Differential Inhibitory Actions of Multitargeted Tyrosine Kinase Inhibitors on Different Ionic Current Types in Cardiomyocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
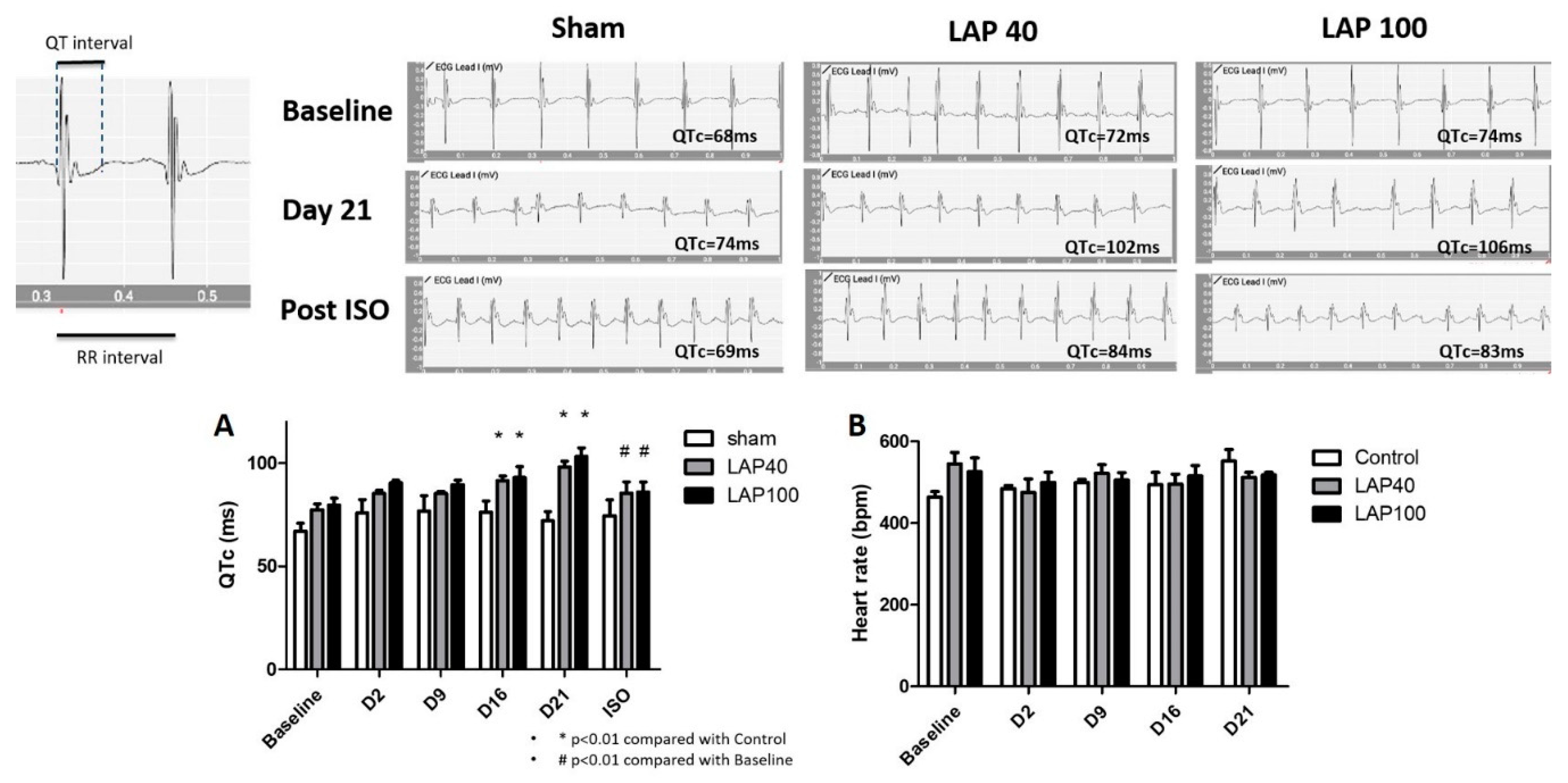
2.1. LAP Caused QTc Prolongation in the Mouse Model
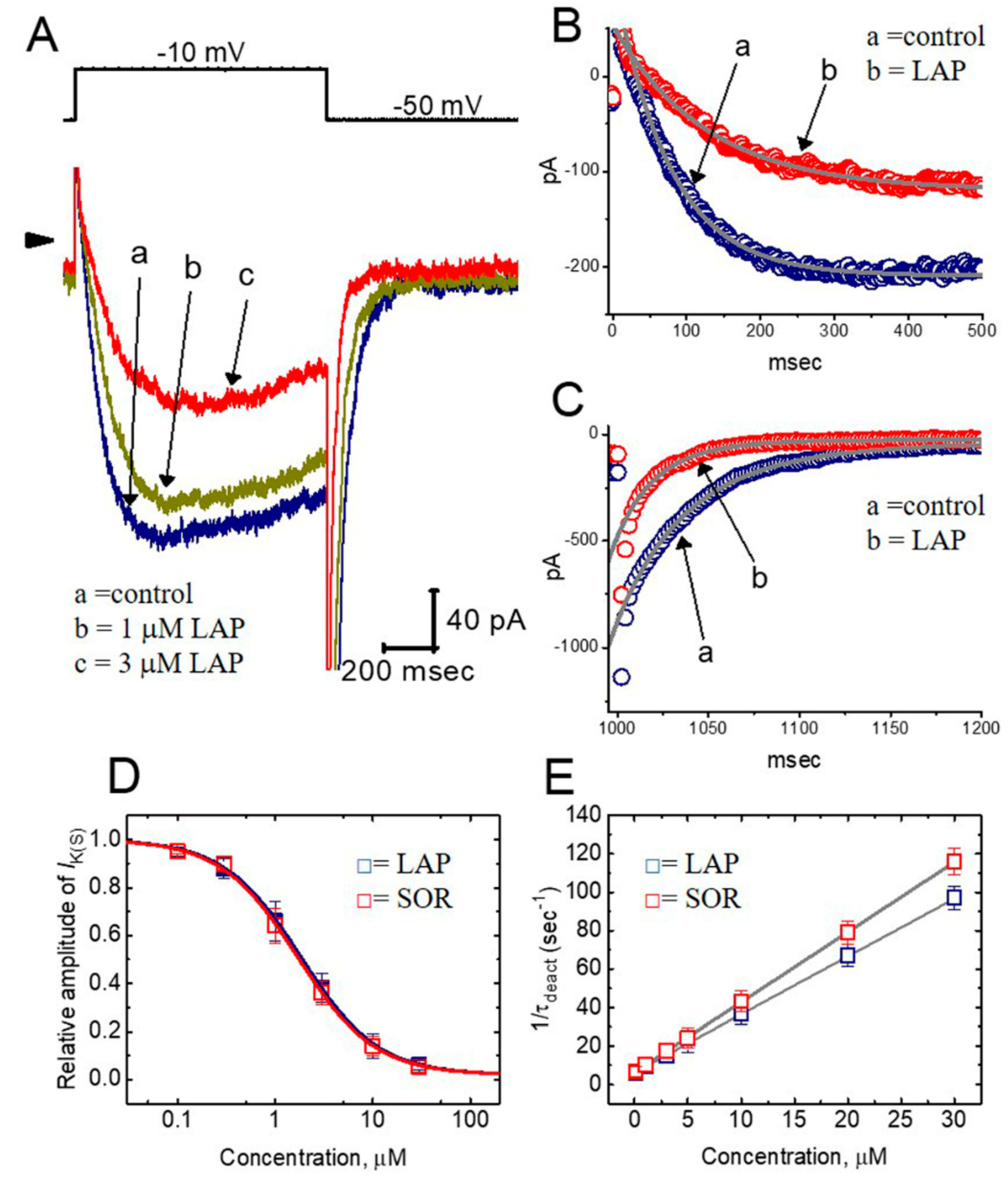
2.2. Effect of LAP on the Whole-Cell Slowly Activating Delayed-Rectifier K+ Current (IK(S)) in Differentiated H9c2 Cells
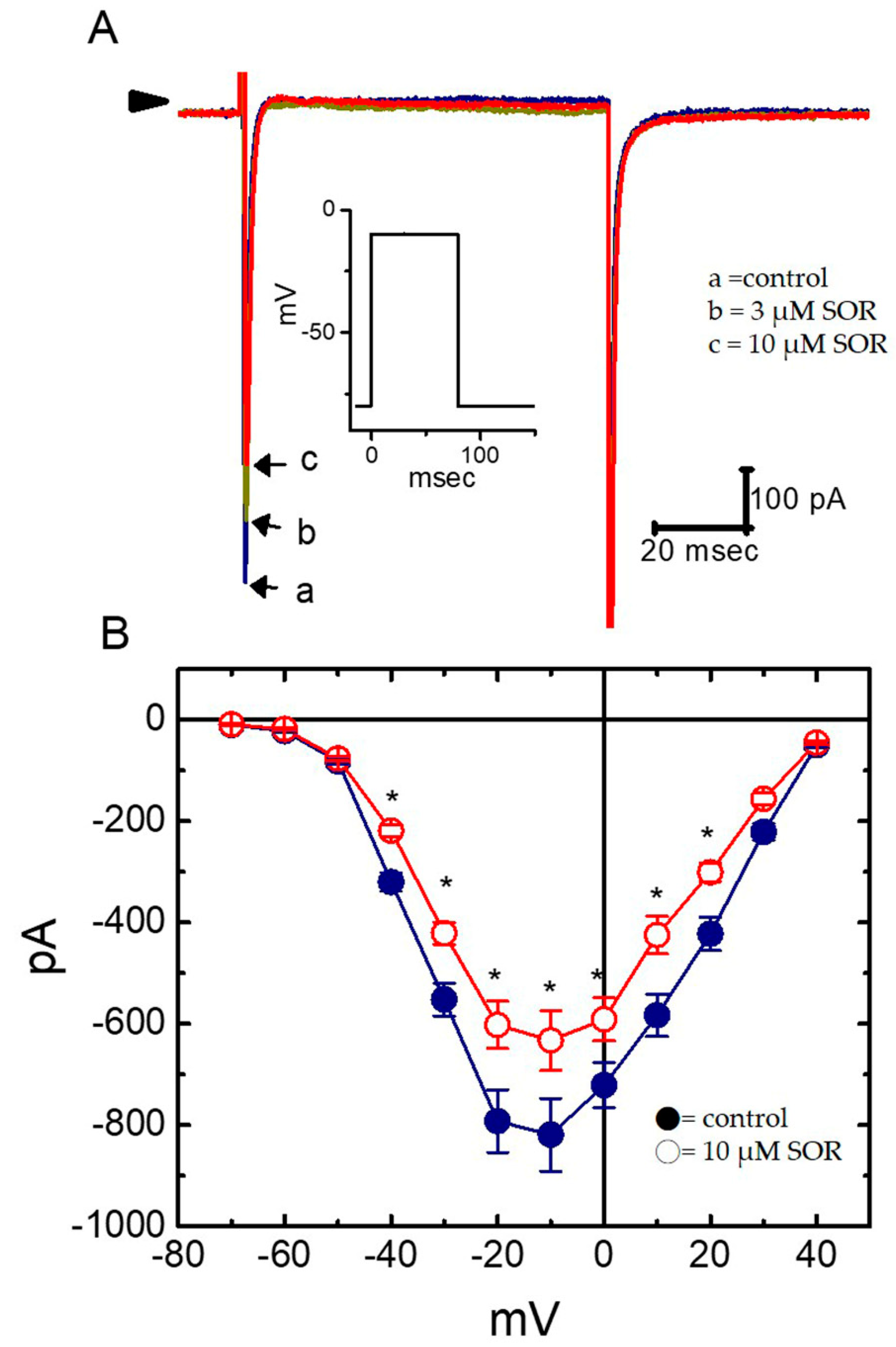
2.3. Concentration-Dependent Effects of LAP or SOR on IK(S) in H9c2 Cells
2.4. Comparisons of the Effects of SOR, LAP, LAP Plus Isoproterenol, LAP Plus Meclofenamic Acid, and LAP Plus diazoxide on the IK(S) Amplitude
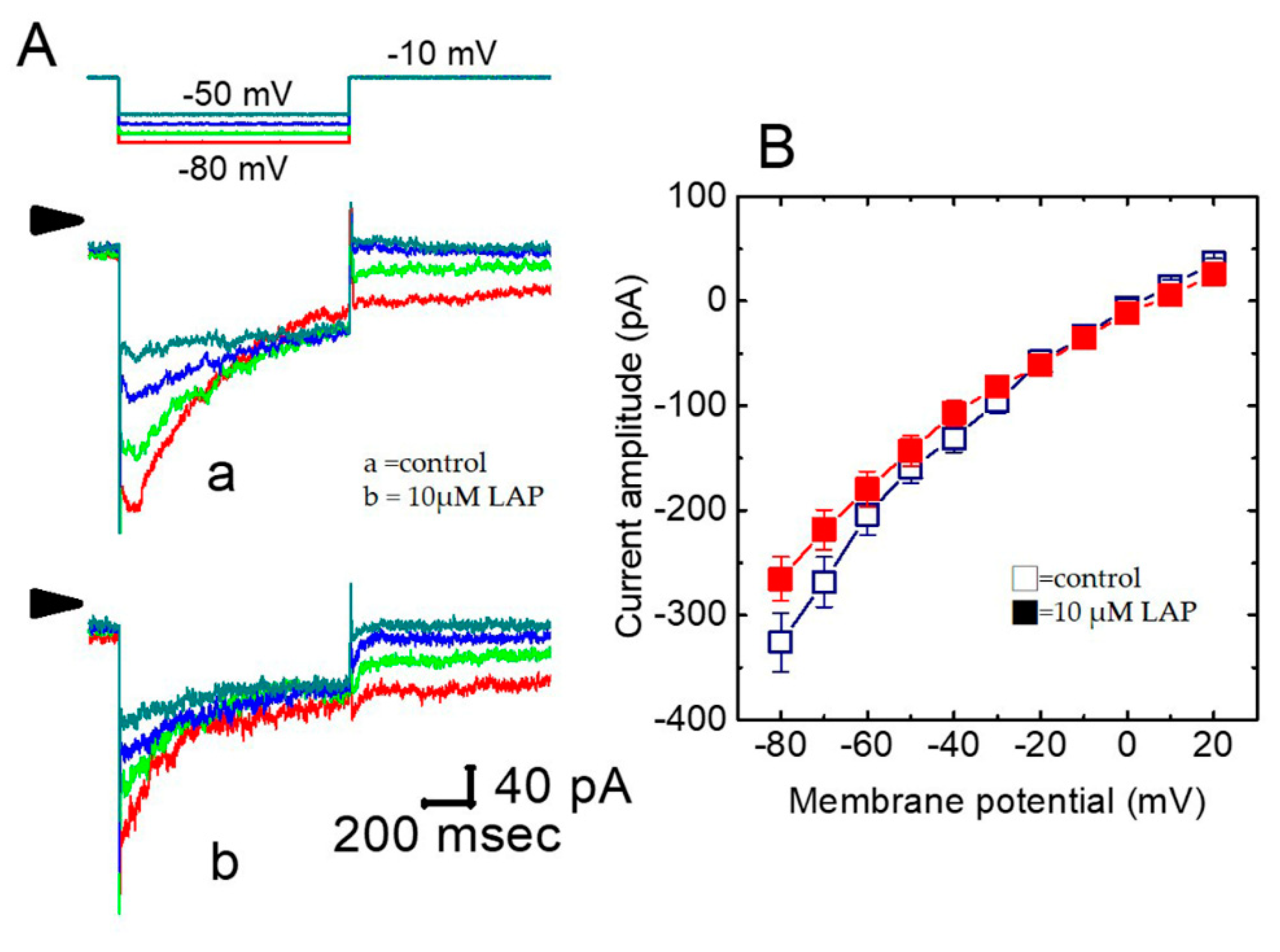
2.5. Effect of LAP on the IK(S) I–V Relationship in H9c2 Cells
2.6. Effect of LAP on the Recovery from IK(S) Deactivation in H9c2 Cells
2.7. Inhibitory Effect of LAP on IK(S) Elicited by a Train of Rapid Repetitive Depolarizations
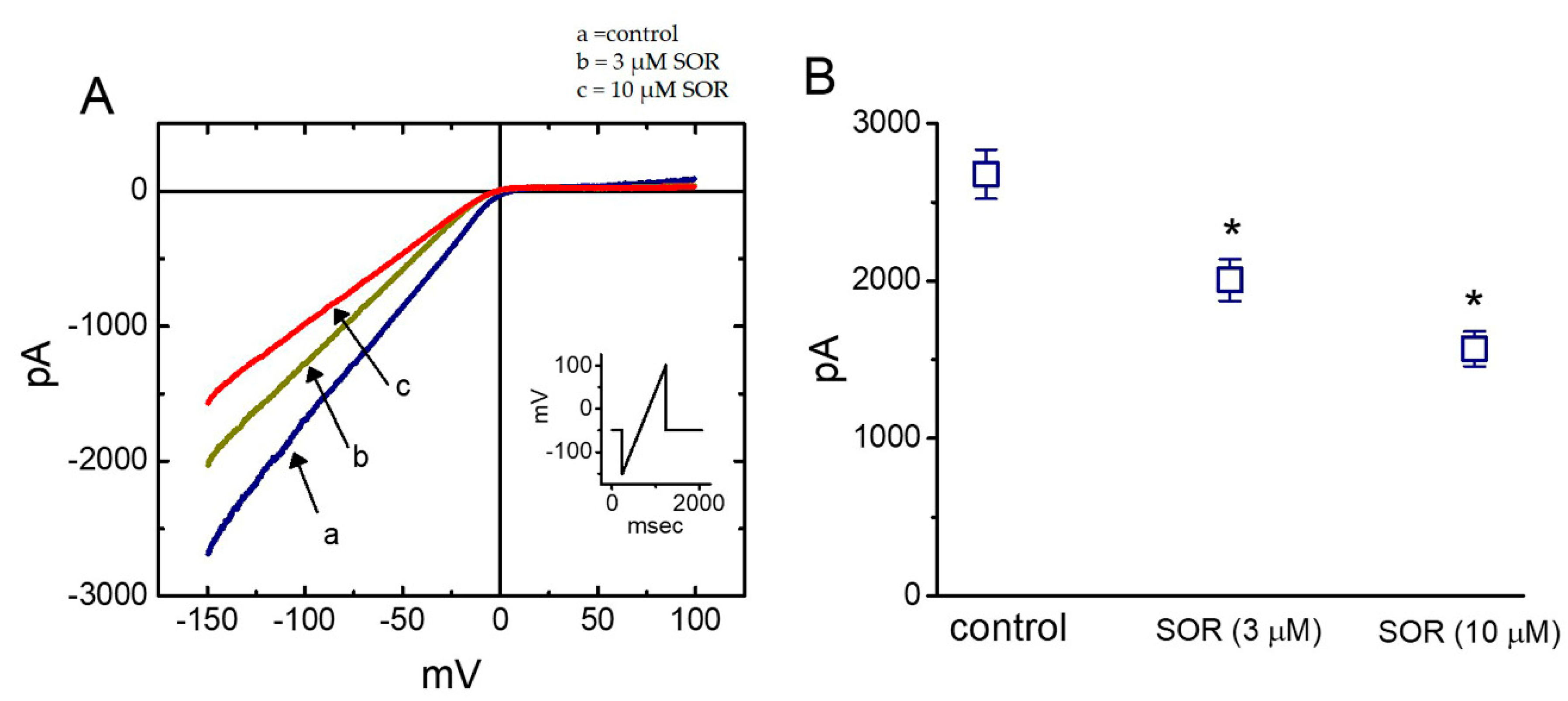
2.8. Effect of LAP in erg-Mediated K+ Current (IK(erg)) in H9c2 Cells
