Repetitive Intermittent Hyperglycemia Drives the M1 Polarization and Inflammatory Responses in THP-1 Macrophages Through the Mechanism Involving the TLR4-IRF5 Pathway

 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Anthropometric Measurements
2.2. Clinical Analyses
2.3. Cell Cultures
2.4. Real-Time qRT-PCR
2.5. Flow Cytometry
2.6. ELISA
2.7. Small Interfering RNA (siRNA) Transfections
2.8. Confocal Microscopy
2.9. Statistical Analysis
3. Results
3.1. Demographic and Clinical Characteristics of the Study Population
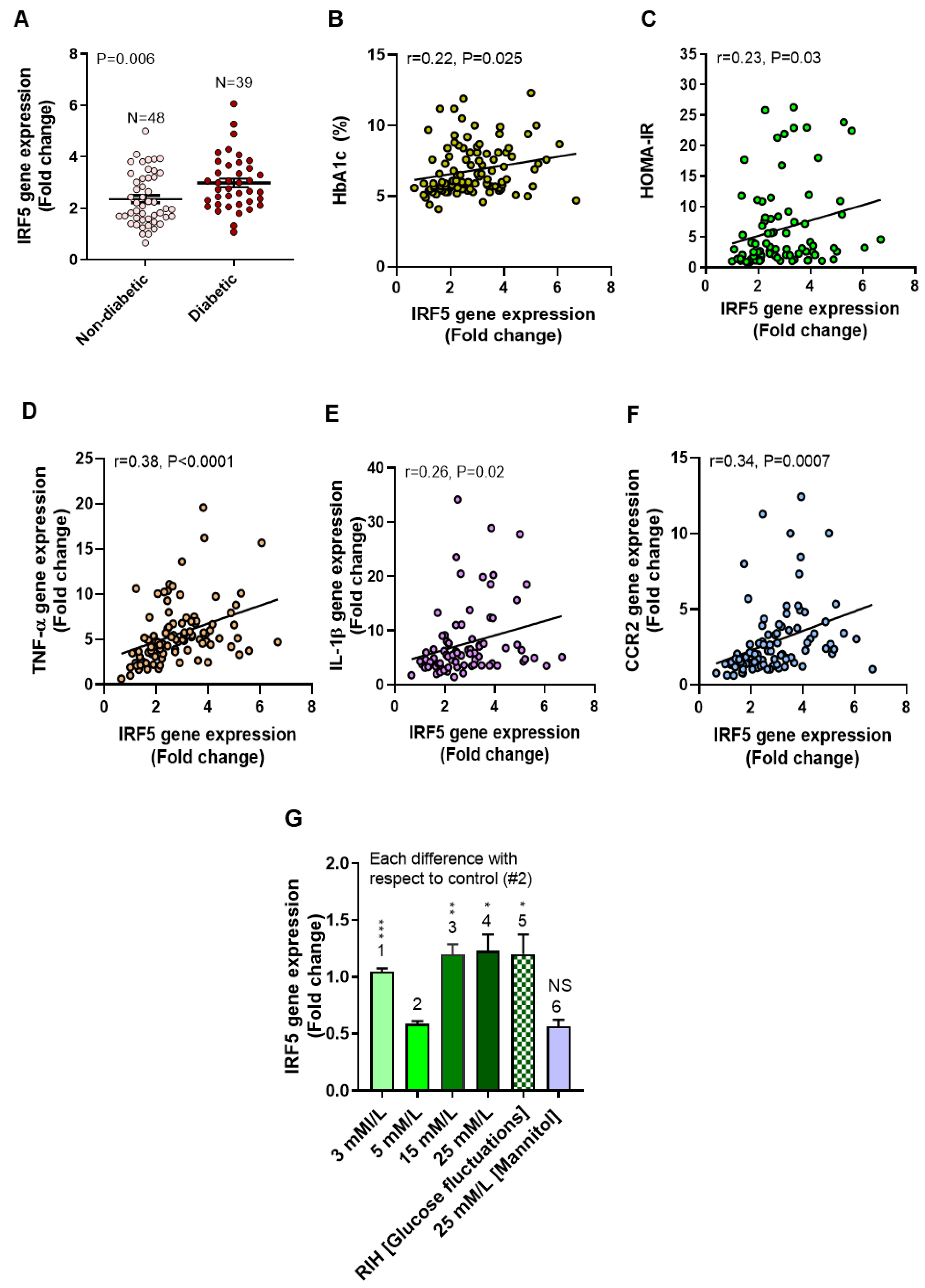
3.2. Elevated Adipose IRF5 Gene Expression in Diabetic Patients as Well as IRF5 Induction in Macrophages by Repetitive Intermittent Hyperglycemia (RIH)
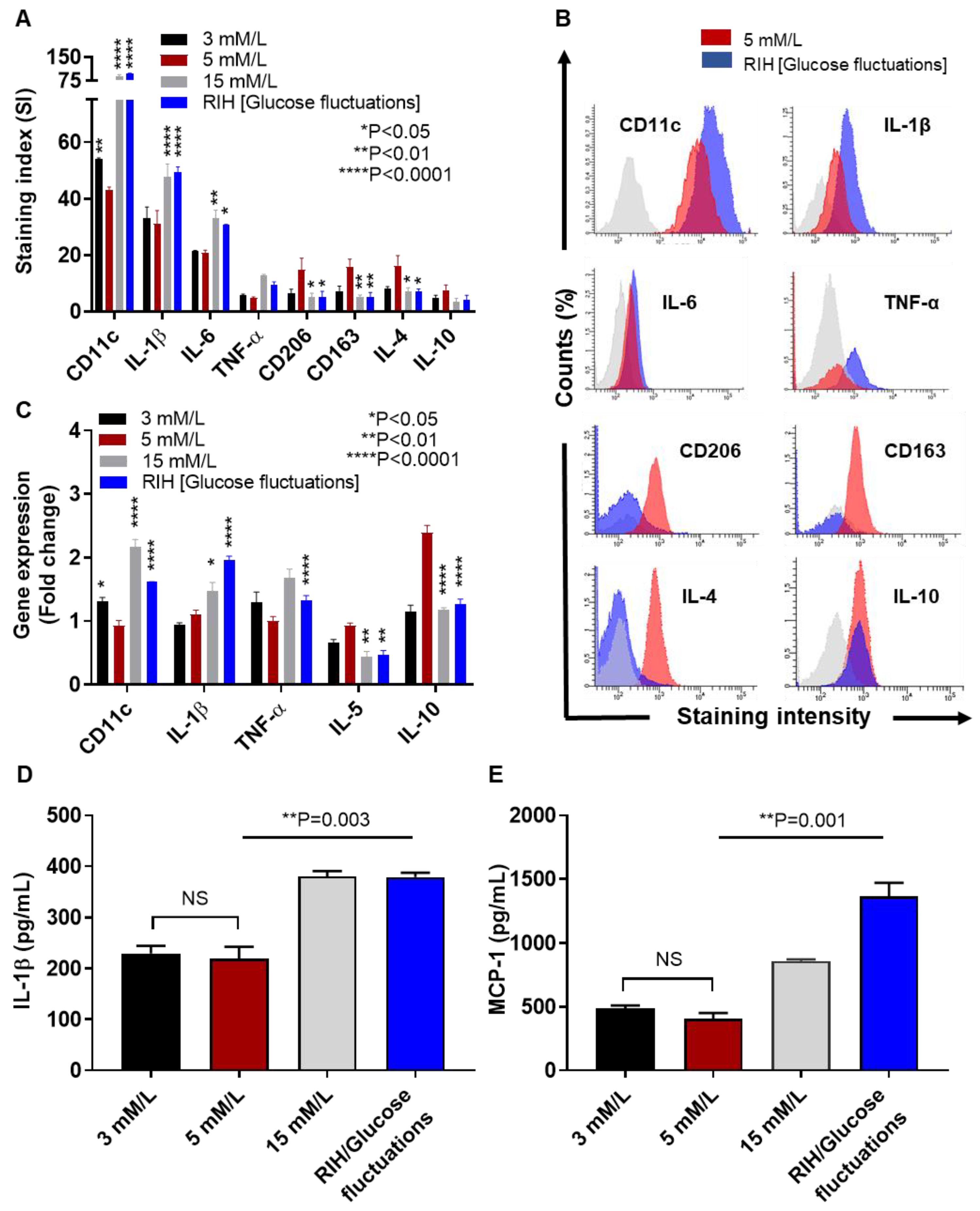
3.3. IRF5 Upregulation Is Paralleled by Expression of M1 Macrophage Polarization Markers and Pro-Inflammatory Cytokines
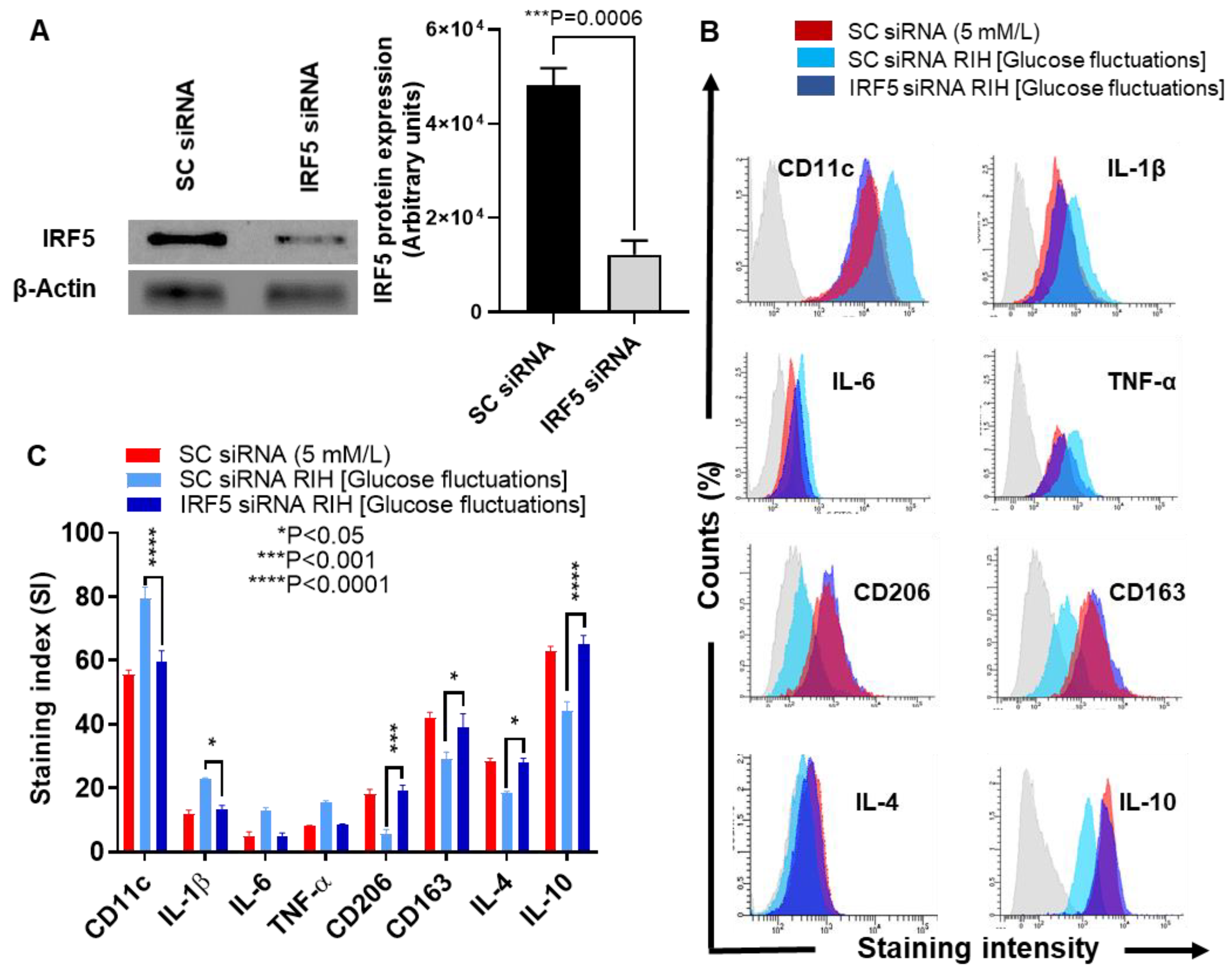
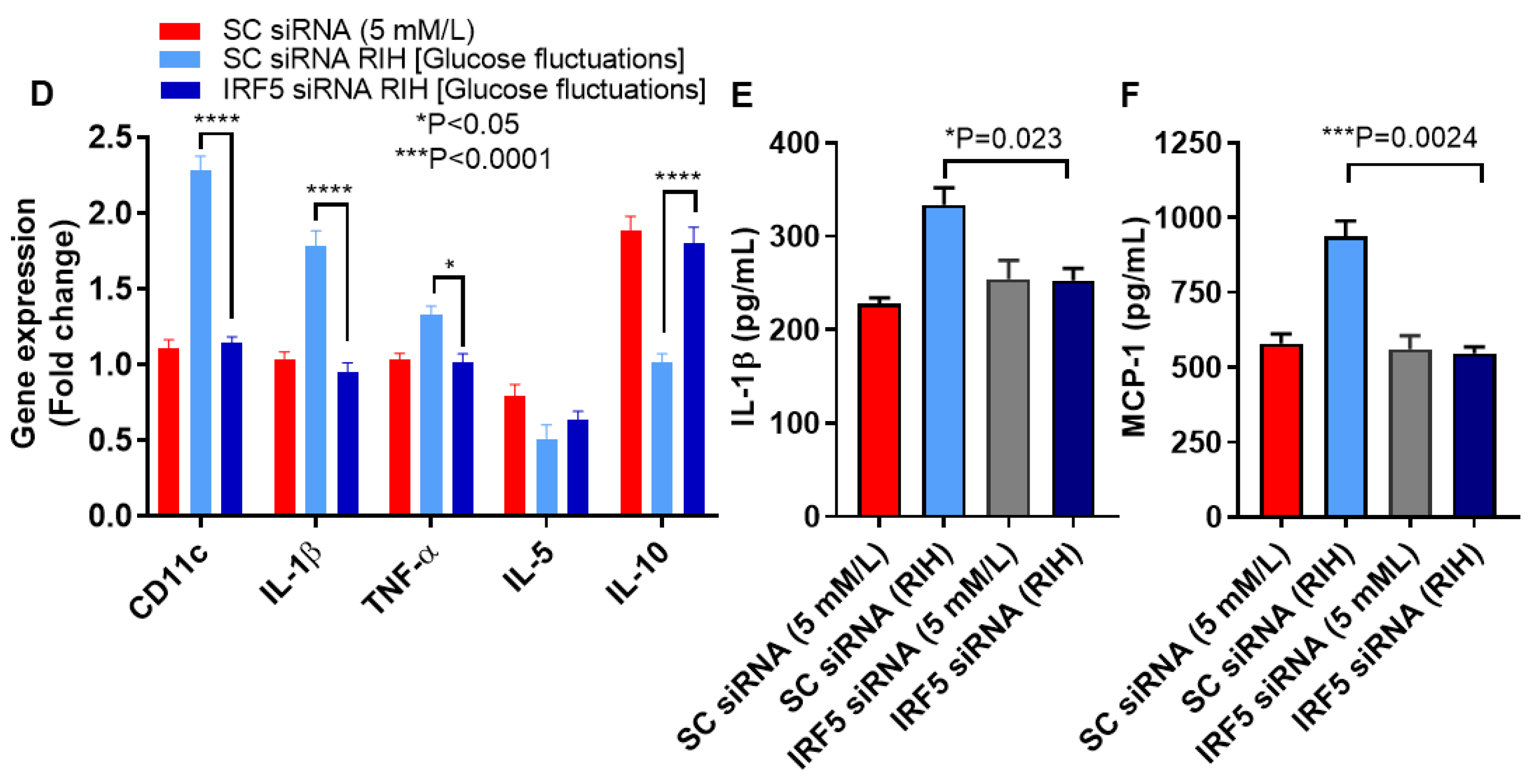
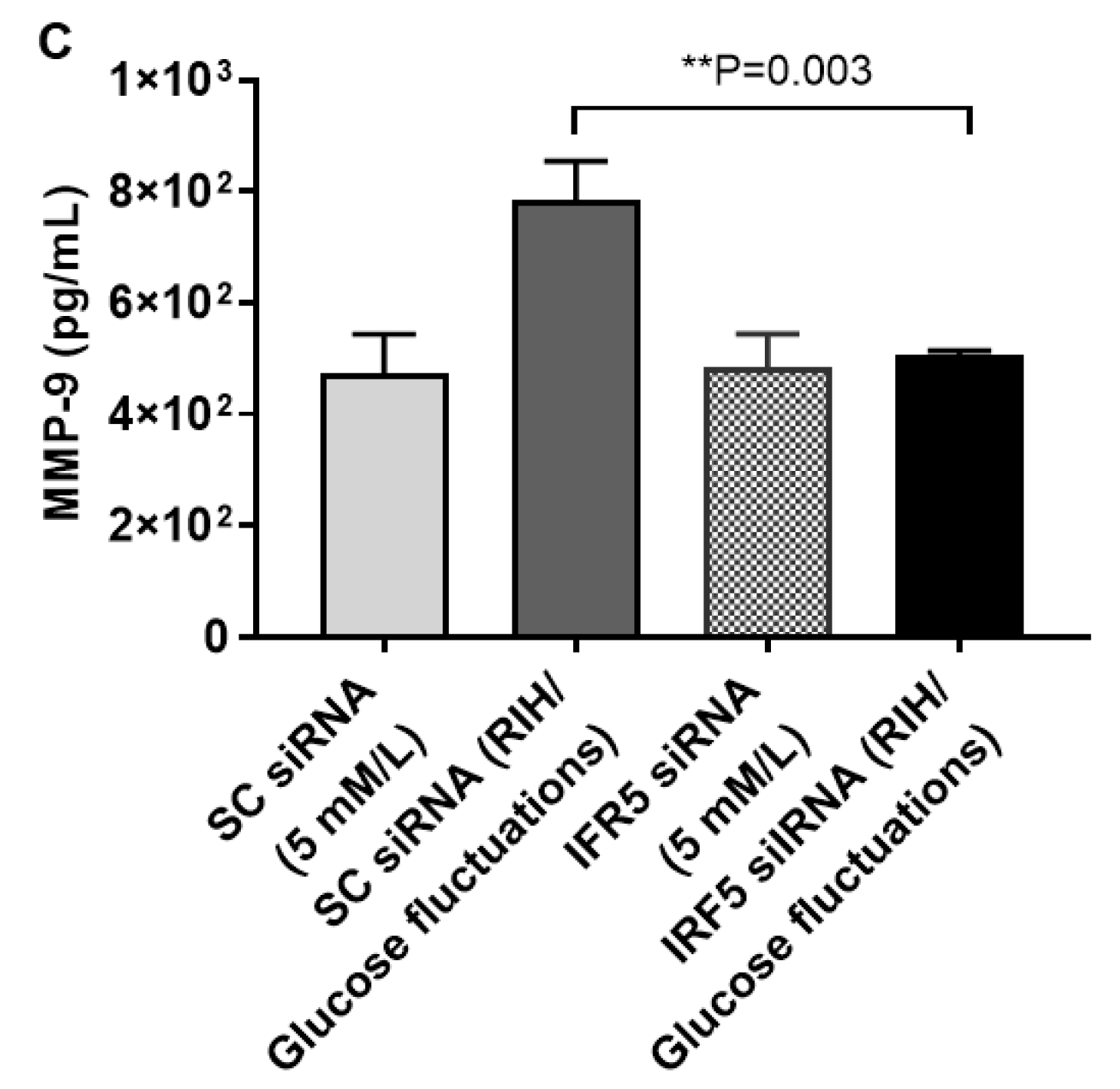
3.4. IRF5 Suppression Prevents the Expression of M1 Markers and Inflammatory Cytokines/Chemokine in Macrophages Cultured under RIH/Glucose Fluctuations
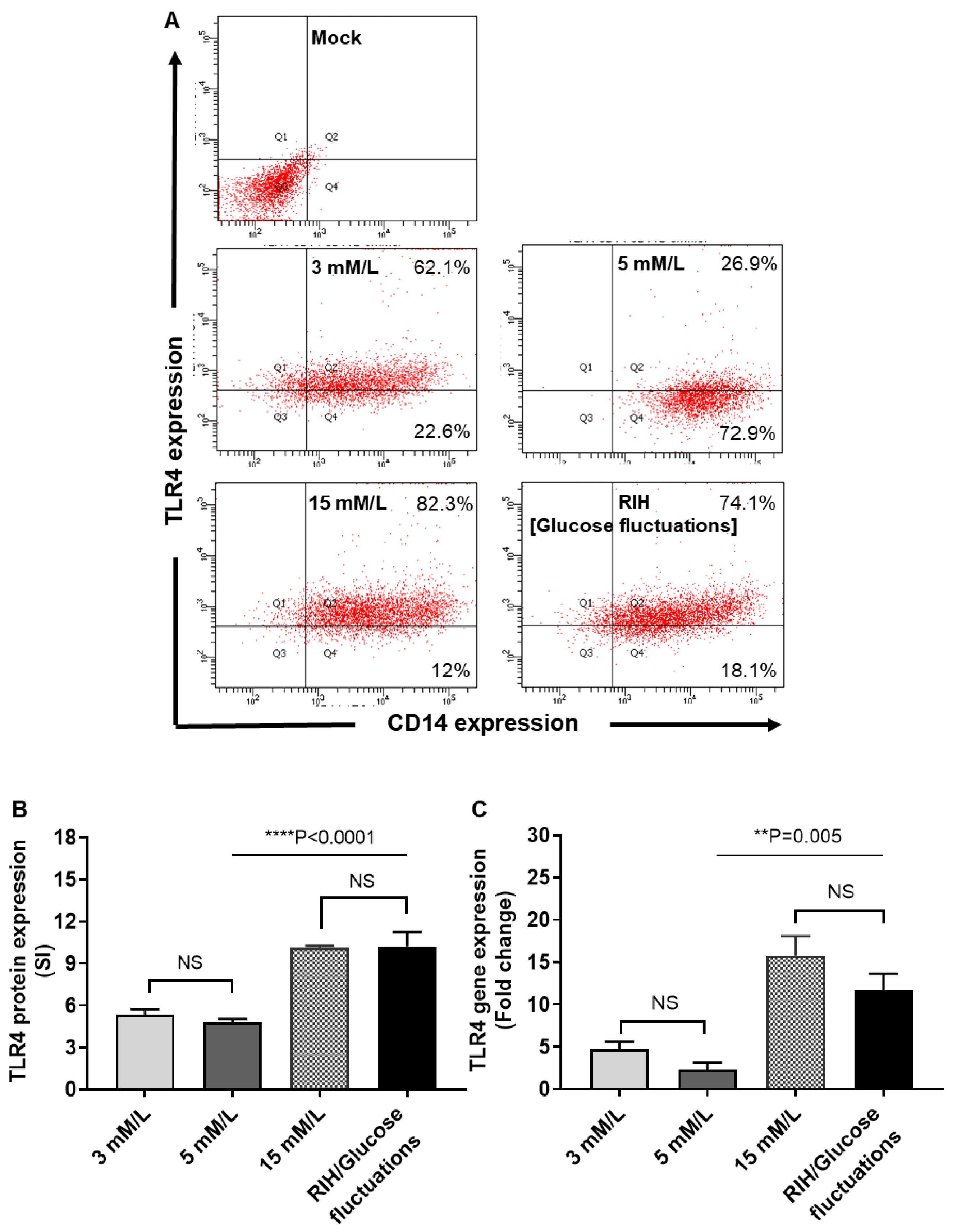
3.5. RIH Promotes the Expression of TLR4 on Monocytes/Macrophages
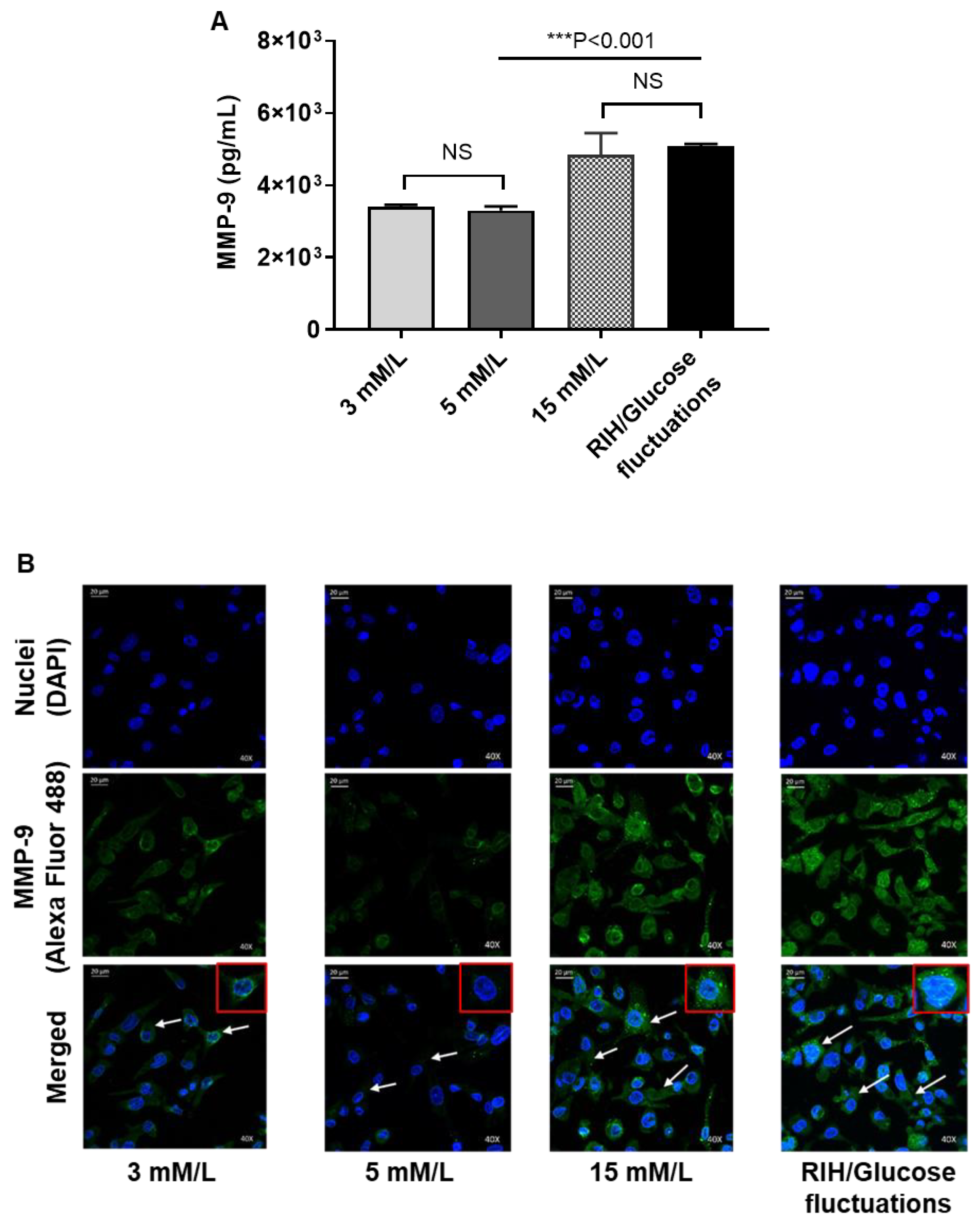
3.6. RIH Induces the Expression of Matrix Metalloproteinase (MMP)-9, Enhancing the Risk for Diabetes-Related Pathologies in a Clinical Setting
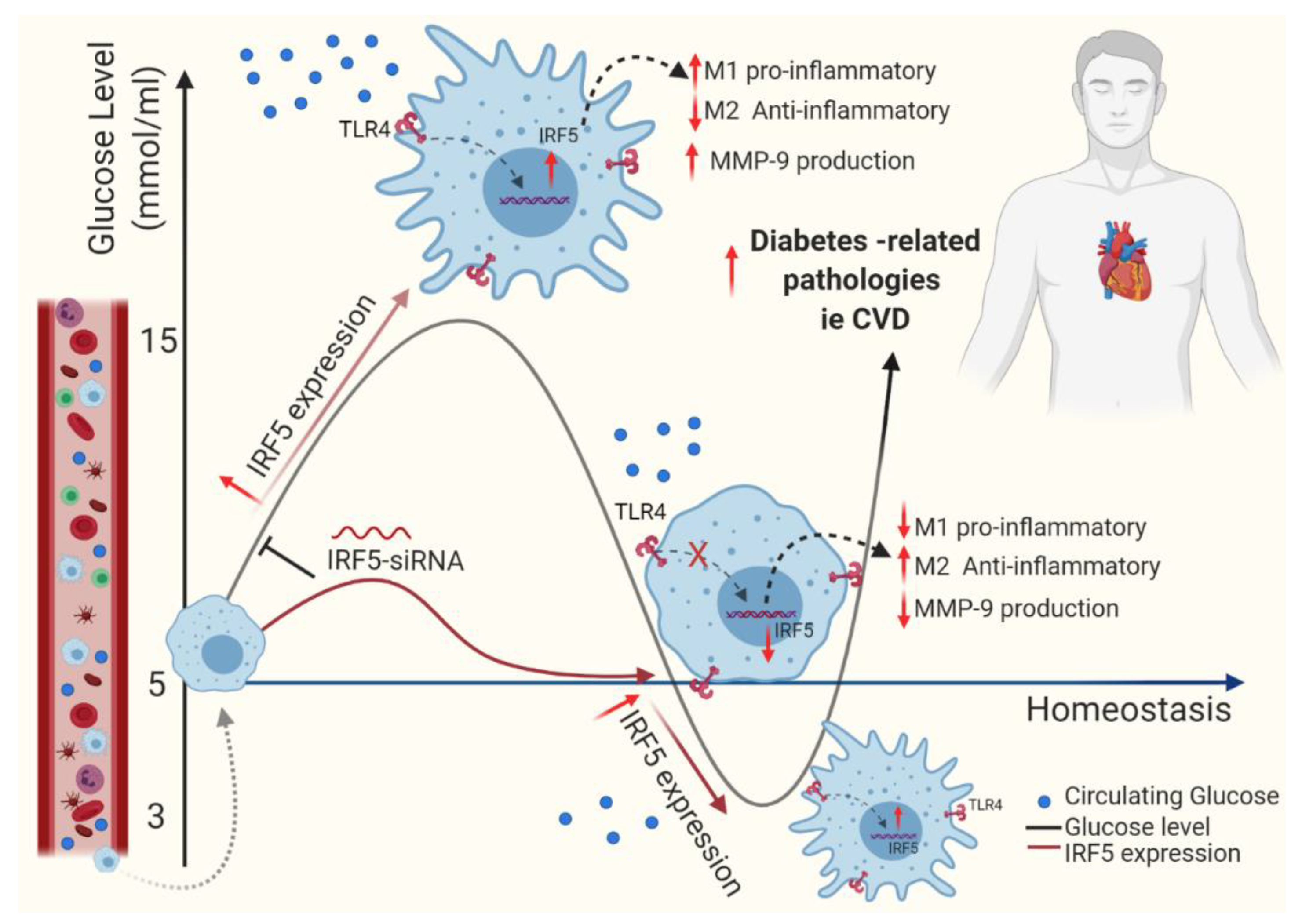
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American type culture collection |
| BMI | Body mass index |
| CCR2 | C-C motif chemokine receptor-2 |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FACS | Fluorescence-activated cell sorting |
| FBG | Fasting blood glucose |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| HbA1c | Glycated hemoglobin |
| HDL | High-density lipoprotein |
| HOMA-IR | Homeostatic model assessment of insulin resistance |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IL-1β | Interleukin-1β |
| IRF5 | Interferon regulatory factor-5 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MMP-9 | Matrix metalloproteinase-9 |
| PMA | Phorbol 12-myristate 13-acetate |
| PRR | Pattern recognition receptors |
| RIH | Repetitive intermittent hyperglycemia |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus-2 |
| siRNA | Small interfering RNA |
| T2D | Type-2 diabetes |
| TLR4 | Toll-like receptor-4 |
| TNF-α | Tumor necrosis factor-α |
| WMA | World medical association |
References
