GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness



Abstract
1. Introduction
1.1. Major Depressive Disorder
1.2. Glycogen Synthase Kinase 3β
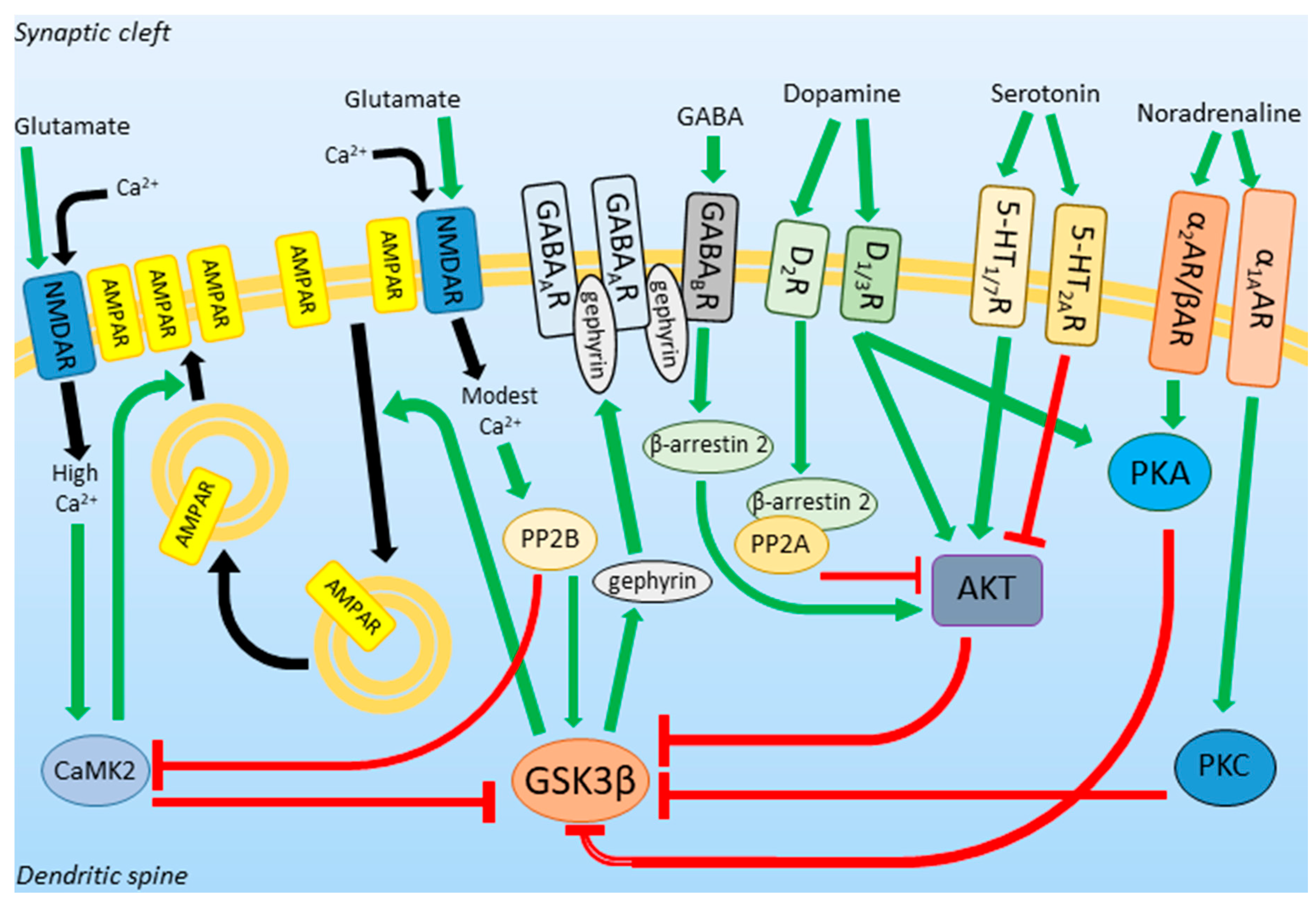
1.3. GSK3β Activity in Neurotransmission and Neuroplasticity
2. GSK3β Expression Profile and Activity in Depression
3. Putative Role of GSK3β in the MDD Pathogenesis
3.1. GSK3β in the Animal Models of Depression
3.2. Inhibitors of GSK3β in Depression
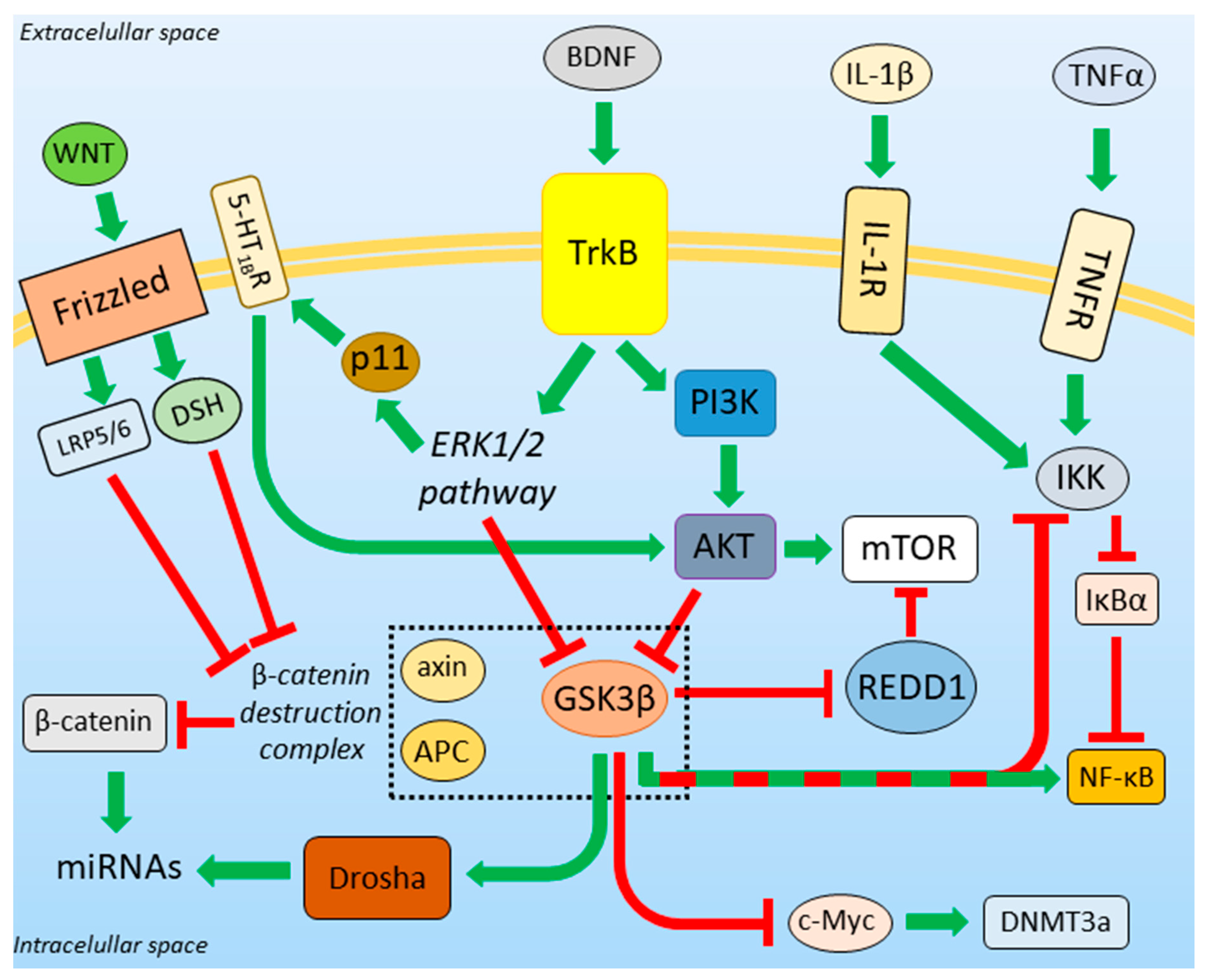
3.3. BDNF-Regulated Action of GSK3β
3.4. GSK3β and the Unfolded Protein Response
3.5. β-Catenin Destruction Complex
3.6. GSK3β-miRNA Interaction
3.7. Role of GSK3β in DNA Methylation
3.8. Neuroinflammation in Depression
4. DA and 5-HT/AKT/GSK3 Pathway Modulation and Its Behavioral Consequences
5. Influence of Anti-Depressants on GSK3β Activity
5.1. Tricyclic Anti-Depressants
5.2. Selective Serotonin Reuptake Inhibitors
5.3. Selective Serotonin and Noradrenaline Reuptake Inhibitors
5.4. α2-Receptor Blockers
5.5. Monoamine Oxidase Inhibitors
5.6. Selective Noradrenaline Reuptake Inhibitors
5.7. Selective Noradrenaline and Dopamine Reuptake Inhibitors
5.8. Melatonin Receptors Agonists
5.9. Trazodone
5.10. Lithium
5.11. Ketamine
5.12. Electroconvulsive Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Kuhn, R. Treatment of depressive states with an iminodibenzyl derivative (G 22355). Schweiz. Med. Wochenschr. 1957, 87, 1135–1140. [Google Scholar] [PubMed]
- Kline, N.S. Clinical experience with iproniazid (marsilid). J. Clin. Exp. Psychopathol. 1958, 19, 72–79. [Google Scholar] [PubMed]
- Schildkraut, J.J. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am. J. Psychiatry 1965, 122, 509–522. [Google Scholar] [CrossRef]
- Jelovac, A.; Kolshus, E.; McLoughlin, D.M. Relapse following successful electroconvulsive therapy for major depression: A meta-analysis. Neuropsychopharmacology 2013, 38, 2467–2474. [Google Scholar] [CrossRef]
- Costemale-Lacoste, J.F.; Guilloux, J.P.; Gaillard, R. The role of GSK-3 in treatment-resistant depression and links with the pharmacological effects of lithium and ketamine: A review of the literature. Encephale 2016, 42, 156–164. [Google Scholar] [CrossRef]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef]
- Yao, H.B.; Shaw, P.C.; Wong, C.C.; Wan, D.C.C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 2002, 23, 291–297. [Google Scholar] [CrossRef]
- Lau, K.F.; Miller, C.C.; Anderton, B.H.; Shaw, P.C. Expression analysis of glycogen synthase kinase-3 in human tissues. J. Pept. Res. 1999, 54, 85–91. [Google Scholar] [CrossRef]
- Hughes, K.; Nikolakaki, E.; Plyte, S.E.; Totty, N.F.; Woodgett, J.R. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993, 12, 803–808. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Hermida, M.A.; Dinesh, K.J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2018, 56, 25–36. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Seger, R.; Vandenheede, J.R.; Krebs, E.G. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J. Biol. Chem. 1995, 270, 987–990. [Google Scholar] [CrossRef]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Wu, C.; Dedhar, S. Integrin-linked kinase (ILK) and its interactors: A new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 2001, 155, 505–510. [Google Scholar] [CrossRef]
- Song, B.; Lai, B.; Zheng, Z.; Zhang, Y.; Luo, J.; Wang, C.; Chen, Y.; Woodgett, J.R.; Li, M. Inhibitory Phosphorylation of GSK-3 by CaMKII Couples Depolarization to Neuronal Survival. J. Biol. Chem. 2010, 285, 41122–41134. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Langa, E.; Cuadros, R.; Avila, J.; Villanueva, N. Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Mol. Cell Biochem. 2010, 344, 211–215. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stefan, M.I.; Le Novère, N. Calcium Input Frequency, Duration and Amplitude Differentially Modulate the Relative Activation of Calcineurin and CaMKII. PLoS ONE 2012, 7, e43810. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, Y.I.; Seo, M.; Kim, S.Y.; Lee, J.E.; Youn, H.D.; Kim, Y.S.; Juhnn, Y.S. Calcineurin dephosphorylates glycogen synthase kinase-3 beta at serine-9 in neuroblast-derived cells. J. Neurochem. 2009, 111, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Bats, C.; Groc, L.; Choquet, D. The Interaction between Stargazin and PSD-95 Regulates AMPA Receptor Surface Trafficking. Neuron 2007, 53, 719–734. [Google Scholar] [CrossRef]
- Wei, J.; Liu, W.; Yan, Z. Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 2010, 285, 26369–26376. [Google Scholar] [CrossRef]
- Choii, G.; Ko, J. Gephyrin: A central GABAergic synapse organizer. Exp. Mol. Med. 2015, 47, e158. [Google Scholar] [CrossRef]
- Tyagarajan, S.K.; Ghosh, H.; Yevenes, G.E.; Nikonenko, I.; Ebeling, C.; Schwerdel, C.; Sidler, C.; Zeilhofer, H.U.; Gerrits, B.; Muller, D.; et al. Regulation of GABAergic synapse formation and plasticity by GSK3β-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. USA 2011, 108, 379–384. [Google Scholar] [CrossRef]
- Lu, F.F.; Su, P.; Liu, F.; Daskalakis, Z.J. Activation of GABA(B) receptors inhibits protein kinase B/glycogen synthase kinase 3 signaling. Mol. Brain 2012, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Barajas, C.; Coronel, I.; Florán, B. Dopamine Receptors and Neurodegeneration. Aging Dis. 2015, 6, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 2012, 37, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, W.; Roh, M.S.; Friedman, A.B.; Rosborough, K.; Jope, R.S. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology 2004, 29, 1426–1431. [Google Scholar] [CrossRef]
- Polter, A.M.; Li, X. Glycogen Synthase Kinase-3 is an Intermediate Modulator of Serotonin Neurotransmission. Front. Mol. Neurosci. 2011, 4, 31. [Google Scholar] [CrossRef]
