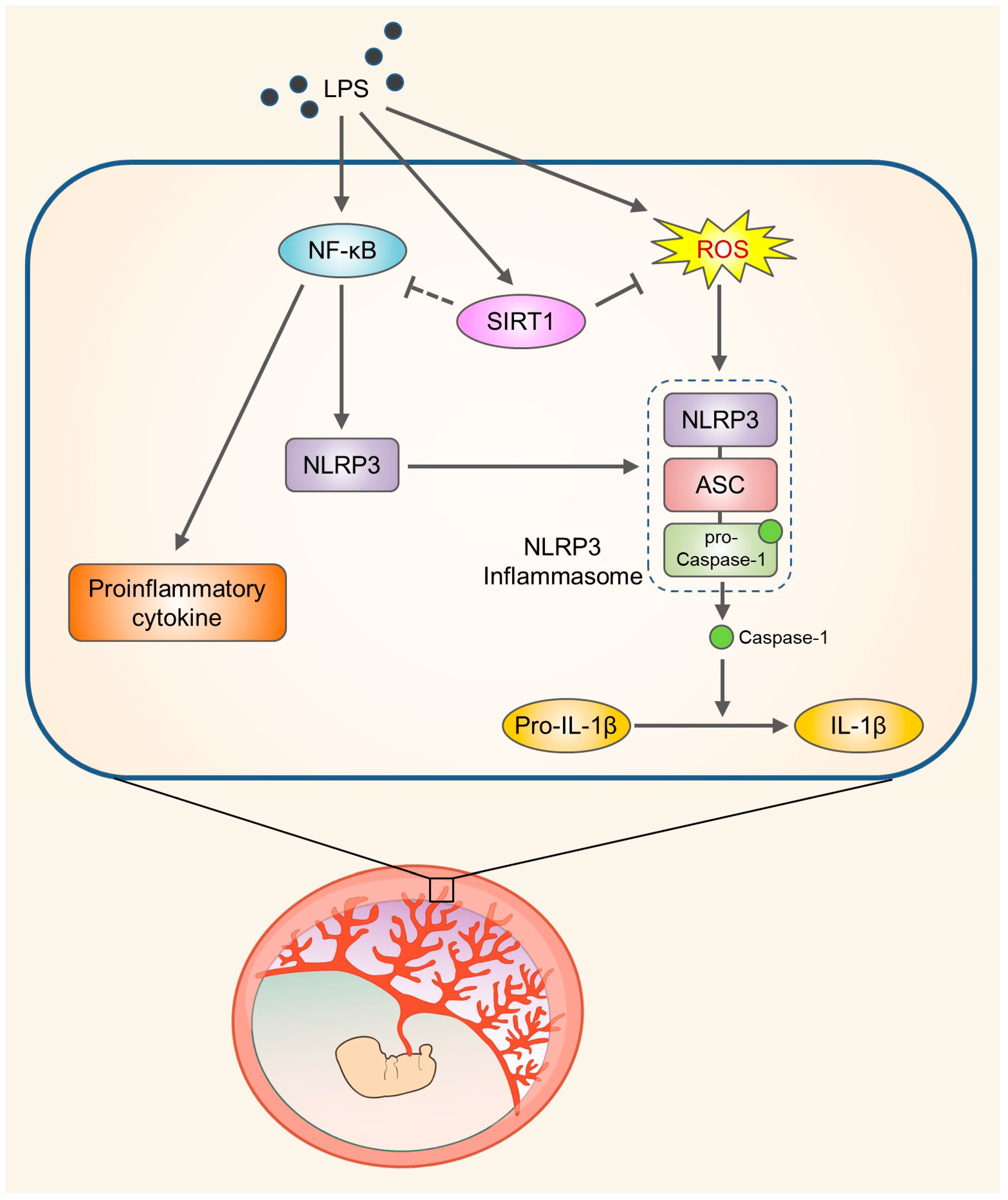
SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts

 ,
, 
Abstract
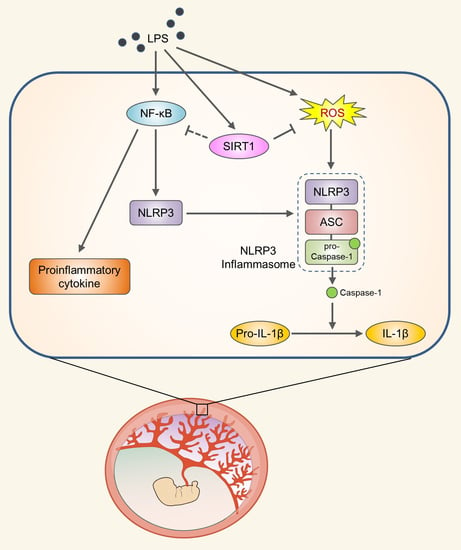
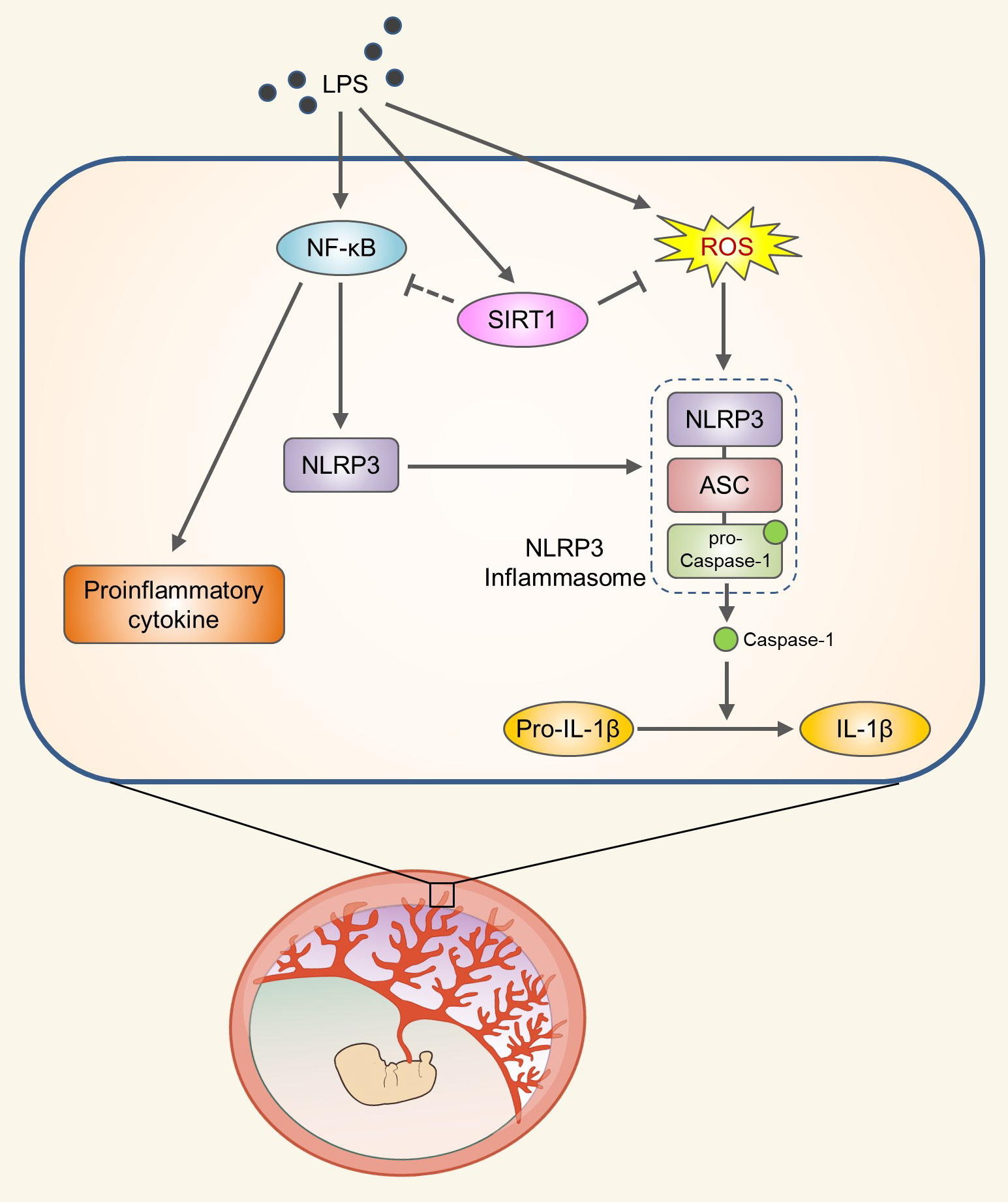{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals and Treatments
2.3. Cell Culture and Treatments
2.4. Plasmids and Viral Production
2.5. Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.6. Luciferase Reporter Assay
2.7. Immunoblotting
2.8. Immunocytochemistry
2.9. Immunohistochemistry
2.10. Detection of Reactive Oxygen Species (ROS)
2.11. Enzyme-Linked Immunosorbent Assay (ELISA)
2.12. Statistical Analysis
3. Results
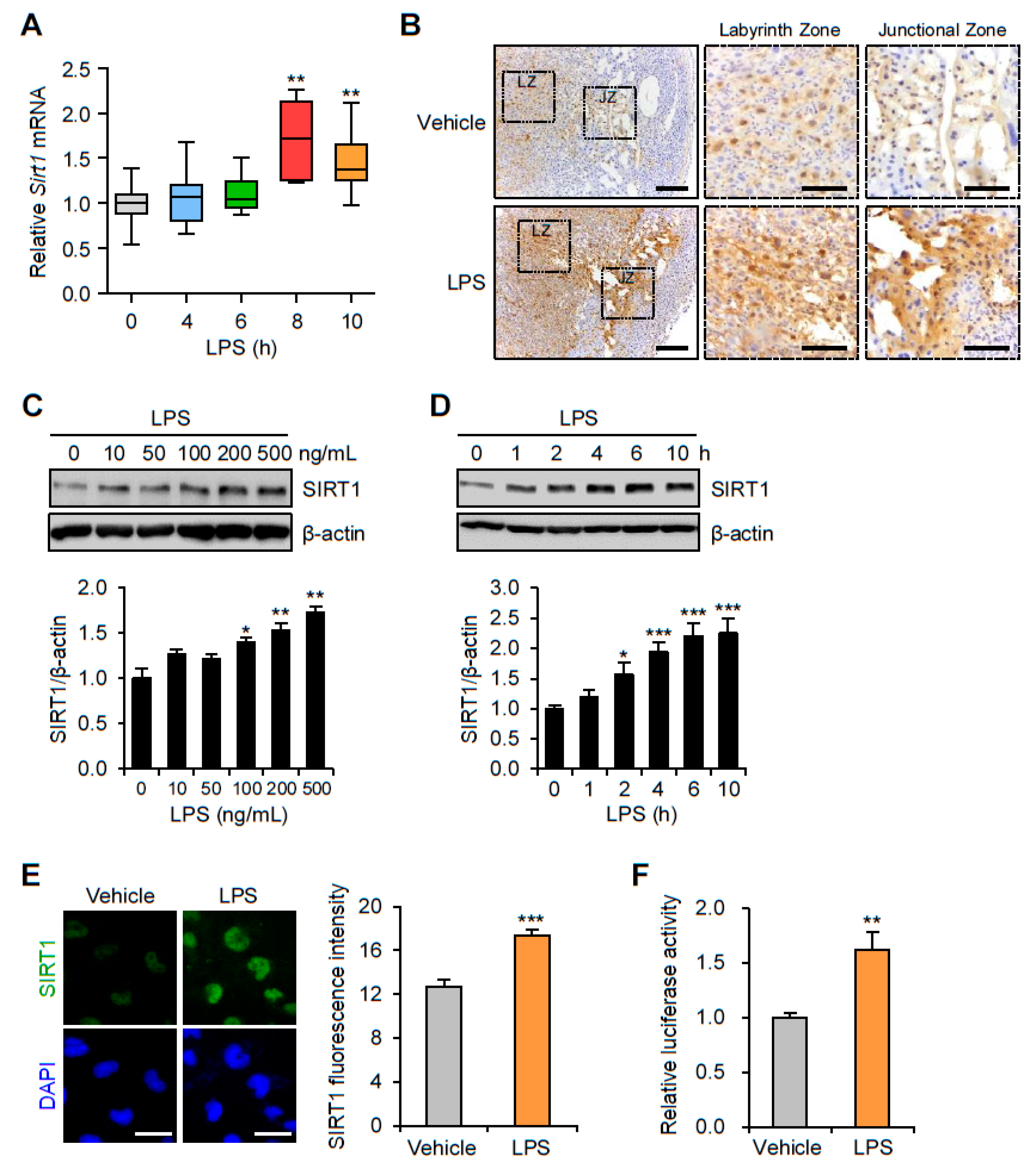
3.1. SIRT1 Is Upregulated in the Placentas and Trophoblast Cells after LPS Exposure
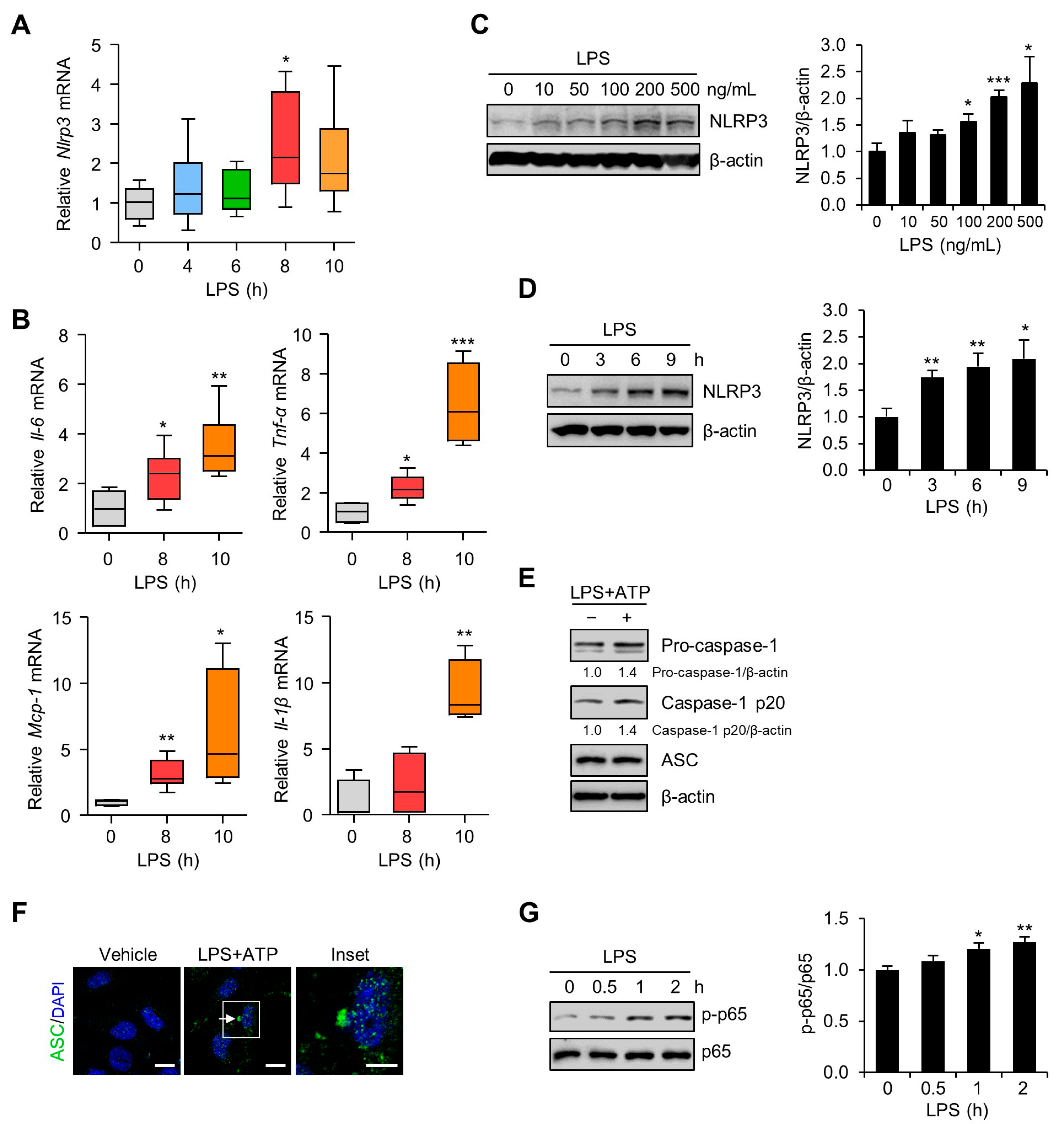
3.2. Inflammation and NLRP3 Inflammasome Activation in the Placentas and Trophoblast Cells after LPS Exposure
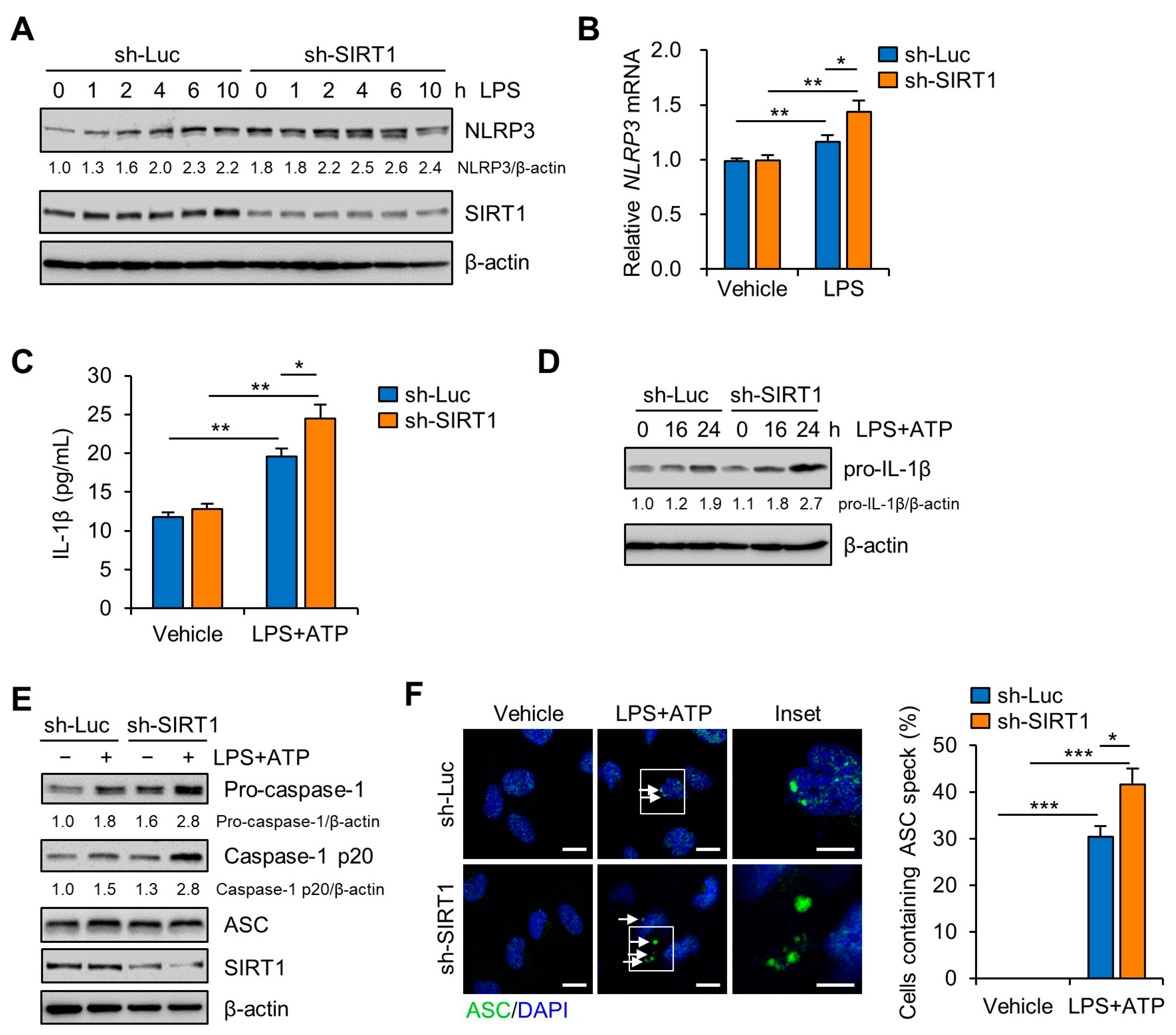
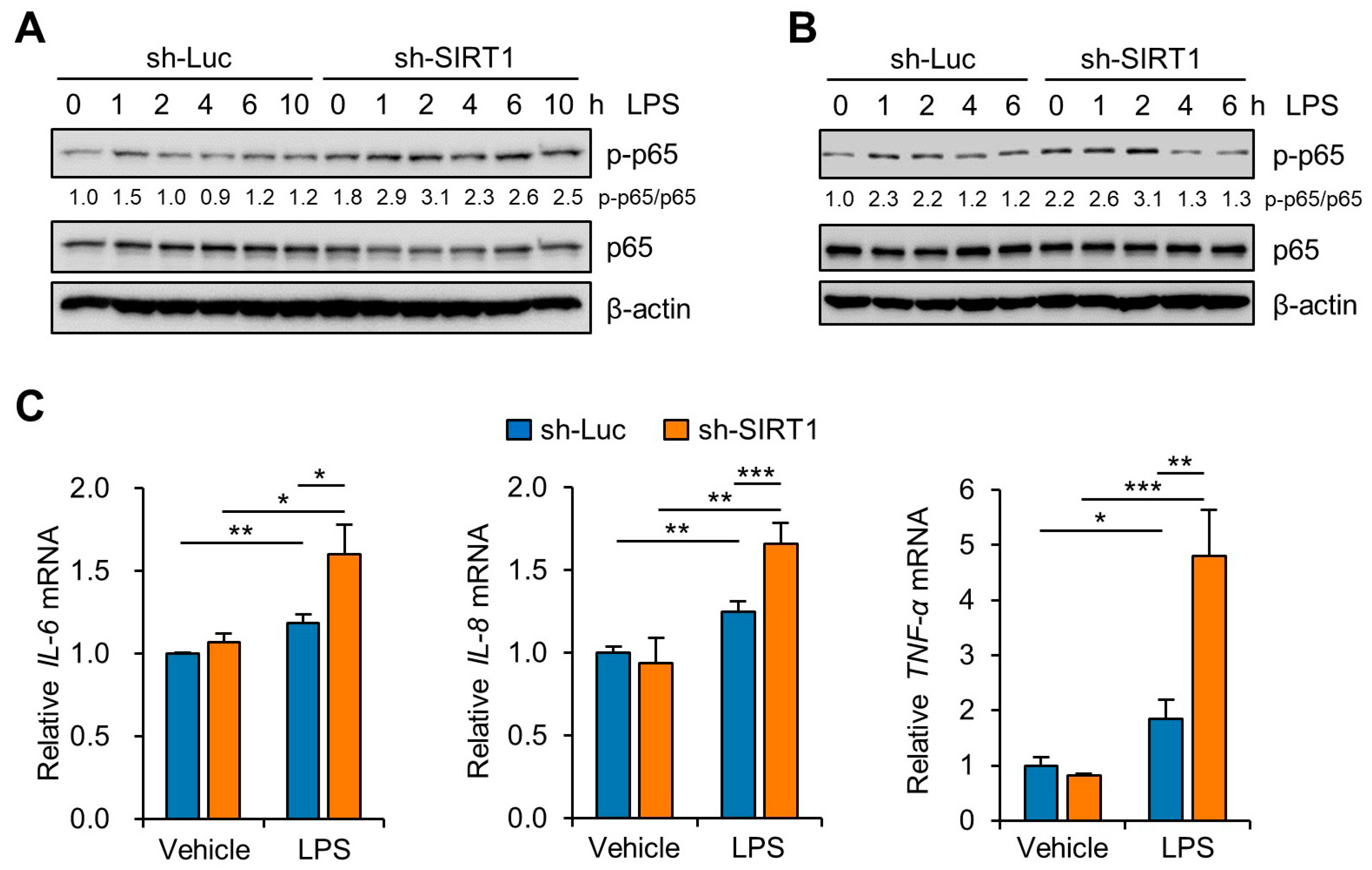
3.3. SIRT1 Attenuates LPS-Induced Inflammation and NLRP3 Inflammasome Activation in Trophoblast Cells
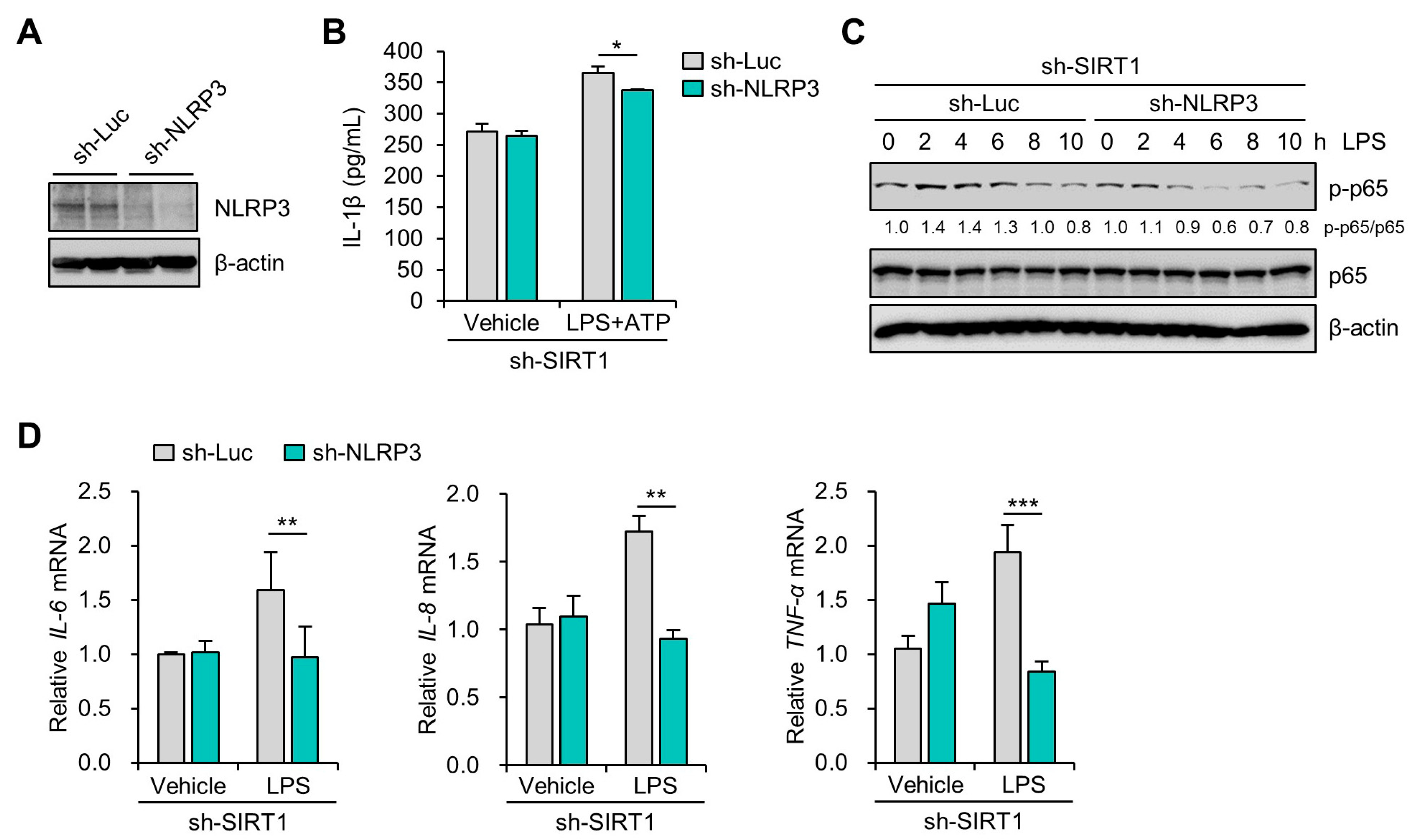
3.4. SIRT1 Suppresses LPS-Induced IL-1β Secretion in Trophoblast Cells in a NLRP3-Dependent Manner
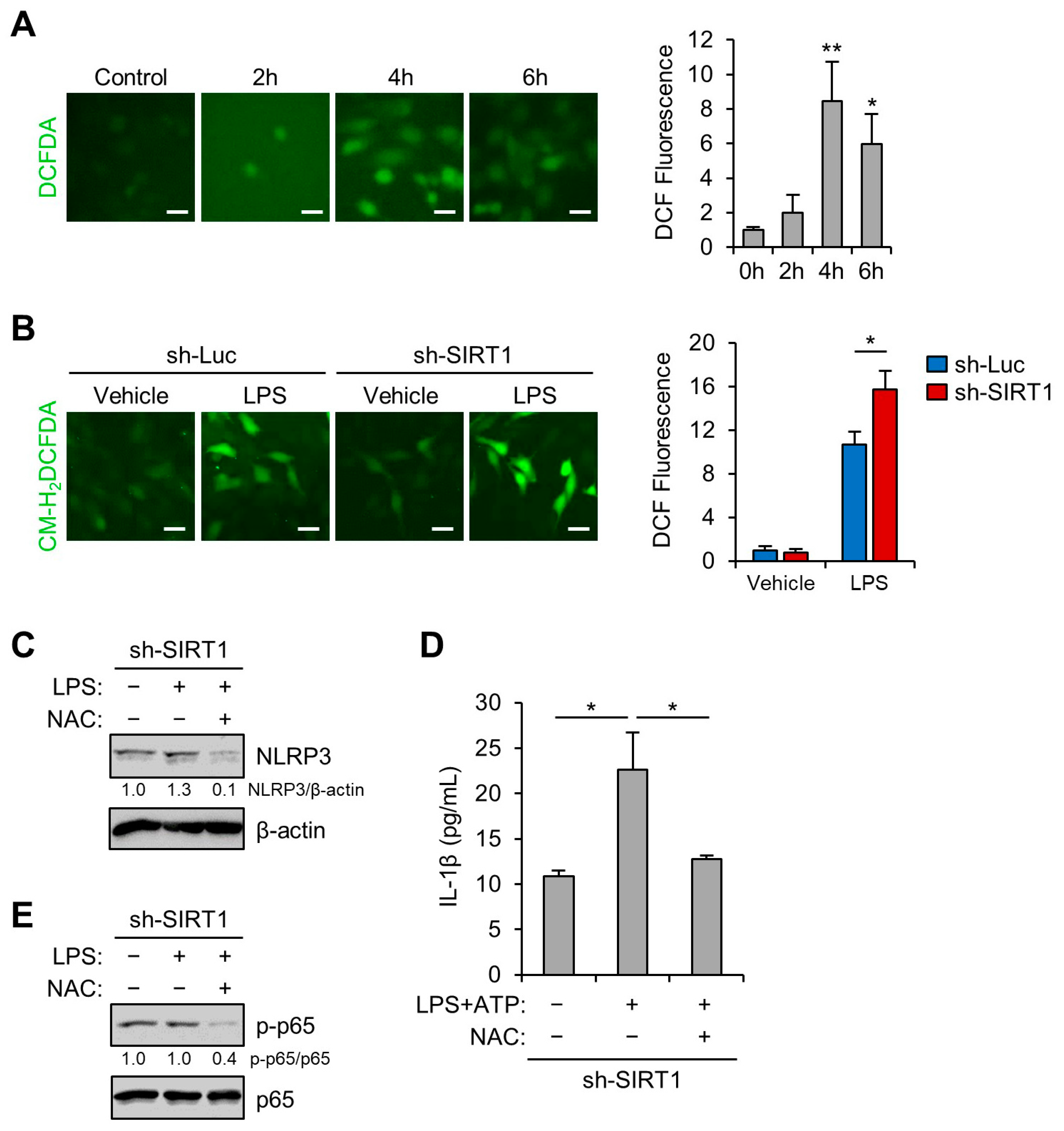
3.5. SIRT1 Inhibits LPS-Induced NLRP3 Inflammasome in Trophoblast Cells Via Reducing Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and Pregnancy: The Role of the Immune System at the Implantation Site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef]
- He, N.; van Iperen, L.; de Jong, D.; Szuhai, K.; Helmerhorst, F.M.; van der Westerlaken, L.A.; Chuva de Sousa Lopes, S.M. Human Extravillous Trophoblasts Penetrate Decidual Veins and Lymphatics before Remodeling Spiral Arteries During Early Pregnancy. PLoS ONE 2017, 12, e0169849. [Google Scholar] [CrossRef]
