Kinin B2 Receptor Activation Prevents the Evolution of Alzheimer’s Disease Pathological Characteristics in a Transgenic Mouse Model

 , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
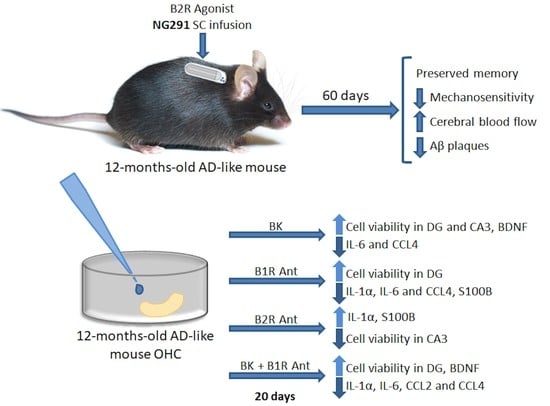
2.1. Kinin B2R Agonist Preserves Spatial Memory of Transgenic Alzheimer’s Disease Mice
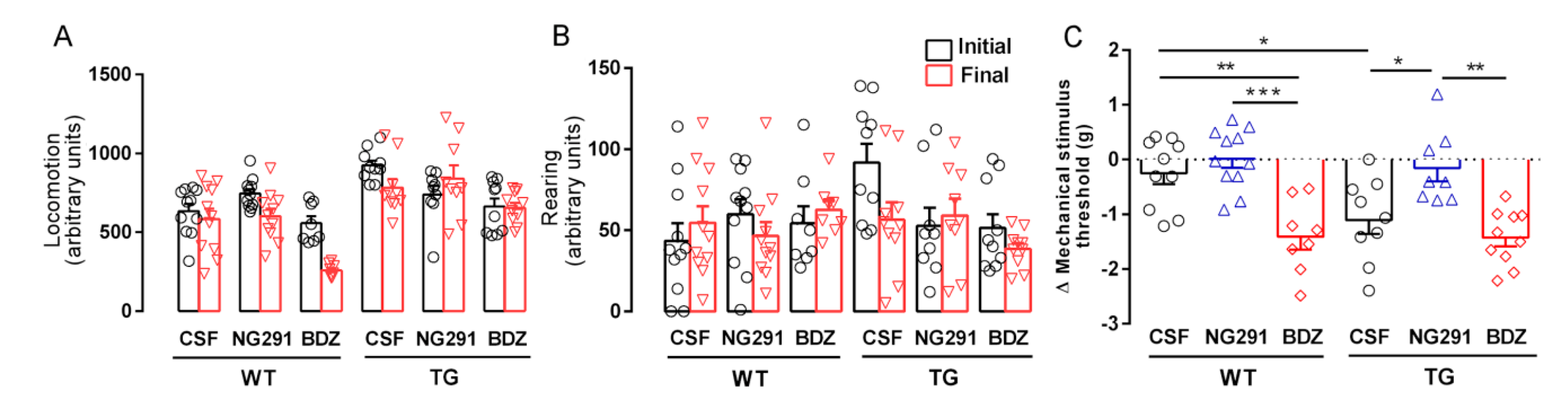
2.2. B2R Does Not Alter Motor Activity but Plays a Role in Animal Sensitivity
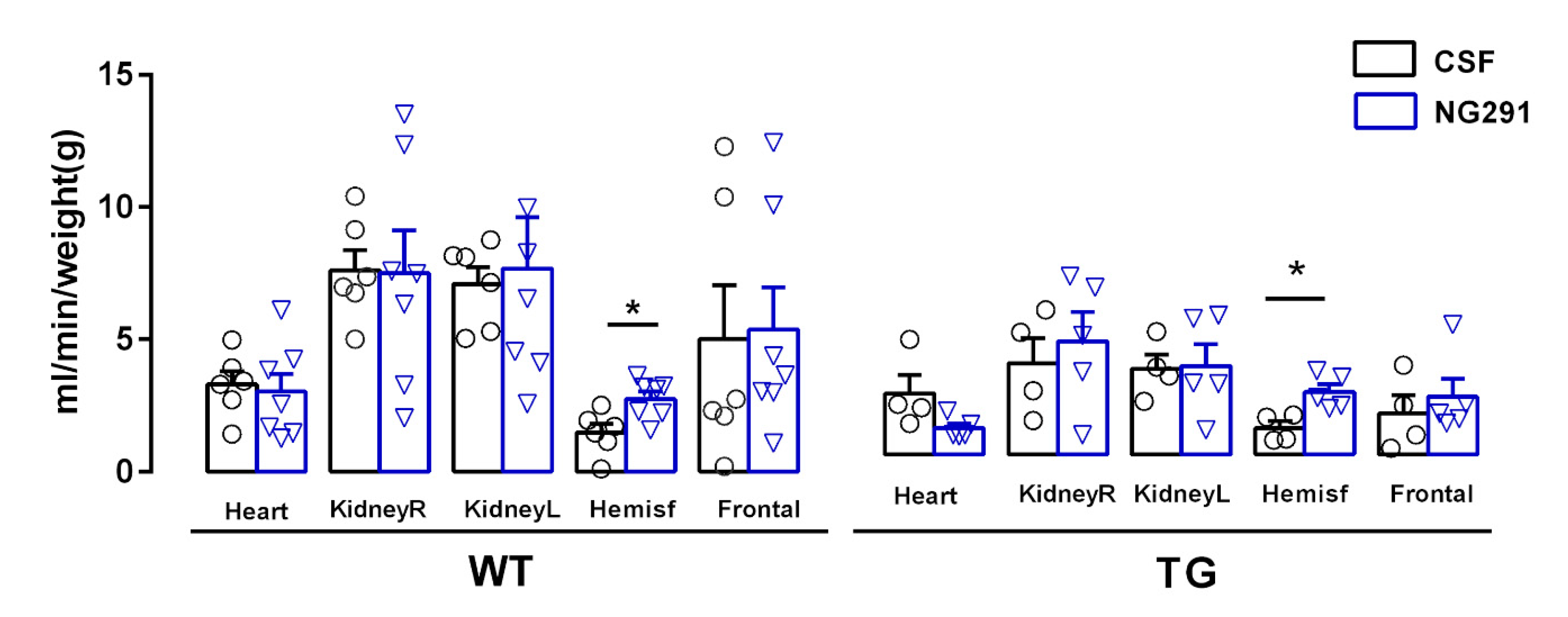
2.3. B2R Agonist Increases Cerebral Blood Flow of WT and TG Animals
2.4. Kinin B2R Agonist Decreased Amyloid Plaques in TG Animals
2.5. Treatment with Different KKS Modulators Increased Cell Viability
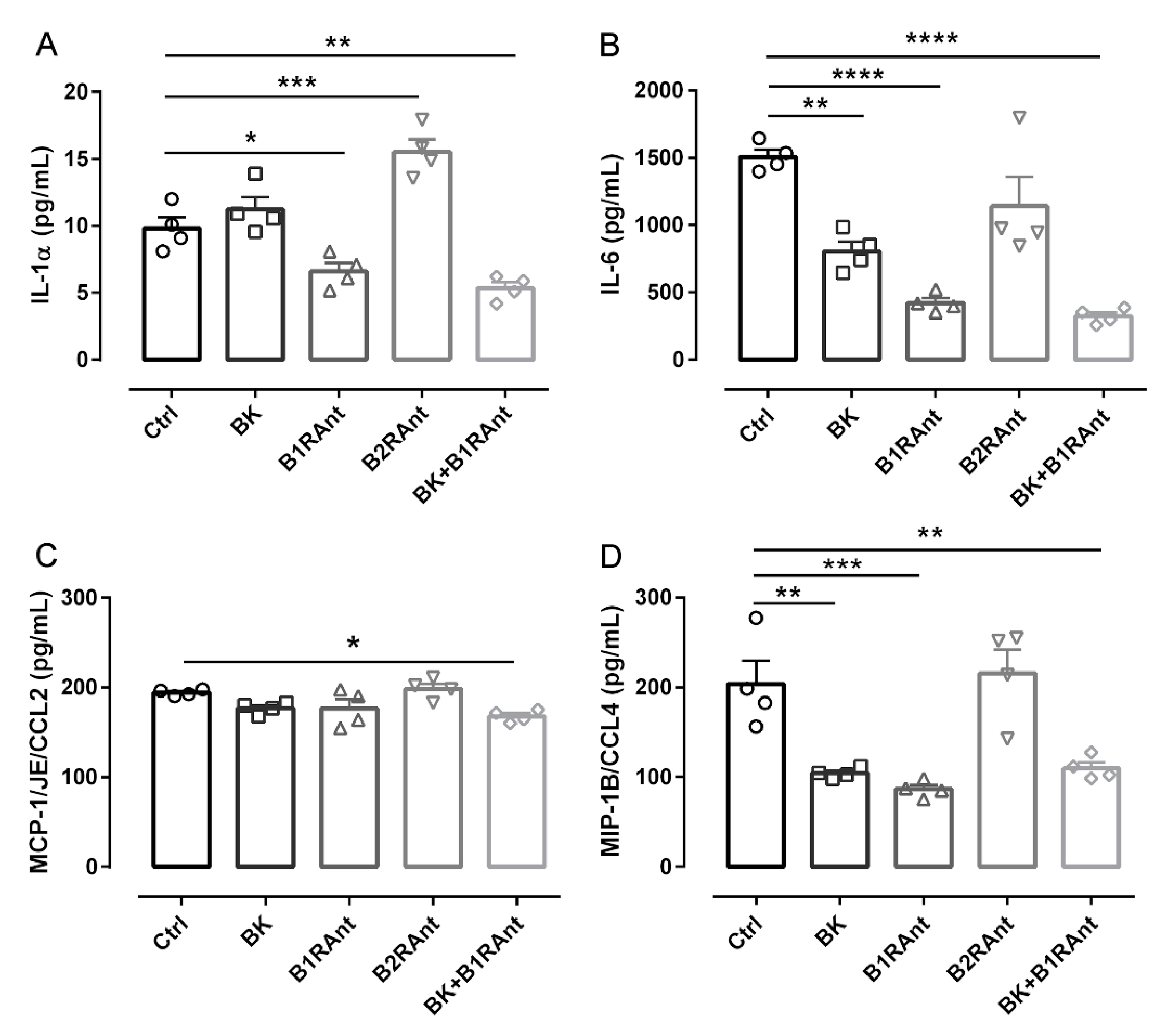
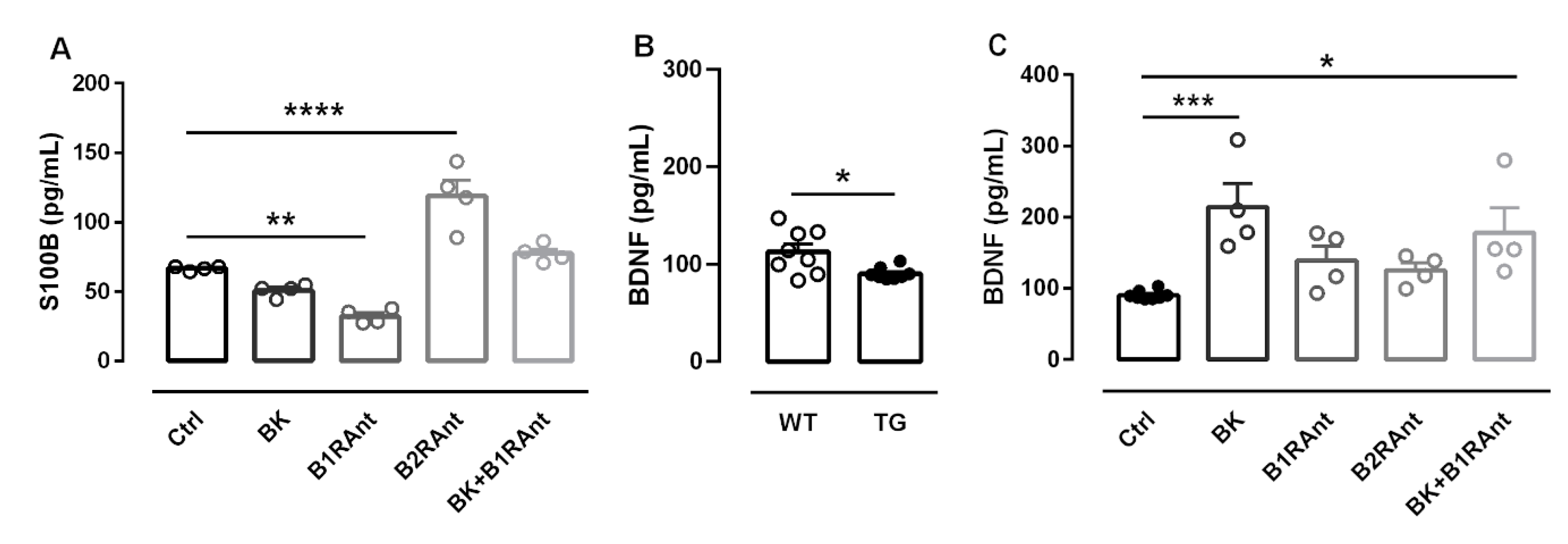
2.6. B2R Activation Decreases the Release of Inflammatory Markers
2.7. B2R Protected Hippocampal Slices of Transgenic Animals from Brain Damage
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Animal Treatment
4.3. Motor Activity
4.4. Spatial Memory
4.5. Nociception
4.6. Blood Flow Analysis
4.7. Quantification of Amyloid-β Plaques
4.8. Preparation of Organotypic Hippocampal Cultures (OHCs)
4.9. OHC Drug Treatment
4.10. Cell Death Evaluation
4.11. Inflammatory Markers Evaluation
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278; discussion 278–284. [Google Scholar] [CrossRef]
- Nunes, M.A.; Schowe, N.M.; Monteiro-Silva, K.C.; Baraldi-Tornisielo, T.; Souza, S.I.; Balthazar, J.; Albuquerque, M.S.; Caetano, A.L.; Viel, T.A.; Buck, H.S. Chronic microdose lithium treatment prevented memory loss and neurohistopathological changes in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 2015, 10, e0142267. [Google Scholar] [CrossRef]
