Liraglutide Protects Against Brain Amyloid-β1–42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation

 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
2. Results
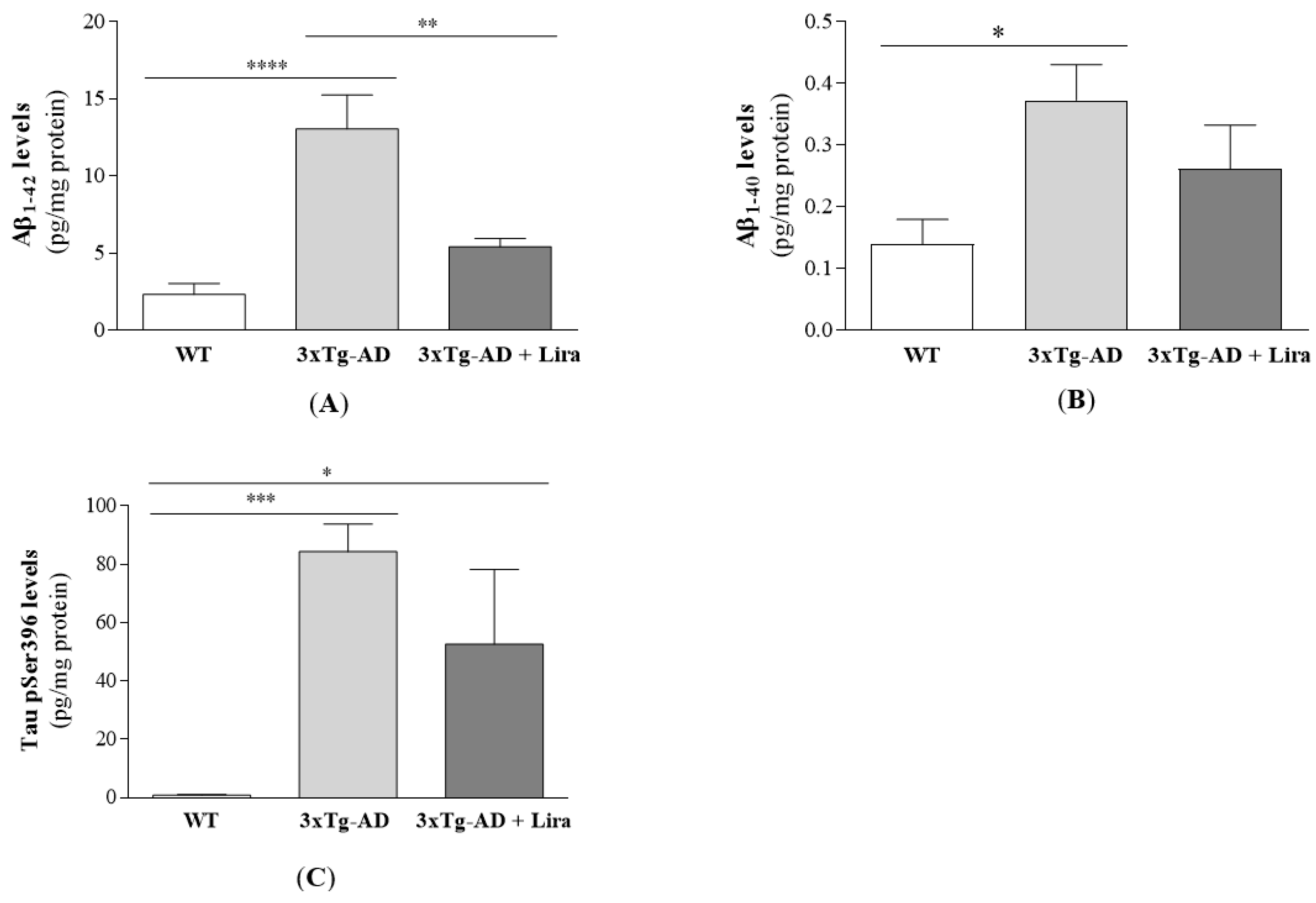
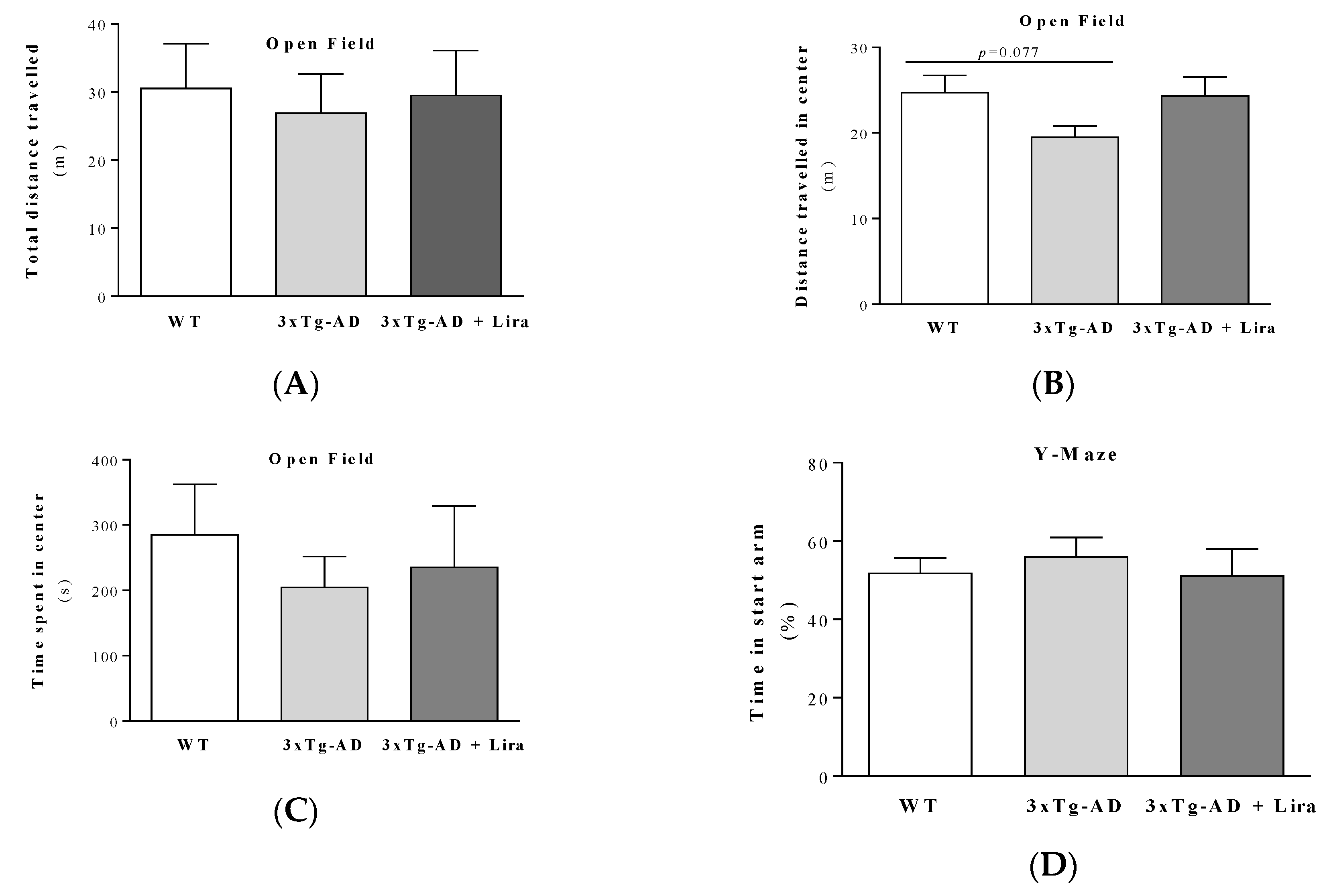
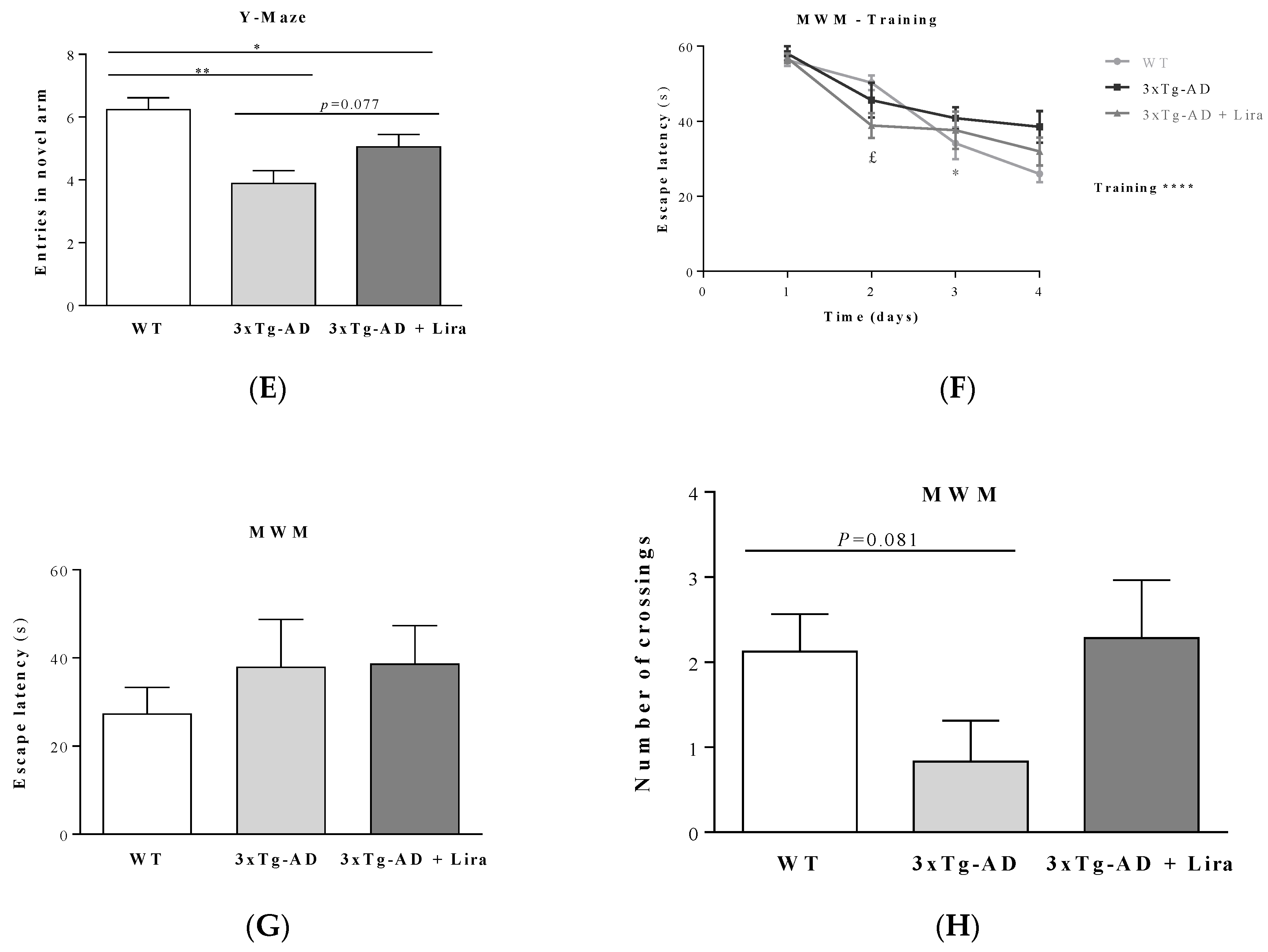
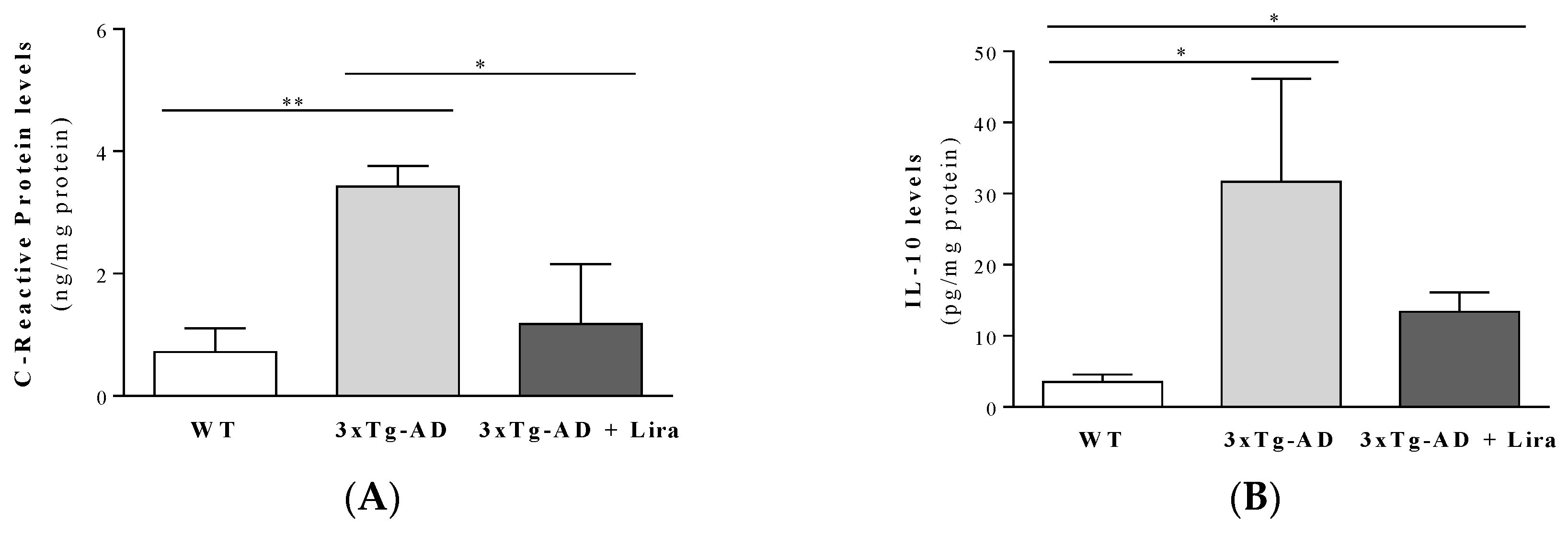
2.1. Effect of Liraglutide Treatment on Brain and Peripheral Features in Female Mice
2.2. Liraglutide Partially Normalizes Brain Levels of Estradiol and GLP-1-Related Signaling in Female Mice with Early AD-Like Pathology
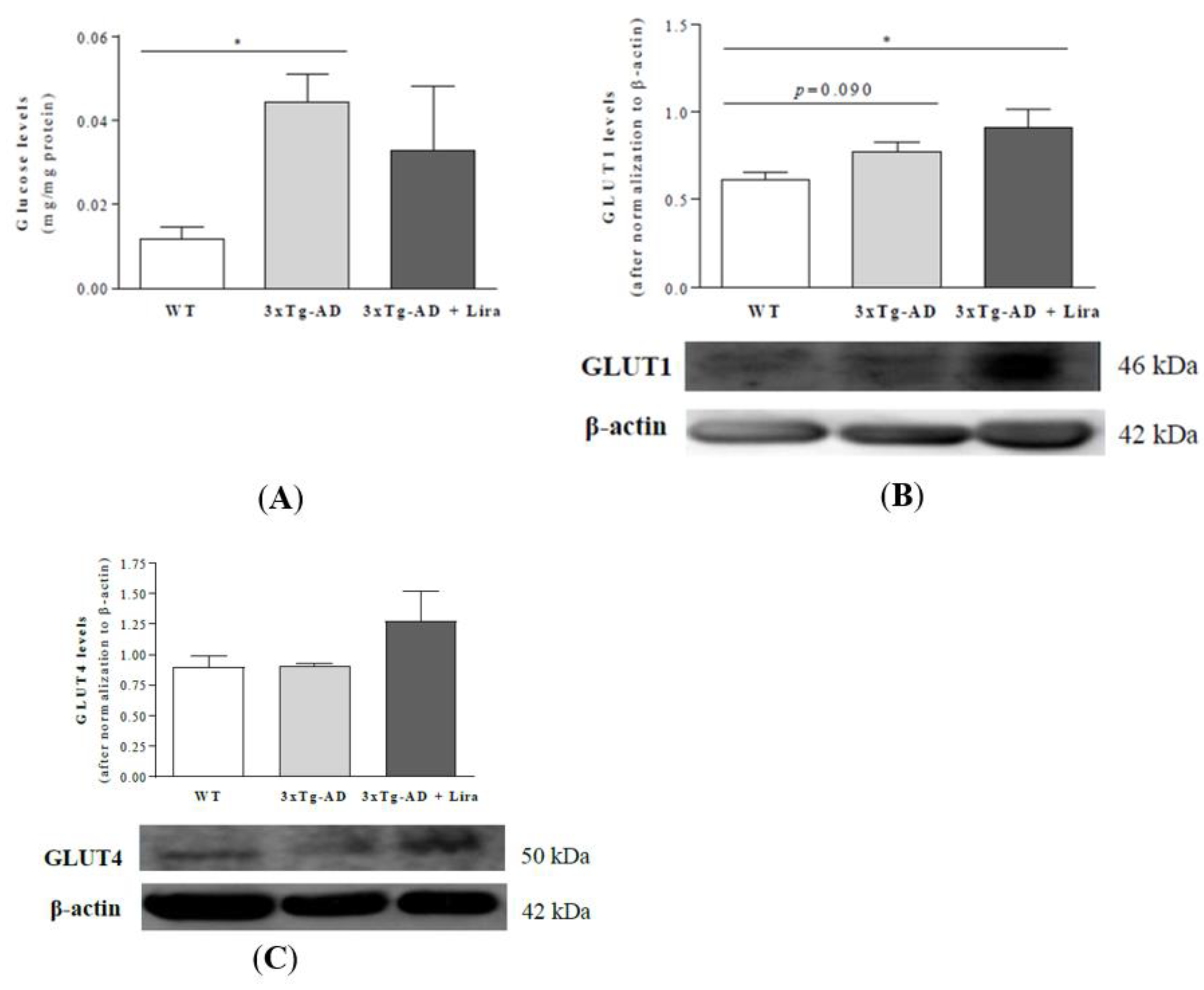
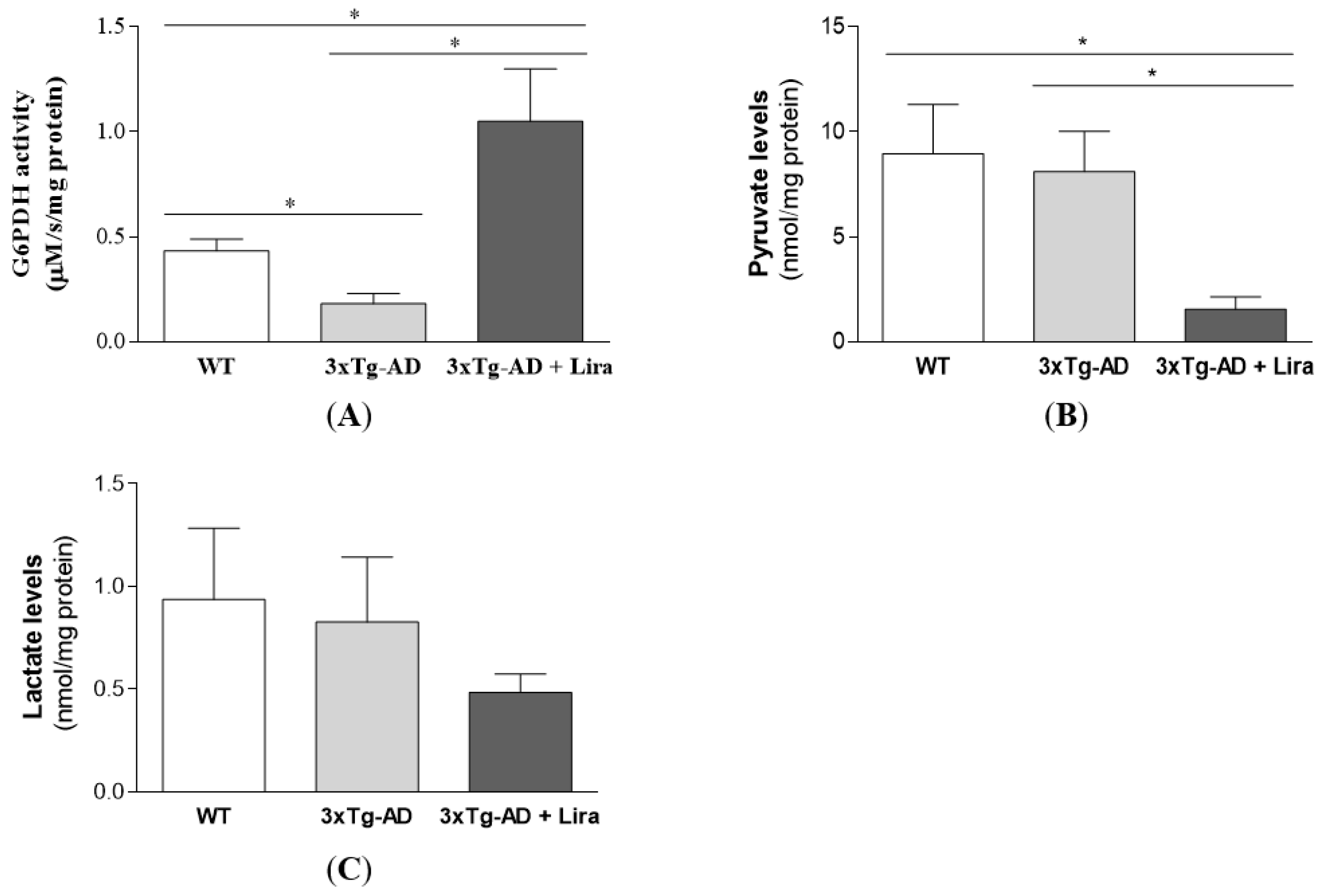
2.3. Liraglutide Promotes Brain Glucose Metabolism via the Oxidative Branch of the Pentose Phosphate Pathway in Female Mice with Early AD-Like Pathology
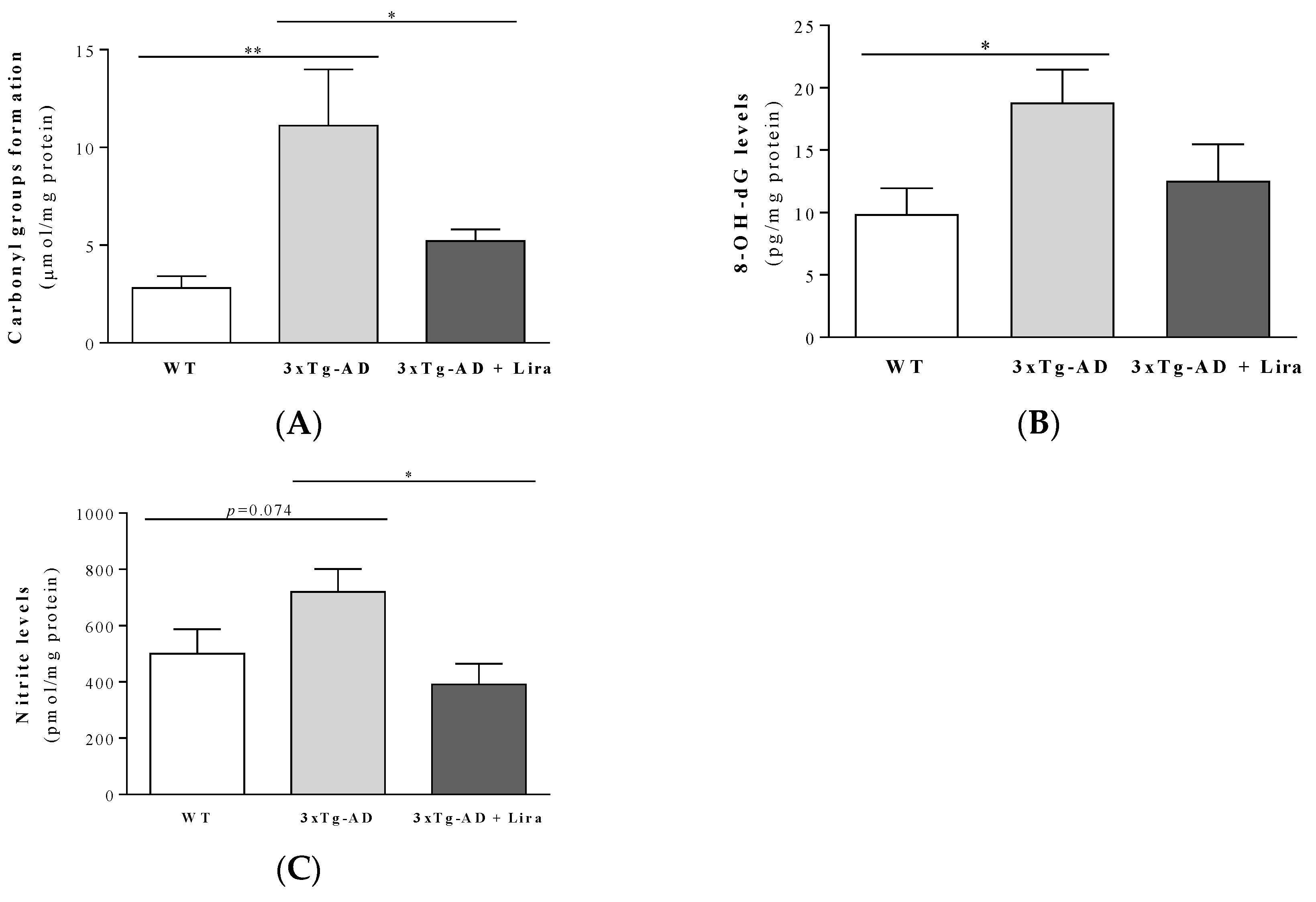
2.4. Liraglutide Partially Rescues Brain Oxidative/Nitrosative Stress Markers in Female Mice with Early AD-Like Pathology
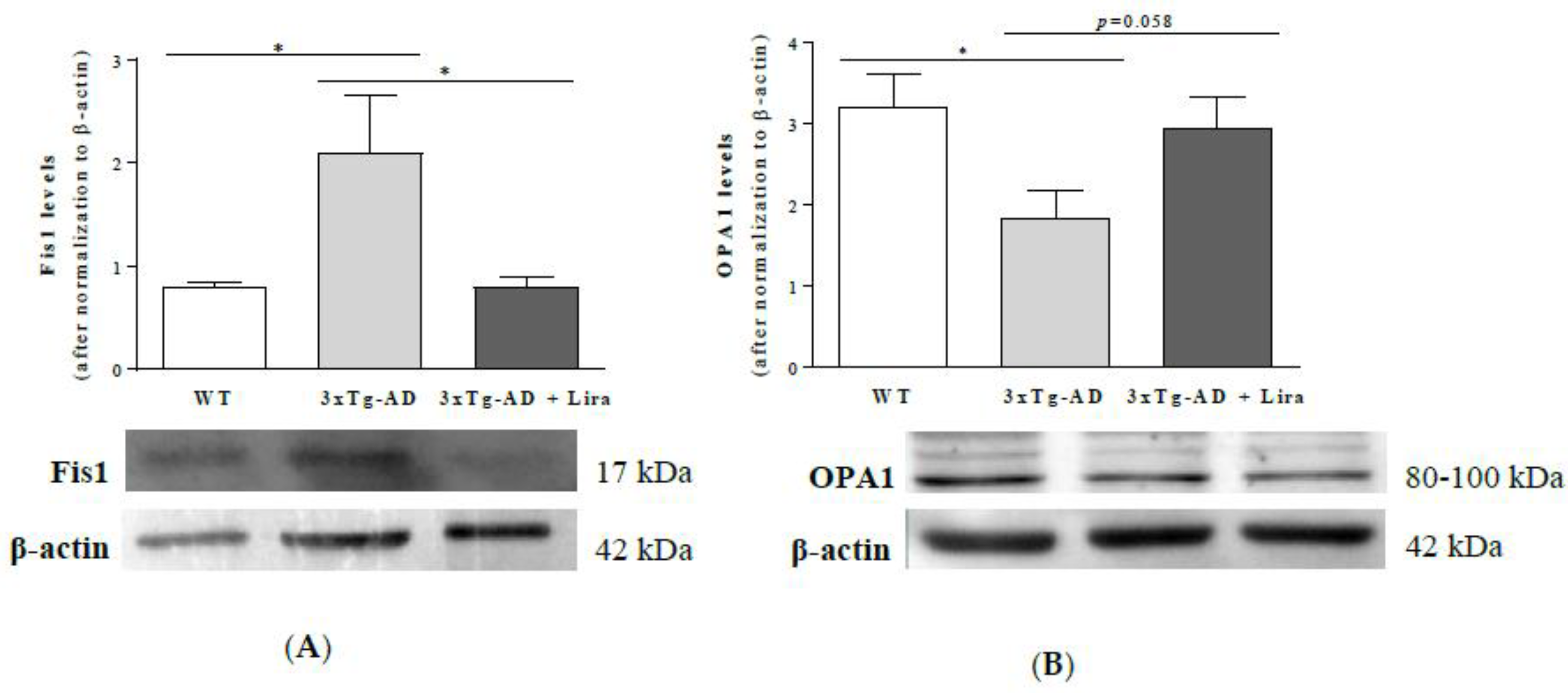
2.5. Liraglutide Partially Attenuates the Altered Mitochondrial Fission/Fusion Proteins in Female Mice with Early AD-Like Pathology
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Animal Housing and Treatment
4.3. Body and Brain Weight
4.4. Collection of Peripheral Blood and Routine Biochemical Analyses
4.5. Isolation and Preparation of Brain Cortical Homogenates
4.6. Evaluation of AD Pathological Hallmarks
4.7. Behavioral Analyses
4.7.1. Open Field Behavior Test
4.7.2. Y-maze Behavior Test
4.7.3. Morris Water Maze Test
4.8. Evaluation of Inflammation Markers
4.9. Evaluation of Brain Cortical Hormones’ Levels
4.10. Assessment of Brain Cortical PKA Activity
4.11. Assessment of Brain Cortical Glucose Levels
4.12. Determination of Brain Markers for Glycolysis and Pentose Phosphate Pathway
4.13. Evaluation of Oxidative/Nitrosative Stress Markers
4.14. Western Blot Analyses
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Lopera, F. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. J. Am. Med. Assoc. 1997, 277, 793–799. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Gray, S.; Kawas, C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 1998, 88, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. J. Am. Med. Assoc. 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Sperling, R.; Mormino, E.; Johnson, K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 2014, 84, 608–622. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Guyara-Quinn, C.; McHugh, P.; Petrongolo, G.; Osorio, R.S.; Connaughty, C.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS ONE 2017, 12, e0185926. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Varsavsky, I.; Osorio, R.S.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging. Neurology 2017, 89, 1382–1390. [Google Scholar] [CrossRef]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef]
- Mosconi, L.; Rahman, A.; Diaz, I.; Wu, X.; Scheyer, O.; Hristov, H.W.; Vallabhajosula, S.; Isaacson, R.S.; de Leon, M.J.; Brinton, R.D. Increased Alzheimer’s risk during the menopause transition: A 3-year longitudinal brain imaging study. PLoS ONE 2018, 13, e0207885. [Google Scholar] [CrossRef]
- Laughlin, G.A.; Kritz-Silverstein, D.; Barrett-Connor, E. Higher endogenous estrogens predict four year decline in verbal fluency in postmenopausal women: The rancho Bernardo study. Clin. Endocrinol. 2010, 72, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Cui, J.; Jothishankar, B.; Shen, J.; He, P.; Shen, Y. Early reproductive experiences in females make differences in cognitive function later in life. J. Alzheimers Dis. 2013, 34, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Heys, M.; Jiang, C.; Cheng, K.K.; Zhang, W.; Au Yeung, S.L.; Lam, T.H.; Leung, G.M.; Schooling, C.M. Lifelong endogenous estrogen exposure and later adulthood cognitive function in a population of naturally postmenopausal women from southern China: The Guangzhou biobank cohort study. Psychoneuroendocrinology 2011, 36, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Cammarata, S.; Assini, A.; Croce, R.; Clerici, F.; Novello, C.; Mazzella, L.; Dagnino, N.; Mariani, C.; Tanganelli, P. The number of pregnancies is a risk factor for Alzheimer’s disease. Eur. J. Neurol. 2006, 13, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Sobow, T.; Kloszewska, I. Parity, number of pregnancies, and the age of onset of Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 120–121. [Google Scholar] [CrossRef]
- Aragno, M.; Mastrocola, R.; Brignardello, E.; Catalano, M.; Robino, G.; Manti, R.; Parola, M.; Danni, O.; Boccuzzi, G. Dehydroepiandrosterone modulates nuclear factor-kappaB activation in hippocampus of diabetic rats. Endocrinology 2002, 143, 3250–3258. [Google Scholar] [CrossRef]
- Ptok, U.; Barkow, K.; Heun, R. Fertility and number of children in patients with Alzheimer’s disease. Arch. Womens Ment. Health 2002, 5, 83–86. [Google Scholar] [CrossRef]
