Effects of GH/IGF on the Aging Mitochondria
by
 , and
, and
Sher Bahadur Poudel
1,† ,
,
Manisha Dixit
1,†,
Maria Neginskaya
1,†,
Karthik Nagaraj
2,†,
Evgeny Pavlov
1,
Haim Werner
2 and
and
Shoshana Yakar
1,* 1
David B. Kriser Dental Center, Department of Molecular Pathobiology, New York University College of Dentistry New York, NY 10010–4086, USA
2
Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(6), 1384; https://doi.org/10.3390/cells9061384
Submission received: 16 April 2020
/
Revised: 25 May 2020
/
Accepted: 26 May 2020
/
Published: 2 June 2020
(This article belongs to the Special Issue Insulin-Like Growth Factors in Development, Cancers and Aging)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The mitochondria are key organelles regulating vital processes in the eukaryote cell. A decline in mitochondrial function is one of the hallmarks of aging. Growth hormone (GH) and the insulin-like growth factor-1 (IGF-1) are somatotropic hormones that regulate cellular homeostasis and play significant roles in cell differentiation, function, and survival. In mammals, these hormones peak during puberty and decline gradually during adulthood and aging. Here, we review the evidence that GH and IGF-1 regulate mitochondrial mass and function and contribute to specific processes of cellular aging. Specifically, we discuss the contribution of GH and IGF-1 to mitochondrial biogenesis, respiration and ATP production, oxidative stress, senescence, and apoptosis. Particular emphasis was placed on how these pathways intersect during aging.
1. Overview of the growth hormone (GH)/insulin-like growth factor (IGF ) Axis
Growth hormone (GH) and the insulin-like growth factor-1 (IGF-1) are part of the somatotropic hypothalamic-pituitary axis that regulates somatic growth and aging. As such, GH and IGF-1 peak during puberty to support lean and fat mass gain as well as to enhance skeletal acquisition and linear growth. During aging, GH and IGF-1 levels are significantly reduced, a state termed somatopause. Somatopause associates with many pathologies, such as osteopenia [1], sarcopenia [2], cardiovascular disorders [3,4], and more. For the sake of this review, it is important to distinguish between congenital somatopause, which results from germline mutations in components of the GH/IGF-1 axis (resulting in life-long epigenetic changes), and age-induced somatopause, which refers to the natural decline in GH and IGF-1 levels during aging.
The GH/IGF-1 axis includes the hypothalamic GH-releasing hormone (GHRH) and its receptor (GHRHR), expressed mainly in the pituitary (Figure 1) [5]. Upon activation, the GHRHR stimulates pituitary secretion of GH, which is released to the circulation and binds to its specific membrane receptor (GHR) on cells of different tissues. Among the many actions of GH in liver, its major role is stimulating the transcription of the IGF-1 gene, the IGF-binding proteins (IGFBPs) [6], and the acid labile subunit (ALS) that carry IGF-1 in circulation [7]. Other members of this axis are the proteases that release the IGF-1 from its complex with IGFBPs, pregnancy-associated plasma protein A (PAPP-A), and the PAPP-A2 [8]. The somatotropic axis is tightly regulated under normal physiological conditions. IGF-1 in serum provides a negative feedback to the pituitary to specifically inhibit GH secretion. Somatostatin (SST), secreted by the hypothalamus, inhibits several pituitary hormones including GH. In addition, SST binds to several SST-receptors (SSTR) on peripheral tissues to inhibit production of pancreatic (glucagon and insulin) and gastric (secretin and gastrin) hormones. Secretion of GH is also regulated by the gastrointenstinal hormone ghrelin. Ghrelin is one of the hormones that regulate food intake [9]. During hunger, ghrelin levels in circulation are increased and activate hypothalamic ghrelin receptor to initiate appetite. In addition, ghrelin acts as a GH secretagogue and can induce pituitary secretion of GH [10].
Receptors to GH and IGF-1 are present in virtually all cells. The IGF-1 receptor (IGF-1R) is a tyrosine kinase receptor that activates multiple pathways including (but not limited to) the Phosphoinositide-3-kinase (PI3K)/Protein kinase B (PKB or AKT), Ras/Raf/Mitogen-activated protein kinase (MAPK), and Shc [11]. These pathways are implicated in cellular growth, proliferation, differentiation, survival, metabolism, gene transcription, and protein translation. Apparently, the PI3K/AKT pathway is central to regulation of cell metabolism and cell fate (apoptosis), both of which involve mitochondrial function and integrity. The GHR is a cytokine-like receptor that, upon ligand binding, activates the Janus kinase 2 (JAK2)/Signal transducer and activator of transcription 5B (STAT5b) pathway to induce transcriptional activation of genes involved in cellular growth (among them is Igf1) and metabolic homeostasis [11].
Among other aspects of cellular homeostasis, GH and IGF-1 have important roles in mitochondrial function. Mitochondria participate in a wide range of processes with vital roles in cellular function. A key function of mitochondria is generation of adenosine triphosphate (ATP) from metabolic processing of carbohydrates, fats, and amino acids. In addition, mitochondria regulate other functions such as calcium homeostasis, cell death programming (apoptosis), inflammation, heat production, and other tissue-specific functions. Given their critical roles in the cell, it is not surprising that mitochondria are considered central regulators of aging. This review summarizes a few aspects of mitochondrial regulation by the GH/IGF-1 axis. We could not possibly include all of the studies that reported how GH/IGF-1 control mitochondrial function, including the effects of this axis on mitochondria in cancer cells (which can be found in [12,13,14]). Instead, our review focuses mostly on different aspects of aging and how GH/IGF-1 affects mitochondria in that context.
2. GH/IGF-1 Effects on Mitochondrial Biogenesis
Mitochondria are dynamic organelles. The number and size of mitochondria change according to the cellular metabolic and physiologic conditions. In addition, mitochondria abundance involves fusion (merging of two originally distinct mitochondria into one), fission (division of a single mitochondrion into two distinct mitochondria), and biogenesis. Mitochondrial biogenesis requires coupling of mitochondrial genome replication and fission.
The mitochondrial genome (mtDNA) encodes 13 proteins, 22 transfer-RNAs, and two ribosomal-RNAs [15]. The 13 proteins encoded by mitochondria take part in oxidative phosphorylation (OXPHOS) including cytochrome b of complex III; ATP synthase subunits 6 and 8 of the F0 ATP-synthase complex; cytochrome c (Cyt c) oxidase subunits 1, 2, and 3; and six subunits of the nicotinamide adenine dinucleotide (NADH) dehydrogenase of complex I (ND1 through ND6). However, there are approximately 1500 proteins encoded by the nucleus which are necessary for mitochondrial biogenesis and function [16,17]. As such, replication of the mitochondrial genome is a complex mechanism that requires activation and translation of a series of nuclear encoded proteins. These mitochondrially targeted proteins are transported to the mitochondria via channels composed of transporters located at the outer mitochondrial membrane (TOMs) [18]. One of the proteins that plays key roles in mitochondrial biogenesis is peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC1α). Activation of PGC1α leads to subsequent activations of the transcriptional regulators, nuclear respiratory factors (NRF1 and 2), and peroxisome proliferator-activated receptors (PPARs), which initiate transcription of nuclear genes involved in mitochondrial biogenesis and function (Figure 2).
The possible roles of the GH/IGF-1 axis in mitochondrial biogenesis were initially recognized in the early 1970s. Following injection of radiolabelled bovine GH (125I-bGH) to Sprague Dawley rats, radioactive signals were detected in the mitochondria, nuclei, and other cytoplasmic compartments of liver and kidney cells [19]. However, a later study found that, in fact, a very negligible amount of radiolabelled 125I-bGH gets transported to the mitochondria compared to other subcellular compartments, including microsomes, endosomes, lysosomes, and Golgi bodies [20], suggesting that GH does not act directly on mitochondria. Instead, it was found that GH affects mitochondrial protein synthesis. Injection of human GH (hGH) to intact and hypophysectomized (hypox) rats revealed that the mitochondrial protein synthetic capacity of liver, measured by radioactive leucine incorporation in vitro as well as in vivo, significantly increased in hGH-treated rats [21]. However, a later study showed that GH is not the only player in mitochondrial biogenesis. Thus, hypox SD rats that were treated with hGH (120 µg/day), triiodothyronine (T3, 10 ng/day), or a combination of both hormones for six days showed that cellular respiration was recovered with T3 treatment but not with hGH alone [20].
Identification of the tight relationship between GH and IGF-1 in control of cellular growth and replication led to numerous studies aimed at assessing the effects of IGF-1 on mitochondrial biogenesis. However, as was found with GH, IGF-1 plays indirect effects on mitochondrial biogenesis, likely in synergy with other growth factors. As such, exogenous IGF-1 increased cell volume but not mitochondria in rat sciatic Schwann cells [22]. However, addition of the mitogenic factor neuregulin (NRG) together with IGF-1 increased mitochondrial mass and DNA replication. In MCF-7 and ZR75.1 breast cancer cells, IGF-1 induced the expression of PGC-1β and PGC-1α-related coactivator (PRC), which are required for mitochondrial biogenesis (Figure 2) [13]. Increased mitochondrial mass in these cells associated with reduced expression of the mitophagy mediators BNIP3 and BNIP3L (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 and L) and impaired mitophagy suggest that IGF-1 indirectly regulates mitochondrial turnover via BNIP3. Increased levels of PGC-1α, Nrf1, Cyt c, and citrate synthase activity were also seen in gastrocnemius muscle cells of old animals treated with GH, likely mediated by IGF-1 (Figure 2) [23]. Age-induced mitochondrial dysfunction in reproductive organs causes reductions in oocyte quality leading to infertility. GH partially rescued the age-induced loss of ovulation stimulation, oocyte formation, and maturation through its effects on mitochondria [24]. Treating young (4 weeks) and aged (32 weeks) female mice with low, medium, or high doses of hGH for 8 weeks resulted in increased oocyte number as well as maturation in aged females that was similar to young females, likely via IGF-1. However, in the latter study, despite these oocyte functional improvements, there was no change in mitochondrial DNA copy number, suggesting again that mitochondrial biogenesis was not affected directly by hGH treatment.
3. GH/ IGF-1 Effects on Mitochondrial Respiration and ATP Production During Aging
Mitochondrial function involves several processes, including cellular respiration, energy production via the tricarboxylic acid (TCA) cycle coupled with OXPHOS, calcium homeostasis, cellular replication, apoptosis, and generation of (and protection from) reactive oxygen species (ROS). These functions are fundamental during growth and development and play major roles during aging. Although age-induced mitochondrial dysfunction and overall reductions in the secretion and action of GH and IGF-1 coincide, a clear causality between both processes has not yet been established.
Congenital (life-long) reductions in the activity of the GH/IGF-1 axis was associated with increased longevity in mice [25], flies [26], and nematodes [27,28]. In the long-lived Ames dwarf mice, the activity and expression of several complexes of the electron transport chain (ETC) in liver and in kidney increased [29]. Interestingly, high activity of complex IV of the ETC in Ames mice was in agreement with enhanced oxygen metabolism found also in the long-lived GH receptor null (GHRKO) mice [30]. Elevated expression of key proteins of the mitochondrial respiration complexes in Ames mice coincided with reduced production of H2O2 in state 3 (ATP production) and state 4 (reflecting mitochondrial leak) of respiration in liver mitochondria [31]. Accordingly, the long-lived GHRKO and Ames mice exhibit improved glucose homeostasis and energy metabolism evidenced by decreased respiratory quotient (RQ) and increased oxygen consumption (VO2). In contrast, bGH mice, with high GH/IGF-1 levels show decreased VO(2) and increased RQ [30,32,33]. Taken together, these animal models suggest that diminished GH/IGF-1 activity improves mitochondrial flexibility and increases the capacity for fat oxidation. Interestingly, a recent study found that housing the GHRKO mice at thermoneutral temperature (30 °C) resulted in decreased expression of thermogenic genes in brown adipose tissue (BAT) and elevated core body temperature [32]. However, these mice still maintained their extended longevity phenotype at 30 °C [32], suggesting an intrinsic advantage of mitochondrial function in the GHRKO mice. Together, congenital decreases in the GH/IGF signaling in genetically modified mice were associated with reduced levels of ROS, upregulated activity of the mitochondrial ETC, and overall enhanced mitochondrial function [31,34].
