The Rosmarinus Bioactive Compound Carnosic Acid Is a Novel PPAR Antagonist That Inhibits the Browning of White Adipocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Stromal Vascular Fraction Preparation
2.2.1. hMADS Cells Culture
2.2.2. Mouse Adipose Tissue SVF Preparation and Culture
2.3. Isolation and Analysis of RNA
2.4. Lipolysis Assays
2.5. Western Blot Analysis
2.6. Cell-Based PPAR Transactivation Assay
2.7. Statistical Analyses
3. Results
3.1. Carnosic Acid Inhibits the Browning Process of White Adipocytes
3.2. Carnosic Acid Inhibits Thermogenic Marker Gene Expression of Human Brite Adipocytes
3.3. Carnosic Acid Is a Potential Competitor of Rosiglitazone
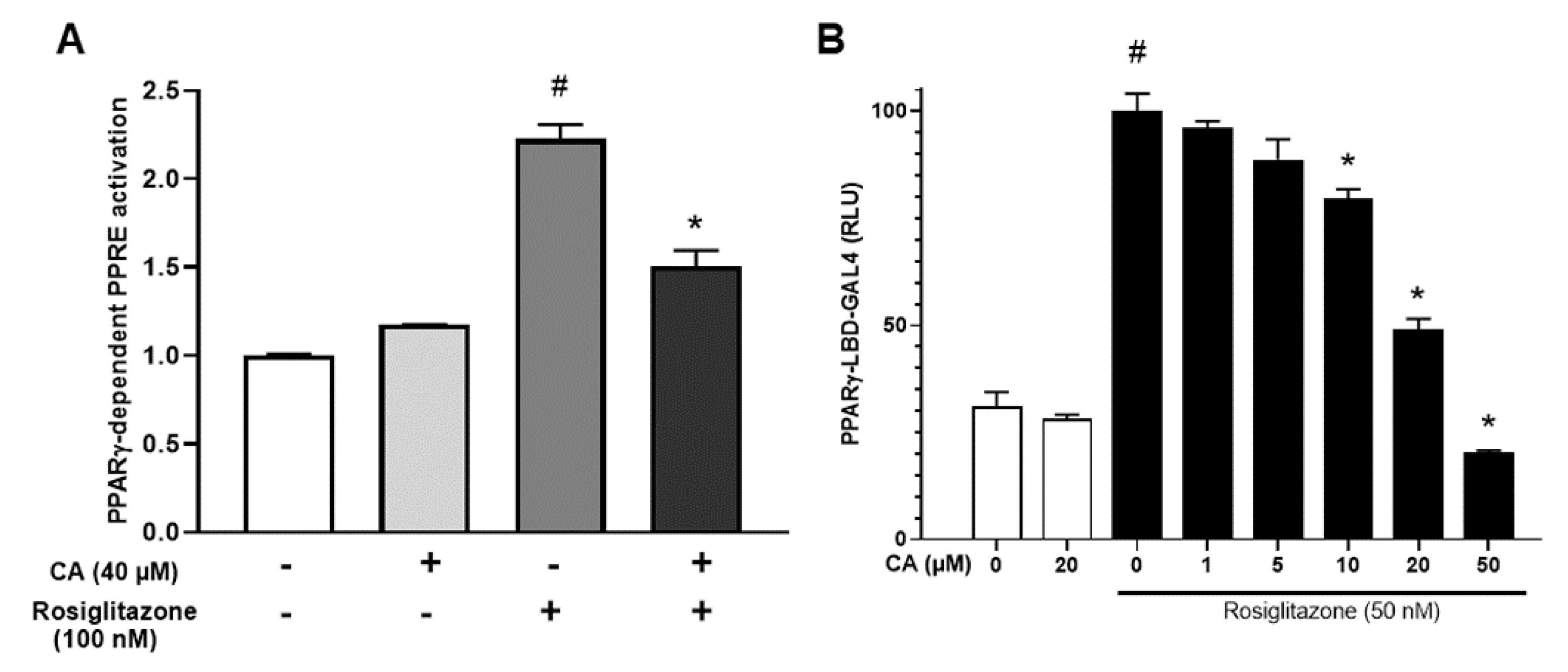
3.4. Carnosic Acid Antagonizes Rosiglitazone-Induced Activation of PPARγ
3.5. The Carnosic Acid Inhibitory Effect Is Normalized upon its Removal
3.6. Effects of Carnosic Acid on Lipolysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cinti, S. Adipose organ development and remodeling. Compr. Physiol. 2018, 8, 1357–1431. [Google Scholar] [PubMed]
- Cinti, S. The adipose organ at a glance. Dis. Model. Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. UCP1 protein: The molecular hub of adipose organ plasticity. Biochimie 2017, 134, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kane, H.; Lynch, L. Innate Immune Control of Adipose Tissue Homeostasis. Trends Immunol. 2019, 40, 857–872. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight. 2020. Available online: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 1 April 2020).