- Camkurt, M.A.; Lavagnino, L.; Zhang, X.Y.; Teixeira, A.L. Liraglutide for psychiatric disorders: Clinical evidence and challenges. Horm. Mol. Biol. Clin. Investig. 2018, 36. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Cedazo-Minguez, A.; Kenigsberg, P.A.; Page, G.; Duarte, A.I.; Giusti, P.; Zusso, M.; Robert, P.; Frisoni, G.B.; Cattaneo, A.; et al. Current and emerging avenues for Alzheimer’s disease drug targets. J. Intern. Med. 2019, 286, 398–437. [Google Scholar] [CrossRef]
- Duarte, A.I.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Brain insulin signalling, glucose metabolism and females’ reproductive aging: A dangerous triad in Alzheimer’s disease. Neuropharmacology 2018, 136, 223–242. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, P.; Chepurny, O.G.; Holz, G.G. Regulation of glucose homeostasis by GLP-1. Prog. Mol. Biol. Transl. Sci. 2014, 121, 23–65. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Patterson, S.; Porter, D.; Gault, V.A.; Holscher, C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J. Neurosci. Res. 2011, 89, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Z.F.; Holscher, C.; Gao, C.; Jiang, Y.H.; Liu, Y.Z. (Val(8)) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur. J. Pharmacol. 2012, 674, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Gault, V.A.; Holscher, C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur. J. Pharmacol. 2008, 587, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, J.; Zhao, G.; Guo, A.; Chen, Y.; Fu, R.; Deng, Y. Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of Tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem. Res. 2017, 42, 2326–2335. [Google Scholar] [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, J.Z.; Xu, X.Y.; Hu, J.; Xu, T.; Jin, S.; Yang, S.J.; Wang, J.Z. Liraglutide ameliorates hyperhomocysteinemia-induced Alzheimer-like pathology and memory deficits in rats via multi-molecular targeting. Neurosci. Bull. 2019, 35, 724–734. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- Han, W.N.; Holscher, C.; Yuan, L.; Yang, W.; Wang, X.H.; Wu, M.N.; Qi, J.S. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol. Aging 2013, 34, 576–588. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.; Holscher, C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, S.; Coley, N.; Lovestone, S.; Aisen, P.S.; Vellas, B. Prevention of sporadic Alzheimer’s disease: Lessons learned from clinical trials and future directions. Lancet Neurol. 2015, 14, 926–944. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Pierpoint, L. Photodynamic therapy based on 5-aminolevulinic acid and its use as an antimicrobial Agent. Med. Res. Rev. 2012, 32, 1292–1327. [Google Scholar] [CrossRef]
- Stuss, D.T.; Knight, R.T. Principles of Frontal Lobe Function, 2nd ed.; Oxford University Press: New York, NY, USA, 2013. [Google Scholar]
- Carvalho, C.; Machado, N.; Mota, P.C.; Correia, S.C.; Cardoso, S.; Santos, R.X.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Type 2 diabetic and Alzheimer’s disease mice present similar behavioural, cognitive, and vascular anomalies. J. Alzheimers Dis. 2013, 35, 623–635. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold. Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- De Luigi, A.; Pizzimenti, S.; Quadri, P.; Lucca, U.; Tettamanti, M.; Fragiacomo, C.; De Simoni, M.G. Peripheral inflammatory response in Alzheimer’s disease and multiinfarct dementia. Neurobiol. Dis. 2002, 11, 308–314. [Google Scholar] [CrossRef]
- Yang, J.T.; Wang, Z.J.; Cai, H.Y.; Yuan, L.; Hu, M.M.; Wu, M.N.; Qi, J.S. Sex differences in neuropathology and cognitive behaviour in APP/PS1/tau triple-transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 2018, 34, 736–746. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Gillette-Guyonnet, S.; Nourhashemi, F.; Andrieu, S.; de Glisezinski, I.; Ousset, P.J.; Riviere, D.; Albarede, J.L.; Vellas, B. Weight loss in Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 637S–642S. [Google Scholar] [CrossRef] [PubMed]
- Raji, C.A.; Lopez, O.L.; Kuller, L.H.; Carmichael, O.T.; Becker, J.T. Age, Alzheimer disease, and brain structure. Neurology 2009, 73, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, C.; Hernandez, B.; Burgos, C.F.; Fuentes, E.; Palomo, I.; Alarcon, M. The cAMP/PKA pathway inhibits beta-amyloid peptide release from human platelets. Neuroscience 2019, 397, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Glucose transporters in brain in health and in Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1. [Google Scholar] [CrossRef]
- Resende, R.; Moreira, P.I.; Proenca, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S401–S412. [Google Scholar] [CrossRef]