In humans, GH and IGF-1 levels peak during puberty, decline during aging [35], and coincide with unfavorable effects on mitochondrial metabolism in brain (cognition [36]), muscle (sarcopenia [2]), and skeletal tissue (osteopenia [1]). IGF-1 haploinsufficiency (IGF-1+/−) in mice was associated with mitochondrial dysfunction accompanied by increased level of lipid peroxidation, protein carboxylation, and intramitochondrial ROS in hepatocytes [37]. The reductions in mitochondrial membrane potential and OXPHOS in the IGF-1+/− mice were resolved with IGF-1 treatment. IGF-1 administration to old Wister rats (103 weeks) for 30 days rescued mitochondrial membrane potential, oxygen consumption rate, proton leak, ATP production, cytochrome oxidase, and ATPase complexes activities in isolated mitochondria from livers [38]. Additionally, the age-associated increases in mitochondrial ROS production, reduced antioxidants activities, and increased apoptosis in isolated mitochondria were resolved by IGF-1 treatment, suggesting cytoprotective effects of IGF-1.
IGF-1 plays essential roles in the function of mitochondria in the central nervous system [39]. Hippocampal mitochondria of 18-month-old mice with adult-induced liver IGF-1 deficiency (iLID) (at 5 months of age) showed lower level of OXPHOS and increased mitochondrial uncoupling, lower level of ATP production, and increased level of oxidative damage, as compared to aged controls [39]. This compromised mitochondrial function in the iLID mice manifested as impaired spatial acquisition and reversal learning. Similarly, specific ablation of IGF-1R in astrocytes [40] caused impaired mitochondrial energy metabolism, OXPHOS, and decreased glucose and amyloid β uptake. Overall, data suggest that increases in IGF-1 signaling in astrocytes may rescue from age-related mitochondrial dysfunction and cognitive decline.
Sarcopenia, age-associated reductions in skeletal muscle-mass, is thought to result from reduced level of GH. Low doses of GH administration to 22-month-old Wistar rats for 8 weeks increased circulating IGF-1 levels, enhanced the synthesis of mitochondrial proteins and antioxidant enzyme activities (catalase, glutathione peroxidase, and glucose-6-phosphate dehydrogenase (G6PDH)), and reduced oxidative damage (measured by the levels of 8-OHdG) in the skeletal muscle [23]. GH administration induced the activation of the anabolic AKT, mTOR, p70S6K, and Myf-5 factors while inhibiting p21, p38, and muscle RING finger-1 (MuRF-1) catabolic signals. Similarly, it was reported that a decline in GH/IGF-1 signaling in muscles of old rats associated with mitochondrial dysfunction. Following treatment with antioxidants, GH and IGF-1 levels in serum increased and associated with improvement of cristae structure and clustering of muscle mitochondria [41]. Likewise, IGF-1 alleviated dysfunctional mitochondria of cardiomyocytes from obese mice [42]. Glucose uptake, ATP production, and aconitase activity increased while lipid peroxidation, ROS production, protein carbonyl content, and apoptosis decreased in transgenic mice with overexpression of cardiomyocyte-specific IGF-1 that were fed a high fat diet. Furthermore, cardiomyocyte-specific IGF-1 induced the expression of Cyt c, PGC-1α, and UCP2 as well as the essential intracellular Ca2+ regulatory proteins SERCA2a and the Na+/Ca2+ exchanger.
Mitochondrial dysfunction was also reported for cortical bone osteocytes in GHRKO mice, which exhibited reduced mitochondrial membrane potential, decreased ATP production, and reduced maximal respiration in both young and old mice [43].
Overall, it seems that GH/IGF-1 signaling involvement in mitochondrial function is multifaceted, with important tissue-, organ-, and age-dependent features. Importantly, there are fundamental differences between the effects of age-induced reductions in GH/IGF-1 on mitochondrial function and those of life-long, congenital, ablations of members of the GH/IGF-1 axis on mitochondrial function.
4. GH/IGF-1 Effects on Oxidative Stress During Aging
According to the free radical theory of aging, ROSs generated by the ETC in the mitochondria or via nitric oxide metabolism in the cytosol have the potential to result in oxidative damage to DNA, proteins, and lipids and thus to accelerate aging (Figure 3) [44,45,46,47]. Superoxide anion (O2−) and H2O2 are produced in mitochondria as byproducts of OXPHOS. Further, H2O2 can be converted into a dangerous hydroxyl radical (HO) during Fenton’s reaction in the presence of Fe2+ [48]. Antioxidant defense includes enzymatic activation of superoxide dismutases (SODs), which are metalloproteins that convert superoxide to hydrogen peroxide and molecular oxygen. There are three types of SODs: Cu/Zn-SOD, predominantly located in the cytosolic fractions; Mn-SOD, located in the mitochondria; and EC-SOD, which is found in the extracellular space [49]. Catalase is a heme protein located predominantly in peroxisomes and the inner mitochondrial membrane.
There are numerous reports showing that GH/IGF-1 signaling controls the expression and activity of antioxidant enzymes and thus regulates the level of oxidative stress. Congenital IGF-1R haploinsufficiency in mice (Igf1r+/−) alters sensitivity to oxidative stress. Embryonic fibroblasts from Igf1r+/− mice are more resistant to hydrogen peroxide-induced cell death [25]. Accordingly, we found that IGF-1 null mice that exclusively express hepatic IGF-1 transgene (KO-HIT mice) [50] show increased levels of lipid peroxidation products in serum and increased mortality rate at 18 months of age in both sexes, suggesting that elevations in serum IGF-1 are harmful. Mutations that affect pituitary development (Prop1 and Pit1) and consequently lead to decreases in GH, thyroid stimulating hormone, or prolactin are associated with resistance to oxidative stress. Ames dwarf mice have very low serum IGF-1 levels and increased activities of catalase and Cu/Zn SOD [51]. These mice show reduced levels of DNA and protein oxidation in liver [52] and reduced serum and liver F2-isoprostanes, which are a stable lipid peroxidation product [53]. Similarly, the GH-deficient Snell and lit/lit dwarfs show resistance to oxidative stress [54,55]. Supplementation of GH to Ames dwarf mice for 7 days increased plasma IGF-1 levels and body and liver weights. However, mitochondrial glutathione S-transferase (GST) proteins (GSTK1 and GSTM4) significantly reduced with treatment [56,57]. Furthermore, glutaredoxins, which are localized in the mitochondria and sense cellular oxidative stress, significantly reduced with GH administration. In line with these data, it was reported that GHRKO mice exhibit increases in glutathione (GSH) and methionine (MET) metabolism in several tissues, making them more resistant to oxidative damage and delayed aging [58]. However, GHRKO mice, although resistant to oxidative stress, do not show improved free radical scavenging in the liver or kidney [59], though it is possible that other tissues such as muscle respond differently. Although informative, data from congenital models of the GH/IGF-1 axis cannot be directly extrapolated to normal aging in other animal models or in humans. These models exhibit altered developmental programming that affect their aging.
GHRKO mice resemble the human Laron syndrome (LS), which is caused by a deletion or an inactivating mutation of the GHR gene [60,61,62,63]. Similar to the GHRKO mice, LS patients have short stature, increased body adiposity, and low IGF-1 in serum [63,64]. Genome-wide microarray studies conducted on lymphocytes from LS patients identified a series of genes that are differentially expressed in various pathways, including oxidative stress, apoptosis, metabolism, Jak-STAT, and PI3K-AKT signaling. Among the overexpressed genes in LS, thioredoxin-interacting protein (TXNIP) was identified as a new target for IGF-1 and insulin action. TXNIP belongs to the α-arrestin family [65]. It binds to the catalytic active-center of reduced thioredoxin (TRX) and inhibits its expression and activity, highlighting the key role of TXNIP in redox regulation. Oxidative stress leads to TXNIP shuttling from the nucleus into the mitochondria. TXNIP inhibits proliferation via activation of the apoptosis signal regulating kinase 1 (ASK1) [66] and functions as a tumor suppressor, being commonly silenced in cancer cells [67,68,69,70,71]. Similar to LS patients, GHRKO mice show reduced tumor incidence in experimental models of cancer [72]. In accordance with its enhanced expression in LS-derived cells, qPCR revealed that TXNIP expression increased approximately two-fold in livers of GHRKO mice, while it decreased in HIT mice overexpressing IGF-1 in the liver.
LS lymphocytes were shown to display protection from oxidative stress [73]. Accordingly, induction of oxidative stress in lymphocytes from LS patient (Figure 4A) leads to upregulation of TXNIP. The capacity of IGF-1 to downregulate the oxidative stress-induced TXNIP upregulation (Figure 4B) indicates that IGF-1 could rescue the cells by downregulating TXNIP. TXNIP acts as an oxidative stress mediator by inhibiting TRX activity or by limiting its bioactivity [74]. The redox-related protein complex TRX/TXNIP, or “redoxisome,” is a critical regulator of ROS signalling and is involved in the pathogenesis of various diseases, including autoimmune and degenerative conditions [68]. The finding that TXNIP levels are increased in response to oxidation in LS patient-derived but not control lymphoblastoid cells is of major translational relevance.
Apart from oxidative stress, TXNIP was reported to function as a strong glucose sensor as its expression increased upon high-glucose stress [75]. TXNIP knockout mice (TXNIP KO) show impaired metabolic homeostasis, including adipogenesis and reduced gluconeogenesis [76], and decreased glucose uptake [77]. Similarly, a recently described human mutation demonstrated that diminished TXNIP function is linked to inefficient utilization of glucose [78]. Accordingly, glucose stress (hyperglycemia) upregulated TXNIP levels in 3T3LY adipocytes and was downregulated by IGF-1 and insulin (Figure 5A) [79]. Overall, these studies show that oxidative and glucose stresses induced TXNIP levels at both the transcriptional and translational levels (Figure 5B) and that IGF-1 indirectly regulates cellular TXNIP, protecting the cells from apoptosis (Figure 5C) [79]. The potential involvement of epigenetic mechanisms, particularly DNA methylation and histone acetylation, in inhibitory regulation of TXNIP gene expression by IGF-1 is currently unknown.
Notably, there are numerous studies indicating that administration of GH/IGF-1 in vivo or in vitro protects from oxidative stress, specifically during aging. Old Wistar rats treated with GH showed increased circulating IGF-1 levels and reductions in age-associated oxidative stress in skeletal muscle [23]. This was accompanied by increased levels of the antioxidant enzymes catalase, glutathione peroxidase, and G6PDH. Similarly, supplementation of IGF-1 to aged rats associated with reduced oxidative damage and restored levels of SOD, glutathione peroxidase, and catalase in hippocampus [34]. Hepatic tissue level of catalase was also restored with IGF-1 treatment in old rats, suggesting antioxidant properties of IGF-1 in brain and liver [34].
IGF-1 is reported to be protective against oxidative stress in cardiac and skeletal muscle. Pathologic left ventricular remodeling and functional loss following myocardial infarction were more severe in dwarf rats (dw/dw) with significantly reduced GH/IGF-1 [80]. Using an ex vivo murine model of myocardial ischemia/reperfusion injury, it was found that IGF-1 protects ischemic myocardium from further reperfusion injury, likely via maintenance of mitochondrial to nuclear DNA ratio within heart tissue [81] and by activation of the PI3K-Akt and/or Erk 1/2 kinase cascades [82]. Likewise, hearts of IGF-1 transgenic mice were protected from ischemia and reperfusion [83], and streptozotocin-induced diabetic cardiomyopathy in mice resulted in accumulation of nitrotyrosine (a reactive oxygen product) in vivo and the formation of H2O2 in myocytes in vitro that were rescued in IGF-1 transgenic mice [84]. Additionally, IGF-1 protected from 2:4 dinitrophenol-induced oxidative stress in rat muscle in vitro [85] and myoblasts protected from H2O2 stress-induced apoptosis [86].
The protective effects of the GH/IGF-1 axis from oxidative stress was demonstrated for many cell types. Endothelial cells from Ames dwarf mice show elevated levels of H2O2, increased mitochondrial ROS, and decreased antioxidative enzymes such as SODs and glutathione peroxidase. These observations were in accordance with in vitro findings using cultured aortic segments and human coronary arterial endothelial cells. Treatment of these cultures with GH and IGF-1 lead to elevations in the level of antioxidants and reduced prooxidant levels [87]. In astrocytes, downregulation of the IGF-1R increased mitochondrial ROS production and reduced resistance to external oxidative damage [40]. IGF-1 promoted the survival of rat primary cerebellar neurons and of immortalized hypothalamic rat GT1-7 cells following H2O2-induced oxidative stress [88]. There is a long list of studies utilizing numerous cell types, showing the protective effects of GH/IGF-1 from oxidative stress that, although important, were not included in our review.