- Ceriello, A.; Ihnat, M.A. ‘Glycaemic variability’: A new therapeutic challenge in diabetes and the critical care setting. Diabet. Med. 2010, 27, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Butler, S.O.; Btaiche, I.F.; Alaniz, C. Relationship Between Hyperglycemia and Infection in Critically Ill Patients. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2005, 25, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazali, N.; O’brien, J.M.; Dungan, K.; Phillips, G.; Marsh, C.B.; Lemeshow, S.; Connors, A.F.; Preiser, J.-C. Glucose variability and mortality in patients with sepsis*. Crit. Care Med. 2008, 36, 2316–2321. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhang, X.; Li, J.; Tang, L.; Jiao, X.; Lv, X. Impact of glucose fluctuation on acute cerebral infarction in type 2 diabetes. Can. J. Neurol. Sci./J. Can. des Sci. Neurol. 2014, 41, 486–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.; Lee, H.-J.; Lee, H.; Ryu, H.G. Association Between Perioperative Hyperglycemia or Glucose Variability and Postoperative Acute Kidney Injury After Liver Transplantation. Anesthesia Analg. 2017, 124, 35–41. [Google Scholar] [CrossRef]
- Kuroda, M.; Shinke, T.; Otake, H.; Sugiyama, D.; Takaya, T.; Takahashi, H.; Terashita, D.; Uzu, K.; Tahara, N.; Kashiwagi, D.; et al. Effects of daily glucose fluctuations on the healing response to everolimus-eluting stent implantation as assessed using continuous glucose monitoring and optical coherence tomography. Cardiovasc. Diabetol. 2016, 15, 79. [Google Scholar] [CrossRef] [Green Version]
- Torimoto, K.; Okada, Y.; Mori, H.; Tanaka, Y. Relationship between fluctuations in glucose levels measured by continuous glucose monitoring and vascular endothelial dysfunction in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2013, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, Y.; Arima, H.; Zoungas, S.; Ninomiya, T.; Cooper, M.; Hamet, P.; Mancia, G.; Poulter, N.; Harrap, S.B.; Woodward, M.; et al. Impact of Visit-to-Visit Glycemic Variability on the Risks of Macrovascular and Microvascular Events and All-Cause Mortality in Type 2 Diabetes: The ADVANCE Trial. Diabetes Care 2014, 37, 2359–2365. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Shen, H.; Wang, Y.; He, B.; Zhang, Y.; Bai, Y.; Du, R.; Du, Q.; Han, P. Role of the PKCβII/JNK signaling pathway in acute glucose fluctuation-induced apoptosis of rat vascular endothelial cells. Acta Diabetol. 2017, 54, 727–736. [Google Scholar] [CrossRef]
- Azuma, K.; Kawamori, R.; Toyofuku, Y.; Kitahara, Y.; Sato, F.; Shimizu, T.; Miura, K.; Mine, T.; Tanaka, Y.; Mitsumata, M.; et al. Repetitive Fluctuations in Blood Glucose Enhance Monocyte Adhesion to the Endothelium of Rat Thoracic Aorta. Arter. Thromb. Vasc. Biol. 2006, 26, 2275–2280. [Google Scholar] [CrossRef] [Green Version]
