20) isolated from Liriope muscari, and the mechanism of its action is associated with the non-muscle myosin heavy chain IIa (NMMHC IIA). In turn, NMMHC IIA is important in the expression of TF and, consequently, venous thrombosis. Using HUVEC cells has been shown that saponin D39 inhibits TF expression and has anticoagulant activity. In addition, saponin D39 inhibited thrombus formation by affecting the Akt/GSK3β and NF-κB signalling pathways by NMMHC IIA. It was suggested that D39 could be a potential therapy for DVT and other cardiovascular diseases [289].
Figure 20.
Chemical structure of saponin D39 (70).
The steroid saponin DT-13 isolated from Liriope muscari has been used in traditional Chinese medicine to treat a variety of diseases such as pharyngitis, bronchitis, pneumonia, cough, and cardiovascular diseases. DT-13 also shows anticoagulant properties as demonstrated in vivo in the classical model of IVC ligation thrombosis in mice and rats. DT-13 inhibits venous thrombosis partly by reducing gene expressions of TF and decreasing IL-6 expression [290].
7.2.3. Steroidal Sapogenin
Ruscogenin (RUS) (71) (Figure 21), a steroidal sapogenin isolated from Ruscus aculeatus, is also the main component of the traditional Chinese herb Radix Ophiopogon japonicus, showing significant anti-inflammatory and anti-coagulant effects [291,292]. In addition, ruscogenin significantly inhibits leukocyte adhesion to the human umbilical vein endothelial line (ECV304). It has been used for thousands of years in the treatment of acute and chronic inflammatory and cardiovascular diseases [293].
Figure 21.
Chemical structure of ruscogenin (RUS) (71).
Diosgenin and other steroidal saponins isolated from this plant are demonstrated to have anti-thrombotic activity. From few isolated saponins, especially Diosgenyl saponin, which has the linkage of the sugar chains at the C-3 position of diosgenin, had anti-thrombotic activity and significantly inhibited platelet aggregation in vitro and in vivo. This saponin did not act via the extrinsic coagulation pathway but rather by inhibiting molecular targets in intrinsic coagulation pathways. Diosgenyl saponin protected mice against pulmonary thromboembolism induced by collagen and epinephrine. It demonstrates anti-thrombotic activity through inhibiting FVIII activities and platelet aggregation [294].
7.2.4. Other Saponins
Polygala fallax Hesml root saponin extract (PTS) significantly prolonged coagulation time in mice, reduce thrombus length and inhibited carrageenan-induced inflammation without affecting thrombus formation. PTS had anticoagulant and anticoagulant effects, probably via the endogenous coagulation system [295]. Seeds of chestnut (Aesculus hippocastanum L., Sapindaceae) contain approx. 3–5% saponins and flavonoids: quercetin and derivatives of kaempferol, proanthocyanidins, and sterols. They are used to isolate escin, a saponin used to treat chronic venous insufficiency. Escin has a positive effect on venous tone, rheological properties, and blood clotting. In addition, it has anti-inflammatory and anti-oedema properties [296]. This chapter describes the different classes of saponins found in plants and their antithrombotic properties.
7.3. Stevioside and Derivertes
Plant-derived diterpenoids (Figure 22) demonstrate a wide range of biological activity and exhibit antitumor, antibacterial, antihyperglycemic, anti-inflammatory and immunomodulatory, antifungal, antiparasitic, antiviral, antiallergic, antispasmodic, antihyperglycemic, and other effects [297]. Stevioside (72) is diterpenoid found in significant amounts in the leaves of the herb Stevia (Stevia rebaudiana Bertoni) [298], which is native to the region of South America. Stevia belongs to the Asteraceae chrysanthemum family. Stevioside is a sweet non-toxic and non-mutagenic drug used in patients with metabolic problems in type II diabetes, phenylketonuria, and obesity [299]. Stevioside (Figure 22) reduces the influx of calcium, lowers blood pressure, increases the insulin signal, increases the antioxidant potential in the aorta, and inhibits the process of atherosclerosis. The antioxidant properties and the reduction of oxidative damage were previously well documented in many studies [300,301]. Recently, the mechanism of its action has been explained, which is associated with the enhancement of the endogenous related pathway with nuclear factor erythroid 2-related factor 2 (Nrf2), which is one of the key transcription factors in the human body. Nrf2 also regulates the expression of enzymes to counteract oxidative stress [302].
Figure 22.
Chemical structure of diterpenoids: stevioside (72), steviol (73), and isosteviol (74).
In contrast, isosteviol (74) caused an increase in vasodilatation, a decrease in angiotensin II, and cell proliferation, as well as a decrease in the concentration of reactive oxygen and endothelin-1. A decrease in extracellular signal-regulated kinase (ERK), hyperplasia, and hypertrophy were also observed in vascular smooth muscle cells [303]. Stevioside has been shown to inhibit the expression of IL-6, IL-1β and TNF-α and LPS-induced initiation, NF-κB, inhibitor of kappa B (IκBα) degradation, c-Jun N-terminal kinase (JNK), p38 and ERK phosphorylation in a dose-dependent manner. In addition, it was shown that the anti-inflammatory activity can be exerted by inhibiting NF-κB activation and MAPK and expression of pro-inflammatory cytokines [304]. Stevioside inhibited the release of proinflammatory cytokines and inhibited LPS-induced COX-2 and iNOS expression in mice in vivo [305].
Steviol (73), similar to stevioside, significantly inhibited TNF-α induced IL-8 expression in human colon cancer cells, Caco-2, T84 10 and HT29 [155]. Steviol’s immunomodulatory activity was associated with NF-κB signalling [306]. In turn, Boonkaewwan et al. [304] conducting studies on the colon carcinoma Caco-2 cell line showed that stevioside and steviol inhibited LPS-induced release of IL-6, TNF-α and IL-1β by affecting the expression of the cytokine gene through the signal pathway IκBα/NF-κB.
Isosteviol (ent-16-oxobeyran-19-acid) is a tetracyclic skeletal diterpenoid of the Beyerene type, obtained by acid hydrolysis of stevioside [307]. Isosteviol has biological activity, including anti-inflammatory, antihypertensive, controlling blood lipids, immunoregulation, antiviral, antibacterial, and others [308]. It has been reported that steviol significantly inhibited proliferation of cancer cells by induction of apoptosis. In addition, steviol leads to the generation of ROS in cancer cells. Isosteviol also showed strong antithrombotic properties by selectively inhibiting FXa against a serine protease panel and anticoagulant activity by significantly prolonging PT and aPTT time as well as inhibiting ADP-induced platelet aggregation [309].
8. Conclusions
This paper reviews the pathophysiology of clot formation involving blood cells and cell components, critical factors released from activated cells and damaged cells as well as all already known drugs and methods to prevent or cure thromboembolism in all stages.
A new insight into the physiology of platelets and their involvement in inflammatory response and activation of clotting has been presented. These two processes have an important role in defending the host from infection. Their role in innate immunity in recognising pathogens, signal transduction, and cytokine/chemokine release is essential in thrombus formation, preventing the spread of microorganisms. In addition to platelets, other blood morphotic components such as leukocytes, erythrocytes, and vascular endothelial cells, along with activation of the coagulation system, have a significant role in the development of blood clots.
We considered the specific and key role of ECs in signalling mechanisms in which cyclophilin A (CypA) is secreted and ROS are generated. Both factors are sufficient to initiate vasculitis, secret MMPs, and induce changes in the structure of the vessel wall. The studies presented thus far show the role of platelets in the stages of inflammation, which indicates that they can be the target of an appropriate group of drugs. Undoubtedly, antiplatelet agents are effective in the treatment of thrombosis, but the problem is an increased risk of bleeding. However, a more accurate understanding of the processes associated with thrombosis and haemostasis has allowed the adoption of new strategies that are tested on animal models. Those are antiplatelet agents that inhibit thrombosis but do not affect haemostasis. In addition to antiplatelet drugs, there are also new strategies targeting clotting factors. An example is FXI, the inhibition of which may prove to be safer than the use of FXa inhibitors or thrombin. The FXI factor is important for the stabilisation and growth of the thrombus, but less important for haemostasis.
Even though modern medicine has a wide range of anticoagulant drugs, thrombosis still leads to cardiovascular diseases, myocardial infarction, and stroke. While significant advances have been made in understanding the mechanisms of thrombus formation and the pathophysiology of thrombosis, current prevention or treatment drugs that have been used for many years have been replaced by newer ones that show greater potential for the prevention and treatment of thrombosis with modest but gradual improvement. High hopes are associated with compounds of natural origin, which show a strong anticoagulant effect both at the experimental and clinical stage and can be used in the prophylactic or supportive methods of treatment of thrombotic diseases.
Author Contributions
Conceptualization, A.L.; data curation, E.M.S. and K.G.; formal analysis, A.L.; funding acquisition, E.M.S. and K.G.; investigation, A.L., E.M.S., and K.G.; methodology, A.L.; project administration, A.L.; supervision, A.L., E.M.S., and K.G.; validation, A.L., E.M.S., and K.G.; visualization, A.L.; writing—original draft, A.L., E.M.S., and K.G.; writing—review and editing, A.L., E.M.S., and K.G. All authors have read and agreed to the published version of the manuscript.