Antioxidant administration to old animals can elevate circulating GH and IGF-1 levels. Thus, treatment of Wistar rats or senescence-accelerated OXYS rats with antioxidants and, in particular, with mitochondrial antioxidants prevented the age-associated decrease in serum levels of GH and IGF-1 [41,89]. These were accompanied by improvements in pathologies such as retinopathy and cataract, learning ability and memory, and immune system decline [89,90]. Treatments of aged rats with mitochondrial antioxidants also led to improvement of mitochondrial structure-disorganization developed with age in muscle tissues. Disorders of muscles tissue mitochondrial apparatus in old rats could be driven by a decline in GH/IGF-1 signaling, as improvement of cristae structure and clustering of muscle mitochondria was correlated with an increase of GH and IGF-1 levels in serum after treatment with mitochondrial antioxidants [41].
In summary, insufficient levels of GH/IGF-1 are associated with organ-specific impairment of free radical scavenging systems. GH/IGF-1 plays significant roles in regulating oxidative stress, which is clearly only one of many mechanisms affecting mitochondrial function during aging. Finally, there are fundamental differences in handling oxidative stress between age-associated decline in GH/IGF-1 (somatopause) and congenital impairments in the GH/IGF-1 axis.
5. GH/IGF-1 Effects on Cellular Senescence
Cellular senescence is characterized by an irreversible block of the cell cycle. This mechanism was initially thought to function as protection against cancer [91], but later studies have found that senescence is linked to aging and age-related diseases. Senescent cells undergo numerous phenotypic and metabolic modifications. They show increased cells size [92], dysfunctional mitochondria and telomeres, impaired DNA damage response, increased secretory functions, formation of heterochromatic foci, and a senescence-associated secretory phenotype (SASP). In fact, the SASP is a main feature of senescent cells, which secrete pro-inflammatory cytokines and chemokines, growth factors, and proteases, forming a toxic and harmful microenvironment to non-senescent cells [93,94,95,96,97].
Mitochondria undergo drastic changes in morphology and function in senescent cells. Given the complex functions of mitochondria, it is hard to dissect the specific mitochondria-mediated mechanisms leading to cell senescence. Several potential mechanisms, mediated by mitochondria, that may lead to cell senescence include increased ROS production and elevations in damage-associated molecular patterns (DAMPs) leading to DNA damage response (DDR), which locks the cells in a senescence mode, leading to SASP. Senescent cells exhibit increased mitochondrial mass but, at the same time, show reduced OXPHOS and rely mainly on ATP from glycolysis. Increased mitochondrial mass is driven by upregulation of PGC-1β [98]. Accordingly, deletion of PGC-1β in mice delayed several aspects of senescence [98]. In line with these data, it was shown that senescent pancreatic beta cells exhibit increased mitochondrial biogenesis [99].
Pituitary adenomas expressing and secreting GH exhibit a senescent phenotype. Autocrine/paracrine GH acts in pituitary cells as an apoptosis switch for p53-mediated senescence, likely preventing the pituitary adenoma cells from progression to malignancy [100]. Skin fibroblasts from acromegalic patients (with excess GH and IGF-1) exhibit shortened telomeres and cellular senescence [101]. Similarly, senescence-associated gene expression of p16 and IL-6 increased in white adipose tissue of 10-month-old female bGH transgenic mice as compared to controls. In addition, β-galactosidase (β-gal)-positive cells (senescence-associated) were elevated in GH-injected 19-month-old female mice as compared to age-matched saline-injected controls [102]. Accordingly, mice with a specific deletion of the IGF-1R in cardiomyocytes show delayed development of aging-associated myocardial pathologies [103]. Cultured cardiomyocytes treated with IGF-1 exhibited increased senescence, while inhibition of phosphoinositide 3-kinase prevented the IGF-1-mediated increase in interleukin (IL)-1α, IL-1β, receptor activator of nuclear factor-κB ligand, and p21 protein levels [103]. Finally, prolonged IGF-1 treatment of MCF7 cells inhibited SIRT1 deacetylase activity, associated with increased p53 acetylation and activation, and lead to premature cellular senescence [104]. Similarly, prolonged exposure of primary human mesangial cells to glycated albumin (GA) was associated with IGF-1 release, activation of the IGF-1R, and enhanced cellular senescence. GA-induced IGF-1R activation associated with a reduction in the catalase content likely through activation of the Ras and Erk1/2 pathway. Downregulation of the IGF-1R via overexpression of klotho lowered p53 and reversed the senescence phenotype [105]. In vitro, cell senescence is often observed once cells reach confluency. IGF-1 promoted senescence in mouse embryonic fibroblasts and human fibroblasts as well as in rat vascular smooth muscle cells (VSMCs) after attaining confluency. The IGF-1-induced senescence in these cells was associated with elevated cellular ROS, p53, and p21 protein levels and the DNA damage marker γH2AX [106].
On the other hand, there are studies showing that GH/IGF-1 protects from senescence. GH treatment (0.4 mg/d for 7 days) of endothelial progenitor cells from patients with atherosclerosis reversed age-related dysfunction and attenuated senescence (indicated by increased telomerase activity) [107]. These findings were in line with another study in which GH was incorporated into reconstituted high-density lipoprotein (rHDL) and delivered to zebrafish. In this study, GH enhanced anti-atherosclerotic activity and antisenescence activity with inhibition of fructose-mediated glycation [108]. Likewise, IGF-1 gene transfer to CCl4-treated rats (with liver injury and fibrosis) relieved hepatocyte oxidative stress and premature senescence, likely mediated by the p53/progerin pathway [109]. In vitro, IGF-1 treatment of H2O2-exposed hepatocytes reversed oxidative stress-induced premature senescence via enhancing cytoplasmic AKT1–p53 interaction and by subsequently inhibiting nuclear p53–progerin interaction [109].
6. GH/IGF-1 Effects on Mitochondria-Mediated Apoptosis During Aging
Similar to senescence, programmed cell death, or apoptosis, is believed to be a consequence of cellular stress and mitochondrial dysfunction. However, the roles of apoptosis during aging are uncertain. In general, the determinants of whether a cell activates senescence or apoptosis pathways are cell-type, stress-type, and stress-intensity dependent. It is important to note that apoptosis occurs mainly through activation of a mitochondrial intrinsic pathway, which depends on caspase 9 and apoptotic protease activating factor 1 (Apaf1) or via an extrinsic pathway mediated by the Fas ligand and tumor necrosis factor receptor (TNFR) and by activation of caspase 8. Herein, we will describe shortly the roles of the GH/IGF-1 axis in mitochondrial (intrinsic)-mediated apoptosis.
A key step in mitochondria-mediated apoptosis is the release of Cyt c from the inner mitochondrial space (Figure 6). Several pathways are known to trigger Cyt c release including opening of the mitochondrial permeability transition pore (PTP). Opening of the PTP leads to mitochondrial swelling, depolarization of the membrane potential, subsequent rupture of the outer membrane, and nonselective release of intermembrane space proteins. Once released, Cyt c binds to Apaf1 and activates a group of cysteine proteases called caspases that cleave an array of substrates and proteins that are vital to cellular function. The Apaf1-cyt c-caspase 9 form apoptosome complexes. Apoptosome, central to the apoptotic pathway, binds to other pro-enzymes and cleaves them to their active forms.
Cyt c release from mitochondria is a primary signal for B-cell lymphoma 2 (Bcl-2)-regulated inhibition of apoptosis [110,111]. Bcl-2 is localized in the outer mitochondrial membrane (Figure 6). Bcl-2 belongs to a large family of proteins with antiapoptotic properties (i.e., Bcl-XL, Bcl-w, Mcl-1, A1, Bcl-Rambo, Bcl-L10, and Bcl-G) and proapoptotic properties (i.e., Bax, Bak, and Bok) [112]. There are several other proteins that block the antiapoptotic activity of Bcl-2, which are termed proapoptotic BH3-only proteins (i.e., Puma, Noxa, Bid, Bad, Bim, Bik, Hrk, and Bmf) [113]. However, it appears that Bax/Bak are the key regulatory targets where many intracellular signals converge and determine a cell’s fate [114]. It was widely accepted that the role of the antiapoptotic BCL-2-like proteins is to inhibit their proapoptotic counteracting partners, such that the balance between anti- and proapoptotic BCL-2 family proteins determines cell fate. However, this model is oversimplified in view of later discoveries, including the occasional interconversion of anti- and proapoptotic activities of these proteins, and the findings that BCL-2 family members exhibit nonapoptotic functions [115,116].
The insulin and IGF-1 signaling pathway (IIS) are the most evolutionarily conserved pathway of aging. IGF-1 has been recognized as a survival factor of numerous cell types. Activation of the insulin or IGF-1 receptors elicits activation of several pathways; among them are the mitogen-activated protein kinase (MAPK) pathway and the PI3K-AKT pathway. Activation of AKT leads to phosphorylation and inactivation of a group of forkhead box transcription factors of the class O (FOXO) factors by retaining them in the cytoplasm. Phosphorylation of FOXOs and their binding to the regulatory proteins 14-3-3 sequester them from the nucleus, leading to suppression of FOXO-dependent transcription (mostly the BH3-only proteins) [117,118,119]. Thus, the survival factor IGF-1 attenuates apoptosis via activation of the PI3K-AKT pathway and inhibition of FOXO proteins.
FOXOs are the most important transcriptional effectors of the IIS, and as such, they are tightly regulated post-translationally [120]. Mammals have four FOXO genes: FOXO1, FOXO3, FOXO4, and FOXO6. FOXO1-null mice are embryonically lethal, while FOXO3, FOXO4, and FOXO6 show mild phenotypes [121]. Tissue-specific approaches to delete FOXO proteins 1, 3, and 4 have established the importance of these factors in regulation of cell fate. For example, ablation of Foxo1, 3, and 4 proteins in osteoblasts associated with increases in oxidative stress and osteoblast apoptosis [122], while osteoblast-specific overexpression of Foxo3 decreased oxidative stress and apoptosis.
In summary, the GH/IGF-1 axis regulates mitochondrial (intrinsic)-mediated apoptosis mostly via activation of the PI3K-AKT/FOXO pathway and possibly via cell-specific transcriptional regulation of antiapoptotic genes. However, while during development and adult stages apoptosis is required for the normal cell turnover in specific tissues (such as endothelium, intestinal epithelium, etc.), the role of apoptosis during aging is not clear. In fact, apoptosis was shown to decrease in adipose mesenchymal stem cells from healthy young, middle-aged, and aged volunteers [123]; in bone marrow mesenchymal stem cells from old mice [124]; in livers of old rats challenged with DNA-damaging agents as compared to their young controls [125]; and even in response to radiation of peripheral blood lymphocytes from old mice [126]. Accordingly, reductions in markers of apoptosis during normal aging was also found in human serum [127]. Perhaps during aging, mitochondrial dysfunction leads mostly to cell senescence as opposed to apoptosis.
7. GH/IGF-1 Effects on Mitochondrial Function During Inflammation
Inflammation is a defense mechanism against harmful stimuli that damage cellular homeostasis [128]. Inappropriate response to cellular damage may lead to chronic inflammation, including autoimmune diseases, diabetes, allergies, cardiovascular diseases, asthma, cancer, arthritis, and aging [129,130]. Response to various immunological challenges entails particular metabolic configurations for energy generation required for biosynthesis of molecules [131]. Dysfunctional mitochondria with excessive ROS, abnormal calcium and potassium ion mobilization, and unusual ATP or NAD+ levels can trigger immune response [132]. On the other hand, inflammatory cytokines acting upon cells can trigger intracellular signaling cascades altering mitochondrial dynamics and mitophagy and eventually resulting in cell death [133,134].