- Barnes, B.; Lubyova, B.; Pitha, P.M. Review: On the Role of IRF in Host Defense. J. Interf. Cytokine Res. 2002, 22, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, J.; Yan, Q.-W.; Schones, D.E.; Kamal, M.; Hsu, C.-H.; Zhang, M.Q.; Crawford, G.E.; Rosen, E.D. Interferon Regulatory Factors Are Transcriptional Regulators of Adipogenesis. Cell Metab. 2008, 7, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindhu, S.; Thomas, R.; Kochumon, S.; Wilson, A.; Abu-Farha, M.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Increased Adipose Tissue Expression of Interferon Regulatory Factor (IRF)-5 in Obesity: Association with Metabolic Inflammation. Cells 2019, 8, 1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindhu, S.; Kochumon, S.; Thomas, R.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Enhanced Adipose Expression of Interferon Regulatory Factor (IRF)-5 Associates with the Signatures of Metabolic Inflammation in Diabetic Obese Patients. Cells 2020, 9, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef] [Green Version]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Wei, J.; Tang, D.; Lu, C.; Yang, J.; Lu, Y.; Wang, Y.; Jia, L.; Wang, J.; Ru, W.; Lu, Y.; et al. Irf5 deficiency in myeloid cells prevents necrotizing enterocolitis by inhibiting M1 macrophage polarization. Mucosal Immunol. 2019, 12, 888–896. [Google Scholar] [CrossRef]
- Castoldi, A.; De Souza, C.N.; Câmara, N.O.S.; Moraes-Vieira, P.M.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2016, 6, 637. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Anazawa, Y.; Iiizumi, M.; Fukuda, S.; Nakamura, Y.; Arakawa, H. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 2002, 21, 2914–2918. [Google Scholar] [CrossRef] [Green Version]
- Yanai, H.; Chen, H.-M.; Inuzuka, T.; Kondo, S.; Mak, T.W.; Takaoka, A.; Honda, K.; Taniguchi, T. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. USA 2007, 104, 3402–3407. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Yanai, H.; Kondo, S.; Duncan, G.; Negishi, H.; Mizutani, T.; Kano, S.-I.; Honda, K.; Ohba, Y.; Mak, T.W.; et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 2005, 434, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Kochumon, S.; Shenouda, S.; Wilson, A.; Al-Mulla, F.; Ahmad, R. The Cooperative Induction of CCL4 in Human Monocytic Cells by TNF-α and Palmitate Requires MyD88 and Involves MAPK/NF-κB Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, R.; Kochumon, S.; Chandy, B.; Shenouda, S.; Koshy, M.; Hasan, A.; Arefanian, H.; Al-Mulla, F.; Sindhu, S. TNF-α Drives the CCL4 Expression in Human Monocytic Cells: Involvement of the SAPK/JNK and NF-κB Signaling Pathways. Cell. Physiol. Biochem. 2019, 52, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochumon, S.; Al-Roub, A.; Al-Ghanim, M.; Al-Mass, A.; Behbehani, K.; Ahmad, R. TLR2 and AP-1/NF-kappaB are involved in the regulation of MMP-9 elicited by heat killed Listeria monocytogenes in human monocytic THP-1 cells. J. Inflamm. 2015, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Al-Roub, A.; Kochumon, S.; Akther, N.; Thomas, R.; Kumari, M.; Koshy, M.S.; Tiss, A.; Hannun, Y.A.; Tuomilehto, J.; et al. The Synergy between Palmitate and TNF-α for CCL2 Production Is Dependent on the TRIF/IRF3 Pathway: Implications for Metabolic Inflammation. J. Immunol. 2018, 200, 3599–3611. [Google Scholar] [CrossRef]