- Ballou, L.M.; Tian, P.Y.; Lin, H.Y.; Jiang, Y.P.; Lin, R.Z. Dual Regulation of Glycogen Synthase Kinase-3β by the α1A-Adrenergic Receptor. J. Biol. Chem. 2001, 276, 40910–40916. [Google Scholar] [CrossRef]
- Xing, B.; Li, Y.C.; Gao, W.J. Norepinephrine versus dopamine and their interaction in modulating synaptic function in the prefrontal cortex. Brain Res. 2016, 1641, 217–233. [Google Scholar] [CrossRef]
- Morioka, N.; Abe, H.; Araki, R.; Matsumoto, N.; Zhang, F.F.; Nakamura, Y.; Hisaoka-Nakashima, K.; Nakata, Y. A β1/2 Adrenergic Receptor-Sensitive Intracellular Signaling Pathway Modulates CCL2 Production in Cultured Spinal Astrocytes. J. Cell Physiol. 2014, 229, 323–332. [Google Scholar] [CrossRef]
- Jope, R.S. Glycogen Synthase Kinase-3 in the Etiology and Treatment of Mood Disorders. Front. Mol. Neurosci. 2011, 4, 16. [Google Scholar] [CrossRef]
- Karege, F.; Perroud, N.; Burkhardt, S.; Schwald, M.; Ballmann, E.; La Harpe, R.; Malafosse, A. Alteration in Kinase Activity but Not in Protein Levels of Protein Kinase B and Glycogen Synthase Kinase-3β in Ventral Prefrontal Cortex of Depressed Suicide Victims. Biol. Psychiatry 2007, 61, 240–245. [Google Scholar] [CrossRef]
- Karege, F.; Perroud, N.; Burkhardt, S.; Fernandez, R.; Ballmann, E.; La Harpe, R.; Malafosse, A. Protein levels of β-catenin and activation state of glycogen synthase kinase-3β in major depression. A study with postmortem prefrontal cortex. J. Affect. Disord. 2012, 136, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Diniz, B.S.; Talib, L.L.; Giroud, J.H.P.; de Paula, V.R.J.; Gattaz, W.F.; Forlenza, O.V. Platelet GSK3B activity in patients with late-life depression: Marker of depressive episode severity and cognitive impairment? World J. Biol. Psychiatry 2011, 12, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Pláteník, J.; Fišar, Z.; Buchal, R.; Jirák, R.; Kitzlerová, E.; Zvěřová, M.; Raboch, J. GSK3β, CREB, and BDNF in peripheral blood of patients with Alzheimer’s disease and depression. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2014, 50, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Alttoa, A.; Kõiv, K.; Hinsley, T.A.; Brass, A.; Harro, J. Differential gene expression in a rat model of depression based on persistent differences in exploratory activity. Eur. Neuropsychopharmacol 2010, 20, 288–300. [Google Scholar] [CrossRef]
- Höflich, A.; Michenthaler, P.; Kasper, S.; Lanzenberger, R. Circuit Mechanisms of Reward, Anhedonia, and Depression. Int. J. Neuropsychopharmacol 2019, 22, 105–118. [Google Scholar] [CrossRef]
- Wilkinson, M.B.; Dias, C.; Magida, J.; Mazei-Robison, M.; Lobo, M.; Kennedy, P.; Dietz, D.; Covington, H.; Russo, S.; Neve, R.; et al. A novel role of the WNT-dishevelled-GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J. Neurosci. 2011, 31, 9084–9092. [Google Scholar] [CrossRef]
- Crofton, E.J.; Nenov, M.N.; Zhang, Y.; Scala, F.; Page, S.A.; McCue, D.L.; Li, D.; Hommel, J.D.; Laezza, F.; Green, T.A. Glycogen synthase kinase 3 beta alters anxiety-, depression-, and addiction-related behaviors and neuronal activity in the nucleus accumbens shell. Neuropharmacology 2017, 117, 49–60. [Google Scholar] [CrossRef]
- Crofton, E.J.; Zhang, Y.; Green, T.A. Inoculation stress hypothesis of environmental enrichment. Neurosci. Biobehav. Rev. 2015, 49, 19–31. [Google Scholar] [CrossRef]
- Barik, J.; Marti, F.; Morel, C.; Fernandez, S.P.; Lanteri, C.; Godeheu, G.; Tassin, J.P.; Mombereau, C.; Faure, P.; Tronche, F. Chronic stress triggers social aversion via glucocorticoid receptor in dopaminoceptive neurons. Science 2013, 339, 332–335. [Google Scholar] [CrossRef]
- Sugama, S.; Kakinuma, Y. Loss of dopaminergic neurons occurs in the ventral tegmental area and hypothalamus of rats following chronic stress: Possible pathogenetic loci for depression involved in Parkinson’s disease. Neurosci. Res. 2016, 111, 48–55. [Google Scholar] [CrossRef]
- Douma, E.H.; de Kloet, E.R. Stress-induced plasticity and functioning of ventral tegmental dopamine neurons. Neurosci. Biobehav. Rev. 2020, 108, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Wei, Y.M.; Shi, H.S.; Luo, Y.X.; Ding, Z.B.; Xue, Y.X.; Lu, L.; Yu, C.X. Glycogen synthase kinase-3β in the ventral tegmental area mediates diurnal variations in cocaine-induced conditioned place preference in rats. Addict. Biol. 2014, 19, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Han, M.H.; Mazei-Robison, M.; Iñiguez, S.D.; Ables, J.L.; Vialou, V.; Berton, O.; Ghose, S.; Covington, H.E.; Wiley, M.D.; et al. AKT Signaling within the Ventral Tegmental Area Regulates Cellular and Behavioral Responses to Stressful Stimuli. Biol. Psychiatry 2008, 64, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yang, C.; Xu, Y.; Sun, N.; Yang, H.; Liu, J.; Xu, Q.; Shen, Y. Genetic association of the interaction between the BDNF and GSK3B genes and major depressive disorder in a Chinese population. J. Neural Transm. 2010, 117, 393–401. [Google Scholar] [CrossRef]
- Yang, C.; Xu, Y.; Sun, N.; Ren, Y.; Liu, Z.; Cao, X.; Zhang, K. The combined effects of the BDNF and GSK3B genes modulate the relationship between negative life events and major depressive disorder. Brain Res. 2010, 1355, 1–6. [Google Scholar] [CrossRef]
- Saus, E.; Soria, V.; Escaramís, G.; Crespo, J.M.; Valero, J.; Gutiérrez-Zotes, A.; Martorell, L.; Vilella, E.; Menchón, J.M.; Estivill, X.; et al. A haplotype of glycogen synthase kinase 3β is associated with early onset of unipolar major depression. Genes Brain Behav. 2010, 9, 799–807. [Google Scholar] [CrossRef]
- Kwok, J.B.J.; Hallupp, M.; Loy, C.T.; Chan, D.K.Y.; Woo, J.; Mellick, G.D.; Buchanan, D.D.; Silburn, P.A.; Halliday, G.M.; Schofield, P.R. GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann. Neurol. 2005, 58, 829–839. [Google Scholar] [CrossRef]