- Xiong, H.; Zheng, C.; Wang, J.; Song, J.; Zhao, G.; Shen, H.; Deng, Y. The neuroprotection of liraglutide on Alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J. Alzh. Dis. 2013, 37, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, J.; Ma, D.; Zhang, M.; Hu, S.; Shao, S.; Gong, C.X. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J. Alzh. Dis. 2013, 37, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Kongsbak-Wismann, P.; Schlumberger, C.; Jelsing, J.; Terwel, D.; Termont, A.; Pyke, C.; Knudsen, L.B.; et al. Long-term treatment with liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, has no effect on β-amyloid plaque load in two transgenic APP/PS1 mouse models of Alzheimer’s disease. PLoS ONE 2016, 11, e0158205. [Google Scholar] [CrossRef] [PubMed]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Lee, V.M.Y.; Leight, S.; Varga, I.; Otvos, L., Jr. Unique Alzheimer’s disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry 1997, 36, 8114–8124. [Google Scholar] [CrossRef]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef]
- Candeias, E.; Duarte, A.I.; Sebastiao, I.; Fernandes, M.A.; Placido, A.I.; Carvalho, C.; Correia, S.; Santos, R.X.; Seica, R.; Santos, M.S.; et al. Middle-aged diabetic females and males present distinct susceptibility to Alzheimer disease-like pathology. Mol. Neurobiol. 2017, 54, 6471–6489. [Google Scholar] [CrossRef]
- Valencak, T.G.; Osterrieder, A.; Schulz, T.J. Sex matters: The effects of biological sex on adipose tissue biology and energy metabolism. Redox Biol. 2017, 12, 806–813. [Google Scholar] [CrossRef]
- Goyal, M.S.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.; Morris, J.C.; Raichle, M.E.; Vlassenko, A.G. Persistent metabolic youth in the aging female brain. Proc. Natl. Acad. Sci. USA 2019, 116, 3251–3255. [Google Scholar] [CrossRef]
- Yan, H.; Yang, W.; Zhou, F.; Li, X.; Pan, Q.; Shen, Z.; Han, G.; Newell-Fugate, A.; Tian, Y.; Majeti, R.; et al. Estrogen improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor Foxo1. Diabetes 2019, 68, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, S.; Sasahara, K.; Shikimi, H.; Honda, S.-i.; Harada, N.; Tsutsui, K. Estradiol promotes Purkinje dendritic growth, spinogenesis, and synaptogenesis during neonatal life by inducing the expression of BDNF. Cerebellum 2011, 11, 416–417. [Google Scholar] [CrossRef]
- Dave, K.A.; Platel, J.-C.; Huang, F.; Tian, D.; Stamboulian-Platel, S.; Bordey, A. Prostaglandin E2 induces glutamate release from subventricular zone astrocytes. Neuron Glia Biol. 2010, 6, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Yang, L. Neuronal cAMP/PKA signaling and energy homeostasis. Adv. Exp. Med. Biol. 2018, 1090, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Lerche, S.; Egefjord, L.; Brock, B.; Moller, N.; Vang, K.; Rodell, A.B.; Bibby, B.M.; Holst, J.J.; Rungby, J.; et al. Glucagon-like peptide-1 (GLP-1) raises blood-brain glucose transfer capacity and hexokinase activity in human brain. Front. Neuroenergetics 2013, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Egefjord, L.; Lerche, S.; Vang, K.; Bibby, B.M.; Holst, J.J.; Mengel, A.; Moller, N.; Rungby, J.; Brock, B.; et al. Glucagon-like peptide-1 decreases intracerebral glucose content by activating hexokinase and changing glucose clearance during hyperglycemia. J. Cereb. Blood Flow Metab. 2012, 32, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Cova, I.; Clerici, F.; Rossi, A.; Cucumo, V.; Ghiretti, R.; Maggiore, L.; Pomati, S.; Galimberti, D.; Scarpini, E.; Mariani, C.; et al. Weight loss predicts progression of mild cognitive impairment to Alzheimer’s disease. PLoS ONE 2016, 11, e0151710. [Google Scholar] [CrossRef]
- Duarte, A.I.; Sjogren, M.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I.; Bjorkqvist, M. Dual therapy with liraglutide and ghrelin promotes brain and peripheral energy metabolism in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2018, 8, 8961. [Google Scholar] [CrossRef]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Niehoff, M.L.; Morley, J.E.; Jelsing, J.; Pyke, C.; Knudsen, L.B.; Farr, S.A.; Vrang, N. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J. Alzh. Dis. 2015, 46, 877–888. [Google Scholar] [CrossRef]
- Rollins, C.P.E.; Gallino, D.; Kong, V.; Ayranci, G.; Devenyi, G.A.; Germann, J.; Chakravarty, M.M. Contributions of a high-fat diet to Alzheimer’s disease-related decline: A longitudinal behavioural and structural neuroimaging study in mouse models. Neuroimage Clin. 2018, 21, 101606. [Google Scholar] [CrossRef]
- Cardoso, S.; Seica, R.; Moreira, P.I. Diabesity and brain energy metabolism: The case of Alzheimer’s Disease. Adv. Neurobiol. 2017, 19, 117–150. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. The risk of dementia among persons with diabetes mellitus: A population-based cohort study. Ann. N. Y. Acad. Sci. 1997, 826, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; John, C.S.; Wilkins, H.M.; Wang, X.; Weidling, I.; Thyfault, J.P.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Alzheimer’s disease subjects exhibit impaired systemic glucose metabolism following a mixed meal. Alzh. Dement. 2018, 14, 1159–1160. [Google Scholar] [CrossRef]