- Wray, G.M.; Foster, S.J.; Hinds, C.J.; Thiemermann, C. A cell wall component from pathogenic and non-pathogenic Gram-positive bacteria (peptidoglycan) synergizes with endotoxin to cause the release of tumor necrosis factor-α, nitric oxide production, shock, and multiple organ injury/dysfunction in the rat. Shock 2001, 15, 135–142. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Kochumon, S.; Usmani, S.; Sindhu, S.; Ahmad, R. Pam3CSK4 Induces MMP-9 Expression in Human Monocytic THP-1 Cells. Cell. Physiol. Biochem. 2017, 41, 1993–2003. [Google Scholar] [CrossRef]
- Ahmad, R.; El Bassam, S.; Cordeiro, P.; Menezes, J. Requirement of TLR2-mediated signaling for the induction of IL-15 gene expression in human monocytic cells by HSV-1. Blood 2008, 112, 2360–2368. [Google Scholar] [CrossRef] [Green Version]
- Al-Rashed, F.; Alghaith, A.; Azim, R.; AlMekhled, D.; Thomas, R.; Sindhu, S.; Ahmad, R. Increasing the Duration of Light Physical Activity Ameliorates Insulin Resistance Syndrome in Metabolically Healthy Obese Adults. Cells 2020, 9, 1189. [Google Scholar] [CrossRef]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the Human Monocytic Cells is Mediated through the TLR4-MyD88 Dependent Mechanism. Cell. Physiol. Biochem. 2016, 39, 889–900. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Rashed, F.; Akhter, N.; Al-Mulla, F.; Ahmad, R. ACSL1 Regulates TNFα-Induced GM-CSF Production by Breast Cancer MDA-MB-231 Cells. Biomolecules 2019, 9, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, A.; Shai, I.; Bitzur, R.; Kochba, I.; Tekes-Manova, D.; Israeli, E.; Shochat, T.; Rudich, A. Changes in Triglyceride Levels Over Time and Risk of Type 2 Diabetes in Young Men. Diabetes Care 2008, 31, 2032–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stentz, F.B.; Kitabchi, A.E. Hyperglycemia-induced activation of human T-lymphocytes with de novo emergence of insulin receptors and generation of reactive oxygen species. Biochem. Biophys. Res. Commun. 2005, 335, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, G.; Davis, T.; Patterton, H.-G.; Engelbrecht, A. How Does Inflammation-Induced Hyperglycemia Cause Mitochondrial Dysfunction in Immune Cells? BioEssays 2019, 41, e1800260. [Google Scholar] [CrossRef]