Funding
This review was funded by Medical University of Lodz & University of Lodz.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AA | Arachidonic acid |
| ACE | Angiotensin-converting enzyme |
| ADP | Adenosine diphosphate |
| AGEs | Advanced glycation end products |
| Akt | Protein kinase B |
| AMI | Acute myocardial infarction |
| aPTT | Activated partial thromboplastin time |
| apoAI | Apoplipoprotein A-I |
| APS | Antiphospholipid syndrome |
| ASA | Acetylsalicylic acid |
| ASO | Antisense oligonucleotides |
| AT | Antithrombin |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| CAT | Catalase |
| CCL | Chemokine (C-C motif) ligand |
| CD | Cluster of differentiation |
| CDT | Catheter-directed thrombolysis |
| CEP | Centrosome-associated protein |
| CHCL1 | Chemokine induces the infiltration of neutrophils |
| CLEC | C-type lectin-like receptor |
| COX | Cyclooxygenase |
| CT | Computed tomography |
| CX3CL1 | Fractalkine |
| CX3CR1 | CX3C chemokine receptor 1 |
| CXCL | Chemokine CXC ligand-1 |
| CYP | Cytochrome P450 |
| CypA | Cyclophilin A |
| DNA | Deoxyribonucleic acid |
| DVT | Deep vein thrombosis |
| eATP | Extracellular ATP |
| ECE | Endothelin converting enzyme |
| ECs | Endothelial cells |
| ED | Endothelial dysfunction |
| EDCF | Endothelium-derived contracting factor |
| EDGF | Endothelium-derived growth factor |
| EDHF | Endothelium-derived hyperpolarizing factor |
| EGF | Epidermal growth factor |
| EMP | Microparticles of endothelial cells |
| EMT | Epithelial-mesenchymal transition |
| eNOS | Endothelial nitric oxide synthase |
| ERK | Extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| FGF | Fibroblast growth factor |
| GP | Glycoprotein |
| GSK | Glycogen synthase kinase |
| H2O | Water |
| H2O2 | Hydrogen peroxide |
| Hb | Haemoglobin |
| HClO | Hypochlorous acid |
| HIF | Hypoxia inducible factor |
| HNE | 4-hydroxy-2-nonenal |
| HO• | Hydroxyl radical |
| HO2• | Hydroperoxyl radical |
| Hp | Haptoglobin |
| HRE | Hormone response element |
| HsCRP | High-sensitivity C-reactive protein |
| 5HT2A | Serotonin receptor 2A |
| HUVEC | Human umbilical vein endothelial cells |
| Hx | Hemopexin |
| IAV | Influenza A virus |
| ICAM | Intercellular adhesion molecule |
| iCypA | Extracellular cyclophilin A |
| IDUs | Intravenous drug users |
| IFN | Interferon |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| iNOS | Induced nitric oxide synthase |
| INR | International normalized ratio |
| IVC | Inferior vena cava |
| IκBα | Inhibitor of kappa B |
| JAK-2 | Janus kinase 2 |
| JNK | C-Jun N-terminal kinase |
| LMWH | Low molecular weight heparin |
| LOX | Lipoxygenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinases |
| MCP | Monocyte chemoattractant protein |
| metHb | Methaemoglobin |
| MHC | Major histocompatibility complex |
| MIDAS | Metal ion-dependent adhesion site |
| MIP-2 | Macrophage inflammatory protein 2 |
| MKP-1 | Mitogen-activated protein kinases phosphatase 1 |
| MMP | Matrix metalloproteinase |
| MPO | Myeloperoxidase |
| MPV | Mean platelets volume |
| mPTP | Mitochondrial permeability transition pore |
| MRI | Magnetic resonance imaging |
| NAD+ | Nicotinamide-adenine dinucleotide phosphate (oxidised form) |
| NADPH | Nicotinamide-adenine dinucleotide phosphate (reduced form) |
| NAPc2 | Nematode anticoagulant protein c2 |
| NETs | Neutrophil extracellular traps |
| NF-κB | Nuclear factor kappa B |
| NLRP3 | Inflammasome is component of the innate immune system |
| NMMHC IIA | Non-muscle myosin heavy chain IIa |
| NO• | Nitric oxide |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLC | Non-small-cell lung carcinoma |
| O2 | Oxygen |
| 1O2 | Singlet oxygen |
| O2•− | Superoxide anion radical |
| ONOO− | Peroxynitrite |
| ox-L | Oxidised lipids |
| ox-P | Oxidised proteins |
| oxyHb | Oxyhaemoglobin |
| PAF | Platelet activating factor |
| PAI-I | Plasminogen activator inhibitor-1 |
| PAR | Protease-activated receptor |
| PAT | Percutaneous aspiration thrombectomy |
| PCDT | Catheter-directed pharmacomechanical thrombolysis |
| PCI | Percutaneous coronary intervention |
| PD-L1 | Programmed death-ligand 1 |
| PDE3 | Phosphodiesterase type 3 |
| PDGF | Platelet-derived growth factor |
| PDI | Protein disulfide-isomerase |
| PE | Pulmonary embolism |
| PF | Platelet factor |
| PGE | Prostaglandin E |
| PGG | Prostaglandin G |
| PGH | Prostaglandin H |
| PGI | Prostacycline |
| PI3Kβ | Phosphatidylinositol 3 kinase β |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLa | Activated platelets |
| PLA | Phospholipase A |
| PLC | Phospholipase C |
| PLCγ2 | Phospholipase Cγ2 |
| PMN | Polymorphonuclear leukocyte |
| PMQ | Pentamethyl quercetin |
| PNS | Saponins Panax notoginseng |
| PPAR-γ | Peroxisome proliferator-activated receptor γ |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| PT | Prothrombin time |
| PTS | Polygala fallax Hesml root saponin extract |
| RBCs | Red blood cells |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SBSI | Simultaneous decrease in prostacyclin synthase |
| SLO | Streptolysin O |
| SLS | Streptolysin S |
| SOD | Superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| STEC | Shigatoxigenic Escherichia coli |
| TF | Tissue factor |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TNF-α | Tumour necrosis factor alfa |
| t-PA | Tissue plasminogen activator |
| TRAP | Thrombin receptor activator peptide |
| TTP | Thrombocytopenic purpura |
| TXA2 | Tromboxan A2 |
| UFH | Unfractionated heparin |
| UV | Ultraviolet |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VKORC1 | Vitamin K epoxide reductase complex |
| VSMCs | Vascular smooth muscle cells |
| VTE | Venous thromboembolism |
| vWF | Von Willebrand factor |
References
- Weitz, J.I.; Chan, N.C. Advances in Antithrombotic Therapy. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 7–12. [Google Scholar] [CrossRef]
- Essien, E.-O.; Rali, P.; Mathai, S.C. Pulmonary Embolism. Med Clin. N. Am. 2019, 103, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Flumignan, C.D.; Flumignan, R.L.; Baptista-Silva, J.C. Antiplatelet agents for the treatment of deep venous thrombosis. Cochrane Database Syst. Rev. 2016, 2016, CD012369. [Google Scholar] [CrossRef]
- Wattanakit, K.; Lutsey, P.L.; Bell, E.J.; Gornik, H.; Cushman, M.; Heckbert, S.R.; Rosamond, W.D.; Folsom, A.R. Association between cardiovascular disease risk factors and occurrence of venous thromboembolism. Thromb. Haemost. 2012, 108, 508–515. [Google Scholar] [PubMed]
- Stone, J.; Hangge, P.; Albadawi, H.; Wallace, A.; Shamoun, F.; Knuttien, M.G.; Naidu, S.; Oklu, R. Deep vein thrombosis: Pathogenesis, diagnosis, and medical management. Cardiovasc. Diagn. Ther. 2017, 7 (Suppl. 3), 276–284. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Denenberg, J.O.; Bergan, J.; Langer, R.D.; Fronek, A. Risk factors for chronic venous disease: The San Diego Population Study. J. Vasc. Surg. 2007, 46, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, W.; Knysz, B.; Gąsiorowski, J.; Witkiewicz, W. Deep vein thrombosis of the lower limbs in intravenous drug users. Postępy Hig. Med. Doświadczalnej 2015, 69, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D. 1 Endothelial cell function and thrombosis. Baillière’s Clin. Haematol. 1994, 7, 441–452. [Google Scholar] [CrossRef]
- Van Hinsbergh, V.W.M. Endothelium—Role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef]
- Wolberg, A.S.; Campbell, R.A. Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci. 2008, 38, 15–23. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell Biochem. 2017, 82, 405–456. [Google Scholar]
- Petrillo, G.; Cirillo, P.; Luana D’Ascoli, G.-; Maresca, F.; Ziviello, F.; Chiariello, M. Tissue Factor/Factor FVII Complex Inhibitors in Cardiovascular Disease. Are Things Going Well? Curr. Cardiol. Rev. 2010, 6, 325–332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Budnik, I.; Brill, A. Immune Factors in Deep Vein Thrombosis Initiation. Trends Immunol. 2018, 39, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2016, 38, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.A.; Key, N.S.; Levy, J.H. Blood Coagulation: Hemostasis and Thrombin Regulation. Anesth. Analg. 2009, 108, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- MCKenzie, P.J. Deep venous thrombosis and anaesthesia. Br. J. Anaesth. 1991, 67, 128. [Google Scholar] [CrossRef][Green Version]
- Branchford, B.R.; Carpenter, S.L. The Role of Inflammation in Venous Thromboembolism. Front. Pediatrics 2018, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci. Rep. 2019, 9, 13397. [Google Scholar] [CrossRef]
- Waldron, B.; Moll, S. A patient’s guide to recovery after deep vein thrombosis or pulmonary embolism. Circulation 2014, 129, 477–479. [Google Scholar] [CrossRef]