- Kilander, L.; Boberg, M.; Lithell, H. Peripheral glucose metabolism and insulin sensitivity in Alzheimer’s disease. Acta Neurol. Scand. 1993, 87, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: Study protocol for a randomised controlled trial (ELAD study). Trials 2019, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Nordberg, A.; Rinne, J.O.; Kadir, A.; Långström, B. The use of PET in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 78–87. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J. Neurochem. 2009, 111, 242–249. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Tsui, W.H.; Herholz, K.; Pupi, A.; Drzezga, A.; Lucignani, G.; Reiman, E.M.; Holthoff, V.; Kalbe, E.; Sorbi, S.; et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J. Nucl. Med. 2008, 49, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Szadujkis-Szadurska, K.; Węclewicz, M.M.; Malinowski, B.; Matusiak, G.; Walczak, M.; Wódkiewicz, E.; Grześk, G.; Pawlak-Osińska, K. The role of Rho-kinase and calcium ions in constriction triggered by ET-1. Microvasc. Res. 2018, 119, 84–90. [Google Scholar] [CrossRef]
- Wiciński, M.; Socha, M.; Walczak, M.; Wódkiewicz, E.; Malinowski, B.; Rewerski, S.; Górski, K.; Pawlak-Osińska, K. Beneficial effects of resveratrol administration—Focus on potential biochemical mechanisms in cardiovascular conditions. Nutrients 2018, 10, 1813. [Google Scholar] [CrossRef]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain glucose transfer in Alzheimer’s disease: Effect of GLP-1 analog treatment. Sci. Rep. 2017, 7, 17490. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Hernandez-Garzón, E.; Fernandez, A.M.; Perez-Alvarez, A.; Genis, L.; Bascuñana, P.; de la Rosa, R.F.; Delgado, M.; Pozo, M.A.; Moreno, E.; McCormick, P.J.; et al. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia 2016, 64, 1962–1971. [Google Scholar] [CrossRef]
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 2016, 165, 882–895. [Google Scholar] [CrossRef]
- Xiao-Yun, X.; Zhao-Hui, M.; Ke, C.; Hong-Hui, H.; Yan-Hong, X. Glucagon-like peptide-1 improves proliferation and differentiation of endothelial progenitor cells via upregulating VEGF generation. Med. Sci. Monit. 2011, 17, BR35–BR41. [Google Scholar] [CrossRef]
- Madadi, G.; Dalvi, P.S.; Belsham, D.D. Regulation of brain insulin mRNA by glucose and glucagon-like peptide 1. Biochem. Biophys. Res. Commun. 2008, 376, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctot, K.L.; Kohler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, M.; Sykora, P.; Scheibye-Knudsen, M.; Dunn, C.; Kasmer, C.; Zhang, Y.; Becker, K.G.; Croteau, D.L.; Bohr, V.A. Sporadic Alzheimer disease fibroblasts display an oxidative stress phenotype. Free Radic. Biol. Med. 2012, 53, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Moreira, P.I.; Harris, P.L.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimers Dis. 2007, 12, 195–206. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic. Biol. Med. 2008, 45, 81–85. [Google Scholar] [CrossRef]
- Calabrese, V.; Sultana, R.; Scapagnini, G.; Guagliano, E.; Sapienza, M.; Bella, R.; Kanski, J.; Pennisi, G.; Mancuso, C.; Stella, A.M.; et al. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid. Redox Signal. 2006, 8, 1975–1986. [Google Scholar] [CrossRef]
- Baker, S.K.; Chen, Z.L.; Norris, E.H.; Revenko, A.S.; MacLeod, A.R.; Strickland, S. Blood-derived plasminogen drives brain inflammation and plaque deposition in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E9687–E9696. [Google Scholar] [CrossRef]
- Choi, S.; Won, J.S.; Carroll, S.L.; Annamalai, B.; Singh, I.; Singh, A.K. Pathology of nNOS-expressing GABAergic neurons in mouse model of Alzheimer’s disease. Neuroscience 2018, 384, 41–53. [Google Scholar] [CrossRef]
- Placido, A.I.; Oliveira, C.R.; Moreira, P.I.; Pereira, C.M. Enhanced amyloidogenic processing of amyloid precursor protein and cell death under prolonged endoplasmic reticulum stress in brain endothelial cells. Mol. Neurobiol. 2015, 51, 571–590. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. Reimagining Alzheimer’s disease—An age-based hypothesis. J. Neurosci. 2010, 30, 16755–16762. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Diniz, B.S.; Talib, L.L.; Mendonca, V.A.; Ojopi, E.B.; Gattaz, W.F.; Teixeira, A.L. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 507–512. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm. 2008, 5, 7. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behaviour. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. IL10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef]
- He, W.; Tian, X.; Lv, M.; Wang, H. Liraglutide protects neurite outgrowth of cortical neurons under oxidative stress through activating the Wnt pathway. J. Stroke Cerebrovasc. Dis. 2018, 27, 2696–2702. [Google Scholar] [CrossRef]
