Co-Immobilization and Co-Localization of Oxidases and Catalases: Catalase from Bordetella Pertussis Fused with the Zbasic Domain



Abstract
:1. Introduction
- an enzyme with a very high specific activity, in order to reduce the necessary excess of catalase/oxidase. In this way, a high amount of the main enzyme (oxidase) could be immobilized per gram of solid support.
- a small protein, in order to cover a small percentage of the internal surface of solid support. Due to the tetrameric structure of catalases, the selection will depend on the complete coverage of mesoporous support (e.g., 10% cross-linked—agarose gels) with around 200 mg of enzyme per gram of support.
- an enzyme that can also be easily immobilized to achieve a very rapid immobilization of a high amount of catalase per gram of activated support. Glyoxyl-agarose supports, where most of the enzymes are highly stabilized by multipoint covalent immobilization [21,22,23], could be an interesting option. Furthermore, it could be interesting to have an affinity tag that could also serve as an “immobilization tag”.
2. Results
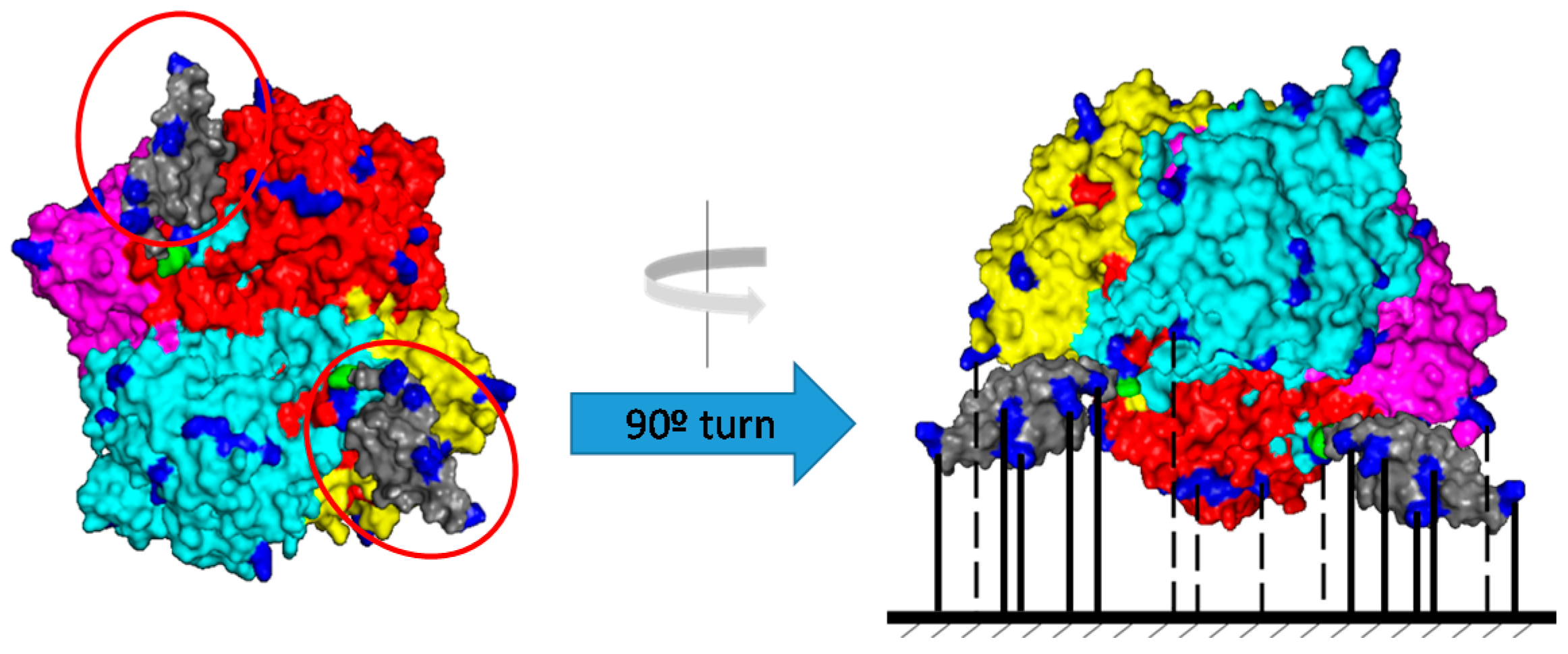
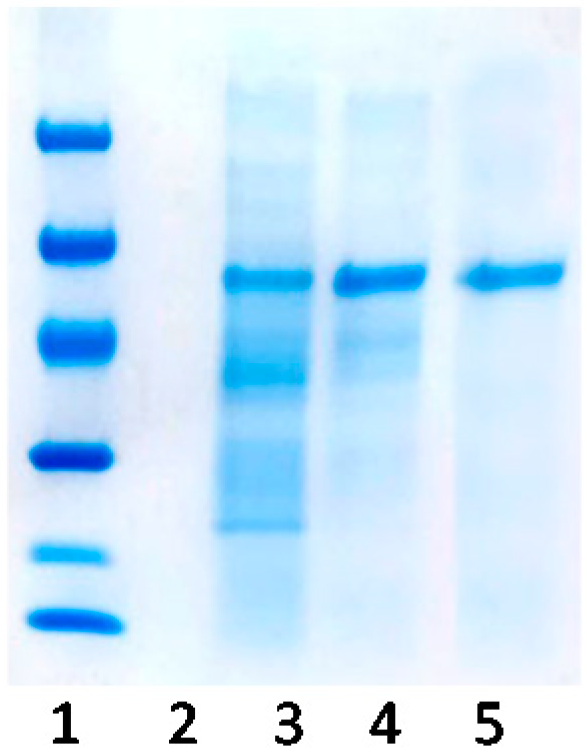
2.1. Purification of Bordetella pertussis Catalase by Ionic Exchange Chromatography on Sulfopropyl-Sepharose Supports
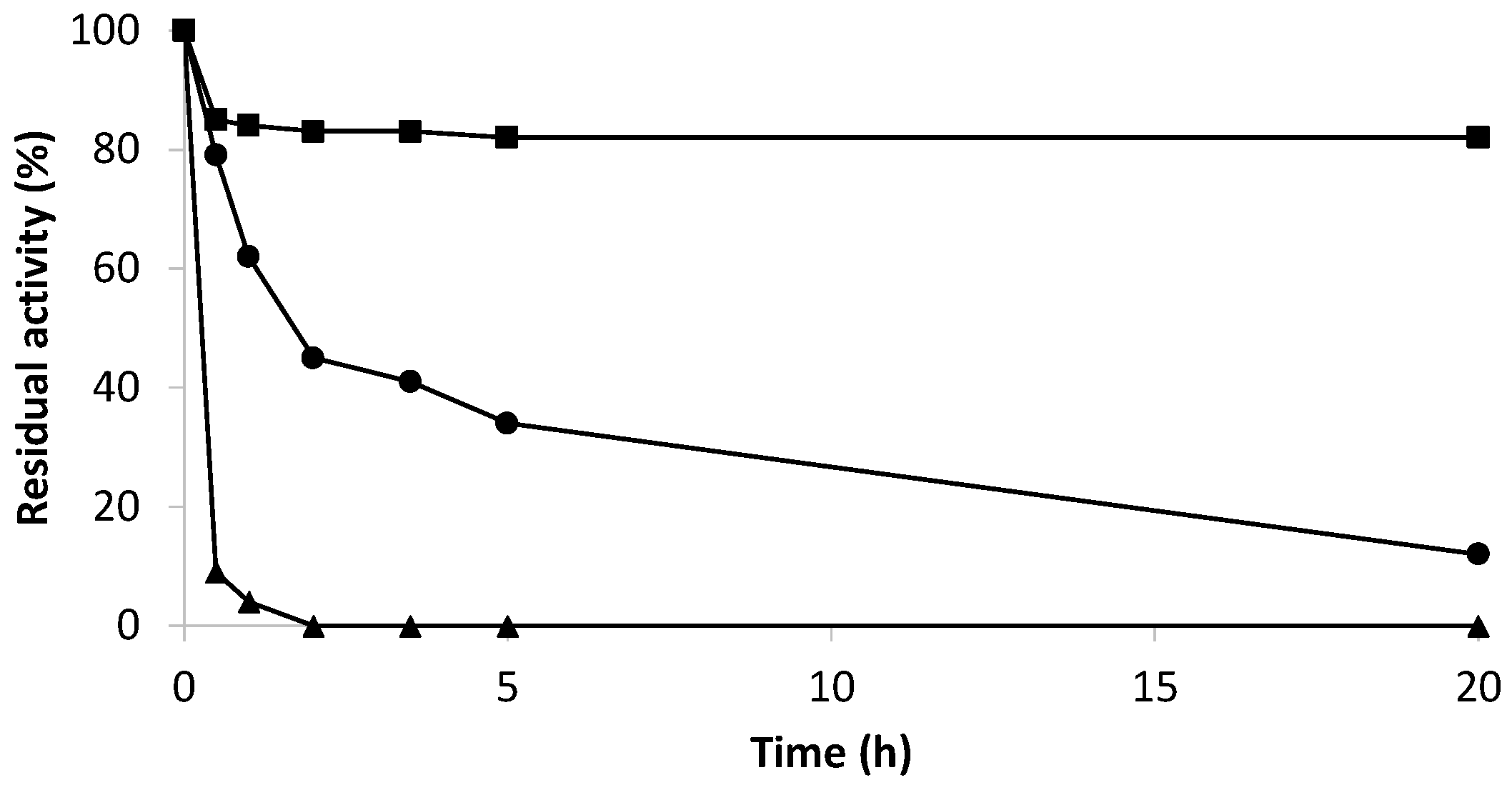