- Graham, C.H.; Lala, P.K. Mechanisms of Placental Invasion of the Uterus and Their Control. Biochem. Cell Biol. 1992, 70, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Benyo, D.F.; Smarason, A.; Redman, C.W.; Sims, C.; Conrad, K.P. Expression of Inflammatory Cytokines in Placentas from Women with Preeclampsia. J. Clin. Endocrinol. Metab. 2001, 86, 2505–2512. [Google Scholar] [CrossRef]
- Boutsikou, T.; Mastorakos, G.; Kyriakakou, M.; Margeli, A.; Hassiakos, D.; Papassotiriou, I.; Kanaka-Gantenbein, C.; Malamitsi-Puchner, A. Circulating Levels of Inflammatory Markers in Intrauterine Growth Restriction. Mediat. Inflamm. 2010, 2010, 790605. [Google Scholar] [CrossRef]
- Sharma, A.; Satyam, A.; Sharma, J.B. Leptin, IL-10 and Inflammatory Markers (TNF-Alpha, IL-6 and IL-8) in Pre-Eclamptic, Normotensive Pregnant and Healthy Non-Pregnant Women. Am. J. Reprod. Immunol. 2007, 58, 21–30. [Google Scholar] [CrossRef]
- Svinarich, D.M.; Bitonti, O.M.; Romero, R.; Gonik, B. Induction and Posttranslational Expression of Cytokines in a First-Trimester Trophoblast Cell Line by Lipopolysaccharide. Am. J. Obstet. Gynecol. 1996, 175, 970–973. [Google Scholar] [CrossRef]
- Zaga-Clavellina, V.; Garcia-Lopez, G.; Flores-Herrera, H.; Espejel-Nunez, A.; Flores-Pliego, A.; Soriano-Becerril, D.; Maida-Claros, R.; Merchant-Larios, H.; Vadillo-Ortega, F. In Vitro Secretion Profiles of Interleukin (IL)-1beta, IL-6, IL-8, IL-10, and TNF Alpha after Selective Infection with Escherichia Coli in Human Fetal Membranes. Reprod. Biol. Endocrinol. 2007, 5, 46. [Google Scholar] [CrossRef]
- Arechavaleta-Velasco, F.; Koi, H.; Strauss, J.F., 3rd; Parry, S. Viral Infection of the Trophoblast: Time to Take a Serious Look at Its Role in Abnormal Implantation and Placentation? J. Reprod. Immunol. 2002, 55, 113–121. [Google Scholar] [CrossRef]