- Wiciński, M.; Socha, M.; Malinowski, B.; Wódkiewicz, E.; Walczak, M.; Górski, K.; Słupski, M.; Pawlak-Osińska, K. Liraglutide and its neuroprotective properties—Focus on possible biochemical mechanisms in Alzheimer’s disease and cerebral ischemic events. Int. J. Mol. Sci. 2019, 20, 1050. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, A.; Oyama, J.i.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Hölscher, C.; Xue, G.F.; Li, G.; Li, D. A novel dual-glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide receptor agonist is neuroprotective in transient focal cerebral ischemia in the rat. Neuroreport 2016, 27, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Shi, Z.; Lu, D.; Li, T.; Ding, Y.; Ruan, Y.; Xu, A. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci. Rep. 2016, 6, 26859. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, S.-Y.; Yin, T.; Tian, F.; Wang, S.; Zhang, Y.; Chen, Y.D. [Liraglutide Promotes Proliferation and Migration of Cardiac Microvascular Endothelial Cells Through PI3K/Akt and MAPK/ERK Signaling Pathways]. J. South. Med. Univ. 2015, 35, 1221–1226. [Google Scholar]
- Zhou, H.; Li, D.; Shi, C.; Xin, T.; Yang, J.; Zhou, Y.; Hu, S.; Tian, F.; Wang, J.; Chen, Y. Effects of exendin-4 on bone marrow mesenchymal stem cell proliferation, migration and apoptosis in vitro. Sci. Rep. 2015, 5, 12898. [Google Scholar] [CrossRef]
- Briyal, S.; Shah, S.; Gulati, A. Neuroprotective and anti-apoptotic effects of liraglutide in the rat brain following focal cerebral ischemia. Neuroscience 2014, 281, 269–281. [Google Scholar] [CrossRef]
- Hamamoto, S.; Kanda, Y.; Shimoda, M.; Tatsumi, F.; Kohara, K.; Tawaramoto, K.; Hashiramoto, M.; Kaku, K. Vildagliptin preserves the mass and function of pancreatic β cells via the developmental regulation and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes. Diabetes Obes. Metab. 2013, 15, 153–163. [Google Scholar] [CrossRef]
- Sato, K.; Kameda, M.; Yasuhara, T.; Agari, T.; Baba, T.; Wang, F.; Shinko, A.; Wakamori, T.; Toyoshima, A.; Takeuchi, H.; et al. Neuroprotective effects of liraglutide for stroke model of rats. Int. J. Mol. Sci. 2013, 14, 21513–21524. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Jalewa, J.; Sharma, M.K.; Hölscher, C. Novel incretin analogues improve autophagy and protect from mitochondrial stress induced by rotenone in SH-SY5Y cells. J. Neurochem. 2016, 139, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-F.; Xue, G.-F.; Hölscher, C.; Tian, M.-J.; Feng, P.; Zheng, J.-Y.; Li, D.-F. Post-treatment with the GLP-1 analogue liraglutide alleviate chronic inflammation and mitochondrial stress induced by Status epilepticus. Epilepsy Res. 2018, 142, 45–52. [Google Scholar] [CrossRef]
- Ji, C.; Xue, G.-F.; Lijun, C.; Feng, P.; Li, D.; Li, L.; Li, G.; Hölscher, C. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson’s disease by increasing expression of BNDF. Brain Res. 2016, 1634, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Liu, L.F.; Jou, M.J.; Wang, H.K. The GLP-1 receptor agonists exendin-4 and liraglutide alleviate oxidative stress and cognitive and micturition deficits induced by middle cerebral artery occlusion in diabetic mice. BMC Neurosci. 2016, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Guan, S.; Qu, D.; Wang, L.; Wang, X.; Li, X.; Zhou, S.; Zhou, Y.; Wang, N.; et al. An orally active allosteric GLP-1 receptor agonist is neuroprotective in cellular and rodent models of stroke. PLoS ONE 2016, 11, e0148827. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.K.; Jalewa, J.; Hölscher, C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J. Neurochem. 2014, 128, 459–471. [Google Scholar] [CrossRef]
- Velmurugan, K.; Balamurugan, A.N.; Loganathan, G.; Ahmad, A.; Hering, B.J.; Pugazhenthi, S. Antiapoptotic actions of exendin-4 against hypoxia and cytokines are augmented by CREB. Endocrinology 2012, 153, 1116–1128. [Google Scholar] [CrossRef]
- Lozano, G.M.; Bejarano, I.; Espino, J.; González, D.; Ortiz, A.; García, J.F.; Rodríguez, A.B.; Pariente, J.A. Relationship between caspase activity and apoptotic markers in human sperm in response to hydrogen peroxide and progesterone. J. Reprod. Dev. 2009, 55, 615–621. [Google Scholar] [CrossRef]
- He, W.; Wang, H.; Zhao, C.; Tian, X.; Li, L.; Wang, H. Role of liraglutide in brain repair promotion through Sirt1-mediated mitochondrial improvement in stroke. J. Cell. Physiol. 2020, 235, 2986–3001. [Google Scholar] [CrossRef]
- Tong, W.; Ju, L.; Qiu, M.; Xie, Q.; Chen, Y.; Shen, W.; Sun, W.; Wang, W.; Tian, J. Liraglutide ameliorates non-alcoholic fatty liver disease by enhancing mitochondrial architecture and promoting autophagy through the SIRT1/SIRT3-FOXO3a pathway. Hepatol. Res. 2016, 46, 933–943. [Google Scholar] [CrossRef]
- Deng, C.; Cao, J.; Han, J.; Li, J.; Li, Z.; Shi, N.; He, J. Liraglutide activates the Nrf2/HO-1 antioxidant pathway and protects brain nerve cells against cerebral ischemia in diabetic rats. Comput. Intell. Neurosci. 2018, 2018, 3094504. [Google Scholar] [CrossRef] [PubMed]