- Lemos, M.T.; Amaral, F.A.; Dong, K.E.; Bittencourt, M.F.; Caetano, A.L.; Pesquero, J.B.; Viel, T.A.; Buck, H.S. Role of kinin B1 and B2 receptors in memory consolidation during the aging process of mice. Neuropeptides 2010, 44, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.A.; Lemos, M.T.; Dong, K.E.; Bittencourt, M.F.; Caetano, A.L.; Pesquero, J.B.; Viel, T.A.; Buck, H.S. Participation of kinin receptors on memory impairment after chronic infusion of human amyloid-beta 1-40 peptide in mice. Neuropeptides 2010, 44, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Caetano, A.L.; Dong-Creste, K.E.; Amaral, F.A.; Monteiro-Silva, K.C.; Pesquero, J.B.; Araujo, M.S.; Montor, W.R.; Viel, T.A.; Buck, H.S. Kinin B2 receptor can play a neuroprotective role in Alzheimer’s disease. Neuropeptides 2015, 53, 51–62. [Google Scholar] [CrossRef]
- Viel, T.A.; Lima Caetano, A.; Nasello, A.G.; Lancelotti, C.L.; Nunes, V.A.; Araujo, M.S.; Buck, H.S. Increases of kinin B1 and B2 receptors binding sites after brain infusion of amyloid-beta 1-40 peptide in rats. Neurobiol. Aging 2008, 29, 1805–1814. [Google Scholar] [CrossRef]
- Nokkari, A.; Abou-El-Hassan, H.; Mechref, Y.; Mondello, S.; Kindy, M.S.; Jaffa, A.A.; Kobeissy, F. Implication of the Kallikrein-Kinin system in neurological disorders: Quest for potential biomarkers and mechanisms. Prog. Neurobiol. 2018, 165–167, 26–50. [Google Scholar] [CrossRef]
- Profile, A.R.D. HOE 140, JE 049, JE049. Drugs R D 2004, 5, 343–348. [Google Scholar] [CrossRef]
- Lehmann, A. Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Expert Opin. Biol. Ther. 2008, 8, 1187–1199. [Google Scholar] [CrossRef]
- Lacoste, B.; Tong, X.K.; Lahjouji, K.; Couture, R.; Hamel, E. Cognitive and cerebrovascular improvements following kinin B1 receptor blockade in Alzheimer’s disease mice. J. Neuroinflamm. 2013, 10, 57. [Google Scholar] [CrossRef]
- Noda, M.; Kariura, Y.; Pannasch, U.; Nishikawa, K.; Wang, L.; Seike, T.; Ifuku, M.; Kosai, Y.; Wang, B.; Nolte, C.; et al. Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia. J. Neurochem. 2007, 101, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Toricelli, M.; Evangelista, S.R.; Oliveira, L.R.; Viel, T.A.; Buck, H.S. Neuroprotective effects of kinin B2 receptor in organotypic hippocampal cultures of middle-aged mice. Front. Aging Neurosci. 