- Bauer, S.; Pollheimer, J.; Hartmann, J.; Husslein, P.; Aplin, J.D.; Knofler, M. Tumor Necrosis Factor-Alpha Inhibits Trophoblast Migration through Elevation of Plasminogen Activator Inhibitor-1 in First-Trimester Villous Explant Cultures. J. Clin. Endocrinol. Metab. 2004, 89, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Vicovac, L. Interleukin-6 Stimulates Cell Migration, Invasion and Integrin Expression in HTR-8/Svneo Cell Line. Placenta 2009, 30, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.F. Infection in the Prediction and Antibiotics in the Prevention of Spontaneous Preterm Labour and Preterm Birth. BJOG 2003, 110, 71–75. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A. Inflammasome Activation and IL-1beta and IL-18 Processing During Infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Latz, E. The Inflammasomes: Mechanisms of Activation and Function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef]
- Mulla, M.J.; Myrtolli, K.; Potter, J.; Boeras, C.; Kavathas, P.B.; Sfakianaki, A.K.; Tadesse, S.; Norwitz, E.R.; Guller, S.; Abrahams, V.M. Uric Acid Induces Trophoblast IL-1beta Production Via the Inflammasome: Implications for the Pathogenesis of Preeclampsia. Am. J. Reprod. Immunol. 2011, 65, 542–548. [Google Scholar] [CrossRef]
- Shirasuna, K.; Takano, H.; Seno, K.; Ohtsu, A.; Karasawa, T.; Takahashi, M.; Ohkuchi, A.; Suzuki, H.; Matsubara, S.; Iwata, H.; et al. Palmitic Acid Induces Interleukin-1beta Secretion Via NLRP3 Inflammasomes and Inflammatory Responses through ROS Production in Human Placental Cells. J. Reprod. Immunol. 2016, 116, 104–112. [Google Scholar] [CrossRef]
- Shirasuna, K.; Usui, F.; Karasawa, T.; Kimura, H.; Kawashima, A.; Mizukami, H.; Ohkuchi, A.; Nishimura, S.; Sagara, J.; Noda, T.; et al. Nanosilica-Induced Placental Inflammation and Pregnancy Complications: Different Roles of the Inflammasome Components NLRP3 and ASC. Nanotoxicology 2015, 9, 554–567. [Google Scholar] [CrossRef]
- Abrahams, V.M. The Role of the Nod-Like Receptor Family in Trophoblast Innate Immune Responses. J. Reprod. Immunol. 2011, 88, 112–117. [Google Scholar] [CrossRef]