- Joy, N.G.; Mikeladze, M.; Younk, L.M.; Tate, D.B.; Davis, S.N. Effects of equivalent sympathetic activation during hypoglycemia on endothelial function and pro-atherothrombotic balance in healthy individuals and obese standard treated type 2 diabetes. Metabolism 2016, 65, 1695–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratter, J.; Rooijackers, H.M.; Tack, C.J.; Hijmans, A.G.; Netea, M.G.; De Galan, B.E.; Stienstra, R. Proinflammatory Effects of Hypoglycemia in Humans With or Without Diabetes. Diabetes 2017, 66, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Dalmas, E.; Toubal, A.; Alzaid, F.; Blazek, K.; Eames, H.L.; Lebozec, K.; Pini, M.; Hainault, I.; Montastier, E.; Denis, R.; et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat. Med. 2015, 21, 610–618. [Google Scholar] [CrossRef]
- Dalmas, E.; Venteclef, N. Les macrophages-Nouveaux modulateurs de la répartition de la masse grasse au cours de l’obésité. Med. Sci. (Paris) 2015, 31, 1080–1082. [Google Scholar] [CrossRef] [Green Version]
- Abu El-Asrar, A.M. Role of Inflammation in the Pathogenesis of Diabetic Retinopathy. Middle East Afr. J. Ophthalmol. 2012, 19, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Reusch, J.E.; Wang, C.C.L. Cardiovascular disease in diabetes: Where does glucose fit in? J. Clin. Endocrinol. Metab. 2011, 96, 2367–2376. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Sherman, S.I.; Gorelick, F.S.; Bergenstal, R.M.; Sherwin, R.S.; Buse, J.B. Incretin-Based Therapies for the Treatment of Type 2 Diabetes: Evaluation of the Risks and Benefits. Diabetes Care 2010, 33, 428–433. [Google Scholar] [CrossRef] [Green Version]
- Kalra, S.; Mukherjee, J.J.; Ramachandran, A.; Saboo, B.; Shaikh, S.; Venkataraman, S.; Bantwal, G.; Das, A.K. Hypoglycemia: The neglected complication. Indian J. Endocrinol. Metab. 2013, 17, 819–834. [Google Scholar] [CrossRef]
- Grosick, R.; Alvarado-Vázquez, P.A.; Messersmith, A.R.; Romero-Sandoval, E.A. High glucose induces a priming effect in macrophages and exacerbates the production of pro-inflammatory cytokines after a challenge. J. Pain Res. 2018, 11, 1769–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlou, S.; Lindsay, J.; Ingram, R.J.; Xu, H.; Chen, M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagareddy, P.R.; Kraakman, M.J.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose Tissue Macrophages Promote Myelopoiesis and Monocytosis in Obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentworth, J.M.; Naselli, G.; Brown, W.A.; Doyle, L.; Phipson, B.; Smyth, G.K.; Wabitsch, M.; O’Brien, P.E.; Harrison, L.C. Pro-Inflammatory CD11c+CD206+ Adipose Tissue Macrophages Are Associated With Insulin Resistance in Human Obesity. Diabetes 2010, 59, 1648–1656. [Google Scholar] [CrossRef] [Green Version]
- Torres-Castro, I.; Arroyo-Camarena, Ú.D.; Martínez-Reyes, C.P.; Gómez-Arauz, A.Y.; Dueñas-Andrade, Y.; Hernández, J.; Béjar, Y.L.; Zaga-Clavellina, V.; Morales-Montor, J.; Terrazas, L.I.; et al. Human monocytes and macrophages undergo M1-type inflammatory polarization in response to high levels of glucose. Immunol. Lett. 2016, 176, 81–89. [Google Scholar] [CrossRef]