- Swystun, L.L.; Liaw, P.C. The role of leukocytes in thrombosis. Blood 2016, 128, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Suidan, G.L.; Wagner, D.D. Hypoxia, such as encountered at high altitude, promotes deep vein thrombosis in mice. J. Thromb. Haemost. 2013, 11, 1773–1775. [Google Scholar] [CrossRef]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Malone, P.C. The aetiology of deep venous thrombosis. QJM 2006, 99, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 2009, 23, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, D.J.; Naka, Y.; Liao, H.; Oz, M.C.; Wagner, D.D.; Mayadas, T.N.; Johnson, R.C.; Hynes, R.O.; Heath, M.; Lawson, C.A.; et al. Hypoxia-induced exocytosis of endothelial cell weibel-palade bodies: A mechanism for rapid neutrophil recruitment after cardiac preservation. J. Clin. Investig. 1996, 97, 493–500. [Google Scholar] [CrossRef]
- Michiels, C.; Arnould, T.; Remacle, J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim. Et Biophys. Acta BbaMol. Cell Res. 2000, 1497, 1–10. [Google Scholar] [CrossRef]
- Wang, X.; Liu, K.; Li, B.; Li, Y.; Ye, K.; Qi, J.; Wang, Y. Macrophages Aggravate Hypoxia-Induced Cardiac Microvascular Endothelial Cell Injury via Peroxynitrite: Protection by Tongxinluo. Cell Commun. Adhes. 2015, 22, 39–47. [Google Scholar] [CrossRef]
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; Van De Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial Role for Ecto-5′-Nucleotidase (CD73) in Vascular Leakage during Hypoxia. J. Exp. Med. 2004, 200, 1395–1405. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Abdulla, P.; Hoffman, E.; Hamilton, K.E.; Daniels, D.; Schönfeld, C.; Löffler, M.; Reyes, G.; Duszenko, M.; Karhausen, J.; et al. HIF-1–dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 2005, 202, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Faigle, M.; Grenz, A.; Laucher, S.; Thompson, L.F.; Eltzschig, H.K. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 2008, 111, 2024–2035. [Google Scholar] [CrossRef]
- Blouin, C.C.; Pagé, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1α. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Kruger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef] [PubMed]
- Baldea, I.; Teacoe, I.; Olteanu, D.E.; Vaida-Voievod, C.; Clichici, A.; Sirbu, A.; Filip, G.A.; Clichici, S. Effects of different hypoxia degrees on endothelial cell cultures—Time course study. Mech. Ageing Dev. 2018, 172, 45–50. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Mold, C.; Morris, C.A. Complement activation by apoptotic endothelial cells following hypoxia/reoxygenation. Immunology 2001, 102, 359–364. [Google Scholar] [CrossRef] [PubMed]
- HART, M. Initiation of complement activation following oxidative stress.In vitro and in vivo observations. Mol. Immunol. 2004, 41, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Castello, P.R.; David, P.S.; McClure, T.; Crook, Z.; Poyton, R.O. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006, 3, 277–287. [Google Scholar] [CrossRef]
- Palacios-Callender, M.; Hollis, V.; Mitchison, M.; Frakich, N.; Unitt, D.; Moncada, S. Cytochrome c oxidase regulates endogenous nitric oxide availability in respiring cells: A possible explanation for hypoxic vasodilation. Proc. Natl. Acad. Sci. USA 2007, 104, 18508–18513. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.; Yilmaz, Ö. The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G. Gram-positive and gram-negative bacterial toxins in sepsis. Virulence 2014, 5, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Hynes, W.; Sloan, M. Secreted Extracellular Virulence Factors. In Streptococcus Pyogenes: Basic Biology to Clinical Manifestations [Internet]; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Jürgens, D. Production and characterization of Escherichia coli enterohemolysin and its effects on the structure of erythrocyte membranes. Cell Biol. Int. 2002, 26, 175–186. [Google Scholar] [CrossRef]
- Krause, M.; Barth, H.; Schmidt, H. Toxins of Locus of Enterocyte Effacement-Negative Shiga Toxin-Producing Escherichia coli. Toxins 2018, 10, 241. [Google Scholar] [CrossRef]
- Jiang, N.; Tan, N.S.; Ho, B.; Ding, J.L. Respiratory protein–generated reactive oxygen species as an antimicrobial strategy. Nat. Immunol. 2007, 8, 1114–1122. [Google Scholar] [CrossRef]
- Alayash, A.I.; Patel, R.P.; Cashon, R.E. Redox Reactions of Hemoglobin and Myoglobin: Biological and Toxicological Implications. Antioxid. Redox Signal. 2001, 3, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Coates, C.J.; Decker, H. Immunological properties of oxygen-transport proteins: Hemoglobin, hemocyanin and hemerythrin. Cell. Mol. Life Sci. 2017, 74, 293–317. [Google Scholar] [CrossRef] [PubMed]
- Bahl, N.; Winarsih, I.; Tucker-Kellogg, L.; Ding, J.L. Extracellular haemoglobin upregulates and binds to tissue factor on macrophages: Implications for coagulation and oxidative stress. Thromb. Haemost. 2014, 111, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016, 13, 432–442. [Google Scholar] [CrossRef]
- Barnes, M.; Heywood, A.E.; Mahimbo, A.; Rahman, B.; Newall, A.T.; Macintyre, C.R. Acute myocardial infarction and influenza: A meta-analysis of case–control studies. Heart 2015, 101, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.J.; Voora, D.; Cyr, D.D.; Lucas, J.E.; Zaas, A.K.; Woods, C.W.; Newby, L.K.; Kraus, W.E.; Ginsburg, G.S. Gene Expression Profiles Link Respiratory Viral Infection, Platelet Response to Aspirin, and Acute Myocardial Infarction. PLoS ONE 2015, 10, e0132259. [Google Scholar] [CrossRef] [PubMed]
- Hauguel-Moreau, M.; El Hajjam, M.; De Baynast, Q.; Vieillard-Baron, A.; Lot, A.-S.; Chinet, T.; Mustafic, H.; Bégué, C.; Carlier, R.Y.; Geri, G.; et al. Occurrence of pulmonary embolism related to COVID-19. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Zawilska, K. Fizjologia hemostazy. In Podstawy Hematologii; Dmoszynska, A., Robak, T., Eds.; Wydawnictwo Czelej: Lublin, Poland, 2015; ISBN 978-83-7563-202-6. [Google Scholar]
- Korzonek-Szlacheta, I.; Zubelewicz-Szkodzińska, B.; Gąsior, M. Płytki krwi—Ogniwo łączące zakrzepicę ze stanem zapalnym. Folia Cardiol. 2018, 13, 303–308. [Google Scholar] [CrossRef]
- Violi, F.; Pignatelli, P. Platelet Oxidative Stress and Thrombosis. Thromb. Res. 2012, 129, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Seno, T.; Inoue, N.; Gao, D.; Okuda, M.; Sumi, Y.; Matsui, K.; Yamada, S.; Hirata, K.; Kawashima, S.; Tawa, R.; et al. Involvement of NADH/NADPH Oxidase in Human Platelet ROS Production. Thromb. Res. 2001, 103, 399–409. [Google Scholar] [CrossRef]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Thomas, M.R.; Henderson, I.R.; Watson, S.P.; Cunningham, A.F. Understanding Infection-Induced Thrombosis: Lessons Learned From Animal Models. Front. Immunol. 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2000 2010, 54, 15–44. [Google Scholar] [CrossRef] [PubMed]
- Proulx, F.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Pediatric Res. 2001, 50, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, H.L.; Turitto, V.T. Rheological aspects of thrombosis and haemostasis: Basic principles and applications. ICTH-Report--Subcommittee on Rheology of the International Committee on Thrombosis and Haemostasis. Thromb. Haemost. 1986, 55, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Westein, E.; Niego, B.; Hagemeyer, C.E. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front. Cardiovasc. Med. 2019, 6, 141. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Orje, J.N.; Habermann, R.; Federici, A.B.; Reininger, A.J. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 2006, 108, 1903–1910. [Google Scholar] [CrossRef]
- Spiel, A.O.; Gilbert, J.C.; Jilma, B. Von Willebrand Factor in Cardiovascular Disease. Circulation 2008, 117, 1449–1459. [Google Scholar] [CrossRef]
- Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Diana, V.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 2017, 5620314. [Google Scholar] [CrossRef] [PubMed]
- Gorog, D.A. Coronary angioplasty enhances platelet reactivity through von Willebrand factor release. Heart 2003, 89, 329–330. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blair, T.A.; Moore, S.F.; Williams, C.M.; Poole, A.W.; Vanhaesebroeck, B.; Hers, I. Phosphoinositide 3-Kinases p110α and p110β Have Differential Roles in Insulin-Like Growth Factor-1–Mediated Akt Phosphorylation and Platelet Priming. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, J.J. Flow Effects on Coagulation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1729–1737. [Google Scholar] [CrossRef]
- Fogelson, A.L.; Neeves, K.B. Fluid Mechanics of Blood Clot Formation. Annu. Rev. Fluid Mech. 2015, 47, 377–403. [Google Scholar] [CrossRef] [PubMed]
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Front. Cardiovasc. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Montoro-García, S.; Schindewolf, M.; Stanford, S.; Larsen, O.; Thiele, T. The Role of Platelets in Venous Thromboembolism. Semin. Thromb. Hemost. 2016, 42, 242–251. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef]