- Bigl, M.; Bruckner, M.K.; Arendt, T.; Bigl, V.; Eschrich, K. Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J. Neural. Transm. 1999, 106, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Mehta, J.L.; Chen, M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc. Drugs Ther. 2013, 27, 371–380. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Hölscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- Parthsarathy, V.; Hölscher, C. The type 2 diabetes drug liraglutide reduces chronic inflammation induced by irradiation in the mouse brain. Eur. J. Pharmacol. 2013, 700, 42–50. [Google Scholar] [CrossRef]
- Barreto-Vianna, A.R.C.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Beneficial effects of liraglutide (GLP1 analog) in the hippocampal inflammation. Metab. Brain Dis. 2017, 32, 1735–1745. [Google Scholar] [CrossRef]
- Gao, H.; Zeng, Z.; Zhang, H.; Zhou, X.; Guan, L.; Deng, W.; Xu, L. The Glucagon-Like Peptide-1 analogue liraglutide inhibits oxidative stress and inflammatory response in the liver of rats with diet-induced non-alcoholic fatty liver disease. Biol. Pharm. Bull. 2015, 38, 694–702. [Google Scholar] [CrossRef]
- Wu, L.K.; Liu, Y.C.; Shi, L.L.; Lu, K.D. Glucagon-like peptide-1 receptor agonists inhibit hepatic stellate cell activation by blocking the p38 MAPK signaling pathway. Genet. Mol. Res. 2015, 14, 19087–19093. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: Over-expression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef]
- Alavi, M.V.; Fuhrmann, N. Dominant optic atrophy, OPA1, and mitochondrial quality control: Understanding mitochondrial network dynamics. Mol. Neurodegener. 2013, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011, 21, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissiere, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.P.; Manlandro, C.M.; Picton, L.K.; Tan, A.Z.; Casares, S.; Flanagan, J.M.; Fleming, K.G.; Hill, R.B. A designed point mutant in Fis1 disrupts dimerization and mitochondrial fission. J. Mol. Biol. 2012, 423, 143–158. [Google Scholar] [CrossRef]
- Li, H.; Cao, L.; Ren, Y.; Jiang, Y.; Xie, W.; Li, D. GLP-1 receptor regulates cell growth through regulating IDE expression level in Aβ1-42-treated PC12 cells. Biosci. Rep. 2018, 38, BSR20171284. [Google Scholar] [CrossRef]
- Li, H.; Yang, S.; Wu, J.; Ji, L.; Zhu, L.; Cao, L.; Huang, J.; Jiang, Q.; Wei, J.; Liu, M.; et al. AMP/PKA signaling pathway contributes to neuronal apoptosis via regulating IDE expression in a mixed model of type 2 diabetes and Alzheimer’s disease. J. Cell. Biochem. 2018, 119, 1616–1626. [Google Scholar] [CrossRef]
- Costa, R.; Ferreira-da-Silva, F.; Saraiva, M.J.; Cardoso, I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: A mechanism sensitive to the Kunitz protease inhibitor. PLoS ONE 2008, 3, e2899. [Google Scholar] [CrossRef]
- Costa, R.; Gonçalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide--impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008, 582, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Torres-Aleman, I. Serum insulin-like growth factor I in brain function. Keio J. Med. 2006, 55, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Trejo, J.L.; Gerber, A.; Loetscher, H.; Torrado, J.; Metzger, F.; Torres-Aleman, I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol. Aging 2006, 27, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Mavroudis, I.; Fotiou, D.; Baloyannis, S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr. Danub. 2012, 24, 152–158. [Google Scholar] [PubMed]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef]
- Ayala-Grosso, C.; Ng, G.; Roy, S.; Robertson, G.S. Caspase-cleaved amyloid precursor protein in Alzheimer’s disease. Brain Pathol. 2002, 12, 430–441. [Google Scholar] [CrossRef]
- Koshal, P.; Kumar, P. Neurochemical modulation involved in the beneficial effect of liraglutide, GLP-1 agonist on PTZ kindling epilepsy-induced comorbidities in mice. Mol. Cell. Biochem. 2016, 415, 77–87. [Google Scholar] [CrossRef]
- Koshal, P.; Kumar, P. Effect of Liraglutide on Corneal Kindling Epilepsy Induced Depression and Cognitive Impairment in Mice. Neurochem. Res. 2016, 41, 1741–1750. [Google Scholar] [CrossRef]
- McClean, P.L.; Gault, V.A.; Harriott, P.; Hölscher, C. Glucagon-like peptide-1 analogues enhance synaptic plasticity in the brain: A link between diabetes and Alzheimer’s disease. Eur. J. Pharmacol. 2010, 630, 158–162. [Google Scholar] [CrossRef]