2.9. Suppressive Effect of LAP on the Amplitude of Inwardly-Rectifying K+ Current (IK(IR)) Measured from Cultured NRVMs
2.10. Effect of LAP on Voltage-Gated Na+ Current (INa) in Cultured NRVMs
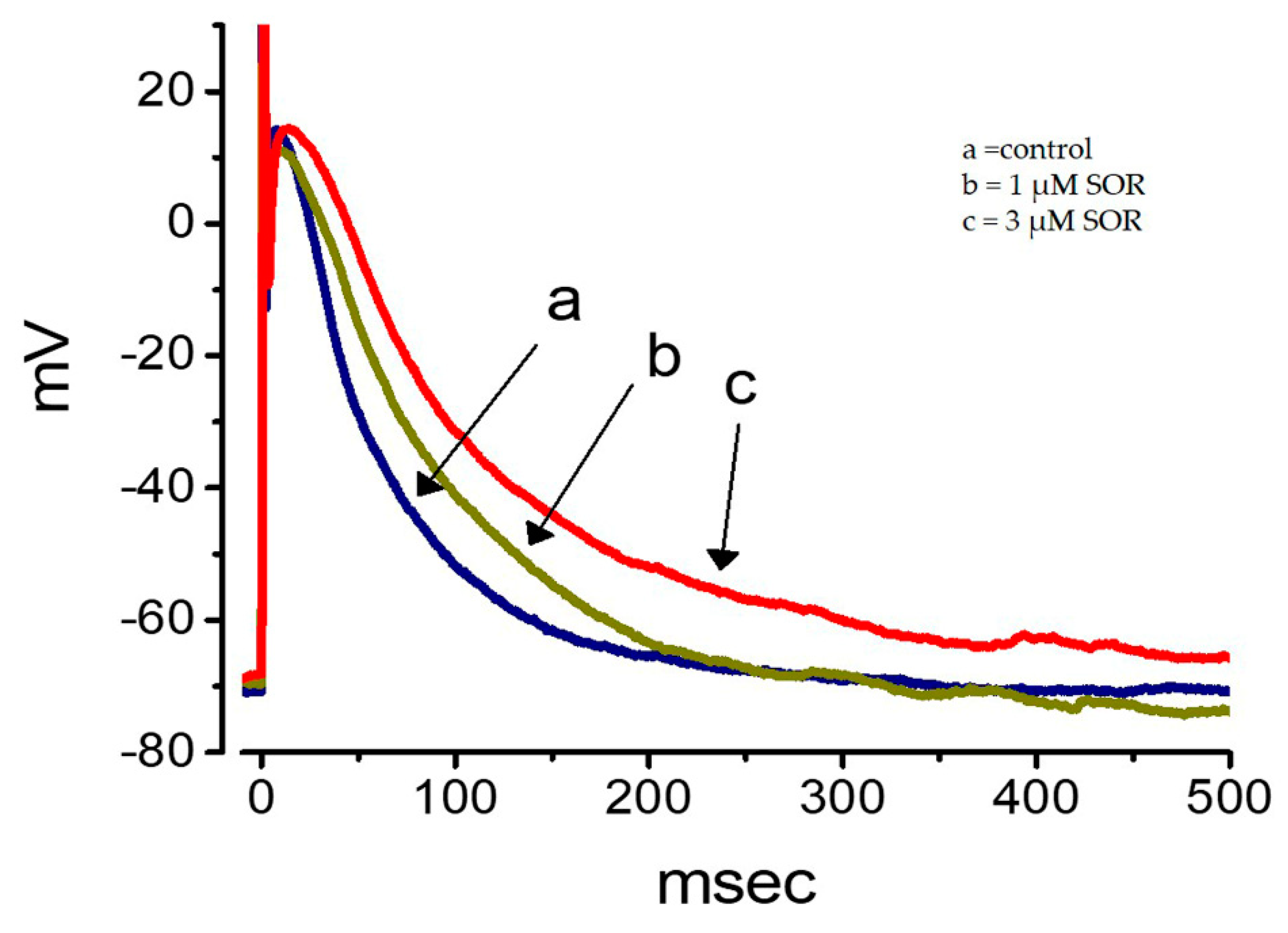
2.11. Effect of LAP on the Membrane Potential Recorded from Cultured NRVMs
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. ECG Recording and QT Specification in Mice
4.3. Isolation and Culture of NRVMs
4.4. Electrophysiological Measurements
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| AP | action potential |
| Chrom | chromanol 293B |
| I-V | current versus voltage |
| IC50 | concentration required for 50% inhibition |
| IK(erg) | erg-mediated K+ current |
| IK(IR) | inwardly-rectifying K+ current |
| IK(S) | slowly activating delayed-rectifier K+ current |
| LAP | lapatinib |
| NRVMs | neonatal rat ventricular myocytes |
| QTc interval | corrected QT interval |
| SOR | sorafenib |
| τact | activating time constant |
| τdeact | deactivating time constant |