- Simmons, G.E., Jr.; Pruitt, W.M.; Pruitt, K. Diverse Roles of Sirt1 in Cancer Biology and Lipid Metabolism. Int. J. Mol. Sci. 2015, 16, 950–965. [Google Scholar] [CrossRef]
- Tang, S.; Huang, G.; Fan, W.; Chen, Y.; Ward, J.M.; Xu, X.; Xu, Q.; Kang, A.; McBurney, M.W.; Fargo, D.C.; et al. Sirt1-Mediated Deacetylation of Crabpii Regulates Cellular Retinoic Acid Signaling and Modulates Embryonic Stem Cell Differentiation. Mol. Cell 2014, 55, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Zainabadi, K.; Bell, E.; Sykes, D.B.; Saez, B.; Lotinun, S.; Baron, R.; Scadden, D.; Schipani, E.; Guarente, L. SIRT1 Regulates Differentiation of Mesenchymal Stem Cells by Deacetylating Beta-Catenin. EMBO Mol. Med. 2013, 5, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental Defects and P53 Hyperacetylation in Sir2 Homolog (SIRT1)-Deficient Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef]
- Lappas, M.; Mitton, A.; Lim, R.; Barker, G.; Riley, C.; Permezel, M. SIRT1 Is a Novel Regulator of Key Pathways of Human Labor. Biol. Reprod. 2011, 84, 167–178. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, Y.; Du, L.; Xu, C.; Cao, J.; Fan, T.; Liu, J.; Su, X.; Fan, S.; Liu, Q.; et al. Resveratrol Inhibits Ionising Irradiation-Induced Inflammation in Mscs by Activating SIRT1 and Limiting NLRP-3 Inflammasome Activation. Int. J. Mol. Sci. 2013, 14, 14105–14118. [Google Scholar] [CrossRef]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an Antiinflammatory and Antiaging Protein, Is Decreased in Lungs of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in Regulation of LPS- or Two Ethanol Metabolites-Induced TNF-Alpha Production in Cultured Macrophage Cell Lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Lin, Q.Q.; Yan, C.F.; Lin, R.; Zhang, J.Y.; Wang, W.R.; Yang, L.N.; Zhang, K.F. SIRT1 Regulates TNF-Alpha-Induced Expression of CD40 in 3T3-L1 Adipocytes Via NF-KappaB Pathway. Cytokine 2012, 60, 447–455. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-KappaB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 Inflammasome Activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Imamura, R.; Kushiyama, H.; Suda, T. NLRP3 Mediates NF-KappaB Activation and Cytokine Induction in Microbially Induced and Sterile Inflammation. PLoS ONE 2015, 10, e0119179. [Google Scholar] [CrossRef] [PubMed]
- Su, W.J.; Zhang, Y.; Chen, Y.; Gong, H.; Lian, Y.J.; Peng, W.; Liu, Y.Z.; Wang, Y.X.; You, Z.L.; Feng, S.J.; et al. NLRP3 Gene Knockout Blocks NF-KappaB and MAPK Signaling Pathway in CUMS-Induced Depression Mouse Model. Behav. Brain Res. 2017, 322, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation through NALP3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 Inflammasome Is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Ilekis, J.V.; Tsilou, E.; Fisher, S.; Abrahams, V.M.; Soares, M.J.; Cross, J.C.; Zamudio, S.; Illsley, N.P.; Myatt, L.; Colvis, C.; et al. Placental Origins of Adverse Pregnancy Outcomes: Potential Molecular Targets: An Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Am. J. Obstet. Gynecol. 2016, 215, S1–S46. [Google Scholar] [CrossRef]
- Koga, K.; Mor, G. Toll-Like Receptors at the Maternal-Fetal Interface in Normal Pregnancy and Pregnancy Disorders. Am. J. Reprod. Immunol. 2010, 63, 587–600. [Google Scholar] [CrossRef]
- Cai, Z.; Pan, Z.L.; Pang, Y.; Evans, O.B.; Rhodes, P.G. Cytokine Induction in Fetal Rat Brains and Brain Injury in Neonatal Rats after Maternal Lipopolysaccharide Administration. Pediatr. Res. 2000, 47, 64–72. [Google Scholar] [CrossRef]
- Cierny, J.T.; Unal, E.R.; Flood, P.; Rhee, K.Y.; Praktish, A.; Olson, T.H.; Goetzl, L. Maternal Inflammatory Markers and Term Labor Performance. Am. J. Obstet. Gynecol. 2014, 210, e1–e447. [Google Scholar] [CrossRef]