- Inkster, B.; Nichols, T.E.; Saemann, P.G.; Auer, D.P.; Holsboer, F.; Muglia, P.; Matthews, P.M. Association of GSK3β Polymorphisms With Brain Structural Changes in Major Depressive Disorder. Arch. Gen. Psychiatry 2009, 66, 721–728. [Google Scholar] [CrossRef]
- Liu, S.; Sun, N.; Xu, Y.; Yang, C.; Ren, Y.; Liu, Z.; Cao, X.; Sun, Y.; Xu, Q.; Zhang, K.; et al. Possible Association of the GSK3β Gene with the Anxiety Symptoms of Major Depressive Disorder and P300 Waveform. Genet. Test Mol. Biomarkers 2012, 16, 1382–1389. [Google Scholar] [CrossRef]
- Levchenko, A.; Losenkov, I.S.; Vyalova, N.M.; Simutkin, G.G.; Bokhan, N.A.; Wilffert, B.; Loonen, A.J.; Ivanova, S.A. The functional variant rs334558 of GSK3B is associated with remission in patients with depressive disorders. Pharmgenomics Pers. Med. 2018, 11, 121–126. [Google Scholar] [CrossRef]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: Beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Cade, J.F. Lithium Salts in the Treatment of Psychotic Excitement. Aust. N. Z. J. Psychiatry 1982, 16, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Polter, A.; Beurel, E.; Yang, S.; Garner, R.; Song, L.; Miller, C.A.; Sweatt, J.D.; McMahon, L.; Bartolucci, A.A.; Li, X.; et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 2010, 35, 1761–1774. [Google Scholar] [CrossRef]
- Pardo, M.; Abrial, E.; Jope, R.S.; Beurel, E. GSK3β isoform-selective regulation of depression, memory and hippocampal cell proliferation. Genes Brain Behav. 2016, 15, 348–355. [Google Scholar] [CrossRef]
- Omata, N.; Chiu, C.T.; Moya, P.R.; Leng, Y.; Wang, Z.; Hunsberger, J.G.; Leeds, P.; Chuang, D.M. Lentivirally mediated GSK-3β silencing in the hippocampal dentate gyrus induces antidepressant-like effects in stressed mice. Int. J. Neuropsychopharmacol 2011, 14, 711–717. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Harper, A.D.; Jové, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 2004, 24, 6791–6798. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Huang, J.; Buccafusca, R.; Garskof, J.; Valvezan, A.J.; Berry, G.T.; Klein, P.S. Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Invest. 2011, 121, 3756–3762. [Google Scholar] [CrossRef]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef]
- Cheng, Y.; Desse, S.; Martinez, A.; Worthen, R.J.; Jope, R.S.; Beurel, E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav. Immun. 2018, 69, 556–567. [Google Scholar] [CrossRef]
- Liu, R.J.; Fuchikami, M.; Dwyer, J.M.; Lepack, A.E.; Duman, R.S.; Aghajanian, G.K. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 2013, 38, 2268–2277. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci. Rep. 2019, 9, 18045. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.G.; Eldar-Finkelman, H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Drevets, W.C.; Price, J.L.; Simpson, J.R.; Todd, R.D.; Reich, T.; Vannier, M.; Raichle, M.E. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 1997, 386, 824–827. [Google Scholar] [CrossRef]
- Rajkowska, G.; Miguel-Hidalgo, J.J.; Wei, J.; Dilley, G.; Pittman, S.D.; Meltzer, H.Y.; Overholser, J.C.; Roth, B.L.; Stockmeier, C.A. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 1999, 45, 1085–1098. [Google Scholar] [CrossRef]
- Hajszan, T.; Dow, A.; Warner-Schmidt, J.L.; Szigeti-Buck, K.; Sallam, N.L.; Parducz, A.; Leranth, C.; Duman, R.S. Remodeling of Hippocampal Spine Synapses in the Rat Learned Helplessness Model of Depression. Biol. Psychiatry 2009, 65, 392–400. [Google Scholar] [CrossRef][Green Version]
- Morales-Medina, J.C.; Juarez, I.; Venancio-García, E.; Cabrera, S.N.; Menard, C.; Yu, W.; Flores, G.; Mechawar, N.; Quirion, R. Impaired structural hippocampal plasticity is associated with emotional and memory deficits in the olfactory bulbectomized rat. Neuroscience 2013, 236, 233–243. [Google Scholar] [CrossRef]
- Videbech, P.; Ravnkilde, B. Hippocampal volume and depression: A meta-analysis of MRI studies. Am. J. Psychiatry 2004, 161, 1957–1966. [Google Scholar] [CrossRef]
- Radley, J.J.; Rocher, A.B.; Miller, M.; Janssen, W.G.M.; Liston, C.; Hof, P.R.; McEwen, B.S.; Morrison, J.H. Repeated Stress Induces Dendritic Spine Loss in the Rat Medial Prefrontal Cortex. Cereb. Cortex 2006, 16, 313–320. [Google Scholar] [CrossRef]
- Izquierdo, A.; Wellman, C.L.; Holmes, A. Brief Uncontrollable Stress Causes Dendritic Retraction in Infralimbic Cortex and Resistance to Fear Extinction in Mice. J. Neurosci. 2006, 26, 5733–5738. [Google Scholar] [CrossRef]
- Vyas, A.; Jadhav, S.; Chattarji, S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience 2006, 143, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Warren, B.L.; Sial, O.K.; Alcantara, L.F.; Greenwood, M.A.; Brewer, J.S.; Rozofsky, J.P.; Parise, E.M.; Bolaños-Guzmán, C.A. Altered Gene Expression and Spine Density in Nucleus Accumbens of Adolescent and Adult Male Mice Exposed to Emotional and Physical Stress. Dev. Neurosci. 2014, 36, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.K.; Huang, K.W.; Grueter, B.A.; Rothwell, P.E.; Malenka, R.C. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 2012, 487, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of depression: Insights from human and rodent studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef]
- Pham, K.; Nacher, J.; Hof, P.R.; McEwen, B.S. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur. J. Neurosci. 2003, 17, 879–886. [Google Scholar] [CrossRef]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef]
- Aznar, S.; Klein, A.B.; Santini, M.A.; Knudsen, G.M.; Henn, F.; Gass, P.; Vollmayr, B. Aging and depression vulnerability interaction results in decreased serotonin innervation associated with reduced BDNF levels in hippocampus of rats bred for learned helplessness. Synapse 2010, 64, 561–565. [Google Scholar] [CrossRef]