- Herder, C.; Müller-Scholze, S.; Rating, P.; Koenig, W.; Thorand, B.; Haastert, B.; Holle, R.; Illig, T.; Rathmann, W.; Seissler, J.; et al. Systemic monocyte chemoattractant protein-1 concentrations are independent of type 2 diabetes or parameters of obesity: Results from the Cooperative Health Research in the Region of Augsburg Survey S4 (KORA S4). Eur. J. Endocrinol. 2006, 154, 311–317. [Google Scholar] [CrossRef]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerová, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2013, 19, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.; Blazek, K.; Byrne, A.J.; Perocheau, D.P.; Udalova, I.A. IRF5 Is a Specific Marker of Inflammatory Macrophages In Vivo. Mediat. Inflamm. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dideberg, V.; Kristjansdottir, G.; Milani, L.; Libioulle, C.; Sigurdsson, S.; Louis, E.; Wiman, A.-C.; Vermeire, S.; Rutgeerts, P.; Belaiche, J.; et al. An insertion deletion polymorphism in the Interferon Regulatory Factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum. Mol. Genet. 2007, 16, 3008–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciesla, M.; Kolarz, B.; Majdan, M.; Darmochwał-Kolarz, D. IRF5 promoter methylation as a new potential marker of rheumatoid arthritis. Pol. Arch. Intern. Med. 2019, 129, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, E.; Jefferies, C.A. IRF5-mediated signaling and implications for SLE. Clin. Immunol. 2014, 153, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; De, S.; Song, S.; Nelson, V. IRF5 hyper-activation is a driver of systemic lupus erythematosus (SLE) onset and severity. J. Immunol. 2018, 200, 45.6. [Google Scholar]
- Corbin, A.L.; Gomez-Vazquez, M.; Berthold, D.L.; Attar, M.; Arnold, I.C.; Powrie, F.; Sansom, S.N.; Udalova, I.A. IRF5 guides monocytes toward an inflammatory CD11c+ macrophage phenotype and promotes intestinal inflammation. Sci. Immunol. 2020, 5, eaax6085. [Google Scholar] [CrossRef]
- Krausgruber, T.; Saliba, D.; Ryzhakov, G.; Lanfrancotti, A.; Blazek, K.; Udalova, I.A. IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 2010, 115, 4421–4430. [Google Scholar] [CrossRef]
- Laviada-Molina, H.A.; Leal-Berumen, I.; Rodriguez-Ayala, E.; Bastarrachea, R.A. Working Hypothesis for Glucose Metabolism and SARS-CoV-2 Replication: Interplay Between the Hexosamine Pathway and Interferon RF5 Triggering Hyperinflammation. Role of BCG Vaccine? Front. Endocrinol. 2020, 11, 514. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Fang, J.; Zhou, H.; Liu, X.; Su, S. High glucose induces and activates Toll-like receptor 4 in endothelial cells of diabetic retinopathy. Diabetol. Metab. Syndr. 