- Tamura, K.; Ishikawa, G.; Yoshie, M.; Ohneda, W.; Nakai, A.; Takeshita, T.; Tachikawa, E. Glibenclamide Inhibits NLRP3 Inflammasome-Mediated IL-1beta Secretion in Human Trophoblasts. J. Pharmacol. Sci. 2017, 135, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Stodle, G.S.; Silva, G.B.; Tangeras, L.H.; Gierman, L.M.; Nervik, I.; Dahlberg, U.E.; Sun, C.; Aune, M.H.; Thomsen, L.C.V.; Bjorge, L.; et al. Placental Inflammation in Pre-Eclampsia by Nod-Like Receptor Protein (NLRP)3 Inflammasome Activation in Trophoblasts. Clin. Exp. Immunol. 2018, 193, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Arul Nambi Rajan, K.; Khater, M.; Soncin, F.; Pizzo, D.; Moretto-Zita, M.; Pham, J.; Stus, O.; Iyer, P.; Tache, V.; Laurent, L.C.; et al. Sirtuin1 Is Required for Proper Trophoblast Differentiation and Placental Development in Mice. Placenta 2018, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhai, Y.; Zhang, W.; Teng, Y.; Yao, K. Single Nucleotide Polymorphisms of the Sirtuin 1 (SIRT1) Gene Are Associated with Age-Related Macular Degeneration in Chinese Han Individuals: A Case-Control Pilot Study. Medicine 2015, 94, e2238. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Falchi, M.; Olsson, B.; Jacobson, P.; Cauchi, S.; Balkau, B.; Marre, M.; Lantieri, O.; Andersson, J.C.; Jernas, M.; et al. Association of Sirtuin 1 (SIRT1) Gene Snps and Transcript Expression Levels with Severe Obesity. Obesity 2012, 20, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Consortium Converge. Sparse Whole-Genome Sequencing Identifies Two Loci for Major Depressive Disorder. Nature 2015, 523, 588–591. [Google Scholar] [CrossRef]
- Kedenko, L.; Lamina, C.; Kedenko, I.; Kollerits, B.; Kiesslich, T.; Iglseder, B.; Kronenberg, F.; Paulweber, B. Genetic Polymorphisms at SIRT1 and Foxo1 Are Associated with Carotid Atherosclerosis in the Saphir Cohort. BMC Med. Genet. 2014, 15, 112. [Google Scholar] [CrossRef]