2.2. Immobilization by Multipoint Covalent Attachment of Bordetella pertussis Catalase on Agarose Supports Highly Activated with Aldehyde Groups
2.3. Co-Immobilization and Co-Localization of Amine Oxidase from Pisum sativum and Catalase from Bordetella pertussis in Glyoxyl-Agarose 6BCL Supports
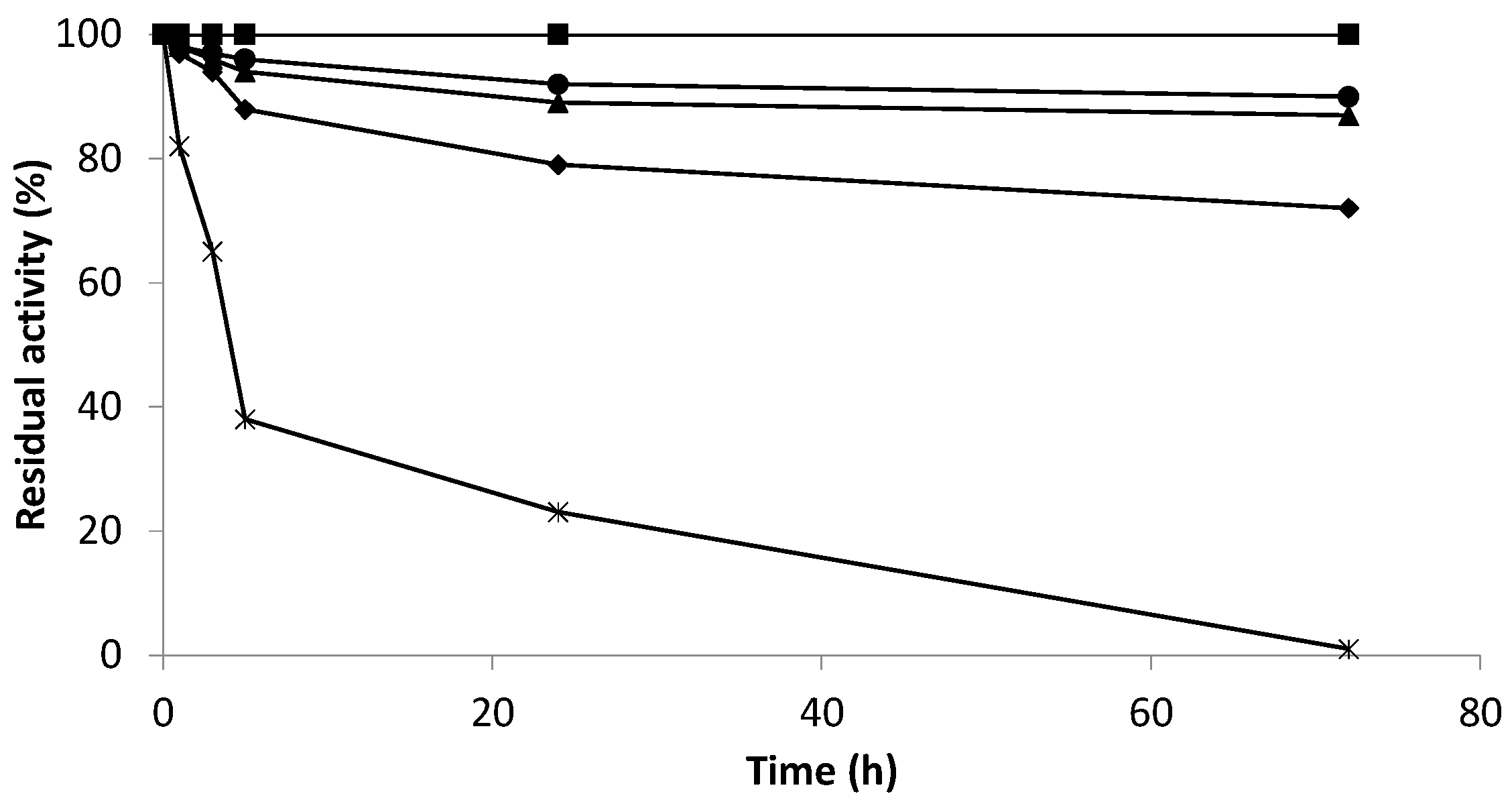
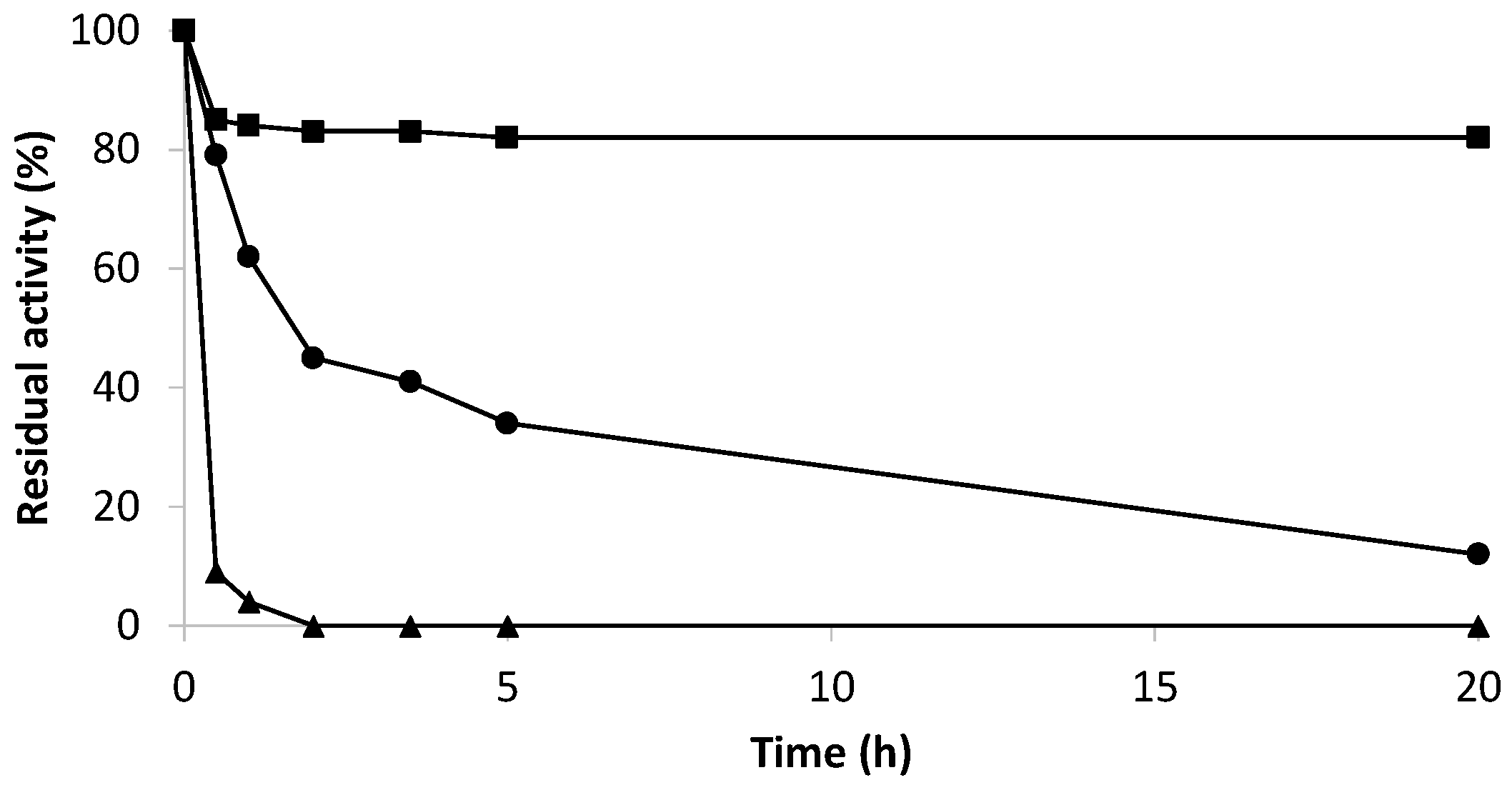
2.4. The Efficiency of Hydrogen Peroxide Elimination by Optimum Biocatalysts
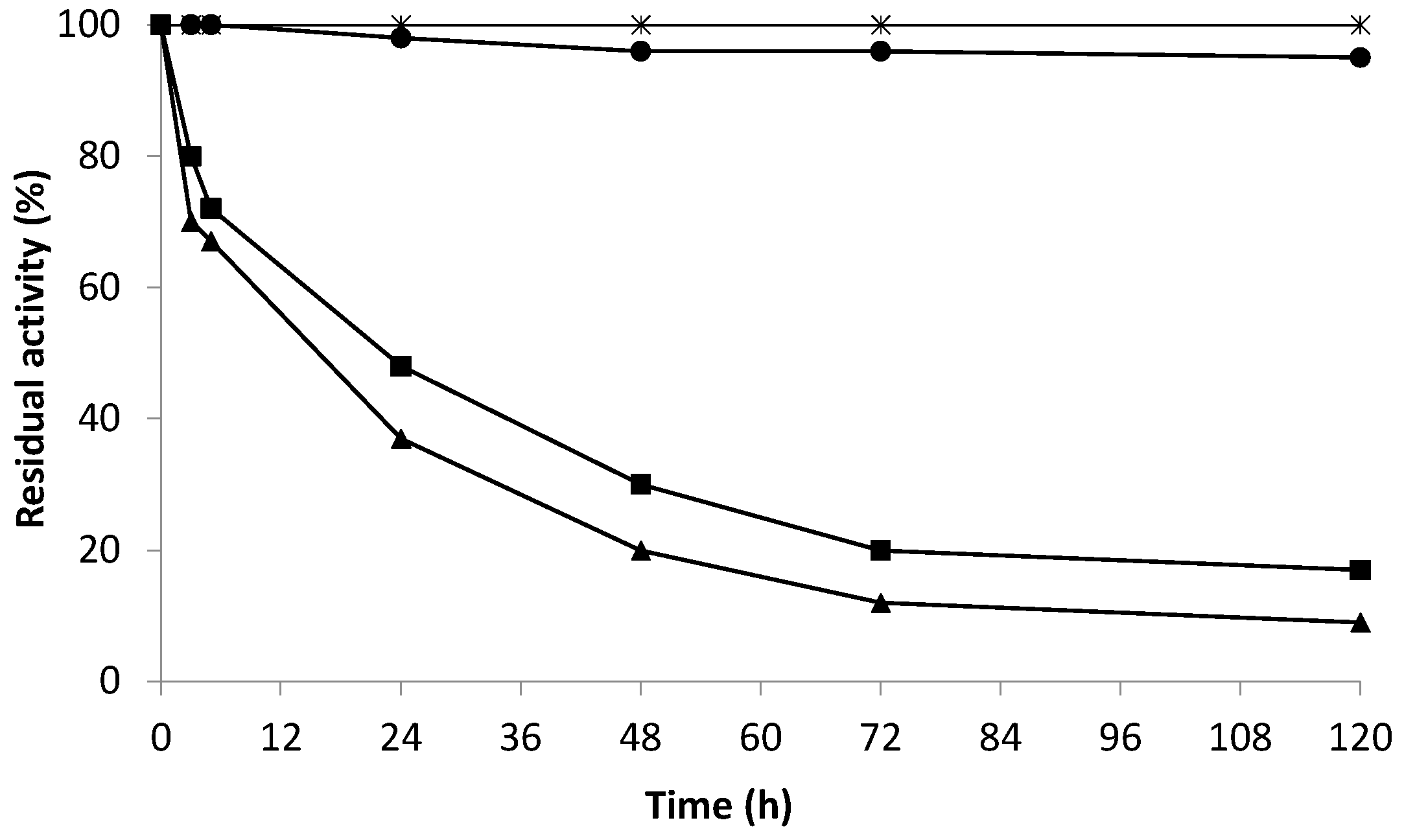
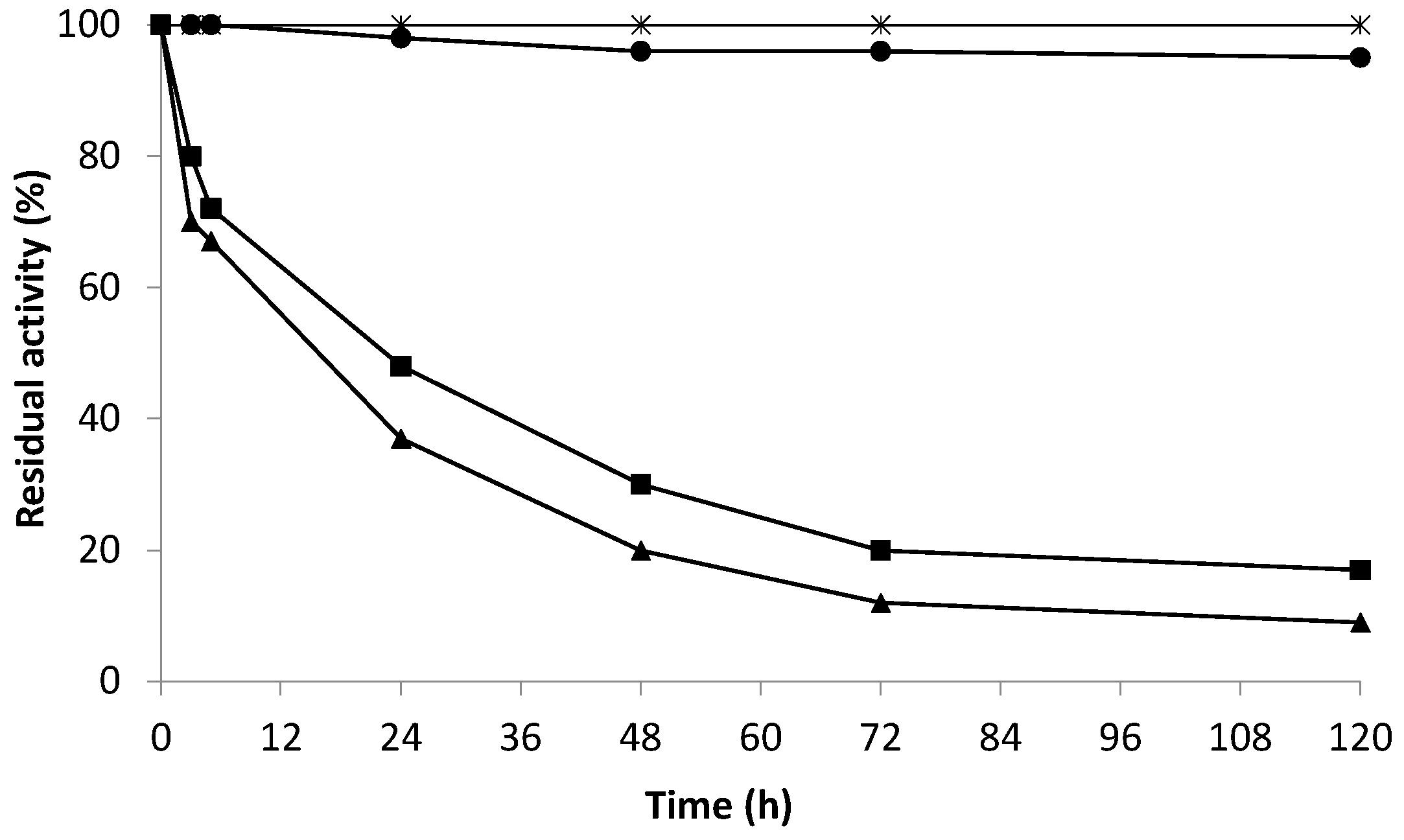