- Lepack, A.E.; Fuchikami, M.; Dwyer, J.M.; Banasr, M.; Duman, R.S. BDNF Release Is Required for the Behavioral Actions of Ketamine. Int. J. Neuropsychopharmacol 2015, 18, pyu033. [Google Scholar] [CrossRef]
- Berton, O.; McClung, C.A.; Dileone, R.J.; Krishnan, V.; Renthal, W.; Russo, S.J.; Graham, D.; Tsankova, N.M.; Bolanos, C.A.; Rios, M.; et al. Essential Role of BDNF in the Mesolimbic Dopamine Pathway in Social Defeat Stress. Science 2006, 311, 864–868. [Google Scholar] [CrossRef]
- Krishnan, V.; Han, M.H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; LaPlant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef]
- Mai, L.; Jope, R.S.; Li, X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J. Neurochem. 2002, 82, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Hsuan, S.L.; Habas, A.; Higgins, M.J.; Xia, Z. ERK1/2 Antagonizes Glycogen Synthase Kinase-3β-induced Apoptosis in Cortical Neurons. J. Biol. Chem. 2002, 277, 49577–49584. [Google Scholar] [CrossRef] [PubMed]
- Cuesto, G.; Jordán-Álvarez, S.; Enriquez-Barreto, L.; Ferrús, A.; Morales, M.; Acebes, Á. GSK3β Inhibition Promotes Synaptogenesis in Drosophila and Mammalian Neurons. PLoS ONE 2015, 10, e0118475. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Myers, K.R.; Yu, K.; Wise, A.; De Blas, A.L.; Hartzell, H.C.; Zheng, J.Q. Activity-dependent regulation of dendritic growth and maintenance by glycogen synthase kinase 3β. Nat. Commun. 2013, 4, 2628. [Google Scholar] [CrossRef] [PubMed]
- Fuster-Matanzo, A.; Llorens-Martín, M.; Sirerol-Piquer, M.S.; García-Verdugo, J.M.; Avila, J.; Hernández, F. Dual effects of increased glycogen synthase kinase-3β activity on adult neurogenesis. Hum. Mol. Genet. 2013, 22, 1300–1315. [Google Scholar] [CrossRef]
- Zhang, K.; Song, X.; Xu, Y.; Li, X.; Liu, P.; Sun, N.; Zhao, X.; Liu, Z.; Xie, Z.; Peng, J. Continuous GSK-3β overexpression in the hippocampal dentate gyrus induces prodepressant-like effects and increases sensitivity to chronic mild stress in mice. J. Affect. Disord. 2013, 146, 45–52. [Google Scholar] [CrossRef]
- Warner-Schmidt, J.L.; Chen, E.Y.; Zhang, X.; Marshall, J.J.; Morozov, A.; Svenningsson, P.; Greengard, P. A role for p11 in the antidepressant action of brain-derived neurotrophic factor. Biol. Psychiatry 2010, 68, 528–535. [Google Scholar] [CrossRef]
- Svenningsson, P.; Greengard, P. p11 (S100A10)—An inducible adaptor protein that modulates neuronal functions. Curr. Opin. Pharmacol. 2007, 7, 27–32. [Google Scholar] [CrossRef]
- Svenningsson, P.; Chergui, K.; Rachleff, I.; Flajolet, M.; Zhang, X.; El Yacoubi, M.; Vaugeois, J.M.; Nomikos, G.G.; Greengard, P. Alterations in 5-HT1B Receptor Function by p11 in Depression-Like States. Science 2006, 311, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.T.; Liu, R.J.; Voleti, B.; Maldonado-Aviles, J.G.; Duric, V.; Iwata, M.; Dutheil, S.; Duman, C.; Boikess, S.; Lewis, D.A.; et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med. 2014, 20, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, C.S.; Goswami, D.B.; Austin, M.C.; Iyo, A.H.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2011, 35, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.; Liu, E.; Knutzen, C.A.; Lang, E.S.; Lombardo, C.R.; Sankar, S.; Toth, J.I.; Petroski, M.D.; Ronai, Z.; Chiang, G.G. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A–DDB1 ubiquitin ligase. EMBO Rep. 2009, 10, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Whitney, M.L.; Jefferson, L.S.; Kimball, S.R. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem. Biophys. Res. Commun. 2009, 379, 451–455. [Google Scholar] [CrossRef]
- Meares, G.P.; Mines, M.A.; Beurel, E.; Eom, T.Y.; Song, L.; Zmijewska, A.A.; Jope, R.S. Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER) stress-induced CHOP expression in neuronal cells. Exp. Cell Res. 2011, 317, 1621–1628. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Timberlake, M., II; Dwivedi, Y. Linking unfolded protein response to inflammation and depression: Potential pathologic and therapeutic implications. Mol. Psychiatry 2019, 24, 987–994. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Bown, C.; Wang, J.F.; MacQueen, G.; Young, L.T. Increased Temporal Cortex ER Stress Proteins in Depressed Subjects Who Died by Suicide. Neuropsychopharmacology 2000, 22, 327–332. [Google Scholar] [CrossRef]
- Nijholt, D.A.T.; Nölle, A.; van Haastert, E.S.; Edelijn, H.; Toonen, R.F.; Hoozemans, J.J.M.; Scheper, W. Unfolded protein response activates glycogen synthase kinase-3 via selective lysosomal degradation. Neurobiol. Aging 2013, 34, 1759–1771. [Google Scholar] [CrossRef]
- Kambe, Y.; Miyata, A. Potential involvement of the mitochondrial unfolded protein response in depressive-like symptoms in mice. Neurosci. Lett. 2015, 588, 166–171. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Feng, J.; Sun, H.; Shao, N.; Mazei-Robison, M.S.; Damez-Werno, D.; Scobie, K.; Bagot, R.; LaBonté, B.; Ribeiro, E.; et al. β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature 2014, 516, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.D.; Einat, H.; O’Donnell, K.C.; Picchini, A.M.; Schloesser, R.J.; Manji, H.K. β-Catenin Overexpression in the Mouse Brain Phenocopies Lithium-Sensitive Behaviors. Neuropsychopharmacology 2007, 32, 2173–2183. [Google Scholar] [CrossRef]
- Kimelman, D.; Xu, W. β-Catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef]
- Metcalfe, C.; Bienz, M. Inhibition of GSK3 by Wnt signalling—Two contrasting models. J. Cell Sci. 2011, 124, 3537–3544. [Google Scholar] [CrossRef]
- Im, H.I.; Kenny, P.J. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012, 35, 325–334. [Google Scholar] [CrossRef]
