Substituted Piperazines as Novel Potential Radioprotective Agents

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Chemistry
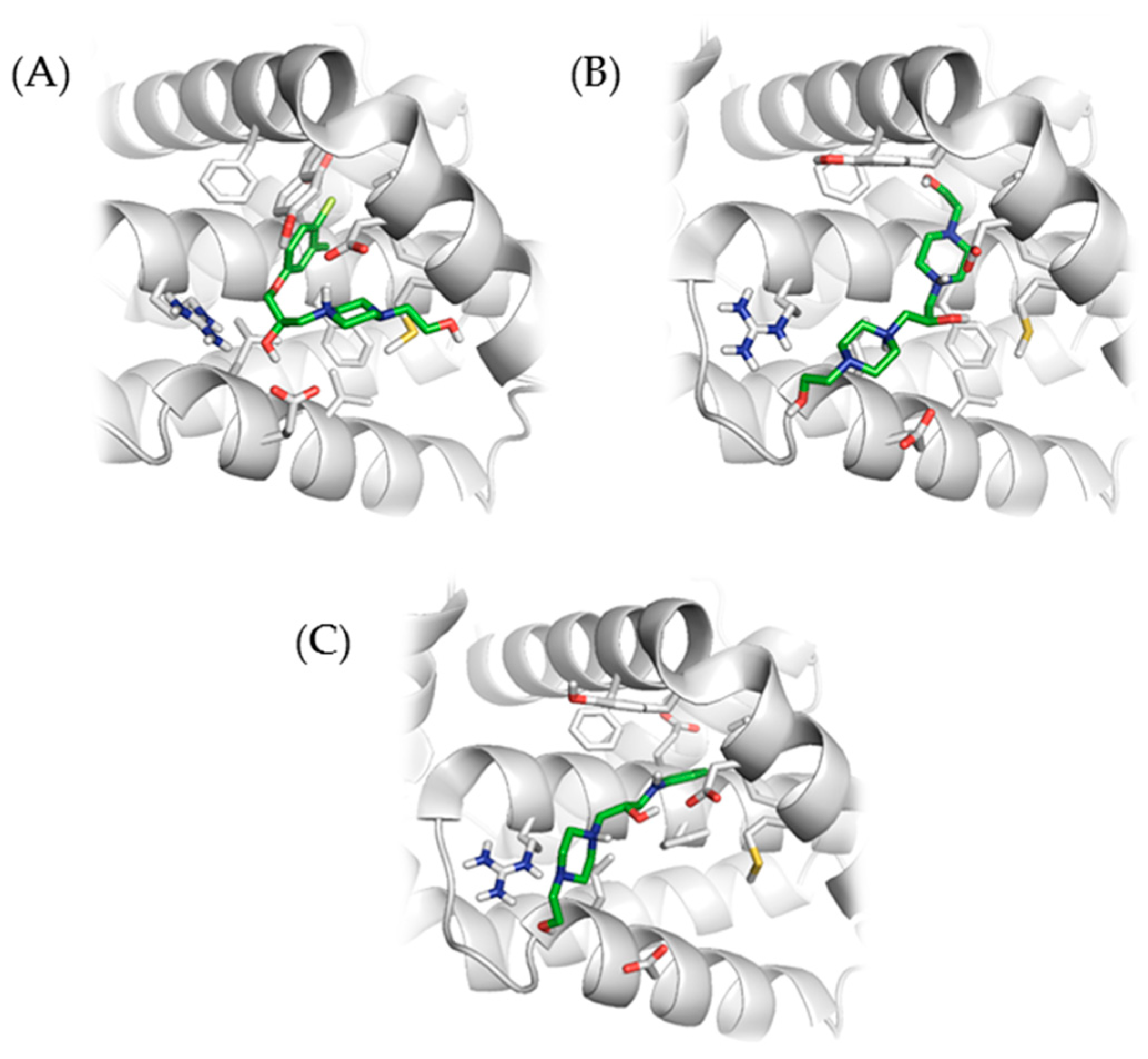
2.2. Molecular Docking with Anti-Apoptotic Protein Bcl-2
2.3. In Vitro Cytotoxicity of Newly Synthesized Compounds 2, 3, 4, 5, 6, 7, 8, 9, and 10
2.4. In Vitro Toxicity Determination of New Inhibitors
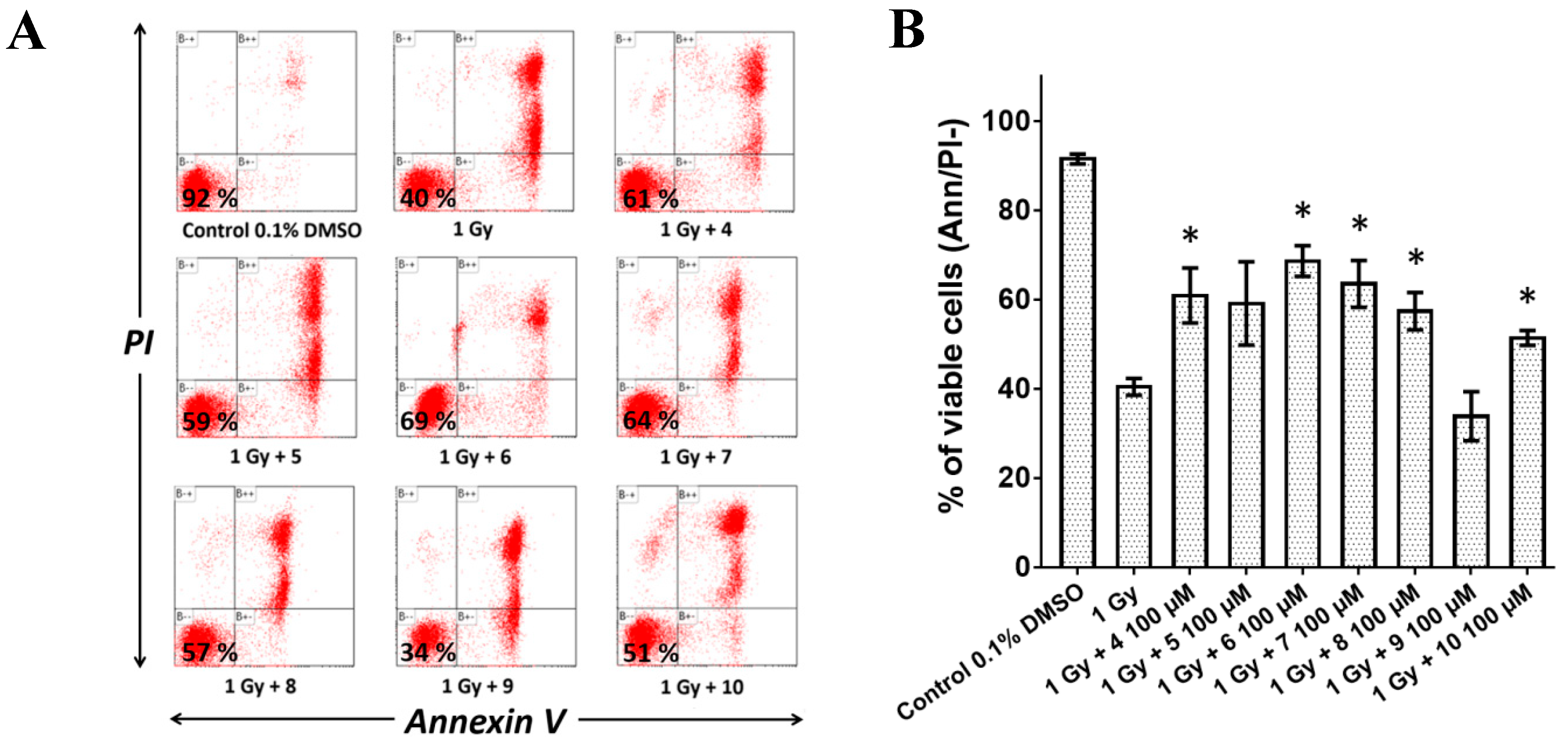
2.5. Pre-Treatment with Compounds 4, 5, 6, 7, 8, and 10 Reduced Radiation-Induced Apoptosis In Vitro
2.6. The Compounds Administered to Mice at MTD Caused No Pathologies
2.7. Pre-Treatment with the Compounds Increased Survival of Whole-Body Irradiated Mice
3. Discussion
4. Experimental Section
4.1. Synthesis and Analysis
4.2. Molecular Docking
4.3. Cell Culture and Treatment with Novel Compounds
4.4. Cytotoxicity of Novel Compounds In Vitro
4.5. Evaluation of Radioprotection In Vitro
4.6. Safe Use Evaluation In Vivo
4.7. Animals and Gamma Radiation
4.8. Preparation of the Novel Compound Solutions and Evaluation of Radioprotection In Vivo
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamamoto, T.; Kinoshita, M. Radioprotective Effect of Vitamin C as an Antioxidant. In Vitamin C.; Hamza, A.H., Ed.; InTech: London, UK, 2017; ISBN 978-953-51-3421-3. [Google Scholar]
- Smith, T.A.; Kirkpatrick, D.R.; Smith, S.; Smith, T.K.; Pearson, T.; Kailasam, A.; Herrmann, K.Z.; Schubert, J.; Agrawal, D.K. Radioprotective agents to prevent cellular damage due to ionizing radiation. J. Trans. Med. 2017, 15, 232. [Google Scholar] [CrossRef]