2015, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Devaraj, S.; Zhao, L.; Hwang, D.H.; Jialal, I. High Glucose Induces Toll-Like Receptor Expression in Human Monocytes: Mechanism of Activation. Diabetes 2008, 57, 3090–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saigusa, R.; Asano, Y.; Taniguchi, T.; Yamashita, T.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; Sugawara, K.; Tsuruta, D.; et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15136–15141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenemeyer, A.; Barnes, B.J.; Mancl, M.E.; Latz, E.; Goutagny, N.; Pitha, P.M.; Fitzgerald, K.A.; Golenbock, D.T. The Interferon Regulatory Factor, IRF5, Is a Central Mediator of Toll-like Receptor 7 Signaling. J. Biol. Chem. 2005, 280, 17005–17012. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Choi, Y.J.; Joung, S.M.; Lee, B.H.; Jung, Y.-S.; Lee, J.Y. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology 2009, 129, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Prados, J.-C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martin-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun-Bryce, S.; Rosenberg, G.A. Matrix Metalloproteinases in Cerebrovascular Disease. Br. J. Pharmacol. 1998, 18, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef]
- Sachwani, G.R.; Jaehne, A.K.; Jayaprakash, N.; Kuzich, M.; Onkoba, V.; Blyden, D.; Rivers, E.P. The association between blood glucose levels and matrix-metalloproteinase-9 in early severe sepsis and septic shock. J. Inflamm. 2016, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Setyopranoto, I.; Malueka, R.G.; Panggabean, A.S.; Widyadharma, I.P.E.; Sadewa, A.H.; Lamsudin, R.; Wibowo, S. Association between Increased Matrix Metalloproteinase-9 (MMP-9) Levels with Hyperglycaemia Incidence in Acute Ischemic Stroke Patients. Open Access Maced. J. Med. Sci. 2018, 6, 2067–2072. [Google Scholar] [CrossRef] [Green Version]
- Setianto, B.Y.; Achmad, A.F.; Purnomo, L.B. Serum matrix metalloproteinase-9 levels in acute coronary syndrome patients with and without hyperglycemia. Acta Med. Indones. 2014, 46, 83–89. [Google Scholar]
- Uemura, S.; Matsushita, H.; Li, W.; Glassford, A.J.; Asagami, T.; Lee, K.H.; Harrison, D.G.; Tsao, P.S. Diabetes mellitus enhances vascular matrix metalloproteinase activity: Role of oxidative stress. Circ. Res. 2001, 88, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Diaz, J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage Polarization in Obesity and Type 2 Diabetes: Weighing Down Our Understanding of Macrophage Function? Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M.; Hirsch, I.B. Glycemic Variability: A Hemoglobin A1c–Independent Risk Factor for Diabetic Complications. JAMA 2006, 295, 1707–1708. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H-oxidase activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Xu, Y.; Sun, S.; Sun, Y.; Wang, X. Intermittent high glucose enhances cell proliferation and VEGF expression in retinal endothelial cells: The role of mitochondrial reactive oxygen species. Mol. Cell. Biochem. 2010, 343, 27–35. [Google Scholar] [CrossRef]