- Tang, X.; Li, M.; Tucker, L.; Ramratnam, B. Glycogen Synthase Kinase 3 Beta (GSK3β) Phosphorylates the RNAase III Enzyme Drosha at S300 and S302. PLoS ONE 2011, 6, e20391. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, F.; Liu, Y.; Liu, X.; Ai, Z.; Guo, Z.; Zhang, Y. GSK3 inhibitors CHIR99021 and 6-bromoindirubin-3′-oxime inhibit microRNA maturation in mouse embryonic stem cells. Sci. Rep. 2015, 5, 8666. [Google Scholar] [CrossRef]
- Gheysarzadeh, A.; Sadeghifard, N.; Afraidooni, L.; Pooyan, F.; Mofid, M.R.; Valadbeigi, H.; Bakhtiari, H.; Keikhavani, S. Serum-based microRNA biomarkers for major depression: MiR-16, miR-135a, and miR-1202. J. Res. Med. Sci. 2018, 23, 69. [Google Scholar] [CrossRef]
- Baudry, A.; Mouillet-Richard, S.; Schneider, B.; Launay, J.M.; Kellermann, O. miR-16 targets the serotonin transporter: A new facet for adaptive responses to antidepressants. Science 2010, 329, 1537–1541. [Google Scholar] [CrossRef]
- Issler, O.; Haramati, S.; Paul, E.D.; Maeno, H.; Navon, I.; Zwang, R.; Gil, S.; Mayberg, H.S.; Dunlop, B.W.; Menke, A.; et al. MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron 2014, 83, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Arborelius, L.; Owens, M.J.; Plotsky, P.M.; Nemeroff, C.B. The role of corticotropin-releasing factor in depression and anxiety disorders. J. Endocrinol. 1999, 160, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Raadsheer, F.C.; Hoogendijk, W.J.G.; Stam, F.C.; Tilders, F.J.H.; Swaab, D.F. Increased Numbers of Corticotropin-Releasing Hormone Expressing Neurons in the Hypothalamic Paraventricular Nucleus of Depressed Patients. Neuroendocrinology 1994, 60, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Ezra-Nevo, G.; Regev, L.; Neufeld-Cohen, A.; Chen, A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 2010, 13, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- LaPlant, Q.; Vialou, V.; Covington, H.E.; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iñiguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kim, J.M.; Lee, J.Y.; Kim, S.Y.; Bae, K.Y.; Kim, S.W.; Shin, I.S.; Kim, H.R.; Shin, M.G.; Yoon, J.S. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord. 2013, 151, 679–685. [Google Scholar] [CrossRef]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011, 505607. [Google Scholar]
- Pyko, I.V.; Nakada, M.; Sabit, H.; Teng, L.; Furuyama, N.; Hayashi, Y.; Kawakami, K.; Minamoto, T.; Fedulau, А.S.; Hamada, J. Glycogen synthase kinase 3β inhibition sensitizes human glioblastoma cells to temozolomide by affecting O 6 -methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis 2013, 34, 2206–2217. [Google Scholar] [CrossRef]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor Antagonist Infliximab for Treatment-Resistant Depression. JAMA Psychiatry 2013, 70, 31. [Google Scholar] [CrossRef]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of Anti-inflammatory Treatment on Depression, Depressive Symptoms, and Adverse Effects. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kubera, M.; Obuchowicz, E.; Goehler, L.; Brzeszcz, J.; Maes, M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2011, 35, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Hodes, G.E.; Pfau, M.L.; Leboeuf, M.; Golden, S.A.; Christoffel, D.J.; Bregman, D.; Rebusi, N.; Heshmati, M.; Aleyasin, H.; Warren, B.L.; et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl. Acad. Sci. USA 2014, 111, 16136–16141. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, J.L.; Tarr, A.J.; Wohleb, E.S.; Godbout, J.P.; Mo, X.; Sheridan, J.F.; Eubank, T.D.; Marsh, C.B. Prolonged Restraint Stress Increases IL-6, Reduces IL-10, and Causes Persistent Depressive-Like Behavior That Is Reversed by Recombinant IL-10. PLoS ONE 2013, 8, e58488. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Russo, S.J.; Ferguson, D.; Nestler, E.J.; Duman, R.S. Nuclear factor- B is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. USA 2010, 107, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; zu Schwabedissen, L.; et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflammation 2011, 8, 94. [Google Scholar] [CrossRef]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of Translocator Protein Density, a Marker of Neuroinflammation, in the Brain During Major Depressive Episodes. JAMA Psychiatry 2015, 72, 268. [Google Scholar] [CrossRef]
- Vallières, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [CrossRef]
- Russo, S.J.; Nestler, E.J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [Google Scholar] [CrossRef]
- Nagy, C.; Suderman, M.; Yang, J.; Szyf, M.; Mechawar, N.; Ernst, C.; Turecki, G. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol. Psychiatry 2015, 20, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; McKim, D.B.; Shea, D.T.; Powell, N.D.; Tarr, A.J.; Sheridan, J.F.; Godbout, J.P. Re-establishment of Anxiety in Stress-Sensitized Mice Is Caused by Monocyte Trafficking from the Spleen to the Brain. Biol. Psychiatry 2014, 75, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Harrington, L.E.; Jope, R.S. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol. Psychiatry 2013, 73, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Harrison, S.C. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef]
- Karin, M. How NF-κB is activated: The role of the IκB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef]
- Cortés-Vieyra, R.; Bravo-Patiño, A.; Valdez-Alarcón, J.J.; Juárez, M.C.; Finlay, B.B.; Baizabal-Aguirre, V.M. Role of glycogen synthase kinase-3 beta in the inflammatory response caused by bacterial pathogens. J. Inflamm. 2012, 9, 23. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflammation 2009, 6, 9. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Cheng, Y.; Pardo, M.; Armini, R.S.; Martinez, A.; Mouhsine, H.; Zagury, J.F.; Jope, R.S.; Beurel, E. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav. Immun. 2016, 53, 207–222. [Google Scholar] [CrossRef]