- Qiang, L.; Sample, A.; Liu, H.; Wu, X.; He, Y.Y. Epidermal Sirt1 Regulates Inflammation, Cell Migration, and Wound Healing. Sci. Rep. 2017, 7, 14110. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, J.; Yan, C.; Zhou, J.; Lin, R.; Lin, Q.; Wang, W.; Zhang, K.; Yang, G.; Bian, X.; et al. SIRT1 Regulates CD40 Expression Induced by TNF-Alpha Via NF-KB Pathway in Endothelial Cells. Cell. Physiol. Biochem. 2012, 30, 1287–1298. [Google Scholar] [CrossRef]
- Li, Y.; Wang, P.; Yang, X.; Wang, W.; Zhang, J.; He, Y.; Zhang, W.; Jing, T.; Wang, B.; Lin, R. SIRT1 Inhibits Inflammatory Response Partly through Regulation of NLRP3 Inflammasome in Vascular Endothelial Cells. Mol. Immunol. 2016, 77, 148–156. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small Molecule Activators of SIRT1 as Therapeutics for the Treatment of Type 2 Diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin Regulates Cigarette Smoke-Induced Proinflammatory Mediator Release Via RelA/P65 NF-KappaB in Macrophages in Vitro and in Rat Lungs In Vivo: Implications for Chronic Inflammation and Aging. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L567–L576. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 Inhibits Inflammatory Pathways in Macrophages and Modulates Insulin Sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.; He, Y.; Wang, W.; Zhang, J.; Zhang, W.; Jing, T.; Wang, B.; Lin, R. Negative Regulation of NLRP3 Inflammasome by SIRT1 in Vascular Endothelial Cells. Immunobiology 2017, 222, 552–561. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive Oxygen Species at the Crossroads of Inflammasome and Inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative Stress in Placental Pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The Crosstalk between SIRT1 and Keap1/NRF2/ARE Anti-Oxidative Pathway Forms a Positive Feedback Loop to Inhibit FN and TGF-Beta1 Expressions in Rat Glomerular Mesangial Cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 Activator, Ameliorates Aging-Related Progressive Renal Injury. Aging (Albany N. Y.) 2018, 10, 83–99. [Google Scholar] [CrossRef]