- Muller, T.D.; Clemmensen, C.; Finan, B.; DiMarchi, R.D.; Tschop, M.H. Anti-obesity therapy: From rainbow pills to polyagonists. Pharmacol. Rev. 2018, 70, 712–746. [Google Scholar] [CrossRef]
- Alipoor, E.; Hosseinzadeh-Attar, M.J.; Rezaei, M.; Jazayeri, S.; Chapman, M. White adipose tissue browning in critical illness: A review of the evidence, mechanisms and future perspectives. Obes. Rev. 2020. [Google Scholar] [CrossRef]
- Petrovic, N.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Thermogenically competent nonadrenergic recruitment in brown preadipocytes by a PPARgamma agonist. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E287–E296. [Google Scholar] [CrossRef]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef]
- Graja, A.; Gohlke, S.; Schulz, T.J. Aging of brown and beige/brite adipose tissue. Handb. Exp. Pharmacol. 2019, 251, 55–72. [Google Scholar]
- Hussain, M.F.; Roesler, A.; Kazak, L. Regulation of adipocyte thermogenesis: Mechanisms controlling obesity. FEBS J. 2020, 287. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.; Wolfrum, C. Feeding brown fat: Dietary phytochemicals targeting non-shivering thermogenesis to control body weight. In Proceedings of the 13th European Nutrition Conference, Dublin, Ireland, 15–18 October 2019; pp. 1–19. [Google Scholar] [CrossRef]
- Lasar, D.; Rosenwald, M.; Kiehlmann, E.; Balaz, M.; Tall, B.; Opitz, L.; Lidell, M.E.; Zamboni, N.; Krznar, P.; Sun, W.; et al. Peroxisome Proliferator Activated Receptor Gamma Controls Mature Brown Adipocyte Inducibility through Glycerol Kinase. Cell Rep. 2018, 22, 760–773. [Google Scholar] [CrossRef]
- Barquissau, V.; Beuzelin, D.; Pisani, D.F.; Beranger, G.E.; Mairal, A.; Montagner, A.; Roussel, B.; Tavernier, G.; Marques, M.A.; Moro, C.; et al. White-to-brite conversion in human adipocytes promotes metabolic reprogramming towards fatty acid anabolic and catabolic pathways. Mol. Metab. 2016, 5, 352–365. [Google Scholar] [CrossRef]
- Mota de Sa, P.; Richard, A.J.; Hang, H.; Stephens, J.M. Transcriptional regulation of adipogenesis. Compr. Physiol. 2017, 7, 635–674. [Google Scholar]
- Matsuda, H.; Nakamura, S.; Yoshikawa, M. Search for new type of PPARgamma agonist-like anti-diabetic compounds from medicinal plants. Biol. Pharm. Bull. 2014, 37, 884–891. [Google Scholar] [CrossRef]
- Neves, J.A.; Neves, J.A.; Oliveira, R.C.M. Pharmacological and biotechnological advances with Rosmarinus officinalis L. Expert Opin. Ther. Pat. 2018, 28, 399–413. [Google Scholar] [CrossRef]
- Naimi, M.; Vlavcheski, F.; Shamshoum, H.; Tsiani, E. Rosemary extract as a potential anti-hyperglycemic agent: Current evidence and future perspectives. Nutrients 2017, 9, 968. [Google Scholar] [CrossRef]
- Bahri, S.; Jameleddine, S.; Shlyonsky, V. Relevance of carnosic acid to the treatment of several health disorders: Molecular targets and mechanisms. Biomed. Pharmacother. 2016, 84, 569–582. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, J.S.; Seo, Y.R.; Park, J.H.; Choi, M.S.; Sung, M.K. Carnosic acid suppresses colon tumor formation in association with antiadipogenic activity. Mol. Nutr. Food Res. 2014, 58, 2274–2285. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Cases, J.; Roller, M.; Chiralt-Boix, A.; Coussaert, A.; Ripoll, C. Carnosic acid-rich rosemary (Rosmarinus officinalis L.) leaf extract limits weight gain and improves cholesterol levels and glycaemia in mice on a high-fat diet. Br. J. Nutr. 2011, 106, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Birtic, S.; Dussort, P.; Pierre, F.X.; Bily, A.C.; Roller, M. Carnosic acid. Phytochemistry 2015, 115, 9–19. [Google Scholar] [CrossRef]