The GHR and IGF-1R are expressed on immune cells and participate in thymic development and differentiation of immune cells such as T-cell, B- cell, and natural killer cells as well as in antigen presentation, antibody production, etc. [135,136]. Additionally, there are studies showing local production of GH and IGF-1 by lymphocytes, suggesting an autocrine/paracrine regulation of immune cells [137,138]. Further, human lymphocytes treated with super physiological levels (abuse) of IGF-1 can undergo cytoskeletal reorganization and overproduction of cytokines, augmenting the inflammatory response [139]. On the other hand, children with chronic inflammatory disorders exhibit excessive pro-inflammatory cytokines production and show growth failure and pubertal abnormalities [140]. Similarly, the elderly, with reduced GH or IGF-1 signals show poor response to immune stimuli [141].
The effects of GH/IGF-1 on mitochondria of inflammatory cells are underexplored. Aging is often associated with metabolic abnormalities accompanied by increased chronic inflammation in many tissues, including the brain. In fact, GH signaling was found to positively modulate brain inflammation in aged mice [142]. Accordingly, GHRKO mice showed reduced inflammation in the hypothalamus. GHRKO and Ames mice displayed lower glial fibrillary acidic protein (GFAP) and tumor necrosis factor (TNF)-α levels in the hypothalamus, indicative of reduced inflammation. Consequently, administration of GH to GHRKO and Ames mice led to increased GFAP and TNF-α and increased brain inflammation [142]. As mentioned above, the GHRKO is a mouse model with congenital ablation of the GH/IGF-1 axis. It is unclear whether this axis plays a role in systemic or central inflammation, particularly during natural aging where a gradual fall in GH/IGF-1 signals happens.
8. Summary
GH and IGF-1 are pleotropic hormones affecting multiple cellular functions, including cell proliferation, differentiation, metabolism, and cell survival. Both hormones activate many signaling cascades implicated in regulation of mitochondrial proteins expression and function. Most evidence indicates that the effects of GH on mitochondrial mass and function are indirect and mostly mediated by IGF-1. IGF-1 affects mitochondrial mass via increased transcriptional activities of key factors involved in mitochondrial biogenesis such as PGC-1α. Additionally, it appears that the effects of IGF-1 on mitochondrial respiration are indirect and coincide with enhanced synthesis of mitochondrial proteins such as Cyt c and UCP. With respect to oxidative stress, the literature is divided and studies showing either positive or negative effects of GH or IGF-1 have been published. While congenital mouse models with life-long decreases in GH/IGF-1 axis signaling indicate mostly protection from oxidative stress, models of age-induced decreases in GH/IGF-1 signals, as seen in humans, associate with increased oxidative stress.
Finally, it is widely accepted that GH/IGF-1 are involved in cell senescence and apoptosis. The molecular mechanisms involved in GH/IGF-1-mediated cellular senescence are still poorly understood. Both hormones exert a dual function and promote, on one hand, cell proliferation and, on the other hand, cellular senescence. Therefore, it is conceivable that the dose and duration of GH/IGF-1 exposure might regulate senescence, and that the effects of GH/IGF-1 on senescence are tissue- and cell type-specific. On the other hand, the protective roles of IGF-1 from mitochondrial-mediated apoptosis have been better defined. Studies of numerous cell types and animal models have shown that IGF-1-mediated activation of the PI3K-AKT/FOXO pathway upregulates transcription of antiapoptotic genes.
Funding
Financial support was received from National Institutes of Health Grant R01AG056397 to S.Y. and from the United States-Israel Binational Science Foundation grant (2013282) to S.Y. and H.W.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| iLID | Adult-induced liver IGF-1 deficiency |
| ALS | Acid labile subunit |
| ATP | Adenosine triphosphate |
| ASK1 | Apoptosis signal regulating kinase 1 |
| Bcl-2 | B-cell lymphoma 2 |
| β-gal | β-galactosidase |
| bGH | Bovine growth hormone |
| BNIP3 and BNIP3L | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 and L |
| BAT | Brown adipose tissue |
| CS | Cockayne syndrome protein |
| Cyt c | Cytochrome c |
| DAMPs | Damage-associated molecular patterns |
| DDR | DNA damage response |
| ETC | Electron transport chain |
| FOXO | Forkhead box transcription factors of the class O |
| GH | Growth hormone |
| GHR | Growth hormone receptor |
| GHRKO | GH receptor null |
| GHRH | GH-releasing hormone |
| GHRHR | GH-releasing hormone receptor |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| GST | Glutathione S-transferase |
| GSH | Glutathione |
| GA | Glycated albumin |
| HO | Heme oxygenase |
| HIT | Hepatic IGF-1 transgene |
| hGH | Human GH |
| H2O2 | Hydrogen peroxide |
| hypx | Hypophysectomized |
| IGF-1 | Insulin-like growth factor-1 |
| IGFBPs | IGF-binding proteins |
| IGF-1R | Insulin-like growth factor-1 receptor |
| IIS | Insulin and IGF-1 signaling pathway |
| IL | Interleukin |
| JAK2 | Janus kinase 2 |
| LS | Laron syndrome |
| MET | Methionine |
| mtDNA | Mitochondrial genome |
| MuRF-1 | Muscle RING finger-1 |
| ND1 through ND6 | NADH dehydrogenase of complex I |
| TNF | Necrosis factor |
| NRG | Neuregulin |
| NO | Nitric oxide |
| NRF1 and 2 | Nuclear respiratory factors |
| OXPHOS | Oxidative phosphorylation |
| PTP | Permeability transition pore |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1 α |
| PPARs | Peroxisome proliferator-activated receptors |
| PRC | PGC-1α-related coactivator |
| PI3K/AKT | Phosphoinositide-3-kinase (PI3K)/Protein kinase B (PKB, or Akt) |
| MAPK | Rat sarcoma GTPase protein (Ras)/Rapidly accelerated fibrosarcoma protein kinase (Raf)/Mitogen-activated protein kinase |
| ROS | Reactive oxygen species |
| RQ | Respiratory quotient |
| rHDL | Reconstituted high-density lipoprotein |
| SST | Somatostatin |
| SSTR | SST-receptors |
| Shc | Src homology 2 domain containing transforming protein |
| STAT5b | Signal transducer and activator of transcription 5B |
| SERCA2a | Sarco/endoplasmic reticulum Ca2+-ATPase-2a |
| SASP | Senescence-associated secretory phenotype |
| TF | Transcription factors |
| TXNIP | Thioredoxin-interacting protein |
| TRX | Thioredoxin |
| T3 | Triiodothyronine |
| TOMs | Transporters located at the outer mitochondrial membrane |
| TCA | Tricarboxylic acid |
| TNFa | Tumor necrosis factor-alpha |
| VSMCs | Vascular smooth muscle cells |
References
- Niu, T.; Rosen, C.J. The insulin-like growth factor-I gene and osteoporosis: A critical appraisal. Gene 2005, 361, 38–56. [Google Scholar] [CrossRef] [PubMed]
- Barclay, R.D.; Burd, N.A.; Tyler, C.; Tillin, N.A.; Mackenzie, R.W. The Role of the IGF-1 Signaling Cascade in Muscle Protein Synthesis and Anabolic Resistance in Aging Skeletal Muscle. Front. Nutr. 2019, 6, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, K.; Hanayama, Y.; Obika, M.; Itoshima, K.; Okada, K.; Otsuka, F. Clinical relevance of insulin-like growth factor-1 to cardiovascular risk markers. Aging Male 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.R.; Evans, P.; Thomas, N.E.; Davies, B.; Baker, J.S. Changes in endothelial dysfunction and associated cardiovascular disease morbidity markers in GH-IGF axis pathology. Am. J. Cardiovasc. Drugs 2009, 9, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Milman, S.; Huffman, D.M.; Barzilai, N. The Somatotropic Axis in Human Aging: Framework for the Current State of Knowledge and Future Research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Haywood, N.J.; Slater, T.A.; Matthews, C.J.; Wheatcroft, S.B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 2019, 19, 86–96. [Google Scholar]
- Domene, H.M.; Hwa, V.; Jasper, H.G.; Rosenfeld, R.G. Acid-labile subunit (ALS) deficiency. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 101–113. [Google Scholar] [CrossRef]
- Banaszak-Ziemska, M.; Niedziela, M. PAPP-A2 a new key regulator of growth. Endokrynol. Pol. 2017, 68, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Broglio, F.; Arvat, E.; Benso, A.; Papotti, M.; Muccioli, G.; Deghenghi, R.; Ghigo, E. Ghrelin: Endocrine and non-endocrine actions. J. Pediatr. Endocrinol. Metab. 2002, 15, 1219–1227. [Google Scholar] [CrossRef]
- Arvat, E.; Broglio, F.; Aimaretti, G.; Benso, A.; Giordano, R.; Deghenghi, R.; Ghigo, E. Ghrelin and synthetic GH secretagogues. Best Pract. Res. Clin. Endocrinol. Metab. 2002, 16, 505–517. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riis, S.; Murray, J.B.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3beta-Nrf2-BNIP3 Pathway. Cells 2020, 9, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurmasheva, R.T.; Houghton, P.J. IGF-I mediated survival pathways in normal and malignant cells. Biochim. Biophys. Acta 2006, 1766, 1–22. [Google Scholar] [CrossRef]
- Craigen, W.J. Mitochondrial DNA mutations: an overview of clinical and molecular aspects. Methods Mol. Biol. 2012, 837, 3–15. [Google Scholar] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef]
- Phu, L.; Rose, C.M.; Tea, J.S.; Wall, C.E.; Verschueren, E.; Cheung, T.K.; Kirkpatrick, D.S.; Bingol, B. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 2020, 77, e1107–e1123. [Google Scholar] [CrossRef]
- Groves, W.E.; Houts, G.E.; Bayse, G.S. Subcellular distribution of 125 I-labeled bovine growth hormone in rat liver and kidney. Biochim. Biophys. Acta 1972, 264, 472–480. [Google Scholar] [CrossRef]
- Mutvei, A.; Husman, B.; Andersson, G.; Nelson, B.D. Thyroid hormone and not growth hormone is the principle regulator of mammalian mitochondrial biogenesis. Acta Endocrinol. (Copenh) 1989, 121, 223–228. [Google Scholar] [CrossRef]
- Maddaiah, V.T.; Sharma, R.K.; Balachandar, V.; Rezvani, I.; Collipp, P.J.; Chen, S.Y. Effect of growth hormone on mitochondrial protein synthesis. J. Biol. Chem. 1973, 248, 4263–4268. [Google Scholar] [PubMed]