- Chen, J.; Lai, J.; Yang, L.; Ruan, G.; Chaugai, S.; Ning, Q.; Chen, C.; Wang, D.W. Trimetazidine Prevents Macrophage-Mediated Septic Myocardial Dysfunction Via Activation of the Histone Deacetylase Sirtuin 1. Br. J. Pharmacol. 2016, 173, 545–561. [Google Scholar] [CrossRef]
- Yoon, D.S.; Choi, Y.; Lee, J.W. Cellular Localization of NRF2 Determines the Self-Renewal and Osteogenic Differentiation Potential of Human MSCS Via the P53-SIRT1 Axis. Cell Death Dis. 2016, 7, e2093. [Google Scholar] [CrossRef]
- Zhou, Z.; You, Z. Mesenchymal Stem Cells Alleviate LPS-Induced Acute Lung Injury in Mice by MiR-142a-5p-Controlled Pulmonary Endothelial Cell Autophagy. Cell. Physiol. Biochem. 2016, 38, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. NRF2 Inhibits NLRP3 Inflammasome Activation through Regulating Trx1/Txnip Complex in Cerebral Ischemia Reperfusion Injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Shin, J.; Bae, J.; Han, D.; Park, S.-R.; Shin, J.; Lee, S.K.; Park, H.-W. SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells 2020, 9, 728. https://doi.org/10.3390/cells9030728
Park S, Shin J, Bae J, Han D, Park S-R, Shin J, Lee SK, Park H-W. SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells. 2020; 9(3):728. https://doi.org/10.3390/cells9030728
Chicago/Turabian StylePark, Sumi, Jiha Shin, Jeongyun Bae, Daewon Han, Seok-Rae Park, Jongdae Shin, Sung Ki Lee, and Hwan-Woo Park. 2020. "SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts" Cells 9, no. 3: 728. https://doi.org/10.3390/cells9030728
APA StylePark, S., Shin, J., Bae, J., Han, D., Park, S.-R., Shin, J., Lee, S. K., & Park, H.-W. (2020). SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells, 9(3), 728. https://doi.org/10.3390/cells9030728