- Foster, J.; McVey Neufeld, K.A. Gut–brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Urs, N.M.; Snyder, J.C.; Jacobsen, J.P.R.; Peterson, S.M.; Caron, M.G. Deletion of GSK3β in D2R-expressing neurons reveals distinct roles for β-arrestin signaling in antipsychotic and lithium action. Proc. Natl. Acad. Sci. USA 2012, 109, 20732–20737. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Sotnikova, T.D.; Gainetdinov, R.R.; Caron, M.G. Paradoxical Striatal Cellular Signaling Responses to Psychostimulants in Hyperactive Mice. J. Biol. Chem. 2006, 281, 32072–32080. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Lao, C.L.; Chen, J.C. Dual alteration of limbic dopamine D1 receptor-mediated signalling and the Akt/GSK3 pathway in dopamine D3 receptor mutants during the development of methamphetamine sensitization. J. Neurochem. 2007, 100, 225–241. [Google Scholar] [CrossRef]
- Bychkov, E.; Ahmed, M.R.; Dalby, K.N.; Gurevich, E.V. Dopamine depletion and subsequent treatment with l-DOPA, but not the long-lived dopamine agonist pergolide, enhances activity of the Akt pathway in the rat striatum. J. Neurochem. 2007, 102, 699–711. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/β-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Sotnikova, T.D.; Yao, W.D.; Kockeritz, L.; Woodgett, J.R.; Gainetdinov, R.R.; Caron, M.G. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. USA 2004, 101, 5099–5104. [Google Scholar] [CrossRef]
- Gould, T.D.; Einat, H.; Bhat, R.; Manji, H.K. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol 2004, 7, 387–390. [Google Scholar] [CrossRef]
- Chang, P.K.; Chu, J.; Tsai, Y.T.; Lai, Y.H.; Chen, J.C. Dopamine D3 receptor and GSK3β signaling mediate deficits in novel object recognition memory within dopamine transporter knockdown mice. J. Biomed. Sci. 2020, 27, 16. [Google Scholar] [CrossRef]
- Prickaerts, J.; Moechars, D.; Cryns, K.; Lenaerts, I.; van Craenendonck, H.; Goris, I.; Daneels, G.; Bouwknecht, J.A.; Steckler, T. Transgenic Mice Overexpressing Glycogen Synthase Kinase 3beta: A Putative Model of Hyperactivity and Mania. J. Neurosci. 2006, 26, 9022–9029. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Zhang, X.; Rodriguiz, R.M.; Sotnikova, T.D.; Cools, M.J.; Wetsel, W.C.; Gainetdinov, R.R.; Caron, M.G. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. USA 2008, 105, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.S.; Eom, T.Y.; Zmijewska, A.A.; De Sarno, P.; Roth, K.A.; Jope, R.S. Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: Protection by mood stabilizers and imipramine. Biol. Psychiatry 2005, 57, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar] [CrossRef]
- Cusack, B.; Nelson, A.; Richelson, E. Binding of antidepressants to human brain receptors: Focus on newer generation compounds. Psychopharmacology 1994, 114, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Solich, J.; Kolasa, M.; Kuśmider, M.; Faron-Górecka, A.; Pabian, P.; Szafran, K.; Żurawek, D.; Dziedzicka-Wasylewska, M. Effect of desipramine on gene expression in the mouse frontal cortex—Microarray study. Pharm. Rep. 2015, 67, 345–348. [Google Scholar] [CrossRef]
- Cong, W.N.; Chadwick, W.; Wang, R.; Daimon, C.M.; Cai, H.; Amma, J.; Wood, W.H.; Becker, K.G.; Martin, B.; Maudsley, S. Amitriptyline Improves Motor Function via Enhanced Neurotrophin Signaling and Mitochondrial Functions in the Murine N171-82Q Huntington Disease Model. J. Biol. Chem. 2015, 290, 2728–2743. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Chan, C.B.; Weinshenker, D.; Hall, R.A.; Xiao, G.; Ye, K. Amitriptyline is a TrkA and TrkB Receptor Agonist that Promotes TrkA/TrkB Heterodimerization and Has Potent Neurotrophic Activity. Chem. Biol. 2009, 16, 644–656. [Google Scholar] [CrossRef]
- Reisi, P.; Eidelkhani, N.; Rafiee, L.; Kazemi, M.; Radahmadi, M.; Alaei, H. Effects of doxepin on gene expressions of Bcl-2 family, TNF-α, MAP kinase 14, and Akt1 in the hippocampus of rats exposed to stress. Res. Pharm. Sci. 2017, 12, 15. [Google Scholar] [CrossRef]
- Eidelkhani, N.; Radahmadi, M.; Kazemi, M.; Rafiee, L.; Alaei, H.; Reisi, P. Effects of doxepin on brain-derived neurotrophic factor, tumor necrosis factor alpha, mitogen-activated protein kinase 14, and AKT1 genes expression in rat hippocampus. Adv. Biomed. Res. 2015, 4, 203. [Google Scholar]
- Bu, J.; Zu, H. Mechanism underlying the effects of doxepin on β-amyloid -induced memory impairment in rats. Iran J. Basic Med. Sci. 2017, 20, 1044–1049. [Google Scholar] [PubMed]
- Rao, T.; Cler, J.; Mick, S.; Dilworth, V.; Contrepas, P.; Iyengar, S.; Wood, P. Neurochemical characterization of dopaminergic effects of opipramol, a potent sigma receptor ligand, in vivo. Neuropharmacology 1990, 29, 1191–1197. [Google Scholar] [CrossRef]
- Möller, H.J.; Volz, H.P.; Reimann, I.W.; Stoll, K.D. Opipramol for the treatment of generalized anxiety disorder: A placebo-controlled trial including an alprazolam-treated group. J. Clin. Psychopharmacol. 2001, 21, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Shinoda, Y.; Yamamoto, Y.; Sasaki, Y.; Miyajima, K.; Tagashira, H.; Fukunaga, K. Stimulation of the Sigma-1 Receptor by DHEA Enhances Synaptic Efficacy and Neurogenesis in the Hippocampal Dentate Gyrus of Olfactory Bulbectomized Mice. PLoS ONE 2013, 8, e60863. [Google Scholar] [CrossRef]
- Albayrak, Y.; Hashimoto, K. Sigma-1 Receptor Agonists and Their Clinical Implications in Neuropsychiatric Disorders. Adv. Exp. Med. Biol. 2017, 964, 153–161. [Google Scholar]