- Fan, L.M.; Douglas, G.; Bendall, J.K.; McNeill, E.; Crabtree, M.J.; Hale, A.B.; Mai, A.; Li, J.-M.; McAteer, M.A.; Schneider, J.E.; et al. Endothelial Cell–Specific Reactive Oxygen Species Production Increases Susceptibility to Aortic Dissection. Circulation 2014, 129, 2661–2672. [Google Scholar] [CrossRef]
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. J. Vasc. Surg. Venous Lymphat. Disord. 2018, 6, 552–553. [Google Scholar] [CrossRef]
- Echeverría, C.; Montorfano, I.; Hermosilla, T.; Armisén, R.; Velásquez, L.A.; Cabello-Verrugio, C.; Varela, D.; Simon, F. Endotoxin Induces Fibrosis in Vascular Endothelial Cells through a Mechanism Dependent on Transient Receptor Protein Melastatin 7 Activity. PLoS ONE 2014, 9, e94146. [Google Scholar] [CrossRef] [PubMed]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and Cytokine Receptors in Advanced Heart Failure. Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [PubMed]
- Fioranelli, M.; Bottaccioli, A.G.; Bottaccioli, F.; Bianchi, M.; Rovesti, M.; Roccia, M.G. Stress and Inflammation in Coronary Artery Disease: A Review Psychoneuroendocrineimmunology-Based. Front. Immunol. 2018, 9, 2031. [Google Scholar] [CrossRef] [PubMed]
- Wheway, J.; Latham, S.L.; Combes, V.; Grau, G.E.R. Endothelial Microparticles Interact with and Support the Proliferation of T Cells. J. Immunol. 2014, 193, 3378–3387. [Google Scholar] [CrossRef]
- Allen, R.C.; Stephens, J.T. Myeloperoxidase Selectively Binds and Selectively Kills Microbes. Infect. Immun. 2011, 79, 474–485. [Google Scholar] [CrossRef]
- Case, A. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, 82. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Cabelli, D.E. Superoxide and Hydroxyl Radical Chemistry in Aqueous Solution. In Active Oxygen in Chemistry; Springer: Dordrecht, The Netherlands, 1995; pp. 66–104. [Google Scholar]
- Li, Y.; Zhu, H.; Kuppusamy, P.; Zweier, J.; Trush, M. Mitochondrial Electron Transport Chain-Derived Superoxide Exits Macrophages: Implications for Mononuclear Cell-Mediated Pathophysiological Processes. React. Oxyg. Species 2016, 1, 81–98. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta Bba Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leukoc. Biol. 2004, 76, 760–781. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.D.; Hsu, A.T.; Ellson, C.D.; Y. Miyazawa, B.; Kong, Y.-W.; Greenwood, J.D.; Dhara, S.; Neal, M.D.; Sperry, J.L.; Park, M.S.; et al. Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clin. Exp. Immunol. 2018, 194, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Galley, J.C.; Straub, A.C. Redox Control of Vascular Function. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Aldosari, S.; Awad, M.; Harrington, E.; Sellke, F.; Abid, M. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 2010, 163, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Arvieux, J.; Regnault, V.; Hachulla, E.; Darnige, L.; Berthou, F.; Youinou, P. Oxidation of beta2-glycoprotein I (beta2GPI) by the hydroxyl radical alters phospholipid binding and modulates recognition by anti-beta2GPI autoantibodies. Arthritis Res. Ther. 2001, 3, P082. [Google Scholar] [CrossRef][Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J.R. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [PubMed]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef]
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat. Res. Rev. Mutat. Res. 2012, 751, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.; Singh, S. Inducible nitric oxide synthase: An asset to neutrophils. J. Leukoc. Biol. 2019, 105, 49–61. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [PubMed]
- Dikshit, M.; Kumari, R.; Srimal, R.C. Pulmonary thromboembolism-induced alterations in nitric oxide release from rat circulating neutrophils. J. Pharmacol. Exp. Ther. 1993, 265, 1369–1373. [Google Scholar] [PubMed]
- Mitsuke, Y.; Lee, J.-D.; Shimizu, H.; Uzui, H.; Iwasaki, H.; Ueda, T. Nitric oxide synthase activity in peripheral polymorphonuclear leukocytes in patients with chronic congestive heart failure. Am. J. Cardiol. 2001, 87, 183–187. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, T.; Möller, S.; Klinger, M.; Solbach, W.; Laskay, T.; Behnen, M. The Impact of Various Reactive Oxygen Species on the Formation of Neutrophil Extracellular Traps. Mediat. Inflamm. 2012, 2012, 849136. [Google Scholar] [CrossRef] [PubMed]
- Paiva, C.N.; Bozza, M.T. Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.-C.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule Protein Processing and Regulated Secretion in Neutrophils. Front. Immunol. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed]
- San José, G.; Bidegain, J.; Robador, P.A.; Díez, J.; Fortuño, A.; Zalba, G. Insulin-induced NADPH oxidase activation promotes proliferation and matrix metalloproteinase activation in monocytes/macrophages. Free Radic. Biol. Med. 2009, 46, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Sowden, M.P.; Berk, B.C. Extracellular and Intracellular Cyclophilin A, Native and Post-Translationally Modified, Show Diverse and Specific Pathological Roles in Diseases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef]
- Yu, W.; Hu, X.; Wang, M. Pterostilbene inhibited advanced glycation end products (AGEs)-induced oxidative stress and inflammation by regulation of RAGE/MAPK/NF-κB in RAW264.7 cells. J. Funct. Foods 2018, 40, 272–279. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 1–14. [Google Scholar]
- Alvarez, B.; Radi, R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids 2003, 25, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, N.M. Toxicity of nitrogen dioxide: An introduction. Toxicology 1994, 89, 161–174. [Google Scholar] [CrossRef]
- Persinger, R.L.; Poynter, M.E.; Ckless, K.; Janssen-Heininger, Y.M.W. Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung. Mol. Cell. Biochem. 2002, 234, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Carmichael, A.J. Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance. Biochem. J. 1992, 281, 795–802. [Google Scholar] [CrossRef]
- Crow, J.P.; Beckman, J.S. Reactions between Nitric Oxide, Superoxide, and Peroxynitrite: Footprints of Peroxynitrite in Vivo. Adv. Pharmacol. 1995, 34, 17–43. [Google Scholar]
- Nauser, T.; Koppenol, W.H. The Rate Constant of the Reaction of Superoxide with Nitrogen Monoxide: Approaching the Diffusion Limit. J. Phys. Chem. A 2002, 106, 4084–4086. [Google Scholar] [CrossRef]
- Münzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.J.; Luchsinger, B.P.; Pawloski, J.R.; Singel, D.J.; Stamler, J.S. The oxyhemoglobin reaction of nitric oxide. Proc. Natl. Acad. Sci. USA 1999, 96, 9027–9032. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatric Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Brzeszczynska, J.; Gwozdzinski, K. Nitric oxide induced oxidative changes in erythrocyte membrane components. Cell Biol. Int. 2008, 32, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Pendurthi, U.R.; Rao, L.V.M. The lipid peroxidation product 4-hydroxy-2-nonenal induces tissue factor decryption via ROS generation and the thioredoxin system. Blood Adv. 2017, 1, 2399–2413. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive Oxygen Species in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 1918. [Google Scholar] [CrossRef] [PubMed]
- Golino, P.; Ragni, M.; Cirillo, P.; Avvdimento, V.E.; Feliciello, A.; Esposito, N.; Scognamiglio, A.; Trimarco, B.; Iaccarino, G.; Condorelli, M.; et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat. Med. 1996, 2, 35–40. [Google Scholar] [CrossRef]
- Cadroy, Y.; Dupouy, D.; Boneu, B.; Plaisancié, H. Polymorphonuclear Leukocytes Modulate Tissue Factor Production by Mononuclear Cells: Role of Reactive Oxygen Species. J. Immunol. 2000, 164, 3822–3828. [Google Scholar] [CrossRef] [PubMed]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Görlach, A. NADPH Oxidase Mediates Tissue Factor—Dependent Surface Procoagulant Activity by Thrombin in Human Vascular Smooth Muscle Cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Lichota, A.; Gwozdzinski, L.; Gwozdzinski, K. Therapeutic potential of natural compounds in inflammation and chronic venous insufficiency. Eur. J. Med. Chem. 2019, 176, 68–91. [Google Scholar] [CrossRef]
- Kovacs, M.J.; Kahn, S.R.; Wells, P.S.; Anderson, D.A.; Chagnon, I.; Le Gal, G.; Solymoss, S.; Crowther, M.; Perrier, A.; Ramsay, T.; et al. Patients with a first symptomatic unprovoked deep vein thrombosis are at higher risk of recurrent venous thromboembolism than patients with a first unprovoked pulmonary embolism. J. Thromb. Haemost. 2010, 8, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Goktay, A.Y.; Senturk, C. Endovascular Treatment of Thrombosis and Embolism. Adv. Exp. Med. Biol. 2017, 906, 195–213. [Google Scholar] [PubMed]
- Shatila, W.; Almanfi, A.; Massumi, M.; Dougherty, K.G.; Parekh, D.R.; Strickman, N.E. Endovascular Treatment of Superior Vena Cava Syndrome via Balloon-in-Balloon Catheter Technique with a Palmaz Stent. Tex. Heart Inst. J. 2016, 43, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Ringleb, P.A. Thrombolytics, Anticoagulants, and Antiplatelet Agents. Stroke 2006, 37, 312–313. [Google Scholar] [CrossRef]