| TKIs | tyrosine kinase inhibitors |
References
- Molife, L.R.; Dean, E.J.; Blanco-Codesido, M. A phase i, dose-escalation study of the multitargeted receptor tyrosine kinase inhibitor, golvatinib, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 6284–6294. [Google Scholar] [CrossRef]
- Hoff, P.M.; Wolff, R.A.; Bogaard, K.; Waldrum, S.; Abbruzzese, J.L. A phase i study of escalating doses of the tyrosine kinase inhibitor semaxanib (su5416) in combination with irinotecan in patients with advanced colorectal carcinoma. Jpn. J. Clin. Oncol. 2006, 36, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Kim, T.M.; Yokota, T. Phase I dose-escalation study of the c-met tyrosine kinase inhibitor sar125844 in asian patients with advanced solid tumors, including patients with met-amplified gastric cancer. Oncotarget 2017, 8, 79546–79555. [Google Scholar] [CrossRef] [PubMed]
- Kloth, J.S.; Pagani, A.; Verboom, M.C. Incidence and relevance of QTc-interval prolongation caused by tyrosine kinase inhibitors. Br. J. Cancer 2015, 112, 1011–1016. [Google Scholar] [CrossRef]
- Calistri, L.; Cordopatri, C.; Nardi, C.; Gianni, E.; Marra, F.; Colagrande, S. Sudden cardiac death in a patient with advanced hepatocellular carcinoma with good response to sorafenib treatment: A case report with literature analysis. Mol. Clin. Oncol. 2017, 6, 389–396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.J.; Chen, B.S.; Lin, M.W. Time-dependent block of ultrarapid-delayed rectifier k+ currents by aconitine, a potent cardiotoxin, in heart-derived h9c2 myoblasts and in neonatal rat ventricular myocytes. Toxicol. Sci. 2008, 106, 454–463. [Google Scholar] [CrossRef]
- Robbins, J. KCNQ potassium channels: Physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 2001, 90, 1–19. [Google Scholar] [CrossRef]
- Ding, W.G.; Toyoda, F.; Matsuura, H. Blocking action of chromanol 293B on the slow component of delayed rectifier K+ current in guinea-pig sino-atrial node cells. Br. J. Pharmacol. 2002, 137, 253–262. [Google Scholar] [CrossRef]
- Taylor, K.C.; Sanders, C.R. Regulation of kCNQ/KV7 family voltage-gated K+ channels by lipids. Biochim. Biophys. Acta 2017, 1859, 586–597. [Google Scholar] [CrossRef]