- Polter, A.M.; Yang, S.; Jope, R.S.; Li, X. Functional significance of glycogen synthase kinase-3 regulation by serotonin. Cell. Signal. 2012, 24, 265–271. [Google Scholar] [CrossRef]
- Chen, Y.C.; Tan, Q.R.; Dang, W.; Wang, H.N.; Zhang, R.B.; Li, Z.Y.; Lin, H.; Liu, R. The effect of citalopram on chronic stress-induced depressive-like behavior in rats through GSK3β/β-catenin activation in the medial prefrontal cortex. Brain Res. Bull. 2012, 88, 338–344. [Google Scholar] [CrossRef]
- Ren, Q.G.; Wang, Y.J.; Gong, W.G.; Xu, L.; Zhang, Z.J. Escitalopram Ameliorates Tau Hyperphosphorylation and Spatial Memory Deficits Induced by Protein Kinase A Activation in Sprague Dawley Rats. J. Alzheimer’s Dis. 2015, 47, 61–71. [Google Scholar] [CrossRef]
- Muneer, A. Wnt and GSK3 Signaling Pathways in Bipolar Disorder: Clinical and Therapeutic Implications. Clin. Psychopharmacol. Neurosci. 2017, 15, 100. [Google Scholar] [CrossRef]
- Gassen, N.C.; Hartmann, J.; Zannas, A.S.; Kretzschmar, A.; Zschocke, J.; Maccarrone, G.; Hafner, K.; Zellner, A.; Kollmannsberger, L.K.; Wagner, K.V.; et al. FKBP51 inhibits GSK3β and augments the effects of distinct psychotropic medications. Mol. Psychiatry 2016, 21, 277–289. [Google Scholar] [CrossRef]
- Coppell, A.L.; Pei, Q.; Zetterström, T.S.C. Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology 2003, 44, 903–910. [Google Scholar] [CrossRef]
- Reddy, K.K.; Lefkove, B.; Chen, L.B.; Govindarajan, B.; Carracedo, A.; Velasco, G.; Carrillo, C.O.; Bhandarkar, S.S.; Owens, M.J.; Mechta-Grigoriou, F.; et al. The antidepressant sertraline downregulates Akt and has activity against melanoma cells. Pigment. Cell Melanoma Res. 2008, 21, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Robert, F.; Sukarieh, R.; Michnick, S.; Pelletier, J. The Antidepressant Sertraline Inhibits Translation Initiation by Curtailing Mammalian Target of Rapamycin Signaling. Cancer Res. 2010, 70, 3199–3208. [Google Scholar] [CrossRef] [PubMed]
- Jesinkey, S.R.; Korrapati, M.C.; Rasbach, K.A.; Beeson, C.C.; Schnellmann, R.G. Atomoxetine prevents dexamethasone-induced skeletal muscle atrophy in mice. J. Pharmacol. Exp. Ther. 2014, 351, 663–673. [Google Scholar] [CrossRef]
- Shadfar, S.; Kim, Y.G.; Katila, N.; Neupane, S.; Ojha, U.; Bhurtel, S.; Srivastav, S.; Jeong, G.S.; Park, P.H.; Hong, J.T.; et al. Neuroprotective Effects of Antidepressants via Upregulation of Neurotrophic Factors in the MPTP Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, B.M.; Motaghinejad, M.; Motevalian, M.; Gholami, M. Duloxetine by Modulating the Akt/GSK3 Signaling Pathways Has Neuroprotective Effects against Methamphetamine-Induced Neurodegeneration and Cognition Impairment in Rats. Iran J. Med. Sci. 2019, 44, 146–154. [Google Scholar]
- Anttila, S.A.; Leinonen, E.V. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001, 7, 249–264. [Google Scholar] [CrossRef]
- Engel, D.; Zomkowski, A.D.E.; Lieberknecht, V.; Rodrigues, A.L.; Gabilan, N.H. Chronic administration of duloxetine and mirtazapine downregulates proapoptotic proteins and upregulates neurotrophin gene expression in the hippocampus and cerebral cortex of mice. J. Psychiatr. Res. 2013, 47, 802–808. [Google Scholar] [CrossRef]
- Couch, Y.; Anthony, D.C.; Dolgov, O.; Revischin, A.; Festoff, B.; Santos, A.I.; Steinbusch, H.W.; Strekalova, T. Microglial activation, increased TNF and SERT expression in the prefrontal cortex define stress-altered behaviour in mice susceptible to anhedonia. Brain Behav. Immun. 2013, 29, 136–146. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Gainetdinov, R.R.; Caron, M.G. Akt/GSK3 Signaling in the Action of Psychotropic Drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 327–347. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, J.G.; Seo, M.K.; Lee, C.H.; Cho, H.Y.; Lee, B.J.; Seol, W.; Kim, Y.H. Differential effects of antidepressant drugs on mTOR signalling in rat hippocampal neurons. Int. J. Neuropsychopharmacol. 2014, 17, 1831–1846. [Google Scholar] [CrossRef]
- Chen, M.J.; Russo-Neustadt, A.A. Running exercise- and antidepressant-induced increases in growth and survival-associated signaling molecules are IGF-dependent. Growth Factors 2007, 25, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Liu, Y.; Wang, H.; Yuan, L.; Luo, Z. Moclobemide up-regulates proliferation of hippocampal progenitor cells in chronically stressed mice. Acta Pharmacol. Sin. 2004, 25, 1408–1412. [Google Scholar] [PubMed]
- Mines, M.A.; Jope, R.S. Brain region differences in regulation of Akt and GSK3 by chronic stimulant administration in mice. Cell. Signal. 2012, 24, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Hadj Ayed Tka, K.; Mahfoudh, B.A.; Zaouali, M.A.; Kammoun, R.; Bejaoui, M.; Ghoul, M.S.; Rosello, C.J.; Ben, A.H. Melatonin Modulates Endoplasmic Reticulum Stress and Akt/GSK3-Beta Signaling Pathway in a Rat Model of Renal Warm Ischemia Reperfusion. Anal. Cell. Pathol. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, S.L.; Warren, C. Agomelatine, a melatonin agonist with antidepressant properties. Expert Opin. Investig. Drugs 2009, 18, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Zappelli, E.; Martini, C. Trazodone regulates neurotrophic/growth factors, mitogen-activated protein kinases and lactate release in human primary astrocytes. J. Neuroinflammation 2015, 12, 225. [Google Scholar] [CrossRef]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1668. [Google Scholar] [CrossRef]
- Ryves, W.J.; Harwood, A.J. Lithium Inhibits Glycogen Synthase Kinase-3 by Competition for Magnesium. Biochem. Biophys. Res. Commun. 2001, 280, 720–725. [Google Scholar] [CrossRef]
- Chalecka-Franaszek, E.; Chuang, D.M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 8745–8750. [Google Scholar] [CrossRef]