2019, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, K.; Gama, V.M.; Farah, V.A.; Irigoyen, M.C. Blood flow measurements in rats using four color microspheres during blockade of different vasopressor systems. Braz. J. Med. Biol. Res. 2005, 38, 119–125. [Google Scholar] [CrossRef]
- Dong-Creste, K.E.; Baraldi-Tornisielo, T.; Caetano, A.L.; Gobeil, F.; Montor, W.R.; Viel, T.A.; Buck, H.S. Kinin B1 receptor mediates memory impairment in the rat hippocampus. Biol. Chem. 2016, 397, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Iores-Marcal, L.M.; Viel, T.A.; Buck, H.S.; Nunes, V.A.; Gozzo, A.J.; Cruz-Silva, I.; Miranda, A.; Shimamoto, K.; Ura, N.; Araujo, M.S. Bradykinin release and inactivation in brain of rats submitted to an experimental model of Alzheimer’s disease. Peptides 2006, 27, 3363–3369. [Google Scholar] [CrossRef]
- Couture, R.; Harrisson, M.; Vianna, R.M.; Cloutier, F. Kinin receptors in pain and inflammation. Eur. J. Pharmacol. 2001, 429, 161–176. [Google Scholar] [CrossRef]
- Pela, I.R.; Rosa, A.L.; Silva, C.A.; Huidobro-Toro, J.P. Central B2 receptor involvement in the antinociceptive effect of bradykinin in rats. Br. J. Pharmacol. 1996, 118, 1488–1492. [Google Scholar] [CrossRef][Green Version]
- Girouard, H.; Iadecola, C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. 2006, 100, 328–335. [Google Scholar] [CrossRef]
- Roher, A.E.; Debbins, J.P.; Malek-Ahmadi, M.; Chen, K.; Pipe, J.G.; Maze, S.; Belden, C.; Maarouf, C.L.; Thiyyagura, P.; Mo, H.; et al. Cerebral blood flow in Alzheimer’s disease. Vasc. Health Risk Manag. 2012, 8, 599–611. [Google Scholar] [CrossRef]
- Chao, L.L.; Buckley, S.T.; Kornak, J.; Schuff, N.; Madison, C.; Yaffe, K.; Miller, B.L.; Kramer, J.H.; Weiner, M.W. ASL perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis. Assoc. Disord. 2010, 24, 19–27. [Google Scholar] [CrossRef]