- Echave, P.; Machado-da-Silva, G.; Arkell, R.S.; Duchen, M.R.; Jacobson, J.; Mitter, R.; Lloyd, A.C. Extracellular growth factors and mitogens cooperate to drive mitochondrial biogenesis. J. Cell Sci. 2009, 122, 4516–4525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brioche, T.; Kireev, R.A.; Cuesta, S.; Gratas-Delamarche, A.; Tresguerres, J.A.; Gomez-Cabrera, M.C.; Vina, J. Growth hormone replacement therapy prevents sarcopenia by a dual mechanism: Improvement of protein balance and of antioxidant defenses. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1186–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, H.Y.; Wang, X.; Yu, Q.; Li, H.Y.; Li, S.J.; Tang, R.Y.; Guo, Z.X.; Chen, Y.Q.; Hu, C.X.; Yang, Z.J.; et al. Evidence that growth hormone can improve mitochondrial function in oocytes from aged mice. Reproduction 2018, 157, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.D.; Tissenbaum, H.A.; Liu, Y.; Ruvkun, G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 1997, 277, 942–946. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Brown-Borg, H.M.; Johnson, W.T.; Rakoczy, S.G. Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age (Dordr) 2012, 34, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, R.; Bonkowski, M.S.; Strader, A.D.; Bartke, A. Alterations in oxygen consumption, respiratory quotient, and heat production in long-lived GHRKO and Ames dwarf mice, and short-lived bGH transgenic mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Brown-Borg, H.; Johnson, W.T.; Rakoczy, S.; Romanick, M. Mitochondrial oxidant generation and oxidative damage in Ames dwarf and GH transgenic mice. J. Am. Aging Assoc. 2001, 24, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; McFadden, S.; Darcy, J.; Hascup, E.R.; Hascup, K.N.; Bartke, A. Lifespan of long-lived growth hormone receptor knockout mice was not normalized by housing at 30 degrees C since weaning. Aging Cell 2020, e13123. [Google Scholar] [CrossRef]
- Darcy, J.; McFadden, S.; Fang, Y.; Berryman, D.E.; List, E.O.; Milcik, N.; Bartke, A. Increased environmental temperature normalizes energy metabolism outputs between normal and Ames dwarf mice. Aging (Albany NY) 2018, 10, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, M.; Delgado, G.; Puche, J.E.; Gonzalez-Baron, S.; Castilla Cortazar, I. Low doses of insulin-like growth factor I improve insulin resistance, lipid metabolism, and oxidative damage in aging rats. Endocrinology 2008, 149, 2433–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadik, Z.; Chalew, S.A.; McCarter, R.J.Jr.; Meistas, M.; Kowarski, A.A. The influence of age on the 24-hour integrated concentration of growth hormone in normal individuals. J. Clin. Endocrinol. Metab. 1985, 60, 513–516. [Google Scholar] [CrossRef]
- Colon, G.; Saccon, T.; Schneider, A.; Cavalcante, M.B.; Huffman, D.M.; Berryman, D.; List, E.; Ikeno, Y.; Musi, N.; Bartke, A.; et al. The enigmatic role of growth hormone in age-related diseases, cognition, and longevity. Geroscience 2019, 41, 759–774. [Google Scholar] [CrossRef]
- Olleros Santos-Ruiz, M.; Sadaba, M.C.; Martin-Estal, I.; Munoz, U.; Sebal Neira, C.; Castilla-Cortazar, I. The single IGF-1 partial deficiency is responsible for mitochondrial dysfunction and is restored by IGF-1 replacement therapy. Growth Horm. IGF Res. 2017, 35, 21–32. [Google Scholar] [CrossRef]
- Puche, J.E.; Garcia-Fernandez, M.; Muntane, J.; Rioja, J.; Gonzalez-Baron, S.; Castilla Cortazar, I. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology 2008, 149, 2620–2627. [Google Scholar] [CrossRef] [Green Version]
- Pharaoh, G.; Owen, D.; Yeganeh, A.; Premkumar, P.; Farley, J.; Bhaskaran, S.; Ashpole, N.; Kinter, M.; Van Remmen, H.; Logan, S. Disparate Central and Peripheral Effects of Circulating IGF-1 Deficiency on Tissue Mitochondrial Function. Mol. Neurobiol. 2020, 57, 1317–1331. [Google Scholar] [CrossRef] [Green Version]
- Logan, S.; Pharaoh, G.A.; Marlin, M.C.; Masser, D.R.; Matsuzaki, S.; Wronowski, B.; Yeganeh, A.; Parks, E.E.; Premkumar, P.; Farley, J.A.; et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-beta uptake in astrocytes. Mol. Metab. 2018, 9, 141–155. [Google Scholar] [CrossRef]
- Vays, V.B.; Eldarov, C.M.; Vangely, I.M.; Kolosova, N.G.; Bakeeva, L.E.; Skulachev, V.P. Antioxidant SkQ1 delays sarcopenia-associated damage of mitochondrial ultrastructure. Aging (Albany NY) 2014, 6, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yuan, M.; Bradley, K.M.; Dong, F.; Anversa, P.; Ren, J. Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: Role of insulin signaling and mitochondrial function. Hypertension 2012, 59, 680–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Solesio, M.E.; Schaffler, M.B.; Frikha-Benayed, D.; Rosen, C.J.; Werner, H.; Kopchick, J.J.; Pavlov, E.V.; Abramov, A.Y.; Yakar, S. Mitochondrial Function Is Compromised in Cortical Bone Osteocytes of Long-Lived Growth Hormone Receptor Null Mice. J. Bone Miner. Res. 2019, 34, 106–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Harman, D. The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Fridovich, I. Biological effects of the superoxide radical. Arch. Biochem. Biophys. 1986, 247, 1–11. [Google Scholar] [CrossRef]
- Elis, S.; Wu, Y.; Courtland, H.W.; Sun, H.; Rosen, C.J.; Adamo, M.L.; Yakar, S. Increased serum IGF-1 levels protect the musculoskeletal system but are associated with elevated oxidative stress markers and increased mortality independent of tissue igf1 gene expression. Aging Cell 2011, 10, 547–550. [Google Scholar] [CrossRef] [Green Version]
- Hauck, S.J.; Bartke, A. Effects of growth hormone on hypothalamic catalase and Cu/Zn superoxide dismutase. Free Radic. Biol. Med. 2000, 28, 970–978. [Google Scholar] [CrossRef]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choksi, K.B.; Roberts, L.J., 2nd; DeFord, J.H.; Rabek, J.P.; Papaconstantinou, J. Lower levels of F2-isoprostanes in serum and livers of long-lived Ames dwarf mice. Biochem. Biophys. Res. Commun. 2007, 364, 761–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, A.B.; Murakami, S.; Bartke, A.; Kopchick, J.; Yasumura, K.; Miller, R.A. Fibroblast cell lines from young adult mice of long-lived mutant strains are resistant to multiple forms of stress. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E23–E29. [Google Scholar] [CrossRef] [Green Version]
- Bartke, A.; Brown-Borg, H. Life extension in the dwarf mouse. Curr. Top. Dev. Biol. 2004, 63, 189–225. [Google Scholar]
- Rojanathammanee, L.; Rakoczy, S.; Brown-Borg, H.M. Growth hormone alters the glutathione S-transferase and mitochondrial thioredoxin systems in long-living Ames dwarf mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Brown-Borg, H.M.; Bode, A.M.; Bartke, A. Antioxidative mechanisms and plasma growth hormone levels: Potential relationship in the aging process. Endocrine 1999, 11, 41–48. [Google Scholar] [CrossRef]
- Brown-Borg, H.M.; Rakoczy, S.G.; Sharma, S.; Bartke, A. Long-living growth hormone receptor knockout mice: Potential mechanisms of altered stress resistance. Exp. Gerontol. 2009, 44, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Hauck, S.J.; Aaron, J.M.; Wright, C.; Kopchick, J.J.; Bartke, A. Antioxidant enzymes, free-radical damage, and response to paraquat in liver and kidney of long-living growth hormone receptor/binding protein gene-disrupted mice. Horm. Metab. Res. 2002, 34, 481–486. [Google Scholar] [CrossRef]
- Laron, Z. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Amselem, S.; Duquesnoy, P.; Attree, O.; Novelli, G.; Bousnina, S.; Postel-Vinay, M.C.; Goossens, M. Laron dwarfism and mutations of the growth hormone-receptor gene. N. Engl. J. Med. 1989, 321, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Godowski, P.J.; Leung, D.W.; Meacham, L.R.; Galgani, J.P.; Hellmiss, R.; Keret, R.; Rotwein, P.S.; Parks, J.S.; Laron, Z.; Wood, W.I. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc. Natl. Acad. Sci. USA 1989, 86, 8083–8087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laron, Z.; Pertzelan, A.; Mannheimer, S. Genetic pituitary dwarfism with high serum concentation of growth hormone—a new inborn error of metabolism? Isr. J. Med. Sci. 1966, 2, 152–155. [Google Scholar] [PubMed]
- Laron, Z.; Pertzelan, A.; Karp, M.; Kowadlo-Silbergeld, A.; Daughaday, W.H. Administration of growth hormone to patients with familial dwarfism with high plasma immunoreactive growth hormone: measurement of sulfation factor, metabolic and linear growth responses. J. Clin. Endocrinol. Metab. 1971, 33, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A. Minireview: Thioredoxin-interacting protein: regulation and function in the pancreatic beta-cell. Mol. Endocrinol. 2014, 28, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [Green Version]
- Baldan, F.; Mio, C.; Lavarone, E.; Di Loreto, C.; Puglisi, F.; Damante, G.; Puppin, C. Epigenetic bivalent marking is permissive to the synergy of HDAC and PARP inhibitors on TXNIP expression in breast cancer cells. Oncol. Rep. 2015, 33, 2199–2206. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chng, W.J. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion 2013, 13, 163–169. [Google Scholar] [CrossRef]
- Minn, A.H.; Pise-Masison, C.A.; Radonovich, M.; Brady, J.N.; Wang, P.; Kendziorski, C.; Shalev, A. Gene expression profiling in INS-1 cells overexpressing thioredoxin-interacting protein. Biochem. Biophys. Res. Commun. 2005, 336, 770–778. [Google Scholar] [CrossRef]
- Han, S.H.; Jeon, J.H.; Ju, H.R.; Jung, U.; Kim, K.Y.; Yoo, H.S.; Lee, Y.H.; Song, K.S.; Hwang, H.M.; Na, Y.S. VDUP1 upregulated by TGF-beta1 and 1,25-dihydorxyvitamin D3 inhibits tumor cell growth by blocking cell-cycle progression. Oncogene 2003, 22, 4035–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Prins, G.S.; Coschigano, K.T.; Kopchick, J.J.; Green, J.E.; Ray, V.H.; Hedayat, S.; Christov, K.T.; Unterman, T.G.; Swanson, S.M. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology 2005, 146, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R.; Laron, Z.; Werner, H. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.Y.; Hagen, T. 2-Deoxyglucose induces the expression of thioredoxin interacting protein (TXNIP) by increasing O-GlcNAcylation—Implications for targeting the Warburg effect in cancer cells. Biochem. Biophys. Res. Commun. 2015, 465, 838–844. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem. 2008, 283, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Katsu-Jimenez, Y.; Vazquez-Calvo, C.; Maffezzini, C.; Halldin, M.; Peng, X.; Freyer, C.; Wredenberg, A.; Gimenez-Cassina, A.; Wedell, A.; Arner, E.S.J. Absence of TXNIP in Humans Leads to Lactic Acidosis and Low Serum Methionine Linked to Deficient Respiration on Pyruvate. Diabetes 2019, 68, 709–723. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Cittadini, A.; Grossman, J.D.; Stromer, H.; Katz, S.E.; Morgan, J.P.; Douglas, P.S. Importance of an intact growth hormone/insulin-like growth factor 1 axis for normal post-infarction healing: Studies in dwarf rats. Endocrinology 2001, 142, 332–338. [Google Scholar] [CrossRef]