2.5. Effect of Hydrogen Peroxide on the Enzymatic Activity of the Co-Localized Biocatalysts
2.6. Optimization of Co-Localized Biocatalysts for Biogenic Amine Degradation
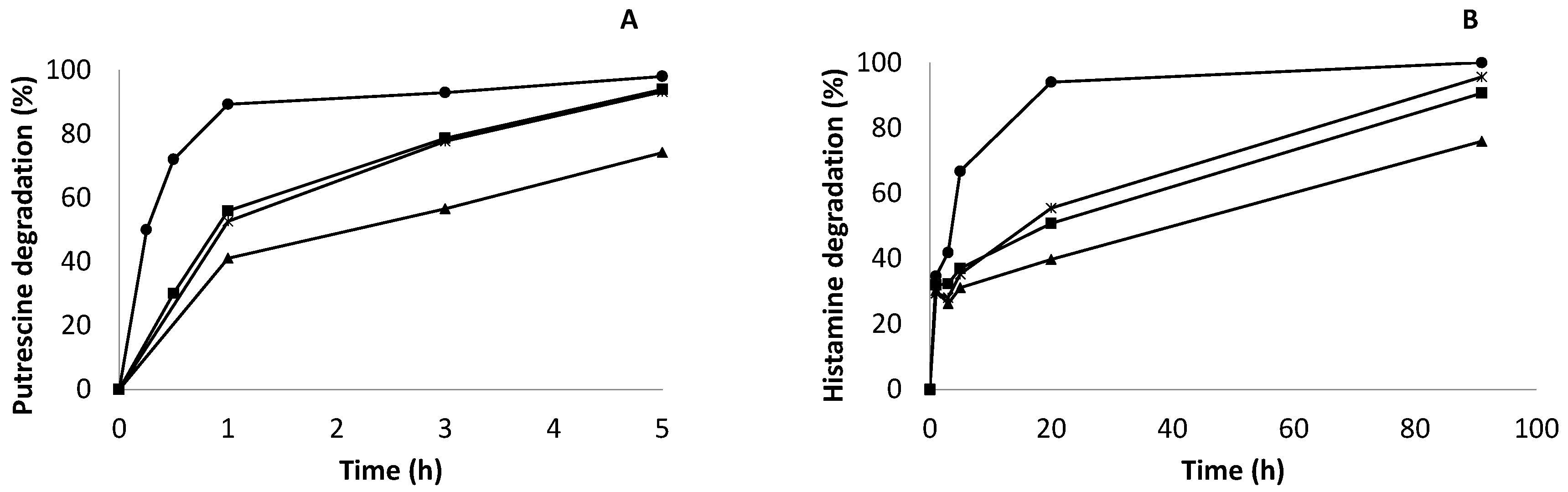
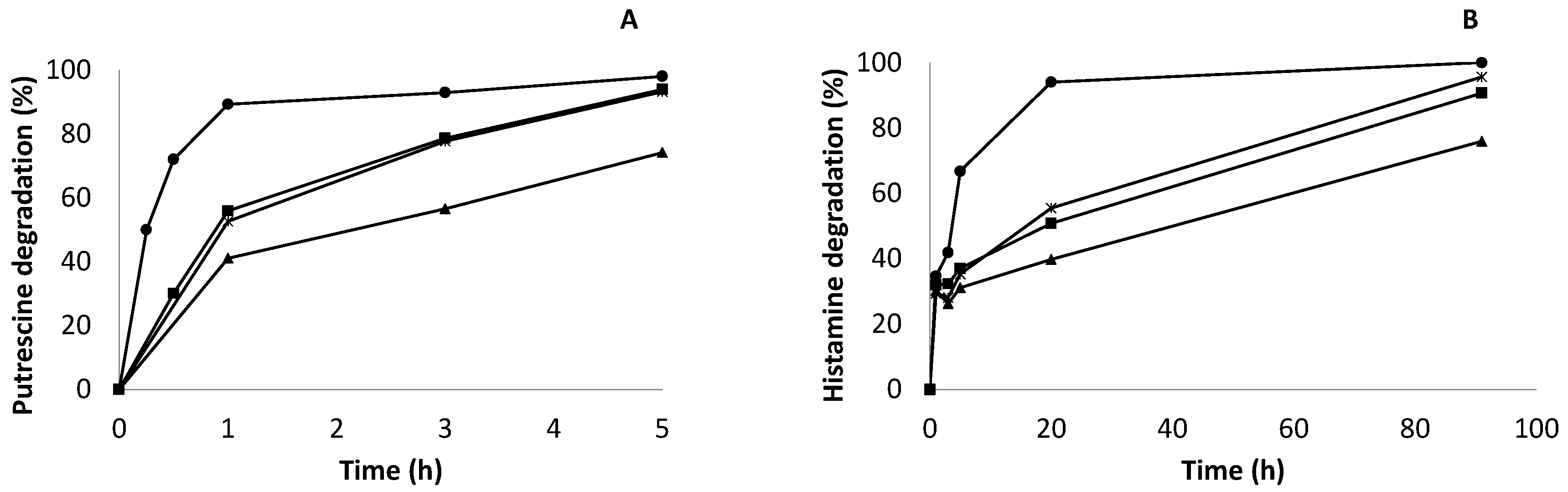
2.7. Enzymatic Degradation of Biogenic Amines (Putrescine and Histamine) in Synthetic Wine by Co-Localized Derivatives
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Spectrophotometric Assays
- a.