- Xekardaki, A.; Kovari, E.; Gold, G.; Papadimitropoulou, A.; Giacobini, E.; Herrmann, F.; Giannakopoulos, P.; Bouras, C. Neuropathological changes in aging brain. Adv. Exp. Med. Biol. 2015, 821, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Raz, L.; Bhaskar, K.; Weaver, J.; Marini, S.; Zhang, Q.; Thompson, J.F.; Espinoza, C.; Iqbal, S.; Maphis, N.M.; Weston, L.; et al. Hypoxia promotes tau hyperphosphorylation with associated neuropathology in vascular dysfunction. Neurobiol. Dis. 2019, 126, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.T.; Paulson, O.B.; Hoj Nielsen, A.; Strandgaard, S. Bradykinin antagonist counteracts the acute effect of both angiotensin-converting enzyme inhibition and of angiotensin receptor blockade on the lower limit of autoregulation of cerebral blood flow. J. Cereb. Blood Flow Metab. 2014, 34, 467–471. [Google Scholar] [CrossRef]
- Bangen, K.J.; Clark, A.L.; Edmonds, E.C.; Evangelista, N.D.; Werhane, M.L.; Thomas, K.R.; Locano, L.E.; Tran, M.; Zlatar, Z.Z.; Nation, D.A.; et al. Cerebral blood flow and amyloid-beta interact to affect memory performance in cognitively normal older Adults. Front. Aging Neurosci. 2017, 9, 181. [Google Scholar] [CrossRef]
- Fazlollahi, A.; Calamante, F.; Liang, X.; Bourgeat, P.; Raniga, P.; Dore, V.; Fripp, J.; Ames, D.; Masters, C.L.; Rowe, C.C.; et al. Increased cerebral blood flow with increased amyloid burden in the preclinical phase of Alzheimer’s disease. J. Magn. Reson. Imaging 2020, 51, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Bicca, M.A.; Figueiredo, C.P.; Piermartiri, T.C.; Meotti, F.C.; Bouzon, Z.L.; Tasca, C.I.; Medeiros, R.; Calixto, J.B. The selective and competitive N-methyl-D-aspartate receptor antagonist, (-)-6-phosphonomethyl-deca-hydroisoquinoline-3-carboxylic acid, prevents synaptic toxicity induced by amyloid-beta in mice. Neuroscience 2011, 192, 631–641. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Innate immunity, local inflammation, and degenerative disease. Sci. Aging Knowl. Environ. 2002, 2002, re3. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar] [CrossRef]