- Sen, C.K.; Roy, S. Wound healing. In Plastic Surgery; Rodriguez, E., Losee, J.E., Neligan, P.C., Eds.; Saunders: Philadelphia, PA, USA; pp. 240–266. ISBN 978-14-557-4047-5. 2012. [Google Scholar]
- Kearon, C.; Akl, E.A.; Ornelas, J.; Blaivas, A.; Jimenez, D.; Bounameaux, H.; Huisman, M.; King, C.S.; Morris, T.A.; Sood, N.; et al. Antithrombotic Therapy for VTE Disease. Chest 2016, 149, 315–352. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, H.; Kim, M.-A.; Choi, J.; Kim, K.-M.; Hwang, J.S.; Na, M.; Bae, J.-S. Evaluation of novel factor Xa inhibitors from Oxya chinensis sinuosa with anti-platelet aggregation activity. Sci. Rep. 2017, 7, 7934. [Google Scholar] [CrossRef]
- Fischer, P.M. Design of Small-Molecule Active-Site Inhibitors of the S1A Family Proteases as Procoagulant and Anticoagulant Drugs. J. Med. Chem. 2018, 61, 3799–3822. [Google Scholar] [CrossRef]
- Zacconi, F.C. FXa Direct Synthetic Inhibitors. In Anticoagulant Drugs; InTech: London, UK, 2018. [Google Scholar]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic Review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef]
- Bratsos, S. Pharmacokinetic Properties of Rivaroxaban in Healthy Human Subjects. Cureus 2019, 11, e5484. [Google Scholar] [CrossRef]
- Rao, P.S.S.; Burkart, T. Advances in oral anticoagulation therapy—What’s in the pipeline? Blood Rev. 2017, 31, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Outes, A.; Terleira-Fernández, A.I.; Calvo-Rojas, G.; Suárez-Gea, M.L.; Vargas-Castrillón, E. Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups. Thrombosis 2013, 2013, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, C.; McCullough, L.D.; Awad, I.A.; Chireau, M.V.; Fedder, W.N.; Furie, K.L.; Howard, V.J.; Lichtman, J.H.; Lisabeth, L.D.; Piña, I.L.; et al. Guidelines for the Prevention of Stroke in Women. Stroke 2014, 45, 1545–1588. [Google Scholar] [CrossRef] [PubMed]
- Yee, J.; Kaide, C. Emergency Reversal of Anticoagulation. West. J. Emerg. Med. 2019, 20, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Meng, J.; Guo, T.; Zhao, J.; Wang, Y.; Wang, J.; Qiu, Y.; Ding, H. Comparison of Apixaban and Low Molecular Weight Heparin in Preventing Deep Venous Thrombosis after Total Knee Arthroplasty in Older Adults. Yonsei Med. J. 2019, 60, 626. [Google Scholar] [CrossRef] [PubMed]
- Skelley, J.W.; Thomason, A.R.; Nolen, J.C. Betrixaban (Bevyxxa): A Direct-Acting Oral Anticoagulant Factor Xa Inhibitor. Pharm. Ther. 2018, 43, 85–88. [Google Scholar]
- Iwatsuki, Y.; Sato, T.; Moritani, Y.; Shigenaga, T.; Suzuki, M.; Kawasaki, T.; Funatsu, T.; Kaku, S. Biochemical and pharmacological profile of darexaban, an oral direct factor Xa inhibitor. Eur. J. Pharmacol. 2011, 673, 49–55. [Google Scholar] [CrossRef]
- Shiraga, T.; Yajima, K.; Suzuki, K.; Suzuki, K.; Hashimoto, T.; Iwatsubo, T.; Miyashita, A.; Usui, T. Identification of UDP-Glucuronosyltransferases Responsible for the Glucuronidation of Darexaban, an Oral Factor Xa Inhibitor, in Human Liver and Intestine. Drug Metab. Dispos. 2012, 40, 276–282. [Google Scholar] [CrossRef]
- Hirayama, F.; Koshio, H.; Ishihara, T.; Hachiya, S.; Sugasawa, K.; Koga, Y.; Seki, N.; Shiraki, R.; Shigenaga, T.; Iwatsuki, Y.; et al. Discovery of N-[2-Hydroxy-6-(4-methoxybenzamido)phenyl]-4- (4-methyl-1,4-diazepan-1-yl)benzamide (Darexaban, YM150) as a Potent and Orally Available Factor Xa Inhibitor. J. Med. Chem. 2011, 54, 8051–8065. [Google Scholar] [CrossRef]
- Clayville, L.R.; Vogel Anderson, K.; Miller, S.A.; Onge, E.L.S. New Options in Anticoagulation for the Prevention Of Venous Thromboembolism and Stroke. PT 2011, 36, 86–99. [Google Scholar]
- Shirley, M.; Dhillon, S. Edoxaban: A Review in Deep Vein Thrombosis and Pulmonary Embolism. Drugs 2015, 75, 2025–2034. [Google Scholar] [CrossRef]
- Brown, D.G.; Wilkerson, E.C.; Love, W.E. A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons. J. Am. Acad. Dermatol. 2015, 72, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Székely, O.; Miyazawa, K.; Lip, G.Y.H. Current and emerging pharmacotherapy for ischemic stroke prevention in patients with atrial fibrillation. Expert Opin. Pharmacother. 2018, 19, 1999–2009. [Google Scholar] [CrossRef]
- Feng, W.; Wu, K.; Liu, Z.; Kong, G.; Deng, Z.; Chen, S.; Wu, Y.; Chen, M.; Liu, S.; Wang, H. Oral direct factor Xa inhibitor versus enoxaparin for thromboprophylaxis after hip or knee arthroplasty: Systemic review, traditional meta-analysis, dose–response meta-analysis and network meta-analysis. Thromb. Res. 2015, 136, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.M.; Weitz, J.I. New anticoagulants: Beyond heparin, low-molecular-weight heparin and warfarin. Br. J. Pharmacol. 2005, 144, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Kamikubo, Y.; Mendolicchio, G.L.; Zampolli, A.; Marchese, P.; Rothmeier, A.S.; Orje, J.N.; Gale, A.J.; Krishnaswamy, S.; Gruber, A.; Østergaard, H.; et al. Selective factor VIII activation by the tissue factor–factor VIIa–factor Xa complex. Blood 2017, 130, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F. Hirudin As Alternative Anticoagulant- A Historical Review. Semin. Thromb. Hemost. 2002, 28, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Ren, J.-X.; Wang, J.-J.; Ding, L.-S.; Zhao, J.-J.; Liu, S.-Y.; Gao, H.-M. Chinese Medicinal Leech: Ethnopharmacology, Phytochemistry, and Pharmacological Activities. Evid. Based Complementary Altern. Med. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Z.; Li, J.; Liu, L. Effects of Natural Hirudin and Low Molecular Weight Heparin in Preventing Deep Venous Thrombosis in Aged Patients with Intertrochanteric Fracture. Sci. Rep. 2018, 8, 8847. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.L. Bivalirudin as an Alternative to Heparin for Anticoagulation in Infants and Children. J. Pediatric Pharmacol. Ther. 2015, 20, 408–417. [Google Scholar] [CrossRef]
- Kimmelstiel, C.; Zhang, P.; Kapur, N.K.; Weintraub, A.; Krishnamurthy, B.; Castaneda, V.; Covic, L.; Kuliopulos, A. Bivalirudin Is a Dual Inhibitor of Thrombin and Collagen-Dependent Platelet Activation in Patients Undergoing Percutaneous Coronary Intervention. Circ. Cardiovasc. Interv. 2011, 4, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, W.; Ali, Z.; Amjad, W.; Alirhayim, Z.; Farooq, H.; Qadir, S.; Khalid, F.; Al-Mallah, M.H. Venous Thromboembolism in Cancer: An Update of Treatment and Prevention in the Era of Newer Anticoagulants. Front. Cardiovasc. Med. 2016, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.W.; Schiering, N.; Melkko, S.; Ewert, S.; Salter, J.; Zhang, Y.; McCormack, P.; Yu, J.; Huang, X.; Chiu, Y.-H.; et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood 2019, 133, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Frigerio, B.; Spadarella, G.; Ravani, A.; Sansaro, D.; Amato, M.; Kitzmiller, J.P.; Pepi, M.; Tremoli, E.; Baldassarre, D. Old and new oral anticoagulants: Food, herbal medicines and drug interactions. Blood Rev. 2017, 31, 193–203. [Google Scholar] [CrossRef]
- Samuelson, B.T.; Cuker, A. Measurement and reversal of the direct oral anticoagulants. Blood Rev. 2017, 31, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Cesarman-Maus, G.; Ruiz-Argüelles, G.J. News in the Indications of Direct Oral Anticoagulants According to the American College of Chest Physicians 2016 Guidelines. Curr. Drug Metab. 2017, 18, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J.E.; Bakhru, S.H.; Laulicht, B.E.; Steiner, S.S.; Grosso, M.A.; Brown, K.; Dishy, V.; Lanz, H.J.; Mercuri, M.F.; Noveck, R.J.; et al. Single-dose ciraparantag safely and completely reverses anticoagulant effects of edoxaban. Thromb. Haemost. 2017, 117, 238–245. [Google Scholar] [CrossRef]
- Okumura, K.; Lip, G.Y.H.; Akao, M.; Tanizawa, K.; Fukuzawa, M.; Abe, K.; Akishita, M.; Yamashita, T. Edoxaban for the management of elderly Japanese patients with atrial fibrillation ineligible for standard oral anticoagulant therapies: Rationale and design of the ELDERCARE-AF study. Am. Heart J. 2017, 194, 99–106. [Google Scholar] [CrossRef]
- Thomson, P.; Brocklebank, C.; Semchuk, W. Treatment Dosing of Low-Molecular-Weight Heparins and the Dose Cap Dilemma: Considerations for Patients in Canada. Can. J. Hosp. Pharm. 2009, 62, 367–374. [Google Scholar] [CrossRef]
- Nadar, S.K.; Goyal, D.; Shantsila, E.; Banerjee, P.; Lip, G.Y. Fondaparinux: An overview. Expert Rev. Cardiovasc. Ther. 2009, 7, 577–585. [Google Scholar] [CrossRef]
- Eriksson, B.; Agnelli, G.; Gallus, A.; Lassen, M.; Prins, M.; Renfurm, R.; Kashiwa, M.; Turpie, A. Darexaban (YM150) versus enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: A randomised phase IIb dose confirmation study (ONYX-3). Thromb. Haemost. 2014, 111, 213–225. [Google Scholar] [PubMed]