- Determination of amine oxidase activity
- b.
- Determination of catalase activity
3.2.2. Preparation of Immobilization Supports
3.2.3. Bordetella pertussis Zbasic2BpKatA Catalase Production
3.2.4. Purification of Catalase KatA by Ionic-Exchange Chromatography on Sulfopropyl Agarose Supports
3.2.5. SDS-PAGE Analysis
3.2.6. Enzyme Immobilization
- a.
- Immobilization of horseradish peroxidase on cyanogen bromide supports
- b.
- Immobilization of catalase KatA on cyanogen bromide supports
- c.
- Multipoint covalent attachment of catalase KatA on glyoxyl-agarose 10BCL supports
- d.
- Co-immobilization and co-localization of Pisum sativum amine oxidase and bovine liver catalase via multipoint covalent attachment on glyoxyl-agarose 6BCL supports
- e.
- Co-immobilization and co-localization of Pisum sativum amine oxidase and catalase KatA via multipoint covalent attachment on glyoxyl-agarose 6BCL and 10BCL supports
3.2.7. Inactivation Assays of Derivatives
- a.
- Stability at different pH and temperature
- b.
- Stability in wine conditions
- c.
- Stability in wine conditions and the presence of hydrogen peroxide
3.2.8. Evaluation of the Hydrogen Peroxide Released into the Reaction Medium
3.2.9. Enzymatic Degradation of Biogenic Amines by Co-Localized Derivatives in Synthetic Wine
3.2.10. Analysis of Biogenic Amine Degradation by RP-HPLC
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vennestrøm, P.N.R.; Christensen, C.H.; Pedersen, S.; Grunwaldt, J.D.; Woodley, J.M. Next-generation catalysis for renewables: Combining enzymatic with inorganic heterogeneous catalysis for bulk chemical production. ChemCatChem 2010, 2, 249–258. [Google Scholar] [CrossRef]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, D.; Ottolina, G.; Carrea, G.; Riva, S. Redox reactions catalyzed by isolated enzymes. Chem. Rev. 2011, 111, 4111–4140. [Google Scholar] [CrossRef] [PubMed]
- Mafra, A.C.O.; Ulrich, L.G.; Kornecki, J.F.; Fernández-Lafuente, R.; Tardioli, P.W.; Ribeiro, M.; Mafra, O.; Ulrich, G.; Lafuente, F. Combi-CLEAs of glucose oxidase and catalase for conversion of glucose to gluconic acid eliminating the hydrogen peroxide to maintain enzyme activity in a bubble column reactor. Catalysts 2019, 9, 657. [Google Scholar] [CrossRef] [Green Version]
- Jiao, M.; Li, Z.; Li, X.; Zhang, Z.; Yuan, Q.; Vriesekoop, F.; Liang, H.; Liu, J. Solving the H2O2 by-product problem using a catalase-mimicking nanozyme cascade to enhance glycolic acid oxidase. Chem. Eng. J. 2020, 388, 124249. [Google Scholar] [CrossRef]
- Wu, Z.; Shi, L.; Yu, X.; Zhang, S.; Chen, G. Co-immobilization of tri-enzymes for the conversion of hydroxymethylfurfural to 2,5-diformylfuran. Molecules 2019, 24, 3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, L.; Meng, Y.; Wang, R.; Jia, B.; Li, P. Coupling and regulation of porous carriers using plasma and amination to improve the catalytic performance of glucose oxidase and catalase. Front. Bioeng. Biotechnol. 2019, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Morthensen, S.T.; Meyer, A.S.; Jørgensen, H.; Pinelo, M. Significance of membrane bioreactor design on the biocatalytic performance of glucose oxidase and catalase: Free vs. immobilized enzyme systems. Biochem. Eng. J. 2017, 117, 41–47. [Google Scholar] [CrossRef]
- Zhuang, W.; Huang, J.; Liu, X.; Ge, L.; Niu, H.; Wang, Z.; Wu, J.; Yang, P.; Chen, Y.; Ying, H. Co-localization of glucose oxidase and catalase enabled by a self-assembly approach: Matching between molecular dimensions and hierarchical pore sizes. Food Chem. 2019, 275, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Küchler, A.; Adamcik, J.; Mezzenga, R.; Schlüter, A.D.; Walde, P. Enzyme immobilization on silicate glass through simple adsorption of dendronized polymer–enzyme conjugates for localized enzymatic cascade reactions. RSC Adv. 2015, 5, 44530–44544. [Google Scholar] [CrossRef]
- García-García, P.; Rocha-Martin, J.; Fernández-Lorente, G.; Guisan, J.M. Co-localization of oxidase and catalase inside a porous support to improve the elimination of hydrogen peroxide: Oxidation of biogenic amines by amino oxidase from Pisum sativum. Enzym. Microb. Technol. 2018, 115, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Gascon, V.; Marquez-Alvarez, C.; Blanco, R.M.; Nidetzky, B. Oriented coimmobilization of oxidase and catalase on tailor-made ordered mesoporous silica. Langmuir 2017, 33, 5065–5076. [Google Scholar] [CrossRef] [Green Version]