- Gaya, M.; Repetto, V.; Toneatto, J.; Anesini, C.; Piwien-Pilipuk, G.; Moreno, S. Antiadipogenic effect of carnosic acid, a natural compound present in Rosmarinus officinalis, is exerted through the C/EBPs and PPARgamma pathways at the onset of the differentiation program. Biochim. Biophys. Acta 2013, 1830, 3796–3806. [Google Scholar] [CrossRef]
- Park, M.Y.; Sung, M.K. Carnosic acid attenuates obesity-induced glucose intolerance and hepatic fat accumulation by modulating genes of lipid metabolism in C57BL/6J-ob/ob mice. J. Sci. Food Agric. 2015, 95, 828–835. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lim, W.; Sung, M.K. Carnosic acid modulates increased hepatic lipogenesis and adipocytes differentiation in ovariectomized mice fed normal or high-fat diets. Nutrients 2018, 10, 1984. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Elabd, C.; Delteil, F.; Astier, J.; Vernochet, C.; Saint-Marc, P.; Guesnet, J.; Guezennec, A.; Amri, E.Z.; Dani, C.; et al. Adipocyte differentiation of multipotent cells established from human adipose tissue. Biochem. Biophys. Res. Commun. 2004, 315, 255–263. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Pisani, D.; Dechesne, C.A.; Turc-Carel, C.; Kurzenne, J.Y.; Wdziekonski, B.; Villageois, A.; Bagnis, C.; Breittmayer, J.P.; Groux, H.; et al. Transplantation of a multipotent cell population from human adipose tissue induces dystrophin expression in the immunocompetent mdx mouse. J. Exp. Med. 2005, 201, 1397–1405. [Google Scholar] [CrossRef]
- Rodbell, M. Metabolism of Isolated Fat Cells. I. Effects of Hormones on Glucose Metabolism and Lipolysis. J. Biol. Chem. 1964, 239, 375–380. [Google Scholar] [PubMed]
- Murholm, M.; Dixen, K.; Qvortrup, K.; Hansen, L.H.; Amri, E.Z.; Madsen, L.; Barbatelli, G.; Quistorff, B.; Hansen, J.B. Dynamic regulation of genes involved in mitochondrial DNA replication and transcription during mouse brown fat cell differentiation and recruitment. PLoS ONE 2009, 4, e8458. [Google Scholar] [CrossRef] [PubMed]
- Ghandour, R.A.; Giroud, M.; Vegiopoulos, A.; Herzig, S.; Ailhaud, G.; Amri, E.Z.; Pisani, D.F. IP-receptor and PPARs trigger the conversion of human white to brite adipocyte induced by carbaprostacyclin. Biochim. Biophys. Acta 2016, 1861, 285–293. [Google Scholar] [CrossRef]
- Pisani, D.F.; Djedaini, M.; Beranger, G.E.; Elabd, C.; Scheideler, M.; Ailhaud, G.; Amri, E.Z. Differentiation of human adipose-derived stem cells into “Brite” (Brown-in-White) adipocytes. Front. Endocrinol. 2011, 2, 87. [Google Scholar] [CrossRef]
- Bradley, D.C.; Kaslow, H.R. Radiometric assays for glycerol, glucose, and glycogen. Anal. Biochem. 1989, 180, 11–16. [Google Scholar] [CrossRef]