- Jones, S.C.; Saunders, H.J.; Qi, W.; Pollock, C.A. Intermittent high glucose enhances cell growth and collagen synthesis in cultured human tubulointerstitial cells. Diabetologia 1999, 42, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Hermanides, J.; Vriesendorp, T.M.; Bosman, R.J.; Zandstra, D.F.; Hoekstra, J.B.; Devries, J.H. Glucose variability is associated with intensive care unit mortality*. Crit. Care Med. 2010, 38, 838–842. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, W.; Huang, R.; Zhang, X. Postchallenge plasma glucose excursions, carotid intima-media thickness, and risk factors for atherosclerosis in Chinese population with type 2 diabetes. Atherosclerosis 2010, 210, 302–306. [Google Scholar] [CrossRef]
- Buscemi, S.; Re, A.; Batsis, J.A.; Arnone, M.; Mattina, A.; Cerasola, G.; Verga, S. Glycaemic variability using continuous glucose monitoring and endothelial function in the metabolic syndrome and in Type 2 diabetes. Diabet. Med. 2010, 27, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.-P.; Colette, C. Activation of Oxidative Stress by Acute Glucose Fluctuations Compared With Sustained Chronic Hyperglycemia in Patients With Type 2 Diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Rashed, F.; Sindhu, S.; Arefanian, H.; Al Madhoun, A.; Kochumon, S.; Thomas, R.; Al-Kandari, S.; Alghaith, A.; Jacob, T.; Al-Mulla, F.; et al. Repetitive Intermittent Hyperglycemia Drives the M1 Polarization and Inflammatory Responses in THP-1 Macrophages Through the Mechanism Involving the TLR4-IRF5 Pathway. Cells 2020, 9, 1892. https://doi.org/10.3390/cells9081892
Al-Rashed F, Sindhu S, Arefanian H, Al Madhoun A, Kochumon S, Thomas R, Al-Kandari S, Alghaith A, Jacob T, Al-Mulla F, et al. Repetitive Intermittent Hyperglycemia Drives the M1 Polarization and Inflammatory Responses in THP-1 Macrophages Through the Mechanism Involving the TLR4-IRF5 Pathway. Cells. 2020; 9(8):1892. https://doi.org/10.3390/cells9081892
Chicago/Turabian StyleAl-Rashed, Fatema, Sardar Sindhu, Hossein Arefanian, Ashraf Al Madhoun, Shihab Kochumon, Reeby Thomas, Sarah Al-Kandari, Abdulwahab Alghaith, Texy Jacob, Fahd Al-Mulla, and et al. 2020. "Repetitive Intermittent Hyperglycemia Drives the M1 Polarization and Inflammatory Responses in THP-1 Macrophages Through the Mechanism Involving the TLR4-IRF5 Pathway" Cells 9, no. 8: 1892. https://doi.org/10.3390/cells9081892