- Guisán, J. Aldehyde-agarose gels as activated supports for immobilization-stabilization of enzymes. Enzyme Microb. Technol. 1988, 10, 375–382. [Google Scholar] [CrossRef]
- Betancor, L.; Gallego, F.L.; Hidalgo, A.; Alonso-Morales, N.; Dellamora-Ortiz, G.; Guisán, J.M.; Fernández-Lafuente, R. Preparation of a very stable immobilized biocatalyst of glucose oxidase from Aspergillus niger. J. Biotechnol. 2006, 121, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gallego, F.; Fernandez-Lorente, G.; Rocha-Martín, J.; Bolivar, J.M.; Mateo, C.; Guisan, J.M. Multi-point covalent immobilization of enzymes on glyoxyl agarose with minimal physico-chemical modification: Stabilization of industrial enzymes. In Immobilization of Enzymes and Cells; Humana: New York, NY, USA, 2020; pp. 93–107. ISBN 9781071602140. [Google Scholar]
- Lončar, N.; Fraaije, M.W. Catalases as biocatalysts in technical applications: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 3351–3357. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Schelch, S.; Pfeiffer, M.; Nidetzky, B. Intensifying the O2-dependent heterogeneous biocatalysis: Superoxygenation of solid support from H2O2 by a catalase tailor-made for effective immobilization. J. Mol. Catal. B Enzym. 2016, 134, 302–309. [Google Scholar] [CrossRef]
- Fuchs, S.M.; Raines, R.T. Polyarginine as a multifunctional fusion tag. Protein Sci. 2009, 14, 1538–1544. [Google Scholar] [CrossRef] [Green Version]
- Pina, A.; Lowe, C.R.; Roque, A.C.A. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol. Adv. 2014, 32, 366–381. [Google Scholar] [CrossRef]
- Li, Y. Self-cleaving fusion tags for recombinant protein production. Biotechnol. Lett. 2011, 33, 869–881. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.; Hidalgo, A.; Fernandez-Lorente, G.; Fernandez-Lafuente, R.; et al. Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins. Enzym. Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]
- Grazu, V.; Betancor, L.; Montes, T.; Gallego, F.L.; Guisan, J.M.; Fernández-Lafuente, R. Glyoxyl agarose as a new chromatographic matrix. Enzym. Microb. Technol. 2006, 38, 960–966. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Gallego, F.L.; Bolivar, J.M.; Rocha-Martin, J.; Moreno-Perez, S.; Guisán, J.M. Immobilization of proteins on glyoxyl activated supports: Dramatic stabilization of enzymes by multipoint covalent attachment on pre-existing supports. Curr. Org. Chem. 2015, 19, 1. [Google Scholar] [CrossRef]
- Da Silva, M.V.C.; Souza, A.B.; De Castro, H.F.; Aguiar, L.G.; De Oliveira, P.C.; Freitas, L. Synthesis of 2-ethylhexyl oleate catalyzed by Candida antarctica lipase immobilized on a magnetic polymer support in continuous flow. Bioprocess Biosyst. Eng. 2019, 43, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Mannsberger, A.; Thomsen, M.S.; Tekautz, G.; Nidetzky, B. Process intensification for O2-dependent enzymatic transformations in continuous single-phase pressurized flow. Biotechnol. Bioeng. 2019, 116, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesbauer, J.; Bolivar, J.M.; Mueller, M.; Schiller, M.; Nidetzky, B. Oriented immobilization of enzymes made fit for applied biocatalysis: Non-covalent attachment to anionic supports using Zbasic2 Module. ChemCatChem 2011, 3, 1299–1303. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Nidetzky, B. Positively charged mini-protein zbasic2 as a highly efficient silica binding module: Opportunities for enzyme immobilization on unmodified silica supports. Langmuir 2012, 28, 10040–10049. [Google Scholar] [CrossRef]
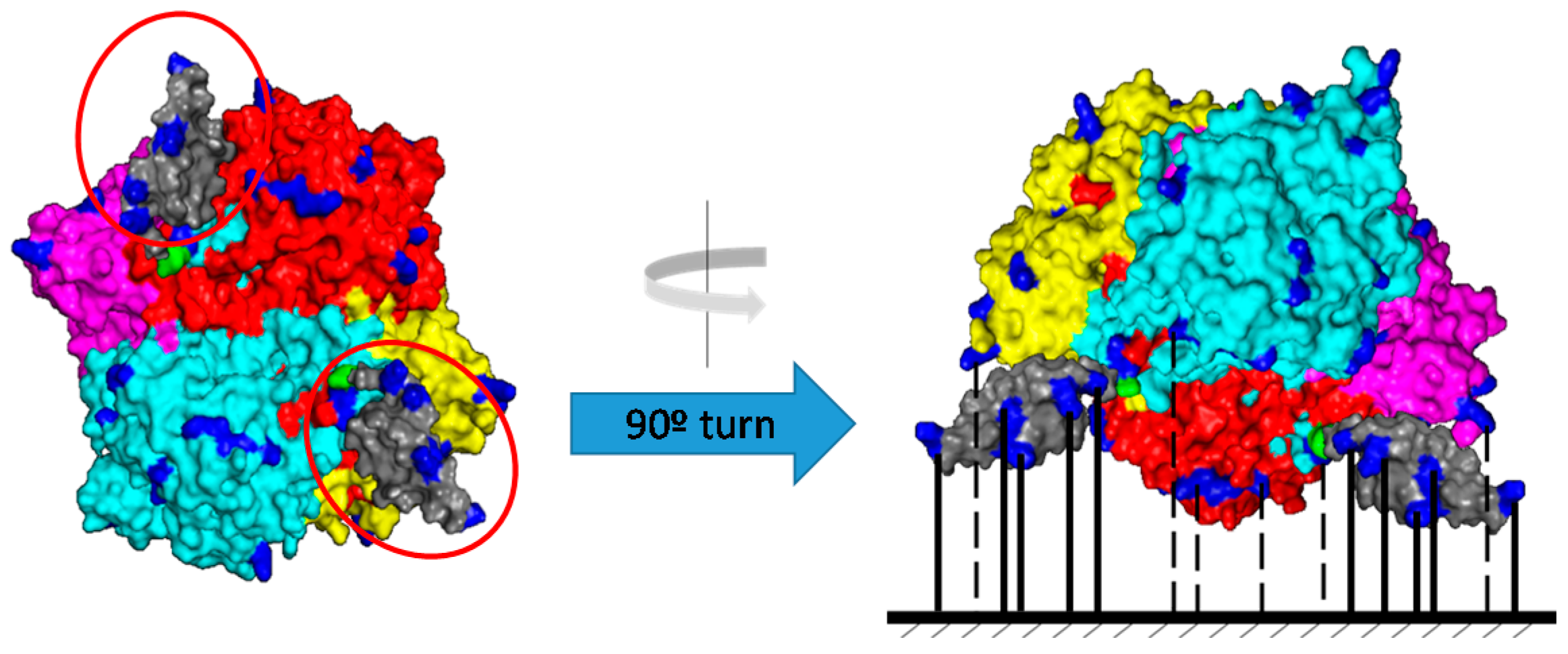
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef]
- Hedhammar, M.; Hober, S. Zbasic—A novel purification tag for efficient protein recovery. J. Chromatogr. A 2007, 1161, 22–28. [Google Scholar] [CrossRef]
- Gräslund, T.; Ehn, M.; Lundin, G.; Hedhammar, M.; Uhlen, M.; Nygren, P.Å.; Hober, S. Strategy for highly selective ion-exchange capture using a charge-polarizing fusion partner. J. Chromatogr. A 2002, 942, 157–166. [Google Scholar] [CrossRef]