- Medeiros, R.; Prediger, R.D.; Passos, G.F.; Pandolfo, P.; Duarte, F.S.; Franco, J.L.; Dafre, A.L.; Di Giunta, G.; Figueiredo, C.P.; Takahashi, R.N.; et al. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: Relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J. Neurosci. 2007, 27, 5394–5404. [Google Scholar] [CrossRef] [PubMed]
- Viel, T.A.; Buck, H.S. Kallikrein-kinin system mediated inflammation in Alzheimer’s disease in vivo. Curr. Alzheimer Res. 2011, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Sanden, C.; Leeb-Lundberg, L.M. Kinin B1 receptor homo-oligomerization is required for receptor trafficking to the cell surface. Int. Immunopharmacol. 2013, 15, 121–128. [Google Scholar] [CrossRef]
- Wang, D.D.; Bordey, A. The astrocyte odyssey. Prog. Neurobiol. 2008, 86, 342–367. [Google Scholar] [CrossRef]
- Park, S.H.; Hwang, S.K. Prognostic value of serum levels of S100 calcium-binding protein B, neuron-specific enolase, and interleukin-6 in pediatric patients with traumatic brain injury. World Neurosurg. 2018, 118, e534–e542. [Google Scholar] [CrossRef] [PubMed]
- Thornton, E.; Ziebell, J.M.; Leonard, A.V.; Vink, R. Kinin receptor antagonists as potential neuroprotective agents in central nervous system injury. Molecules 2010, 15, 6598–6618. [Google Scholar] [CrossRef]
- Kohno, T.; Wang, H.; Amaya, F.; Brenner, G.J.; Cheng, J.K.; Ji, R.R.; Woolf, C.J. Bradykinin enhances AMPA and NMDA receptor activity in spinal cord dorsal horn neurons by activating multiple kinases to produce pain hypersensitivity. J. Neurosci. 2008, 28, 4533–4540. [Google Scholar] [CrossRef]