- Pisani, D.F.; Beranger, G.E.; Corinus, A.; Giroud, M.; Ghandour, R.A.; Altirriba, J.; Chambard, J.C.; Mazure, N.M.; Bendahhou, S.; Duranton, C.; et al. The K+ channel TASK1 modulates beta-adrenergic response in brown adipose tissue through the mineralocorticoid receptor pathway. FASEB J. 2016, 30, 909–922. [Google Scholar] [CrossRef]
- Pisani, D.F.; Ghandour, R.A.; Beranger, G.E.; Le Faouder, P.; Chambard, J.C.; Giroud, M.; Vegiopoulos, A.; Djedaini, M.; Bertrand-Michel, J.; Tauc, M.; et al. The omega6-fatty acid, arachidonic acid, regulates the conversion of white to brite adipocyte through a prostaglandin/calcium mediated pathway. Mol. Metab. 2014, 3, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Elabd, C.; Chiellini, C.; Carmona, M.; Galitzky, J.; Cochet, O.; Petersen, R.; Penicaud, L.; Kristiansen, K.; Bouloumie, A.; Casteilla, L.; et al. Human multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem Cells 2009, 27, 2753–2760. [Google Scholar] [CrossRef]
- Park, M.Y.; Mun, S.T. Carnosic acid inhibits TLR4-MyD88 signaling pathway in LPS-stimulated 3T3-L1 adipocytes. Nutr. Res. Pract 2014, 8, 516–520. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef]
- Laghezza, A.; Piemontese, L.; Cerchia, C.; Montanari, R.; Capelli, D.; Giudici, M.; Crestani, M.; Tortorella, P.; Peiretti, F.; Pochetti, G.; et al. Identification of the First PPARalpha/gamma Dual Agonist Able To Bind to Canonical and Alternative Sites of PPARgamma and To Inhibit Its Cdk5-Mediated Phosphorylation. J. Med. Chem. 2018, 61, 8282–8298. [Google Scholar] [CrossRef]
- Li, J.; Bai, L.; Wei, F.; Zhao, J.; Wang, D.; Xiao, Y.; Yan, W.; Wei, J. Therapeutic Mechanisms of Herbal Medicines Against Insulin Resistance: A Review. Front. Pharmacol. 2019, 10, 661. [Google Scholar] [CrossRef]
- Rau, O.; Wurglics, M.; Dingermann, T.; Abdel-Tawab, M.; Schubert-Zsilavecz, M. Screening of herbal extracts for activation of the human peroxisome proliferator-activated receptor. Pharmazie 2006, 61, 952–956. [Google Scholar]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as nuclear receptors for nutrient and energy metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Park, M.Y.; Sung, M.K. Carnosic Acid Inhibits Lipid Accumulation in 3T3-L1 Adipocytes Through Attenuation of Fatty Acid Desaturation. J. Cancer Prev. 2015, 20, 41–49. [Google Scholar] [CrossRef]
- Wang, T.; Takikawa, Y.; Satoh, T.; Yoshioka, Y.; Kosaka, K.; Tatemichi, Y.; Suzuki, K. Carnosic acid prevents obesity and hepatic steatosis in ob/ob mice. Hepatol Res. 2011, 41, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Song, H.M.; Li, X.; Liu, Y.Y.; Lu, W.P.; Cui, Z.H.; Zhou, L.; Yao, D.; Zhang, H.M. Carnosic acid protects mice from high-fat diet-induced NAFLD by regulating MARCKS. Int. J. Mol. Med. 2018, 42, 193–207. [Google Scholar] [CrossRef]
- Loussouarn, M.; Krieger-Liszkay, A.; Svilar, L.; Bily, A.; Birtic, S.; Havaux, M. Carnosic Acid and Carnosol, Two Major Antioxidants of Rosemary, Act through Different Mechanisms. Plant. Physiol. 2017, 175, 1381–1394. [Google Scholar] [CrossRef]
- Liu, K.L.; Kuo, W.C.; Lin, C.Y.; Lii, C.K.; Liu, Y.L.; Cheng, Y.H.; Tsai, C.W. Prevention of 4-hydroxynonenal-induced lipolytic activation by carnosic acid is related to the induction of glutathione S-transferase in 3T3-L1 adipocytes. Free Radic. Biol. Med. 2018, 121, 1–8. [Google Scholar] [CrossRef]