- Babateen, O.; Korol, S.V.; Jin, Z.; Bhandage, A.K.; Ahemaiti, A.; Birnir, B. Liraglutide modulates GABAergic signaling in rat hippocampal CA3 pyramidal neurons predominantly by presynaptic mechanism. BMC Pharmacol. Toxicol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Gupta, G.; Dahiya, R.; Dua, K.; Chellappan, D.K.; Tiwari, J.; Sharma, G.N.; Singh, S.K.; Mishra, A.; Sharma, R.K.; Agrawal, M. Anticonvulsant effect of liraglutide, GLP-1 agonist by averting a change in GABA and brain glutathione level on picrotoxin-induced seizures. EXCLI J. 2017, 16, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Gilman, C.P.; Perry, T.; Furukawa, K.; Grieg, N.H.; Egan, J.M.; Mattson, M.P. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J. Neurochem. 2003, 87, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, S.; Li, Y. Liraglutide promotes cortical neurite outgrowth via the MEK-ERK pathway. Cell. Mol. Neurobiol. 2015, 35, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lin, W.; Lin, Z.; Hao, M.; Gao, X.; Zhang, Y.; Kuang, H. Liraglutide alleviates H2O2-induced retinal ganglion cells injury by inhibiting autophagy through mitochondrial pathways. Peptides 2017, 92, 1–8. [Google Scholar] [CrossRef]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Wiciński, M.; Malinowski, B.; Węclewicz, M.M.; Grześk, E.; Grześk, G. Anti-atherogenic properties of resveratrol: 4-week resveratrol administration associated with serum concentrations of SIRT1, adiponectin, S100A8/A9 and VSMCs contractility in a rat model. Exp. Ther. Med. 2017, 13, 2071–2078. [Google Scholar] [CrossRef]
- Parthsarathy, V.; Hölscher, C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS ONE 2013, 8, e58784. [Google Scholar] [CrossRef]
- Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol. 2012, 166, 1586–1599. [Google Scholar] [CrossRef]
- Dong, W.; Miao, Y.; Chen, A.; Cheng, M.; Ye, X.; Song, F.; Zheng, G. Delayed administration of the GLP-1 receptor agonist liraglutide improves metabolic and functional recovery after cerebral ischemia in rats. Neurosci. Lett. 2017, 641, 1–7. [Google Scholar] [CrossRef]
- Briyal, S.; Gulati, K.; Gulati, A. Repeated administration of exendin-4 reduces focal cerebral ischemia-induced infarction in rats. Brain Res. 2012, 1427, 23–34. [Google Scholar] [CrossRef]
- Schaar, K.L.; Brenneman, M.M.; Savitz, S.I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2010, 2, 13. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Hsieh, S.H.; Sun, J.H.; Tsai, J.S.; Huang, Y.Y. Glucose variability and β-cell response by GLP-1 analogue added-on CSII for patients with poorly controlled type 2 diabetes. Sci. Rep. 2015, 5, 16968. [Google Scholar] [CrossRef]
- Salehi, M.; Aulinger, B.; Prigeon, R.L.; D’Alessio, D.A. Effect of endogenous GLP-1 on insulin secretion in type 2 diabetes. Diabetes 2010, 59, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Onoviran, O.F.; Li, D.; Toombs Smith, S.; Raji, M.A. Effects of glucagon-like peptide 1 receptor agonists on comorbidities in older patients with diabetes mellitus. Ther. Adv. Chronic Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Harkavyi, A.; Abuirmeileh, A.; Lever, R.; Kingsbury, A.E.; Biggs, C.S.; Whitton, P.S. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 19. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Stephen, C.; Gough, S.C.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef]
- Blackman, A.; Foster, G.D.; Zammit, G.; Rosenberg, R.; Aronne, L.; Wadden, T.; Claudius, B.; Jensen, C.B.; Mignot, E. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: The SCALE Sleep Apnea randomized clinical trial. Int. J. Obes. 2016, 40, 1310–1319. [Google Scholar] [CrossRef]
- Simó, R.; Guerci, B.; Schernthaner, G.; Gallwitz, B.; Rosas-Guzmàn, J.; Dotta, F.; Festa, A.; Zhou, M.; Kiljański, J. Long-term changes in cardiovascular risk markers during administration of exenatide twice daily or glimepiride: Results from the European exenatide study. Cardiovasc. Diabetol. 2015, 14, 116. [Google Scholar] [CrossRef]
- Sun, F.; Wu, S.; Wang, J.; Guo, S.; Chai, S.; Yang, Z.; Li, L.; Zhang, Y.; Ji, L.; Zhan, S. Effect of glucagon-like peptide-1 receptor agonists on lipid profiles among type 2 diabetes: A systematic review and network meta-analysis. Clin. Ther. 2015, 37, 225–241.e8. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.D.; Dao, D.T.; Kovacsics, C.E. The open field test. Neuromethods 2009, 42, 1–20. [Google Scholar] [CrossRef]
- Soares, E.; Prediger, R.D.; Nunes, S.; Castro, A.A.; Viana, S.D.; Lemos, C.; De Souza, C.M.; Agostinho, P.; Cunha, R.A.; Carvalho, E.; et al. Spatial memory impairments in a prediabetic rat model. Neuroscience 2013, 250, 565–577. [Google Scholar] [CrossRef]
- Akwa, Y.; Ladurelle, N.; Covey, D.F.; Baulieu, E.E. The synthetic enantiomer of pregnenolone sulfate is very active on memory in rats and mice, even more so than its physiological neurosteroid counterpart: Distinct mechanisms? Proc. Natl. Acad. Sci. USA 2001, 98, 14033–14037. [Google Scholar] [CrossRef]
- Dellu, F.; Fauchey, V.; Moal, M.L.; Simon, H. Extension of a new two-trial memory task in the rat: Influence of environmental context on recognition processes. Neurobiol. Learn. Mem. 1997, 67, 112–120. [Google Scholar] [CrossRef]
- Dellu, F.; Mayo, W.; Cherkaoui, J.; Le Moal, M.; Simon, H. A two-trial memory task with automated recording: Study in young and aged rats. Brain Res. 1992, 588, 132–139. [Google Scholar] [CrossRef]