- Parpura, V.; Liu, F.; Jeftinija, K.V.; Haydon, P.G.; Jeftinija, S.D. Neuroligand-evoked calcium-dependent release of excitatory amino acids from Schwann cells. J. Neurosci. 1995, 15, 5831–5839. [Google Scholar] [PubMed]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Akita, T.; Shimizu, T.; Sabirov, R.Z.; Okada, Y. Bradykinin-induced astrocyte-neuron signalling: Glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J. Physiol. 2009, 587, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Rauti, R.; Cellot, G.; D’Andrea, P.; Colliva, A.; Scaini, D.; Tongiorgi, E.; Ballerini, L. BDNF impact on synaptic dynamics: Extra or intracellular long-term release differently regulates cultured hippocampal synapses. Mol. Brain 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Blaes, N.; Girolami, J.P. Targeting the ‘Janus face’ of the B2-bradykinin receptor. Expert Opin. Ther. Targets 2013, 17, 1145–1166. [Google Scholar] [CrossRef] [PubMed]
- Bonde, M.M.; Olsen, K.B.; Erikstrup, N.; Speerschneider, T.; Lyngso, C.; Haunso, S.; Nielsen, M.S.; Sheikh, S.P.; Hansen, J.L. The angiotensin II type 1 receptor antagonist Losartan binds and activates bradykinin B2 receptor signaling. Regul. Pept. 2011, 167, 21–25. [Google Scholar] [CrossRef]
- Erdos, E.G.; Tan, F.; Skidgel, R.A. Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension 2010, 55, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Savard, M.; Labonte, J.; Dubuc, C.; Neugebauer, W.; D’Orleans-Juste, P.; Gobeil, F., Jr. Further pharmacological evaluation of a novel synthetic peptide bradykinin B2 receptor agonist. Biol. Chem. 2013, 394, 353–360. [Google Scholar] [CrossRef]
- Burgess, G.M.; Perkins, M.N.; Rang, H.P.; Campbell, E.A.; Brown, M.C.; McIntyre, P.; Urban, L.; Dziadulewicz, E.K.; Ritchie, T.J.; Hallett, A.; et al. Bradyzide, a potent non-peptide B(2) bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br. J. Pharmacol. 2000, 129, 77–86. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S21–S141. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. Paxino’s and Watson’s the Rat Brain in Stereotaxic Coordinates, 7th ed.; Academic Press: Cambridge, MA, USA, 2014; p. 1. [Google Scholar]