- Tsai, C.W.; Liu, K.L.; Lin, Y.R.; Kuo, W.C. The mechanisms of carnosic acid attenuates tumor necrosis factor-alpha-mediated inflammation and insulin resistance in 3T3-L1 adipocytes. Mol. Nutr. Food Res. 2014, 58, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Takikawa, Y.; Tabuchi, T.; Satoh, T.; Kosaka, K.; Suzuki, K. Carnosic acid (CA) prevents lipid accumulation in hepatocytes through the EGFR/MAPK pathway. J. Gastroenterol. 2012, 47, 805–813. [Google Scholar] [CrossRef]
- Poeckel, D.; Greiner, C.; Verhoff, M.; Rau, O.; Tausch, L.; Hornig, C.; Steinhilber, D.; Schubert-Zsilavecz, M.; Werz, O. Carnosic acid and carnosol potently inhibit human 5-lipoxygenase and suppress pro-inflammatory responses of stimulated human polymorphonuclear leukocytes. Biochem. Pharmacol. 2008, 76, 91–97. [Google Scholar] [CrossRef]
- Porter, C.; Herndon, D.N.; Bhattarai, N.; Ogunbileje, J.O.; Szczesny, B.; Szabo, C.; Toliver-Kinsky, T.; Sidossis, L.S. Severe burn injury induces thermogenically functional mitochondria in murine white adipose tissue. Shock 2015, 44, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Qi, P.; Abdullahi, A.; Stanojcic, M.; Chen, P.; Parousis, A.; Amini-Nik, S.; Jeschke, M.G. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep. 2015, 13, 1538–1544. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef]
- Billington, E.O.; Grey, A.; Bolland, M.J. The effect of thiazolidinediones on bone mineral density and bone turnover: Systematic review and meta-analysis. Diabetologia 2015, 58, 2238–2246. [Google Scholar] [CrossRef]
- Brusotti, G.; Montanari, R.; Capelli, D.; Cattaneo, G.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Peiretti, F.; Bonardo, B.; Paiardini, A.; et al. Betulinic acid is a PPARgamma antagonist that improves glucose uptake, promotes osteogenesis and inhibits adipogenesis. Sci. Rep. 2017, 7, 5777. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colson, C.; Batrow, P.-L.; Gautier, N.; Rochet, N.; Ailhaud, G.; Peiretti, F.; Amri, E.-Z. The Rosmarinus Bioactive Compound Carnosic Acid Is a Novel PPAR Antagonist That Inhibits the Browning of White Adipocytes. Cells 2020, 9, 2433. https://doi.org/10.3390/cells9112433
Colson C, Batrow P-L, Gautier N, Rochet N, Ailhaud G, Peiretti F, Amri E-Z. The Rosmarinus Bioactive Compound Carnosic Acid Is a Novel PPAR Antagonist That Inhibits the Browning of White Adipocytes. Cells. 2020; 9(11):2433. https://doi.org/10.3390/cells9112433
Chicago/Turabian StyleColson, Cécilia, Pierre-Louis Batrow, Nadine Gautier, Nathalie Rochet, Gérard Ailhaud, Franck Peiretti, and Ez-Zoubir Amri. 2020. "The Rosmarinus Bioactive Compound Carnosic Acid Is a Novel PPAR Antagonist That Inhibits the Browning of White Adipocytes" Cells 9, no. 11: 2433. https://doi.org/10.3390/cells9112433
APA StyleColson, C., Batrow, P.-L., Gautier, N., Rochet, N., Ailhaud, G., Peiretti, F., & Amri, E.-Z. (2020). The Rosmarinus Bioactive Compound Carnosic Acid Is a Novel PPAR Antagonist That Inhibits the Browning of White Adipocytes. Cells, 9(11), 2433. https://doi.org/10.3390/cells9112433