- Zehelein, J.; Thomas, D.; Khalil, M. Identification and characterisation of a novel kcnq1 mutation in a family with romano-ward syndrome. Biochim. Biophys. Acta 2004, 1690, 185–192. [Google Scholar] [CrossRef][Green Version]
- Nakajima, T.; Wu, S.; Irisawa, H.; Giles, W. Mechanism of acetylcholine-induced inhibition of ca current in bullfrog atrial myocytes. J. Gen. Physiol. 1990, 96, 865–885. [Google Scholar] [CrossRef] [PubMed]
- Giles, W.; Nakajima, T.; Ono, K.; Shibata, E.F. Modulation of the delayed rectifier K+ current by isoprenaline in bull-frog atrial myocytes. J. Physiol. 1989, 415, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Huys, I.; Waelkens, E.; Tytgat, J. Structure-function study of a chlorotoxin-chimer and its activity on Kv1.3 channels. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2004, 803, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Wu, S.N.; Cheng, J.T.; Tsai, J.J.; Huang, C.C. Diazoxide reduces status epilepticus neuron damage in diabetes. Neurotox. Res. 2010, 17, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Hung, T.Y.; Liao, Y.K.; Hsu, M.C.; Wu, S.N. Underlying mechanism of regulatory actions of diclofenac, a nonsteroidal anti-inflammatory agent, on neuronal potassium channels and firing: An experimental and theoretical study. J. Physiol. Pharmacol. 2013, 64, 269–280. [Google Scholar]
- Steffensen, A.B.; Refsgaard, L.; Andersen, M.N. IKs Gain- and Loss-of-Function in Early-Onset Lone Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2015, 26, 715–723. [Google Scholar] [CrossRef]
- Sung, R.J.; Wu, S.N.; Wu, J.S.; Chang, H.D.; Luo, C.H. Electrophysiological mechanisms of ventricular arrhythmias in relation to andersen-tawil syndrome under conditions of reduced IK1: A simulation study. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2597–H2605. [Google Scholar] [CrossRef]
- Testai, L.; Barrese, V.; Soldovieri, M.V. Expression and function of Kv7.4 channels in rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2016, 110, 40–50. [Google Scholar] [CrossRef]