- Dimitropoulos, G.; Rahim, S.M.Z.; Moss, A.S.; Lip, G.Y.H. New anticoagulants for venous thromboembolism and atrial fibrillation: What the future holds. Expert Opin. Investig. Drugs 2018, 27, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Choi, S.; Kim, Y.; Lim, H. Population Pharmacokinetic and Pharmacodynamic Modeling Analysis of GCC-4401C, a Novel Direct Factor Xa Inhibitor, in Healthy Volunteers. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Roffel, A.; van Marle, S.; Choi, S.; Lee, H.; Dueker, S.; Tiessen, R. Metabolism and excretion of GCC-4401C, a factor Xa inhibitor, in humans. Int. J. Pharmacokinet. 2017, 2, 93–104. [Google Scholar] [CrossRef]
- Smythe, M.A.; Priziola, J.; Dobesh, P.P.; Wirth, D.; Cuker, A.; Wittkowsky, A.K. Guidance for the practical management of the heparin anticoagulants in the treatment of venous thromboembolism. J. Thromb. Thrombolysis 2016, 41, 165–186. [Google Scholar] [CrossRef]
- Lippi, G.; Gosselin, R.; Favaloro, E.J. Current and Emerging Direct Oral Anticoagulants: State-of-the-Art. Semin. Thromb. Hemost. 2019, 45, 490–501. [Google Scholar] [CrossRef]
- Capodanno, D.; Ferreiro, J.L.; Angiolillo, D.J. Antiplatelet therapy: New pharmacological agents and changing paradigms. J. Thromb. Haemost. 2013, 11, 316–329. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef]
- Harter, K.; Levine, M.; Henderson, S. Anticoagulation Drug Therapy: A Review. West. J. Emerg. Med. 2015, 16, 11–17. [Google Scholar] [CrossRef]
- Rubboli, A.; Patti, G. What is the Role for Glycoprotein IIB/IIIA Inhibitor Use in the Catheterization Laboratory in the Current Era? Curr. Vasc. Pharmacol. 2018, 16, 451–458. [Google Scholar] [CrossRef]
- Eisert, W.G. Dipyridamole in antithrombotic treatment. Adv. Interv. Cardiol. 2012, 47, 78–86. [Google Scholar]
- Kim, H.-H.; Liao, J.K. Translational Therapeutics of Dipyridamole. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Goto, S. Cilostazol: Potential mechanism of action for antithrombotic effects accompanied by a low rate of bleeding. Atheroscler. Suppl. 2005, 6, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Niu, P.-P.; Guo, Z.-N.; Jin, H.; Xing, Y.-Q.; Yang, Y. Antiplatelet regimens in the long-term secondary prevention of transient ischaemic attack and ischaemic stroke: An updated network meta-analysis. BMJ Open 2016, 6, e009013. [Google Scholar] [CrossRef]
- Steinhubl, S.R.; Badimon, J.J.; Bhatt, D.L.; Herbert, J.-M.; Lüscher, T.F. Clinical evidence for anti-inflammatory effects of antiplatelet therapy in patients with atherothrombotic disease. Vasc. Med. 2007, 12, 113–122. [Google Scholar] [CrossRef]
- Quinn, M.J.; Fitzgerald, D.J. Ticlopidine and Clopidogrel. Circulation 1999, 100, 1667–1672. [Google Scholar] [CrossRef]
- Jiang, M.; You, J.H. Review of pharmacoeconomic evaluation of genotype-guided antiplatelet therapy. Expert Opin. Pharmacother. 2015, 16, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Majithia, A.; Bhatt, D.L. Novel Antiplatelet Therapies for Atherothrombotic Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 546–557. [Google Scholar] [CrossRef]
- Tscharre, M.; Michelson, A.D.; Gremmel, T. Novel Antiplatelet Agents in Cardiovascular Disease. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Crain, E.; Watson, C.; Yang, W.; Wexler, R.; Lam, P.; Rehfuss, R.; Schumacher, W. Differential effects of P2Y1 versus P2Y12 receptor antagonism on thrombosis and bleeding in rabbits. Eur. Heart J. 2013, 34, P1431. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Lai, A.; Qiao, J.X.; Wang, T.C.; Hua, J.; Price, L.A.; Shen, H.; Chen, X.; Wong, P.; et al. Discovery of 4-Aryl-7-Hydroxyindoline-Based P2Y 1 Antagonists as Novel Antiplatelet Agents. J. Med. Chem. 2014, 57, 6150–6164. [Google Scholar] [CrossRef] [PubMed]
- Kolandaivelu, K.; Bhatt, D.L. Novel Antiplatelet Therapies. In Platelets; Elsevier: Amsterdam, The Netherland, 2019; pp. 991–1015. [Google Scholar]
- Bach, P.; Antonsson, T.; Bylund, R.; Björkman, J.-A.; Österlund, K.; Giordanetto, F.; van Giezen, J.J.J.; Andersen, S.M.; Zachrisson, H.; Zetterberg, F. Lead Optimization of Ethyl 6-Aminonicotinate Acyl Sulfonamides as Antagonists of the P2Y 12 Receptor. Separation of the Antithrombotic Effect and Bleeding for Candidate Drug AZD1283. J. Med. Chem. 2013, 56, 7015–7024. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, M.; Furukawa, M.; Goldsberry, S.; Movahed, M.R. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Exp. Clin. Cardiol. 2008, 13, 192–197. [Google Scholar]
- Stoffer, K.; Shah, S. Abciximab; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Bansal, A.B.; Sattar, Y.; Jamil, R.T. Eptifibatide. Drugs 1999, 57, 439–462. [Google Scholar]
- Zhao, C.; Cheng, G.; He, R.; Guo, H.; Li, Y.; Lu, X.; Zhang, Y.; Qiu, C. Effects of different routes of tirofiban injection on the left ventricular function and prognosis of patients with myocardial infarction treated with percutaneous coronary intervention. Exp. Ther. Med. 2015, 9, 2401–2405. [Google Scholar] [CrossRef] [PubMed]
- Beavers, C.J.; Naqvi, I.A. Clopidogrel. Drugs 1997, 54, 745–750. [Google Scholar]
- Hermanides, R.S.; Kilic, S.; van ’t Hof, A.W.J. Optimal pharmacological therapy in ST-elevation myocardial infarction—a review. Neth. Heart J. 2018, 26, 296–310. [Google Scholar] [CrossRef]
- Layne, K.; Ferro, A. Antiplatelet Therapy in Acute Coronary Syndrome. Eur. Cardiol. Rev. 2017, 12, 33. [Google Scholar] [CrossRef]
- Balinski, A.M.; Preuss, C.V. Cilostazol. Drugs Aging 1999, 14, 63–71. [Google Scholar]
- Royston, D. Anticoagulant and Antiplatelet Therapy. In Pharmacology and Physiology for Anesthesia; Elsevier: Amsterdam, The Netherlands, 2019; pp. 870–894. [Google Scholar]
- Desager, J.-P. Clinical Pharmacokinetics of Ticlopidine. Clin. Pharmacokinet. 1994, 26, 347–355. [Google Scholar] [CrossRef]
- Juif, P.; Boehler, M.; Dobrow, M.; Ufer, M.; Dingemanse, J. Clinical Pharmacology of the Reversible and Potent P2Y 12 Receptor Antagonist ACT-246475 After Single Subcutaneous Administration in Healthy Male Subjects. J. Clin. Pharmacol. 2019, 59, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; Gurbel, P.A.; ten Berg, J.; Bernaud, C.; Dangas, G.D.; Frenoux, J.-M.; Gorog, D.A.; Hmissi, A.; Kunadian, V.; James, S.K.; et al. Pharmacodynamics, pharmacokinetics, and safety of single-dose subcutaneous administration of selatogrel, a novel P2Y12 receptor antagonist, in patients with chronic coronary syndromes. Eur. Heart J. 2020, 41, 3132–3140. [Google Scholar] [CrossRef]
- Zhu, J.; Choi, W.-S.; McCoy, J.G.; Negri, A.; Zhu, J.; Naini, S.; Li, J.; Shen, M.; Huang, W.; Bougie, D.; et al. Structure-Guided Design of a High-Affinity Platelet Integrin IIb 3 Receptor Antagonist That Disrupts Mg2+ Binding to the MIDAS. Sci. Transl. Med. 2012, 4, 125ra32. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Vootukuri, S.; Shang, Y.; Negri, A.; Jiang, J.; Nedelman, M.; Diacovo, T.G.; Filizola, M.; Thomas, C.J.; Coller, B.S. RUC-4. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Vootukuri, S.; Li, J.; Nedelman, M.; Thomas, C.; Jiang, J.-K.; Babayeva, M.; Coller, B.S. Preclinical studies of RUC-4, a novel platelet αIIbβ3 antagonist, in non-human primates and with human platelets. J. Clin. Transl. Sci. 2019, 3, 65–74. [Google Scholar] [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Gryka, R.J.; Buckley, L.F.; Anderson, S.M. Vorapaxar: The Current Role and Future Directions of a Novel Protease-Activated Receptor Antagonist for Risk Reduction in Atherosclerotic Disease. Drugs Rd 2017, 17, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Ismat, F.A.; Wang, Z.; Cerra, M.; Narayan, H.; Raftis, J.; Gray, T.J.; Connell, S.; Garonzik, S.; Ma, X.; et al. PAR4 (Protease-Activated Receptor 4) Antagonism With BMS-986120 Inhibits Human Ex Vivo Thrombus Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 448–456. [Google Scholar] [CrossRef]
- Dumas, M.; Nadal-Wollbold, F.; Gaussem, P.; Perez, M.; Mirault, T.; Létienne, R.; Bourbon, T.; Grelac, F.; Le Grand, B.; Bachelot-Loza, C. Antiplatelet and antithrombotic effect of F 16618, a new thrombin proteinase-activated receptor-1 (PAR1) antagonist. Br. J. Pharmacol. 2012, 165, 1827–1835. [Google Scholar] [CrossRef]
- Chieng-Yane, P.; Bocquet, A.; Létienne, R.; Bourbon, T.; Sablayrolles, S.; Perez, M.; Hatem, S.N.; Lompré, A.-M.; Le Grand, B.; David-Dufilho, M. Protease-Activated Receptor-1 Antagonist F 16618 Reduces Arterial Restenosis by Down-Regulation of Tumor Necrosis Factor α and Matrix Metalloproteinase 7 Expression, Migration, and Proliferation of Vascular Smooth Muscle Cells. J. Pharmacol. Exp. Ther. 2011, 336, 643–651. [Google Scholar] [CrossRef]
- Di Serio, C.; Pellerito, S.; Duarte, M.; Massi, D.; Naldini, A.; Cirino, G.; Prudovsky, I.; Santucci, M.; Geppetti, P.; Marchionni, N.; et al. Protease-Activated Receptor 1-Selective Antagonist SCH79797 Inhibits Cell Proliferation and Induces Apoptosis by a Protease-Activated Receptor 1-Independent Mechanism. Basic Clin. Pharmacol. Toxicol. 2007, 101, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, K.; Ohnishi, T.; Miura, N.; Sameshima, H.; Koide, T.; Tanaka, K.A.; Maruyama, I. Antithrombotic effects of PAR1 and PAR4 antagonists evaluated under flow and static conditions. Thromb. Res. 2014, 133, 66–72. [Google Scholar] [CrossRef]