- Davani, E.Y.; Brumme, Z.; Singhera, G.K.; Cote, H.C.; Harrigan, P.R.; Dorscheid, D.R. Insulin-like growth factor-1 protects ischemic murine myocardium from ischemia/reperfusion associated injury. Crit. Care 2003, 7, R176–R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. New directions for protecting the heart against ischaemia-reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, K.; Kajstura, J.; Discher, D.J.; Wasserlauf, B.J.; Bishopric, N.H.; Anversa, P.; Webster, K.A. Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ. Res. 2001, 88, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajstura, J.; Fiordaliso, F.; Andreoli, A.M.; Li, B.; Chimenti, S.; Medow, M.S.; Limana, F.; Nadal-Ginard, B.; Leri, A.; Anversa, P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 2001, 50, 1414–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.D.; al-Jumah, M.; Cwik, V.; Brooke, M.H. Neurotrophic factors decrease the release of creatine kinase and prostaglandin E2 from metabolically stressed muscle. Neuromuscul. Disord. 1998, 8, 7–13. [Google Scholar] [CrossRef]
- Matheny, R.W.Jr.; Adamo, M.L. PI3K p110 alpha and p110 beta have differential effects on Akt activation and protection against oxidative stress-induced apoptosis in myoblasts. Cell Death Differ. 2010, 17, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csiszar, A.; Labinskyy, N.; Perez, V.; Recchia, F.A.; Podlutsky, A.; Mukhopadhyay, P.; Losonczy, G.; Pacher, P.; Austad, S.N.; Bartke, A.; et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1882–H1894. [Google Scholar] [CrossRef] [Green Version]
- Heck, S.; Lezoualc’h, F.; Engert, S.; Behl, C. Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J. Biol. Chem. 1999, 274, 9828–9835. [Google Scholar] [CrossRef] [Green Version]
- Kolosova, N.G.; Stefanova, N.A.; Muraleva, N.A.; Skulachev, V.P. The mitochondria-targeted antioxidant SkQ1 but not N-acetylcysteine reverses aging-related biomarkers in rats. Aging (Albany NY) 2012, 4, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Morley, J.E. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Chesnokova, V.; Zhou, C.; Ben-Shlomo, A.; Zonis, S.; Tani, Y.; Ren, S.G.; Melmed, S. Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3331–E3339. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, R.; Fukuoka, H.; Iguchi, G.; Odake, Y.; Yoshida, K.; Bando, H.; Suda, K.; Nishizawa, H.; Takahashi, M.; Yamada, S.; et al. Accelerated Telomere Shortening in Acromegaly; IGF-I Induces Telomere Shortening and Cellular Senescence. PLoS ONE 2015, 10, e0140189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, M.B.; Tchkonia, T.; Pirtskhalava, T.; Palmer, A.K.; List, E.O.; Berryman, D.E.; Lubbers, E.R.; Escande, C.; Spong, A.; Masternak, M.M.; et al. Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging (Albany NY) 2014, 6, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ock, S.; Lee, W.S.; Ahn, J.; Kim, H.M.; Kang, H.; Kim, H.S.; Jo, D.; Abel, E.D.; Lee, T.J.; Kim, J. Deletion of IGF-1 Receptors in Cardiomyocytes Attenuates Cardiac Aging in Male Mice. Endocrinology 2016, 157, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Del Nogal-Avila, M.; Troyano-Suarez, N.; Roman-Garcia, P.; Cannata-Andia, J.B.; Rodriguez-Puyol, M.; Rodriguez-Puyol, D.; Kuro, O.M.; Ruiz-Torres, M.P. Amadori products promote cellular senescence activating insulin-like growth factor-1 receptor and down-regulating the antioxidant enzyme catalase. Int. J. Biochem. Cell Biol. 2013, 45, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Handayaningsih, A.E.; Takahashi, M.; Fukuoka, H.; Iguchi, G.; Nishizawa, H.; Yamamoto, M.; Suda, K.; Takahashi, Y. IGF-I enhances cellular senescence via the reactive oxygen species-p53 pathway. Biochem. Biophys. Res. Commun. 2012, 425, 478–484. [Google Scholar] [CrossRef]
- Thum, T.; Hoeber, S.; Froese, S.; Klink, I.; Stichtenoth, D.O.; Galuppo, P.; Jakob, M.; Tsikas, D.; Anker, S.D.; Poole-Wilson, P.A.; et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ. Res. 2007, 100, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Lee, E.Y.; Cho, K.H. Incorporation of human growth hormone-2 into proteoliposome enhances tissue regeneration with anti-oxidant and anti-senescence activities. Rejuvenation Res. 2015, 18, 20–29. [Google Scholar] [CrossRef]
- Luo, X.; Jiang, X.; Li, J.; Bai, Y.; Li, Z.; Wei, P.; Sun, S.; Liang, Y.; Han, S.; Li, X.; et al. Insulin-like growth factor-1 attenuates oxidative stress-induced hepatocyte premature senescence in liver fibrogenesis via regulating nuclear p53-progerin interaction. Cell Death Dis. 2019, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Lindsten, T.; Ross, A.J.; MacGregor, G.R.; Thompson, C.B. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001, 15, 1481–1486. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, J.M.; Esteban, M. A poxvirus Bcl-2-like gene family involved in regulation of host immune response: sequence similarity and evolutionary history. Virol. J. 2010, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.S.; Bass, B.P.; Jue, D.; Rodriguez, A.; Abrams, J.M.; McCall, K. Noncanonical cell death pathways act during Drosophila oogenesis. Genesis 2007, 45, 396–404. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef]
- Guo, S.; Rena, G.; Cichy, S.; He, X.; Cohen, P.; Unterman, T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 1999, 274, 17184–17192. [Google Scholar] [CrossRef] [Green Version]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Ambrogini, E.; Almeida, M.; Martin-Millan, M.; Paik, J.H.; Depinho, R.A.; Han, L.; Goellner, J.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A.; et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010, 11, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alt, E.U.; Senst, C.; Murthy, S.N.; Slakey, D.P.; Dupin, C.L.; Chaffin, A.E.; Kadowitz, P.J.; Izadpanah, R. Aging alters tissue resident mesenchymal stem cell properties. Stem Cell Res. 2012, 8, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.; Shehadeh, L.A.; Yu, H.; Webster, K.A. Age-related molecular genetic changes of murine bone marrow mesenchymal stem cells. BMC Genomics 2010, 11, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, Y.; Lee, K.A.; Kim, W.H.; Han, B.G.; Vijg, J.; Park, S.C. Aging alters the apoptotic response to genotoxic stress. Nat. Med. 2002, 8, 3–4. [Google Scholar] [CrossRef]
- Polyak, K.; Wu, T.T.; Hamilton, S.R.; Kinzler, K.W.; Vogelstein, B. Less death in the dying. Cell Death Differ. 1997, 4, 242–246. [Google Scholar] [CrossRef]
- Kavathia, N.; Jain, A.; Walston, J.; Beamer, B.A.; Fedarko, N.S. Serum markers of apoptosis decrease with age and cancer stage. Aging (Albany NY) 2009, 1, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. (Lausanne) 2018, 5, 316. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Breda, C.N.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Camara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships Between Ion Channels, Mitochondrial Functions and Inflammation in Human Aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Kermani, H.; Goffinet, L.; Mottet, M.; Bodart, G.; Morrhaye, G.; Dardenne, O.; Renard, C.; Overbergh, L.; Baron, F.; Beguin, Y.; et al. Expression of the growth hormone/insulin-like growth factor axis during Balb/c thymus ontogeny and effects of growth hormone upon ex vivo T cell differentiation. Neuroimmunomodulation 2012, 19, 137–147. [Google Scholar] [CrossRef]
- Smith, T.J. Insulin-like growth factor-I regulation of immune function: A potential therapeutic target in autoimmune diseases? Pharmacol. Rev. 2010, 62, 199–236. [Google Scholar] [CrossRef] [Green Version]
- Weigent, D.A.; Blalock, J.E. Expression of growth hormone by lymphocytes. Int. Rev. Immunol. 1989, 4, 193–211. [Google Scholar] [CrossRef]
- Rappolee, D.A.; Mark, D.; Banda, M.J.; Werb, Z. Wound macrophages express TGF-alpha and other growth factors in vivo: Analysis by mRNA phenotyping. Science 1988, 241, 708–712. [Google Scholar] [CrossRef]
- Spaziani, S.; Imperlini, E.; Mancini, A.; Caterino, M.; Buono, P.; Orru, S. Insulin-like growth factor 1 receptor signaling induced by supraphysiological doses of IGF-1 in human peripheral blood lymphocytes. Proteomics 2014, 14, 1623–1629. [Google Scholar] [CrossRef]
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the Growth Hormone-Insulin Like Growth Factor 1 Axis in Children With Chronic Inflammation: Current Evidence, Gaps in Knowledge, and Future Directions. Endocr. Rev. 2016, 37, 62–110. [Google Scholar] [CrossRef]
- Masternak, M.M.; Bartke, A. Growth hormone, inflammation and aging. Pathobiol. Aging Age Relat. Dis. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Sadagurski, M.; Landeryou, T.; Cady, G.; Kopchick, J.J.; List, E.O.; Berryman, D.E.; Bartke, A.; Miller, R.A. Growth hormone modulates hypothalamic inflammation in long-lived pituitary dwarf mice. Aging Cell 2015, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic summary of the major molecules composing the somatotropic axis. GHRH—Growth hormone-releasing hormone, SST—Somatostatin, GH—Growth hormone, GHR—Growth hormone receptor, IGF—Insulin-like growth factor, IGFBP—Insulin-like growth factor binding protein, IGF-IR—insulin like growth factor receptor-1, IR—Insulin receptor, ALS—Acid labile subunit, PAPPA2—Pregnancy-associated plasma protein-A2.
Figure 1.
Schematic summary of the major molecules composing the somatotropic axis. GHRH—Growth hormone-releasing hormone, SST—Somatostatin, GH—Growth hormone, GHR—Growth hormone receptor, IGF—Insulin-like growth factor, IGFBP—Insulin-like growth factor binding protein, IGF-IR—insulin like growth factor receptor-1, IR—Insulin receptor, ALS—Acid labile subunit, PAPPA2—Pregnancy-associated plasma protein-A2.
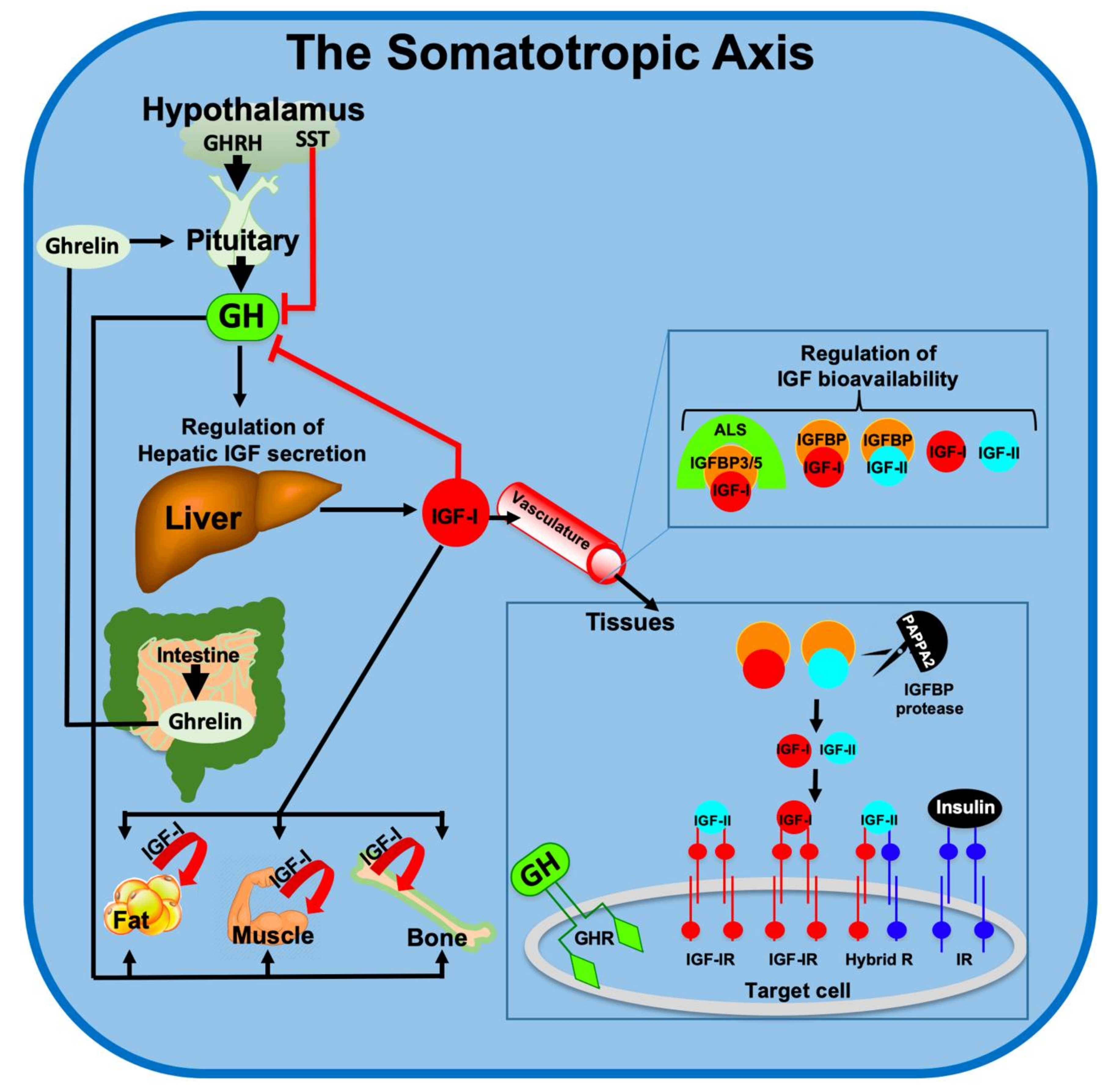
Figure 2.
Schematic summary of the major effects of GH/IGF-1 on mitochondrial gene expression: Upon binding of GH to the GHR, the Janus kinase (JAK)-Signal transducer and activator of transcription 5 (STAT5) signaling pathway is activated, leading mostly to increases in IGF-1 transcription. Binding of IGF-1 to the tyrosine kinase IGF-1R stimulates several signaling pathways including the Phosphoinositide-3-kinase (PI3K)/Protein kinase B (PKB or AKT) and Ras/Raf/Mitogen-activated protein kinase (MAPK), involving phosphorylation and dephosphorylation of candidate proteins. This cascade leads to transcriptional activity of genes involved in mitochondrial biogenesis, control of Reactive oxygen species (ROS), cell survival (antiapoptotic), and genes involved in metabolism.