- García-García, P.; Guisan, J.M.; Fernandez-Lorente, G. A mild intensity of the enzyme-support multi-point attachment promotes the optimal stabilization of mesophilic multimeric enzymes: Amine oxidase from Pisum sativum. J. Biotechnol. 2020. Accepted. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Rocha-Martin, J.; Mateo, C.; Guisan, J.M. Stabilization of a highly active but unstable alcohol dehydrogenase from yeast using immobilization and post-immobilization techniques. Process. Biochem. 2012, 47, 679–686. [Google Scholar] [CrossRef]
- Fernández-Lafuente, R.; Rodríguez, V.; Mateo, C.; Fernandez-Lorente, G.; Arminsen, P.; Sabuquillo, P.; Guisan, J.M. Stabilization of enzymes (d-amino acid oxidase) against hydrogen peroxide via immobilization and post-immobilization techniques. J. Mol. Catal. B: Enzym. 1999, 7, 173–179. [Google Scholar] [CrossRef]
- Hidalgo, A.; Betancor, L.; Gallego, F.L.; Moreno, R.; Berenguer, J.; Fernández-Lafuente, R.; Guisán, J.M. Design of an immobilized preparation of catalase from Thermus thermophilus to be used in a wide range of conditions. Enzyme Microb. Technol. 2003, 33, 278–285. [Google Scholar] [CrossRef]
- Kaddour, S.; Gallego, F.L.; Sadoun, T.; Fernández-Lafuente, R.; Guisan, J.M. Preparation of an immobilized–stabilized catalase derivative from Aspergillus niger having its multimeric structure stabilized: The effect of Zn2+ on enzyme stability. J. Mol. Catal. B Enzym. 2008, 55, 142–145. [Google Scholar] [CrossRef]
- Rocha-Martin, J.; Acosta, A.; Berenguer, J.; Guisán, J.M.; Lopez-Gallego, F. Selective oxidation of glycerol to 1,3-dihydroxyacetone by covalently immobilized glycerol dehydrogenases with higher stability and lower product inhibition. Bioresour. Technol. 2014, 170, 445–453. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Romero, O.; Guisán, J.M.; Illanes, A.; Wilson, L. Reactivation of penicillin acylase biocatalysts: Effect of the intensity of enzyme–support attachment and enzyme load. J. Mol. Catal. B Enzym. 2012, 74, 224–229. [Google Scholar] [CrossRef]
- Cueva, C.; García-Ruiz, A.; González-Rompinelli, E.; Bartolomé, B.; Martin-Alvarez, P.; Salazar, O.; Vicente, M.; Bills, G.; Moreno-Arribas, V. Degradation of biogenic amines by vineyard ecosystem fungi. Potential use in winemaking. J. Appl. Microbiol. 2012, 112, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalase | Specific Activity (U/mg) * | Immobilization Rate ** |
|---|---|---|
| Bovine liver | 4.5 | 0.04 |
| Bordetella pertussis | 23 | 0.29 |
| Incubation Time (h) * | Recovered Activity (%) |
|---|---|
| 0 | 70 |
| 3 | 61 |
| 5 | 48 |
| 24 | 30 |
| Immobilization (%) | Recovered Activity (%) | |||||
|---|---|---|---|---|---|---|
| Ratio AO/KatA | Gx-AO/KatA a | AO | KatA | AO | KatA | Released H2O2 (%) b |
| 1/12 | 5/60 | 90 | 96 | 68 | 35 | 8 |
| 1/8.6 | 7/60 | 95 | 93 | 74 | 33 | 7 |
| 1/6 | 10/60 | 90 | 95 | 70 | 30 | 8 |
| 1/4 | 15/60 | 92 | 94 | 71 | 34 | 12 |
| 1/3 | 20/60 | 90 | 95 | 65 | 34 | 28 |
| Immobilization (%) | Recovered Activity (%) | |||||
|---|---|---|---|---|---|---|
| Ratio AO/KatA | Gx-AO/KatA a | AO | KatA | AO | KatA | Released H2O2 (%) b |
| 1/5 | 30/160 | 95 | 98 | 58 | 20 | 8 |
| Gx-AO/CAT | Ratio AO/Catalase | Specific Activity (U/mL) * |
|---|---|---|
| 1.25/25 | 1/20 | 0.013 |
| 10/60 | 1/6 | 0.099 |
| 30/160 | 1/5 | 0.19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, P.; Rocha-Martin, J.; Guisan, J.M.; Fernandez-Lorente, G. Co-Immobilization and Co-Localization of Oxidases and Catalases: Catalase from Bordetella Pertussis Fused with the Zbasic Domain. Catalysts 2020, 10, 810. https://doi.org/10.3390/catal10070810
García-García P, Rocha-Martin J, Guisan JM, Fernandez-Lorente G. Co-Immobilization and Co-Localization of Oxidases and Catalases: Catalase from Bordetella Pertussis Fused with the Zbasic Domain. Catalysts. 2020; 10(7):810. https://doi.org/10.3390/catal10070810
Chicago/Turabian StyleGarcía-García, Paz, Javier Rocha-Martin, Jose M. Guisan, and Gloria Fernandez-Lorente. 2020. "Co-Immobilization and Co-Localization of Oxidases and Catalases: Catalase from Bordetella Pertussis Fused with the Zbasic Domain" Catalysts 10, no. 7: 810. https://doi.org/10.3390/catal10070810