- Chen, Z.; Li, T.; Kareem, K.; Tran, D.; Griffith, B.P.; Wu, Z.J. The role of PI3K/Akt signaling pathway in non-physiological shear stress-induced platelet activation. Artif. Organs 2019, 43, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, S.A.; Jones, C.; Angus, J.A.; Wright, C.E. Advantages of a selective β-isoform phosphoinositide 3-kinase antagonist, an anti-thrombotic agent devoid of other cardiovascular actions in the rat. Eur. J. Pharmacol. 2008, 587, 209–215. [Google Scholar] [CrossRef]
- Laurent, P.-A.; Séverin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.-P. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef]
- Xu, P.-F.; Yang, J.-A.; Liu, J.-H.; Yang, X.; Liao, J.-M.; Yuan, F.-E.; Liu, B.-H.; Chen, Q.-X. PI3Kβ inhibitor AZD6482 exerts antiproliferative activity and induces apoptosis in human glioblastoma cells. Oncol. Rep. 2019, 41, 125–132. [Google Scholar] [CrossRef]
- Luci, D.; Jameson, J.B.; Yasgar, A.; Diaz, G.; Joshi, N.; Kantz, A.; Markham, K.; Perry, S.; Kuhn, N.; Yeung, J.; et al. Discovery of ML355, a Potent and Selective Inhibitor of Human 12-Lipoxygenase; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Adili, R.; Tourdot, B.E.; Mast, K.; Yeung, J.; Freedman, J.C.; Green, A.; Luci, D.K.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; et al. First Selective 12-LOX Inhibitor, ML355, Impairs Thrombus Formation and Vessel Occlusion In Vivo With Minimal Effects on Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1828–1839. [Google Scholar] [CrossRef]
- Gilbert, J.C.; DeFeo-Fraulini, T.; Hutabarat, R.M.; Horvath, C.J.; Merlino, P.G.; Marsh, H.N.; Healy, J.M.; BouFakhreddine, S.; Holohan, T.V.; Schaub, R.G. First-in-Human Evaluation of Anti–von Willebrand Factor Therapeutic Aptamer ARC1779 in Healthy Volunteers. Circulation 2007, 116, 2678–2686. [Google Scholar] [CrossRef]
- Huang, R.-H.; Fremont, D.H.; Diener, J.L.; Schaub, R.G.; Sadler, J.E. A Structural Explanation for the Antithrombotic Activity of ARC1172, a DNA Aptamer that Binds von Willebrand Factor Domain A1. Structure 2009, 17, 1476–1484. [Google Scholar] [CrossRef]
- Spiel, A.O.; Mayr, F.B.; Ladani, N.; Wagner, P.G.; Schaub, R.G.; Gilbert, J.C.; Jilma, B. The aptamer ARC1779 is a potent and specific inhibitor of von willebrand factor mediated ex vivo platelet function in acute myocardial infarction. Platelets 2009, 20, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Jilma-Stohlawetz, P.; Gilbert, J.; Gorczyca, M.; Knöbl, P.; Jilma, B. A dose ranging phase I/II trial of the von Willebrand factor inhibiting aptamer ARC1779 in patients with congenital thrombotic thrombo—Cytopenic purpura. Thromb. Haemost. 2011, 106, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Yamamoto, H.; Nagano, M.; Arisaka, H.; Kayahara, T.; Yoshimoto, R. Anti-thrombotic effects and bleeding risk of AJvW-2, a monoclonal antibody against human von Willebrand factor. Br. J. Pharmacol. 1997, 122, 165–171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Siller-Matula, J.M.; Merhi, Y.; Tanguay, J.-F.; Duerschmied, D.; Wagner, D.D.; McGinness, K.E.; Pendergrast, P.S.; Chung, J.-K.; Tian, X.; Schaub, R.G.; et al. ARC15105 Is a Potent Antagonist of Von Willebrand Factor Mediated Platelet Activation and Adhesion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, A.; Metjian, A. Caplacizumab in adult patients with acquired thrombotic thrombocytopenic purpura. Ther. Adv. Hematol. 2020, 11. [Google Scholar] [CrossRef]
- Bartunek, J.; Barbato, E.; Heyndrickx, G.; Vanderheyden, M.; Wijns, W.; Holz, J.-B. Novel Antiplatelet Agents: ALX-0081, a Nanobody Directed towards von Willebrand Factor. J. Cardiovasc. Transl. Res. 2013, 6, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Mao, Y.; Abdelgawwad, M.S.; Kocher, N.K.; Li, M.; Dai, X.; Li, B.; Zheng, X.L. Therapeutic efficacy of the platelet glycoprotein Ib antagonist anfibatide in murine models of thrombotic thrombocytopenic purpura. Blood Adv. 2016, 1, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Huo, J.; Xu, W.; Xiong, J.; Li, Y.; Wu, W. A novel anti-platelet aggregation tripeptide from Agkistrodon acutus venom: Isolation and characterization. Toxicon 2009, 54, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb. Haemost. 2014, 111, 279–289. [Google Scholar]
- Li, T.-T.; Fan, M.-L.; Hou, S.-X.; Li, X.-Y.; Barry, D.M.; Jin, H.; Luo, S.-Y.; Kong, F.; Lau, L.-F.; Dai, X.-R.; et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br. J. Pharmacol. 2015, 172, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Yoshida, H.; Satoh, K. Regulation of CX3CL1/Fractalkine Expression in Endothelial Cells. J. Atheroscler. Thromb. 2004, 11, 15–21. [Google Scholar] [CrossRef]
- Stolla, M.; Pelisek, J.; von Brühl, M.-L.; Schäfer, A.; Barocke, V.; Heider, P.; Lorenz, M.; Tirniceriu, A.; Steinhart, A.; Bauersachs, J.; et al. Fractalkine Is Expressed in Early and Advanced Atherosclerotic Lesions and Supports Monocyte Recruitment via CX3CR1. PLoS ONE 2012, 7, e43572. [Google Scholar] [CrossRef] [PubMed]
- Riopel, M.; Vassallo, M.; Ehinger, E.; Pattison, J.; Bowden, K.; Winkels, H.; Wilson, M.; de Jong, R.; Patel, S.; Balakrishna, D.; et al. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol. Metab. 2019, 20, 89–101. [Google Scholar] [CrossRef]
- Gailani, D.; Gruber, A. Factor XI as a Therapeutic Target. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Wallisch, M.; Lorentz, C.U.; Lakshmanan, H.H.S.; Johnson, J.; Carris, M.R.; Puy, C.; Gailani, D.; Hinds, M.T.; McCarty, O.J.T.; Gruber, A.; et al. Antibody inhibition of contact factor XII reduces platelet deposition in a model of extracorporeal membrane oxygenator perfusion in nonhuman primates. Res. Pract. Thromb. Haemost. 2020, 4, 205–216. [Google Scholar] [CrossRef]
- Büller, H.R.; Bethune, C.; Bhanot, S.; Gailani, D.; Monia, B.P.; Raskob, G.E.; Segers, A.; Verhamme, P.; Weitz, J.I. Factor XI Antisense Oligonucleotide for Prevention of Venous Thrombosis. N. Engl. J. Med. 2015, 372, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Thelen, K.; Kraff, S.; Schwers, S.; Schiffer, S.; Unger, S.; Yassen, A.; Boxnick, S. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: First evaluation of safety, pharmacodynamics, and pharmacokinetics. Res. Pract. Thromb. Haemost. 2019, 3, 242–253. [Google Scholar] [CrossRef]
- Woodruff, R.S.; Ivanov, I.; Verhamme, I.M.; Sun, M.-F.; Gailani, D.; Sullenger, B.A. Generation and characterization of aptamers targeting factor XIa. Thromb. Res. 2017, 156, 134–141. [Google Scholar] [CrossRef]
- Hayward, N.J.; Goldberg, D.I.; Morrel, E.M.; Friden, P.M.; Bokesch, P.M. Phase 1a/1b study of EP-7041: A novel, potent, selective, small molecule FXIa inhibitor. Circulation 2017, 136, A13747. [Google Scholar]
- Phang, M.; Lazarus, S.; Wood, L.; Garg, M. Diet and Thrombosis Risk: Nutrients for Prevention of Thrombotic Disease. Semin. Thromb. Hemost. 2011, 37, 199–208. [Google Scholar] [CrossRef]
- Vazhappilly, C.G.; Ansari, S.A.; Al-Jaleeli, R.; Al-Azawi, A.M.; Ramadan, W.S.; Menon, V.; Hodeify, R.; Siddiqui, S.S.; Merheb, M.; Matar, R.; et al. Role of flavonoids in thrombotic, cardiovascular, and inflammatory diseases. Inflammopharmacology 2019, 27, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E. Oxidative Stress and Platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 11–16. [Google Scholar] [CrossRef]
- Faggio, C.; Sureda, A.; Morabito, S.; Sanches-Silva, A.; Mocan, A.; Nabavi, S.F.; Nabavi, S.M. Flavonoids and platelet aggregation: A brief review. Eur. J. Pharmacol. 2017, 807, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Nazmeen, A.; Medda, N.; Patra, R.; Ghosh, T.K. Flavonoids green tea against oxidant stress and inflammation with related human diseases. Clin. Nutr. Exp. 2019, 24, 1–14. [Google Scholar] [CrossRef]
- Kosakowska, O. Experimental Paper. Intrapopulation variability of flavonoid content in roots of Baikal skullcap (Scutellaria baicalensis Georgi). Herba Pol. 2017, 63, 20–31. [Google Scholar] [CrossRef]
- Ryu, R.; Jung, U.J.; Seo, Y.-R.; Kim, H.-J.; Moon, B.S.; Bae, J.-S.; Lee, D.G.; Choi, M.-S. Beneficial effect of persimmon leaves and bioactive compounds on thrombosis. Food Sci. Biotechnol. 2015, 24, 233–240. [Google Scholar] [CrossRef]
- Khan, M.M.; Quoc Tran, B.; Jang, Y.-J.; Park, S.-H.; Fondrie, W.E.; Chowdhury, K.; Hwan Yoon, S.; Goodlett, D.R.; Chae, S.-W.; Chae, H.-J.; et al. Assessment of the Therapeutic Potential of Persimmon Leaf Extract on Prediabetic Subjects. Mol. Cells 2017, 40, 466–475. [Google Scholar]
- Zhang, Y.-X.; Yang, T.-T.; Xia, L.; Zhang, W.-F.; Wang, J.-F.; Wu, Y.-P. Inhibitory Effect of Propolis on Platelet Aggregation In Vitro. J. Healthc. Eng. 2017, 2017, 3050895. [Google Scholar] [CrossRef]
- Lu, W.-J.; Lin, K.-C.; Liu, C.-P.; Lin, C.-Y.; Wu, H.-C.; Chou, D.-S.; Geraldine, P.; Huang, S.-Y.; Hsieh, C.-Y.; Sheu, J.-R. Prevention of arterial thrombosis by nobiletin: In vitro and in vivo studies. J. Nutr. Biochem. 2016, 28, 1–8. [Google Scholar] [CrossRef]