- Toricelli, M.; Evangelista, S.R.; Buck, H.S.; Viel, T.A. Microdose lithium treatment reduced inflammatory factors and neurodegeneration in organotypic hippocampal culture of old SAMP-8 mice. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Happ, D.F.; Tasker, R.A. A method for objectively quantifying propidium iodide exclusion in organotypic hippocampal slice cultures. J. Neurosci. Methods 2016, 269, 1–5. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, M.A.; Toricelli, M.; Schöwe, N.M.; Malerba, H.N.; Dong-Creste, K.E.; Farah, D.M.A.T.; De Angelis, K.; Irigoyen, M.C.; Gobeil, F.; Araujo Viel, T.; et al. Kinin B2 Receptor Activation Prevents the Evolution of Alzheimer’s Disease Pathological Characteristics in a Transgenic Mouse Model. Pharmaceuticals 2020, 13, 288. https://doi.org/10.3390/ph13100288
Nunes MA, Toricelli M, Schöwe NM, Malerba HN, Dong-Creste KE, Farah DMAT, De Angelis K, Irigoyen MC, Gobeil F, Araujo Viel T, et al. Kinin B2 Receptor Activation Prevents the Evolution of Alzheimer’s Disease Pathological Characteristics in a Transgenic Mouse Model. Pharmaceuticals. 2020; 13(10):288. https://doi.org/10.3390/ph13100288
Chicago/Turabian StyleNunes, Marielza Andrade, Mariana Toricelli, Natalia Mendes Schöwe, Helena Nascimento Malerba, Karis Ester Dong-Creste, Daniela Moura Azevedo Tuma Farah, Katia De Angelis, Maria Claudia Irigoyen, Fernand Gobeil, Tânia Araujo Viel, and et al. 2020. "Kinin B2 Receptor Activation Prevents the Evolution of Alzheimer’s Disease Pathological Characteristics in a Transgenic Mouse Model" Pharmaceuticals 13, no. 10: 288. https://doi.org/10.3390/ph13100288
APA StyleNunes, M. A., Toricelli, M., Schöwe, N. M., Malerba, H. N., Dong-Creste, K. E., Farah, D. M. A. T., De Angelis, K., Irigoyen, M. C., Gobeil, F., Araujo Viel, T., & Buck, H. S. (2020). Kinin B2 Receptor Activation Prevents the Evolution of Alzheimer’s Disease Pathological Characteristics in a Transgenic Mouse Model. Pharmaceuticals, 13(10), 288. https://doi.org/10.3390/ph13100288