- De Sarno, P.; Li, X.; Jope, R.S. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 2002, 43, 1158–1164. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Marion, S.; Rodriguiz, R.M.; Medvedev, I.O.; Sotnikova, T.D.; Ghisi, V.; Wetsel, W.C.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. A β-arrestin 2 Signaling Complex Mediates Lithium Action on Behavior. Cell 2008, 132, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Tyler, M.W.; Yourish, H.B.; Ionescu, D.F.; Haggarty, S.J. Classics in Chemical Neuroscience: Ketamine. ACS Chem. Neurosci. 2017, 8, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Li, N.; Liu, R.J.; Duric, V.; Aghajanian, G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012, 62, 35–41. [Google Scholar] [CrossRef]
- Homayoun, H.; Moghaddam, B. NMDA Receptor Hypofunction Produces Opposite Effects on Prefrontal Cortex Interneurons and Pyramidal Neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef]
- Jourdi, H.; Hsu, Y.T.; Zhou, M.; Qin, Q.; Bi, X.; Baudry, M. Positive AMPA Receptor Modulation Rapidly Stimulates BDNF Release and Increases Dendritic mRNA Translation. J. Neurosci. 2009, 29, 8688–8697. [Google Scholar] [CrossRef]
- Réus, G.Z.; Vieira, F.G.; Abelaira, H.M.; Michels, M.; Tomaz, D.B.; dos Santos, M.A.B.; Carlessi, A.S.; Neotti, M.V.; Matias, B.I.; Luz, J.R.; et al. MAPK signaling correlates with the antidepressant effects of ketamine. J. Psychiatr. Res. 2014, 55, 15–21. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Amadei, C.; Downey, K.; Jope, R.S. Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar Disord. 2016, 18, 473–480. [Google Scholar] [CrossRef]
- Beurel, E.; Song, L.; Jope, R.S. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol. Psychiatry 2011, 16, 1068–1070. [Google Scholar] [CrossRef]
- Basar, K.; Eren-Kocak, E.; Ozdemir, H.; Ertugrul, A. Effects of Acute and Chronic Electroconvulsive Shocks on Glycogen Synthase Kinase 3β Level and Phosphorylation in Mice. J. ECT 2013, 29, 261–266. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Anti-Depressants Class | Class Members | Targets | Effects on GSK3β Pathway |
|---|---|---|---|
| Tricyclic anti-depressants (TCA) | Imipramine, desipramine, clomipramine, amitriptyline, protriptyline, doxepin, dosulepin, opipramol | Nonspecific Serotonin transporter inhibitors, noradrenaline transporter inhibitors, anti-serotoninergic, D2R blockers, anti-cholinergic, anti-adrenergic, anti-histaminic, sigma receptors agonists/antagonists | Prevent AKT inhibition through blockade of 5-HT2AR, enhance GSK3β inhibition via PKA, PKC, TrkB/PI3K/AKT, and σ1R |
| Selective serotonin reuptake inhibitors (SSRI) | Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline | Mainly serotonin transporter inhibitors, sigma receptors agonists/antagonists | Enhance GSK3β inhibition via 5-HT1/7R/PI3K/AKT, upregulate BDNF expression |
| Selective serotonin and noradrenaline reuptake inhibitors (SSNRI) | Atomoxetine, desvenlafaxine, duloxetine, levomilnacipran, milnacipran, sibutramine, tramadol, venlafaxine | Serotonin and noradrenaline transporters inhibitors | Increase AKT activity and expression, downregulate GSK3α/β expression |
| α2-receptor blockers | Mianserin, mirtazapine | Anti-adrenergic, anti-serotoninergic, anti-histaminic, noradrenaline transporter inhibitors | Upregulate BDNF expression, downregulate 5-HT2AR expression, activate PI3K/AKT/GSK3β |
| Monoamine oxidase inhibitors (MAOi) | Isocarboxazid, tranylcypromine, moclobemide, toloxatone, rasagiline, selegiline | Reversible or irreversible inhibition of MAO-A and MAO-B | Upregulate BDNF expression, increase AKT activity |
| Selective noradrenaline reuptake inhibitors (SNRI) | Reboxetine, viloxazine, maprotiline | Noradrenaline transporter inhibitors | Upregulate BDNF expression, increase AKT activity |
| Selective noradrenaline and dopamine reuptake inhibitors (SNDRI) | Amineptine, bupropion, dexmethylphenidate, methylphenidate, phenylpiracetam | Noradrenaline and dopamine transporters inhibitors | Increase AKT activity, enhance GSK3β inhibition |
| Melatonin receptor agonists | Ramelteon, agomelatine, tasimelteon | Activate melatonin receptors | Increase AKT activity, enhance GSK3β inhibition, stimulate BDNF release |
| Trazodone | 5-HTR agonist/antagonist, serotonin transporter inhibitor, anti-adrenergic, anti-histaminic | Enhance GSK3β inhibition via 5-HT1AR stimulation and 5-HT2AR blockade | |
| Lithium | A lot of targets Directly activates AKT, directly inhibits GSK3α/β, destabilizes β-arrestin 2/AKT/PP2A complex | Increase AKT activity, enhance GSK3β inhibition | |
| Ketamine | A lot of targets NMDAR antagonist, indirect agonist of AMPAR, opioid receptors antagonist, D2R agonist, sigma receptors agonist, anti-cholinergic, cholinesterase inhibitor, 5-HT/NA/DA reuptake inhibitor, blocker of voltage-dependent sodium and calcium channels, nitric oxide synthase inhibitor | Enhance GSK3β inhibition via TrkB/MEK/ERK and TrkB/PI3K/AKT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duda, P.; Hajka, D.; Wójcicka, O.; Rakus, D.; Gizak, A. GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness. Cells 2020, 9, 727. https://doi.org/10.3390/cells9030727
Duda P, Hajka D, Wójcicka O, Rakus D, Gizak A. GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness. Cells. 2020; 9(3):727. https://doi.org/10.3390/cells9030727
Chicago/Turabian StyleDuda, Przemysław, Daria Hajka, Olga Wójcicka, Dariusz Rakus, and Agnieszka Gizak. 2020. "GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness" Cells 9, no. 3: 727. https://doi.org/10.3390/cells9030727
APA StyleDuda, P., Hajka, D., Wójcicka, O., Rakus, D., & Gizak, A. (2020). GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness. Cells, 9(3), 727. https://doi.org/10.3390/cells9030727