- Ku, S.-K.; Bae, J.-S. Antithrombotic activities of wogonin and wogonoside via inhibiting platelet aggregation. Fitoterapia 2014, 98, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.-L.; Da, X.-W.; He, A.-D.; Yao, G.-Q.; Xie, W.; Liu, G.; Xiang, J.-Z.; Ming, Z.-Y. Pentamethylquercetin (PMQ) reduces thrombus formation by inhibiting platelet function. Sci. Rep. 2015, 5, 11142. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Zhang, H.; Li, Y.; Zhou, Y.; Lu, W.; Yao, L. The effect of Diosmin on the blood proteome in a rat model of venous thrombosis. Int. J. Biol. Macromol. 2017, 104, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.-W.; Wu, M.-P.; Velusamy, M.; Hsia, C.-H.; Chou, D.-S.; Tsai, C.-L.; Hsu, C.-Y.; Jayakumar, T.; Chung, C.-L.; Sheu, J.-R. Novel Therapeutic Agent against Platelet Activation In Vitro and Arterial Thrombosis In Vivo by Morin Hydrate. Int. J. Mol. Sci. 2018, 19, 2386. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Son, Y.K.; Han, Y.N. Tissue factor inhibitory flavonoids from the fruits ofChaenomeles sinensis. Arch. Pharmacal Res. 2002, 25, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Karlíčková, J.; Říha, M.; Filipský, T.; Macáková, K.; Hrdina, R.; Mladěnka, P. Antiplatelet Effects of Flavonoids Mediated by Inhibition of Arachidonic Acid Based Pathway. Planta Med. 2015, 82, 76–83. [Google Scholar] [CrossRef]
- Dasgupta, A. Antiinflammatory Herbal Supplements. In Translational Inflammation; Elsevier: Amsterdam, The Netherlands, 2019; pp. 69–91. [Google Scholar]
- Liakopoulou, C.; Kazazis, C.; Vallianou, N.G. Silimarin and Cancer. Anti-Cancer Agents Med. Chem. 2019, 18, 1970–1974. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Dziedzic, A.; Saluk-Bijak, J. Flavonolignans reduce the response of blood platelet to collagen. Int. J. Biol. Macromol. 2018, 106, 878–884. [Google Scholar] [CrossRef]
- Bijak, M.; Szelenberger, R.; Dziedzic, A.; Saluk-Bijak, J. Inhibitory Effect of Flavonolignans on the P2Y12 Pathway in Blood Platelets. Molecules 2018, 23, 374. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Kang, S.; Moon, H.; Chung, K.H.; Kim, K.R. Antiplatelet and Antithrombotic Effects of the Extract of Lindera obtusiloba Leaves. Biomol. Ther. 2016, 24, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Liu, L.; Li, J.; Qu, L.; Pang, X.; Yu, H.; Zhang, Y.; Wang, T. Bioactive flavonoids from Flos Sophorae. J. Nat. Med. 2017, 71, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, F.-Q.; Zhang, Q.; Wang, F.-Q.; Hu, Y.-J.; Xia, Z.-N. Natural Products for Antithrombosis. Evid. Based Complementary Altern. Med. 2015, 2015, 876426. [Google Scholar] [CrossRef] [PubMed]
- Kregiel, D.; Berlowska, J.; Witonska, I.; Antolak, H.; Proestos, C.; Babic, M.; Babic, L.; Zhang, B. Saponin-Based, Biological-Active Surfactants from Plants. In Application and Characterization of Surfactants; InTech: London, UK, 2017. [Google Scholar]
- Singh, D.; Chaudhuri, P.K. Structural characteristics, bioavailability and cardioprotective potential of saponins. Integr. Med. Res. 2018, 7, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Kwon, H.-W.; Cho, H.-J.; Rhee, M.H.; Park, H.-J. Vasodilator-stimulated phosphoprotein-phosphorylation by ginsenoside Ro inhibits fibrinogen binding to αIIb/β3 in thrombin-induced human platelets. J. Ginseng Res. 2016, 40, 359–365. [Google Scholar] [CrossRef]
- Shen, Q.; Li, J.; Zhang, C.; Wang, P.; Mohammed, A.; Ni, S.; Tang, Z. Panax notoginseng saponins reduce high-risk factors for thrombosis through peroxisome proliferator-activated receptor -γ pathway. Biomed. Pharmacother. 2017, 96, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Meng, X.; Zhai, Y.; Zhou, P.; Ye, T.; Wang, Z.; Sun, G.; Sun, X. Panax Notoginseng Saponins: A Review of Its Mechanisms of Antidepressant or Anxiolytic Effects and Network Analysis on Phytochemistry and Pharmacology. Molecules 2018, 23, 940. [Google Scholar] [CrossRef]
- Thom, V.; Tung, N.; Van Diep, D.; Thuy, D.; Hue, N.; Long, D.; Tung, B.; Huyen, P.; Ly Huong, D. Antithrombotic activity and saponin composition of the roots of Panax bipinnatifidus Seem. growing in Vietnam. Pharmacogn. Res. 2018, 10, 333. [Google Scholar]
- Zhai, K.; Zheng, J.; Tang, Y.; Li, F.; Lv, Y.; Zhang, Y.; Gao, Z.; Qi, J.; Yu, B.; Kou, J. The saponin D39 blocks dissociation of non-muscular myosin heavy chain IIA from TNF receptor 2, suppressing tissue factor expression and venous thrombosis. Br. J. Pharmacol. 2017, 174, 2818–2831. [Google Scholar] [CrossRef]
- Yu, B.; Tian, Y.; Ma, S.; Lin, B.; Kou, J. Anti-thrombotic activity of DT-13, a saponin isolated from the root tuber of Liriope muscari. Indian J. Pharmacol. 2013, 45, 283. [Google Scholar] [CrossRef]
- Kou, J.; Yu, B.; Xu, Q. Inhibitory effects of ethanol extract from Radix Ophiopogon japonicus on venous thrombosis linked with its endothelium-protective and anti-adhesive activities. Vasc. Pharmacol. 2005, 43, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Tian, Y.; Tang, Y.; Yan, J.; Yu, B. Antithrombotic Activities of Aqueous Extract from Radix Ophiopogon japonicus and Its Two Constituents. Biol. Pharm. Bull. 2006, 29, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Huang, Y. Chinese Herbal Medicine on Cardiovascular Diseases and the Mechanisms of Action. Front. Pharmacol. 2016, 7, 469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, B.; Du, D.; Guo, X.; Xin, G.; Xing, Z.; Liang, Y.; Chen, Y.; Chen, Q.; He, Y.; et al. Anti-thrombosis effect of diosgenyl saponins in vitro and in vivo. Steroids 2013, 78, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.P.; Li, J.F.; Yan, J.; Zhu, D.N. Effect of total saponin from root of Polygala fallax Hesml. (PTS) on coagulation and thrombosis. J. China Pharm. Univ. 2003, 34, 63–65. [Google Scholar]
- Yoshikawa, M.; Matsuda, H. Chemical and pharmacological studies on triterpene saponins, escins, from horse chestnut seeds. In Saponins in Food, Feedstuffs and Medicinal Plants; Oleszek, W., Marston, A., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 189–203. ISBN 978-90-481-5341-1. [Google Scholar]
- Wang, M.; Li, H.; Xu, F.; Gao, X.; Li, J.; Xu, S.; Zhang, D.; Wu, X.; Xu, J.; Hua, H.; et al. Diterpenoid lead stevioside and its hydrolysis products steviol and isosteviol: Biological activity and structural modification. Eur. J. Med. Chem. 2018, 156, 885–906. [Google Scholar] [CrossRef]
- Geuns, J.M. Stevioside. Phytochemistry 2003, 64, 913–921. [Google Scholar] [CrossRef]
- Carrera-Lanestosa, A.; Moguel-Ordóñez, Y.; Segura-Campos, M. Stevia rebaudiana Bertoni: A Natural Alternative for Treating Diseases Associated with Metabolic Syndrome. J. Med. Food 2017, 20, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruiz, J.C.; Moguel-Ordoñez, Y.B.; Segura-Campos, M.R. Biological activity of Stevia rebaudiana Bertoni and their relationship to health. Crit. Rev. Food Sci. Nutr. 2017, 57, 2680–2690. [Google Scholar] [CrossRef]
- Salehi, B.; López, M.D.; Martínez-López, S.; Victoriano, M.; Sharifi-Rad, J.; Martorell, M.; Rodrigues, C.F.; Martins, N. Stevia rebaudiana Bertoni bioactive effects: From in vivo to clinical trials towards future therapeutic approaches. Phytother. Res. 2019, 33, 2904–2917. [Google Scholar] [CrossRef]
- KKak, S. Nrf2: A key regulator of redox signaling in liver diseases. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Waltham, MA, USA, 2017; pp. 472–479. ISBN 978-0-12-804274-8. [Google Scholar]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Boonkaewwan, C.; Burodom, A. Anti-inflammatory and immunomodulatory activities of stevioside and steviol on colonic epithelial cells. J. Sci. Food Agric. 2013, 93, 3820–3825. [Google Scholar] [CrossRef] [PubMed]
- Yingkun, N.; Zhenyu, W.; Jing, L.; Xiuyun, L.; Huimin, Y. Stevioside Protects LPS-Induced Acute Lung Injury in Mice. Inflammation 2013, 36, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Boonkaewwan, C.; Ao, M.; Toskulkao, C.; Rao, M.C. Specific Immunomodulatory and Secretory Activities of Stevioside and Steviol in Intestinal Cells. J. Agric. Food Chem. 2008, 56, 3777–3784. [Google Scholar] [CrossRef]
- Bridel, M.; Lavieille, R. The sweet principle in Kaa-he-e (Stevia rebaudiana. Bertoni). II. Hydrolysis of stevioside by enzymes. III. Steviol by enzymic hydrolysis and isosteviol by acid hydrolysis. Bull. Soc. Chim. Biol. 1931, 13, 781–796. [Google Scholar]
- Ullah, A.; Munir, S.; Mabkhot, Y.; Badshah, S. Bioactivity Profile of the Diterpene Isosteviol and its Derivatives. Molecules 2019, 24, 678. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, D.; Li, M.; Wu, Q.; Lam, Y.P.Y.; Guo, Y.; Chen, C.; Bai, N.; Malhotra, S.; Li, W.; et al. Discovery of novel, potent, isosteviol-based antithrombotic agents. Eur. J. Med. Chem. 2019, 183, 111722. [Google Scholar] [CrossRef] [PubMed]






















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).