Figure 2.
Schematic summary of the major effects of GH/IGF-1 on mitochondrial gene expression: Upon binding of GH to the GHR, the Janus kinase (JAK)-Signal transducer and activator of transcription 5 (STAT5) signaling pathway is activated, leading mostly to increases in IGF-1 transcription. Binding of IGF-1 to the tyrosine kinase IGF-1R stimulates several signaling pathways including the Phosphoinositide-3-kinase (PI3K)/Protein kinase B (PKB or AKT) and Ras/Raf/Mitogen-activated protein kinase (MAPK), involving phosphorylation and dephosphorylation of candidate proteins. This cascade leads to transcriptional activity of genes involved in mitochondrial biogenesis, control of Reactive oxygen species (ROS), cell survival (antiapoptotic), and genes involved in metabolism.
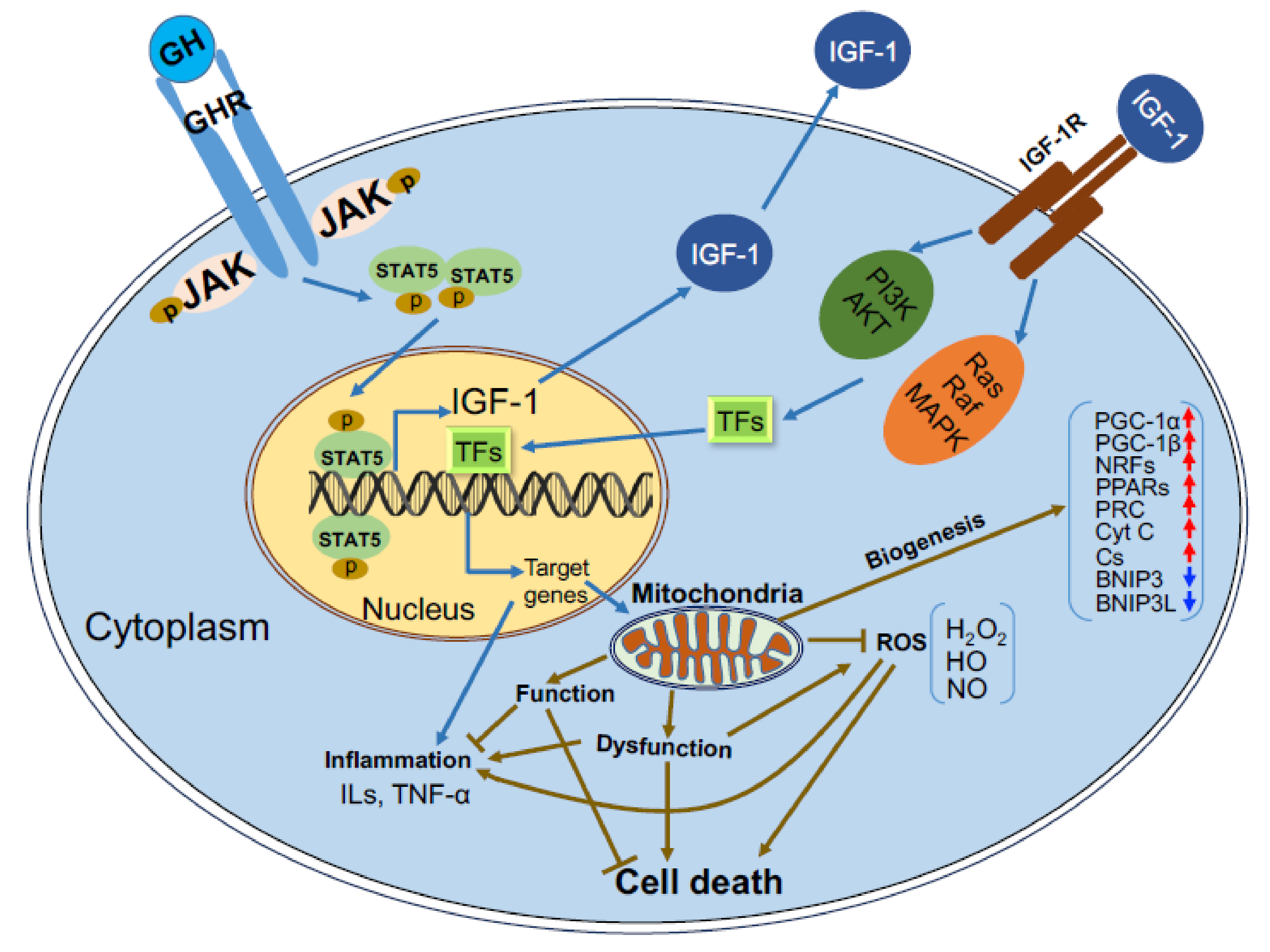
Figure 3.
Mitochondria produce ATP and reactive oxygen species (ROS) as byproducts of oxidative phosphorylation (OXPHOS): In youth, ROSs are neutralized by the antioxidant system. Accumulation of proteins and enzymes damaged by escaped ROS leads to impairment of mitochondrial function during aging. Mitochondrial dysfunction is correlated with the decline in GH/IGF1 signaling and is linked to a variety of age-related diseases.
Figure 3.
Mitochondria produce ATP and reactive oxygen species (ROS) as byproducts of oxidative phosphorylation (OXPHOS): In youth, ROSs are neutralized by the antioxidant system. Accumulation of proteins and enzymes damaged by escaped ROS leads to impairment of mitochondrial function during aging. Mitochondrial dysfunction is correlated with the decline in GH/IGF1 signaling and is linked to a variety of age-related diseases.
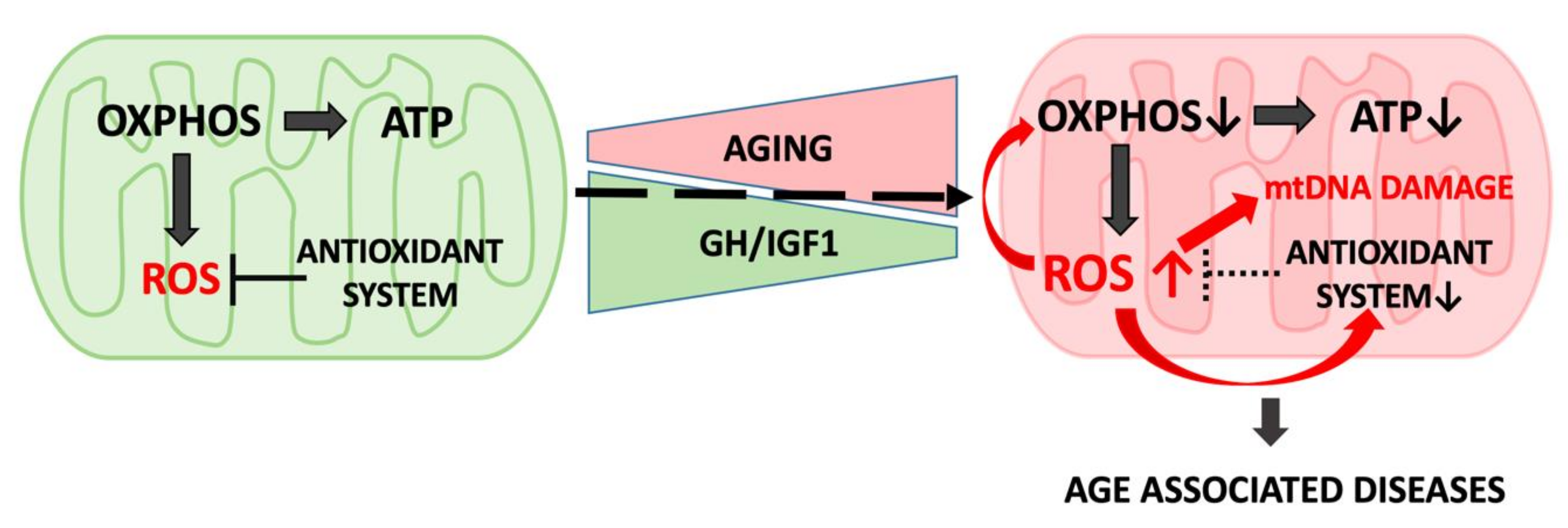
Figure 4.
Effect of oxidative stress on thioredoxin-interacting protein (TXNIP) levels [79]: (A) Effect of oxidative stress on TXNIP levels in Laron syndrome (LS)-derived and control lymphoblastoids. Four individual LS and three control lymphoblastoid cell lines were treated with 300 mM of H2O2 for 2 h, and levels of TXNIP mRNA were measured by RT-QPCR. A value of 1 was given to TXNIP mRNA levels in untreated cells (solid bars). (B) Serum-starved HEK293 cells were treated with H2O2 (100 mM) or IGF1 (50 ng/mL) or both for 2 h. TXNIP and tubulin were detected by Western blotting.
Figure 4.
Effect of oxidative stress on thioredoxin-interacting protein (TXNIP) levels [79]: (A) Effect of oxidative stress on TXNIP levels in Laron syndrome (LS)-derived and control lymphoblastoids. Four individual LS and three control lymphoblastoid cell lines were treated with 300 mM of H2O2 for 2 h, and levels of TXNIP mRNA were measured by RT-QPCR. A value of 1 was given to TXNIP mRNA levels in untreated cells (solid bars). (B) Serum-starved HEK293 cells were treated with H2O2 (100 mM) or IGF1 (50 ng/mL) or both for 2 h. TXNIP and tubulin were detected by Western blotting.
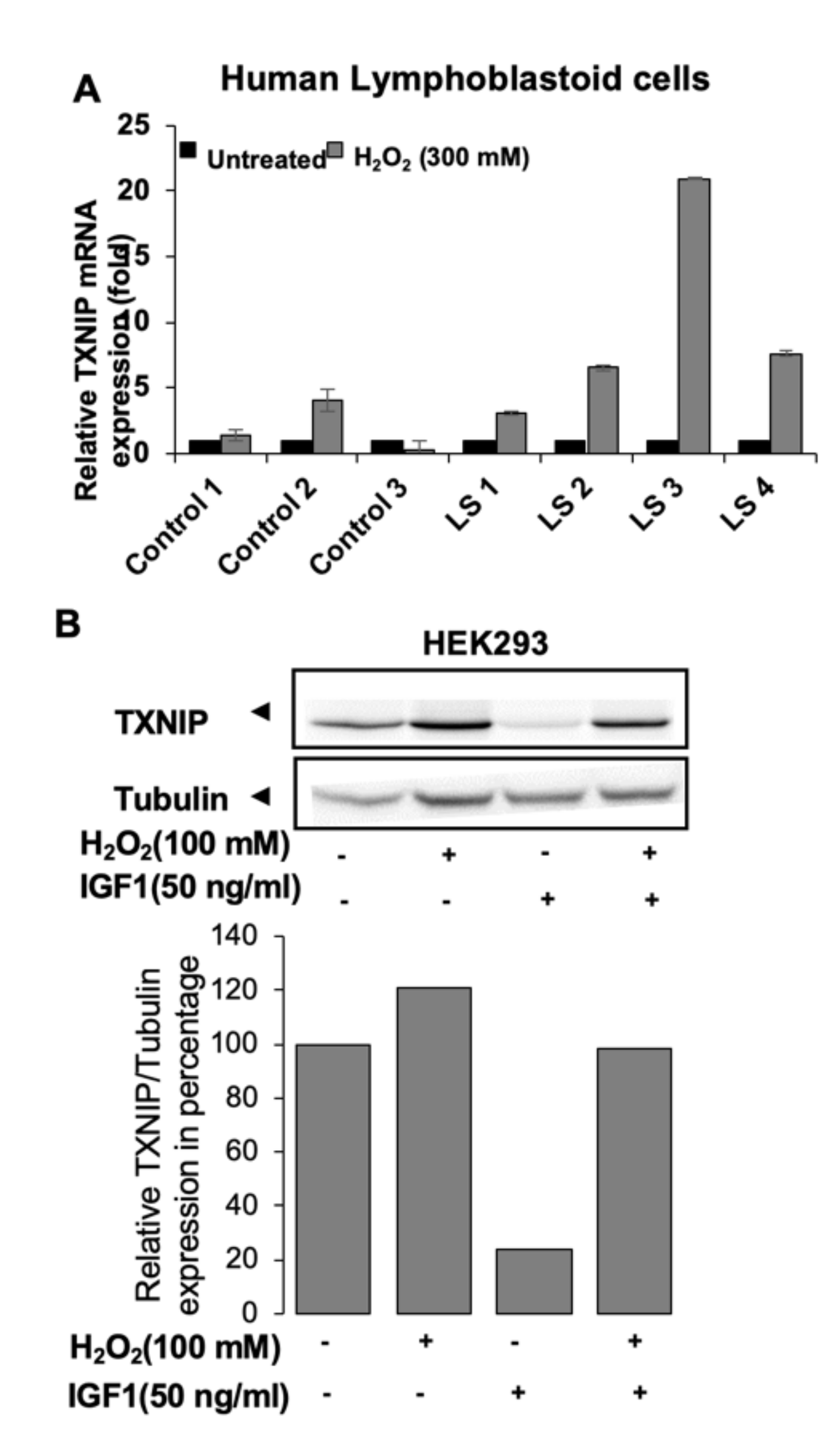
Figure 5.