- Morris, R.G.M.; Garrud, P.; Rawlins, J.N.P.; O’Keefe, J. Place navigation impaired in rats with hippocampal lesions. Nature 1982, 297, 681–683. [Google Scholar] [CrossRef]
- Prediger, R.D.; Batista, L.C.; Medeiros, R.; Pandolfo, P.; Florio, J.C.; Takahashi, R.N. The risk is in the air: Intranasal administration of MPTP to rats reproducing clinical features of Parkinson’s disease. Exp. Neurol. 2006, 202, 391–403. [Google Scholar] [CrossRef]
- García-Nogales, P.; Almeida, A.; Fernández, E.; Medina, J.M.; Bolaños, J.P. Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J. Neurochem. 1999, 72, 1750–1758. [Google Scholar] [CrossRef]
- Fagan, J.M.; Sleczka, B.G.; Sohar, I. Quantitation of oxidative damage to tissue proteins. Int. J. Biochem. Cell Biol. 1999, 31, 751–757. [Google Scholar] [CrossRef]
- Green, L.C.; Ruiz de Luzuriaga, K.; Wagner, D.A.; Rand, W.; Istfan, N.; Young, V.R.; Tannenbaum, S.R. Nitrate biosynthesis in man. Proc. Natl. Acad. Sci. USA 1981, 78, 7764–7768. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | 3xTg-AD | 3xTg-AD + Lira | |
|---|---|---|---|
| Body weight (g) | 29.1 ± 1.2 (n = 10) (95% CI: 26.3–31.8) | 23.3 ± 0.6 **** (n = 12) (95% CI: 22.1–24.6) | 23.3 ± 0.4 **** (n = 14) (95% CI: 22.6–24.1) |
| Brain weight (g) | 0.5 ± 0.01 (n = 7) (95% CI: 0.45–0.51) | 0.4 ± 0.03 (n = 8) (95% CI: 0.36–0.52) | 0.5 ± 0.03 (n = 10) (95% CI: 0.42–0.54) |
| HbA1c (%) | 4.3 ± 0.2 (n = 10) (95% CI: 3.74–4.84) | 4.4 ± 0.1 (n = 11) (95% CI: 4.17–4.65) | 4.4 ± 0.1 (n = 12) (95% CI: 4.13–4.57) |
| Occasional glycemia (mg glucose/dL blood) | 132.8 ± 3.3 (n = 9) (95% CI: 125.2–140.3) | 121.2 ± 7.3 (n = 12) (95% CI: 105.2–137.2) | 128.1 ± 10.5 (n = 14) (95% CI: 105.6–150.7) |
| Fasting glycemia (mg glucose/dL blood) | 126.4 ± 4.7 (n = 9) (95% CI: 115.6–137.7) | 110.3 ± 8.2 (n = 12) (95% CI: 92.4–128.3) | 127.6 ± 6.5 p = 0.073 (n = 14) (95% CI: 113.7–141.6) |
| Fasting insulin levels (ng/mL plasma) | 3.5 ± 1.5 (n = 10) (95% CI: 0.07–6.97) | 2.5 ± 0.8 (n = 11) (95% CI: 0.72–4.23) | 1.3 ± 0.4 (n = 11) (95% CI: 0.56–2.13) |
| HOMA-IR | 30.2 ± 13.1 (n = 10) (95% CI: 0.7–59.8) | 15.2 ± 5.0 (n = 11) (95% CI: 4.05–26.37) | 11.3 ± 3.1 (n = 11) (95% CI: 4.37–18.26) |
| HOMA-β | 217.5 ± 124.23 (n = 8) (95% CI: −76.25–511.26) | 262.1 ± 93.01 (n = 9) (95% CI: 47.60–476.5) | 170 ± 43.33 (n = 10) (95% CI: 72–268) |
| Estradiol levels (pg/mL plasma) | 184.1 ± 15.1 (n = 7) (95% CI: 147.2–220.9) | 230.8 ± 24.3 p = 0.07 (n = 6) (95% CI: 168.3–293.3) | 244.9 ± 9.5 p = 0.023 (n = 6) (95% CI: 220.3–269.4) |
| C-Reactive Protein levels (ng/mL plasma) | 31.9 ± 6.1 (n = 6) (95% CI: 16.25–47.51) | 74.3 ± 17.6 * (n = 6) (95% CI: 29.10–119.4) | 60.8 ± 10.7 (n = 7) (95% CI: 34.53–86.98) |
| IL-10 levels (pg/mL plasma) | 551.5 ± 134.6 (n = 7) (95% CI: 222.1–880.9) | 364.6 ± 81.6 (n = 6) (95% CI: 154.8–574.3) | 494.3 ± 54.5 (n = 7) (95% CI: 361.1–627.6) |
| IL-1β levels (pg/mL plasma) | 43.2 ± 12.3 (n = 6) (95% CI: 11.66–74.66) | 821.6 ± 400.7 ** (n = 6) (95% CI: −208.3–1852) | 355.4 ± 159.3 p = 0.051 (n = 7) (95% CI: −34.38–745.3) |
| WT | 3xTg-AD | 3xTg-AD + Lira | |
|---|---|---|---|
| Estradiol levels (pg/mg protein) | 5.62 ± 1.19 (n = 5) (95% CI: 2.31–8.93) | 15.2 ± 2.7 * (n = 6) (95% CI: 8.27–22.11) | 12.2 ± 3.3 (n = 5) (95% CI: 3.1–21.24) |
| GLP-1 levels (pg/mg protein) | 5.9 ± 2.5 (n = 5) (95% CI: −1.14–12.94) | 21.1 ± 6.5 * (n = 6) (95% CI: 4.52–37.74) | 15.0 ± 2.8 (n = 5) (95% CI: 7.29–22.76) |
| Active PKA kinase (ng active PKA/mg protein) | 0.01 ± 0.004 (n = 6) (95% CI: −0.0005–0.02) | 0.001 ± 0.0004 ** (n = 6) (95% CI: 0.0001–0.002) | 0.009 ± 0.005 (n = 5) (95% CI: −0.0048–0.022) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide Protects Against Brain Amyloid-β1–42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. Int. J. Mol. Sci. 2020, 21, 1746. https://doi.org/10.3390/ijms21051746
Duarte AI, Candeias E, Alves IN, Mena D, Silva DF, Machado NJ, Campos EJ, Santos MS, Oliveira CR, Moreira PI. Liraglutide Protects Against Brain Amyloid-β1–42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. International Journal of Molecular Sciences. 2020; 21(5):1746. https://doi.org/10.3390/ijms21051746
Chicago/Turabian StyleDuarte, Ana I., Emanuel Candeias, Inês N. Alves, Débora Mena, Daniela F. Silva, Nuno J. Machado, Elisa J. Campos, Maria S. Santos, Catarina R. Oliveira, and Paula I. Moreira. 2020. "Liraglutide Protects Against Brain Amyloid-β1–42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation" International Journal of Molecular Sciences 21, no. 5: 1746. https://doi.org/10.3390/ijms21051746
APA StyleDuarte, A. I., Candeias, E., Alves, I. N., Mena, D., Silva, D. F., Machado, N. J., Campos, E. J., Santos, M. S., Oliveira, C. R., & Moreira, P. I. (2020). Liraglutide Protects Against Brain Amyloid-β1–42 Accumulation in Female Mice with Early Alzheimer’s Disease-Like Pathology by Partially Rescuing Oxidative/Nitrosative Stress and Inflammation. International Journal of Molecular Sciences, 21(5), 1746. https://doi.org/10.3390/ijms21051746