- Paech, F.; Bouitbir, J.; Krahenbuhl, S. Hepatocellular toxicity associated with tyrosine kinase inhibitors: Mitochondrial damage and inhibition of glycolysis. Front. Pharmacol. 2017, 8, 367. [Google Scholar] [CrossRef]
- Chien, A.J.; Munster, P.N.; Melisko, M.E. Phase I dose-escalation study of 5-day intermittent oral lapatinib therapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer. J. Clin. Oncol. 2014, 32, 1472–1479. [Google Scholar] [CrossRef]
- Jespersen, T.; Grunnet, M.; Olessen, S.P. The KCNQ1 potassium channel: From gene to physiological function. Physiology (Bethesda) 2005, 20, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Jost, N.; Virág, L.; Comtois, P.; Ordög, B.; Szuts, V.; Seprényi, G.; Bitay, M.; Kohajda, Z.; Koncz, I.; Nagy, N.; et al. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J. Physiol. 2013, 591, 4189–4206. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, W.-T.; Liu, P.-Y.; Lee, K.; Feng, Y.-H.; Wu, S.-N. Differential Inhibitory Actions of Multitargeted Tyrosine Kinase Inhibitors on Different Ionic Current Types in Cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 1672. https://doi.org/10.3390/ijms21051672
Chang W-T, Liu P-Y, Lee K, Feng Y-H, Wu S-N. Differential Inhibitory Actions of Multitargeted Tyrosine Kinase Inhibitors on Different Ionic Current Types in Cardiomyocytes. International Journal of Molecular Sciences. 2020; 21(5):1672. https://doi.org/10.3390/ijms21051672
Chicago/Turabian StyleChang, Wei-Ting, Ping-Yen Liu, Kaisen Lee, Yin-Hsun Feng, and Sheng-Nan Wu. 2020. "Differential Inhibitory Actions of Multitargeted Tyrosine Kinase Inhibitors on Different Ionic Current Types in Cardiomyocytes" International Journal of Molecular Sciences 21, no. 5: 1672. https://doi.org/10.3390/ijms21051672
APA StyleChang, W.-T., Liu, P.-Y., Lee, K., Feng, Y.-H., & Wu, S.-N. (2020). Differential Inhibitory Actions of Multitargeted Tyrosine Kinase Inhibitors on Different Ionic Current Types in Cardiomyocytes. International Journal of Molecular Sciences, 21(5), 1672. https://doi.org/10.3390/ijms21051672