Effect of glucose stress on TXNIP levels [79]: (A) Serum-starved 3T3-L1 cells were maintained in medium with different concentrations of glucose in the presence or absence of IGF1 or insulin for 6 h. TXNIP, phospho-IGF-1R, and tubulin were detected by Western blotting. Schematic representation of the regulation of TXNIP by IGF1 signaling: (B) Under normal serum-free conditions, TXNIP is upregulated upon oxidative and glucose stresses. The activated TXNIP inhibits glucose uptake and is capable of mediating mitochondrial mediated apoptosis. (C) Upon IGF1 stimulation, TXNIP is downregulated even under oxidative and glucose stresses. Suppression of TXNIP leads to inhibition of apoptosis with ensuing increase in cell proliferation.
Figure 5.
Effect of glucose stress on TXNIP levels [79]: (A) Serum-starved 3T3-L1 cells were maintained in medium with different concentrations of glucose in the presence or absence of IGF1 or insulin for 6 h. TXNIP, phospho-IGF-1R, and tubulin were detected by Western blotting. Schematic representation of the regulation of TXNIP by IGF1 signaling: (B) Under normal serum-free conditions, TXNIP is upregulated upon oxidative and glucose stresses. The activated TXNIP inhibits glucose uptake and is capable of mediating mitochondrial mediated apoptosis. (C) Upon IGF1 stimulation, TXNIP is downregulated even under oxidative and glucose stresses. Suppression of TXNIP leads to inhibition of apoptosis with ensuing increase in cell proliferation.
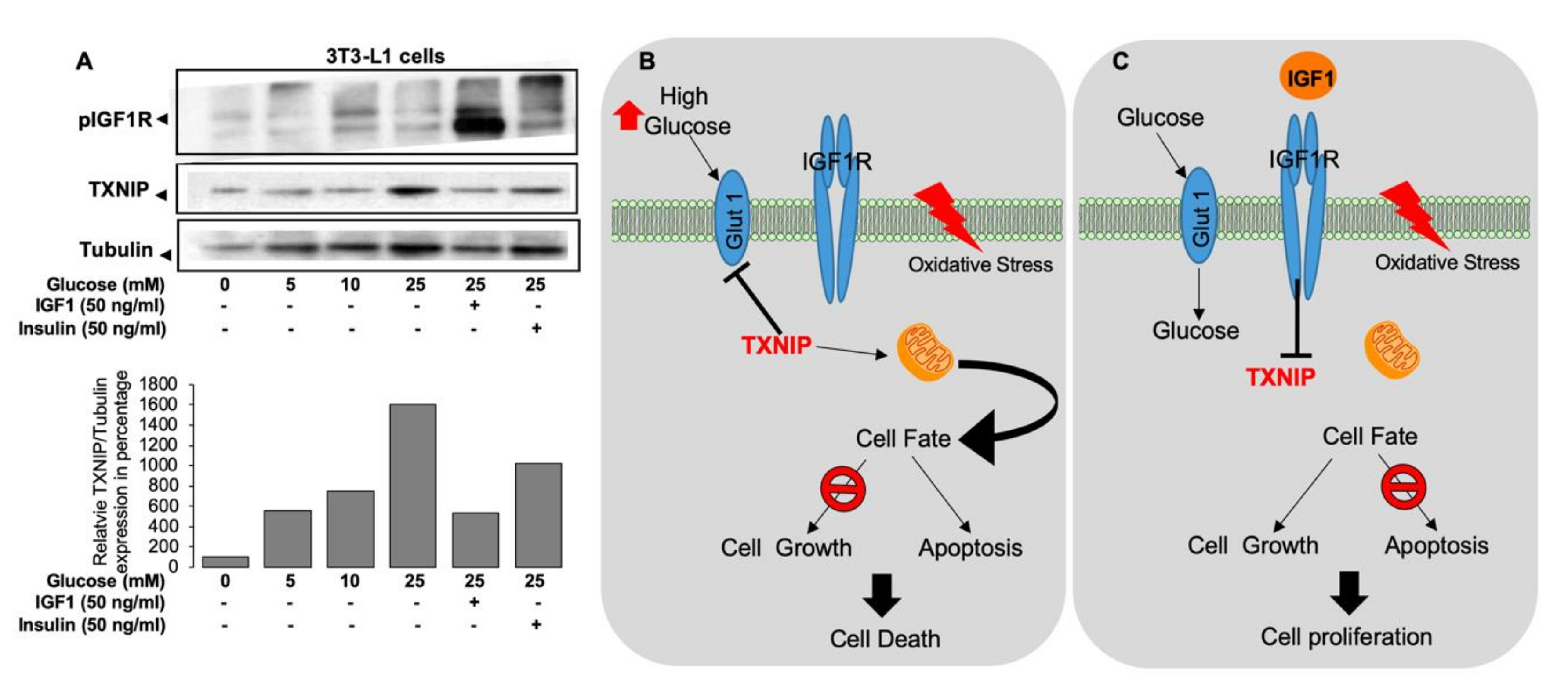
Figure 6.
The effects of IGF-1 on mitochondria-mediated apoptosis: Cellular stress leads to permeability transition pore (PTP) opening, reduction in OXPHOS function, membrane potential, and ATP production, while ROS production is increased, altogether leading to cyt c release. Cytosolic Cyt c activates the apoptosome, leading to cell death. IGF-1R-mediated inhibition of apoptosis occurs via activation of two major signaling pathways that trigger phosphorylation cascade of cytosolic and nuclear proteins regulating transcription and activation of proteins involved in protection from apoptosis. The AKT pathway leads to phosphorylation of Forkhead box transcription factors of the class O (FOXO) proteins and subsequent inhibition of their transcriptional activity. The MAPK pathway, stimulated by IGF-1 binding, activates the Bcl antiapoptotic family of proteins.
Figure 6.
The effects of IGF-1 on mitochondria-mediated apoptosis: Cellular stress leads to permeability transition pore (PTP) opening, reduction in OXPHOS function, membrane potential, and ATP production, while ROS production is increased, altogether leading to cyt c release. Cytosolic Cyt c activates the apoptosome, leading to cell death. IGF-1R-mediated inhibition of apoptosis occurs via activation of two major signaling pathways that trigger phosphorylation cascade of cytosolic and nuclear proteins regulating transcription and activation of proteins involved in protection from apoptosis. The AKT pathway leads to phosphorylation of Forkhead box transcription factors of the class O (FOXO) proteins and subsequent inhibition of their transcriptional activity. The MAPK pathway, stimulated by IGF-1 binding, activates the Bcl antiapoptotic family of proteins.
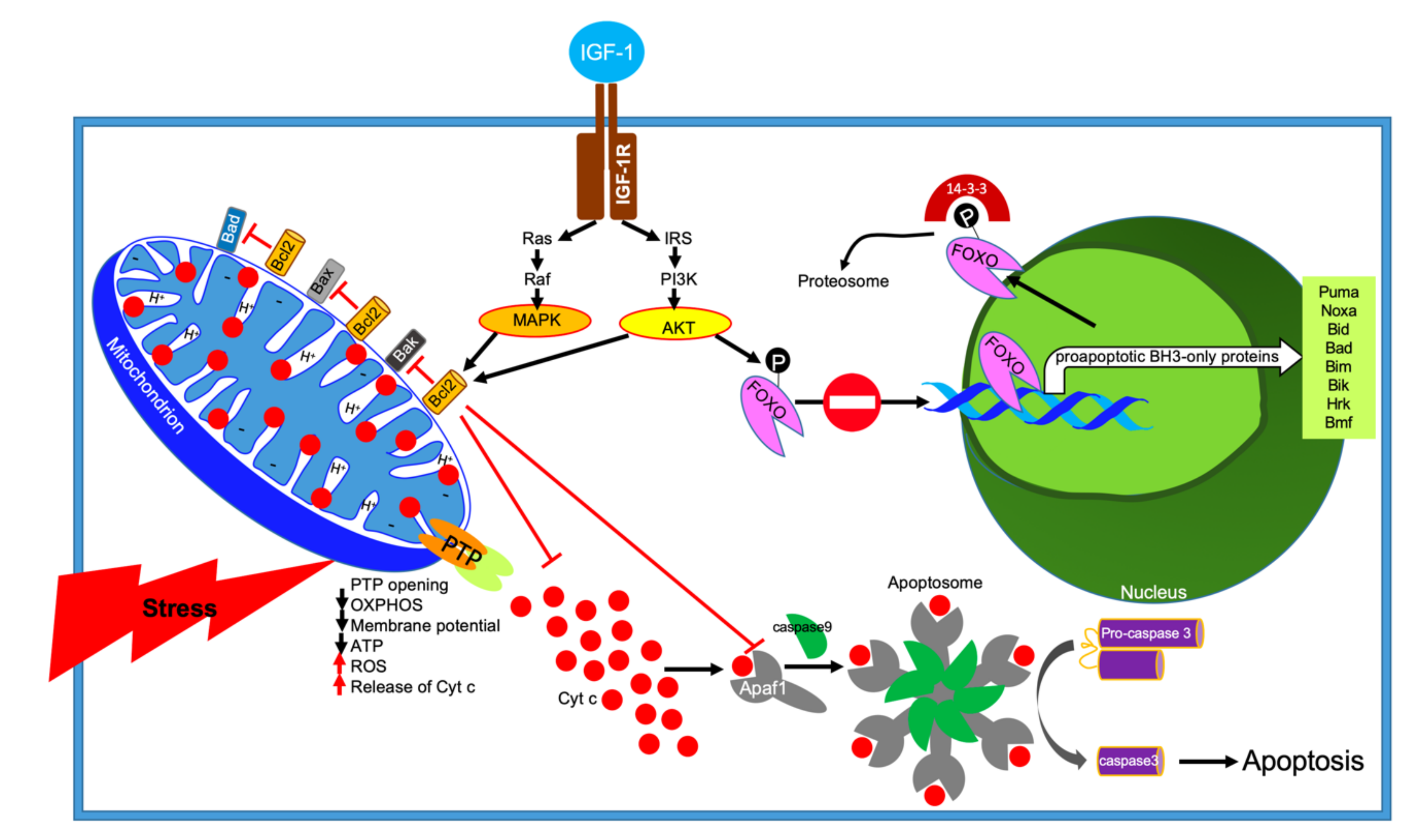
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Poudel, S.B.; Dixit, M.; Neginskaya, M.; Nagaraj, K.; Pavlov, E.; Werner, H.; Yakar, S. Effects of GH/IGF on the Aging Mitochondria. Cells 2020, 9, 1384. https://doi.org/10.3390/cells9061384
AMA Style
Poudel SB, Dixit M, Neginskaya M, Nagaraj K, Pavlov E, Werner H, Yakar S. Effects of GH/IGF on the Aging Mitochondria. Cells. 2020; 9(6):1384. https://doi.org/10.3390/cells9061384
Chicago/Turabian StylePoudel, Sher Bahadur, Manisha Dixit, Maria Neginskaya, Karthik Nagaraj, Evgeny Pavlov, Haim Werner, and Shoshana Yakar. 2020. "Effects of GH/IGF on the Aging Mitochondria" Cells 9, no. 6: 1384. https://doi.org/10.3390/cells9061384
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.