- Jagetia, G.C. Recent Advances in Indian Herbal Drug Research Guest Editor: Thomas Paul Asir Devasagayam Radioprotective Potential of Plants and Herbs against the Effects of Ionizing Radiation. J. Clin. Biochem. Nutr. 2007, 40, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musa, A.E.; Omyan, G.; Esmaely, F.; Shabeeb, D. Radioprotective Effect of Hesperidin: A Systematic Review. Medicina 2019, 55, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging (Albany NY) 2012, 4, 330–349. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Bures, J.; Jirkovska, A.; Sestak, V.; Jansova, H.; Karabanovich, G.; Roh, J.; Sterba, M.; Simunek, T.; Kovarikova, P. Investigation of novel dexrazoxane analogue JR-311 shows significant cardioprotective effects through topoisomerase IIbeta but not its iron chelating metabolite. Toxicology 2017, 392, 1–10. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Trans. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Mustata, G.; Li, M.; Zevola, N.; Bakan, A.; Zhang, L.; Epperly, M.; Greenberger, J.S.; Yu, J.; Bahar, I. Development of small-molecule PUMA inhibitors for mitigating radiation-induced cell death. Curr. Top. Med. Chem. 2011, 11, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Davis, B.; Chiang, Y.-H.; Filichia, E.; Barnett, A.; Greig, N.H.; Hoffer, B.; Luo, Y. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J. Neurochem. 2016, 138, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Gupta, D.; Chawla, R.; Sagar, R.; Sharma, A.; Kumar, R.; Prasad, J.; Singh, S.; Samanta, N.; Sharma, R.K. Radioprotection by plant products: Present status and future prospects. Phytother. Res. 2005, 19, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.G.; Nair, C.K.K. Radioprotective Effects of Gallic Acid in Mice. BioMed Res. Int. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marek, J.; Tichy, A.; Havelek, R.; Seifrtova, M.; Filipova, A.; Andrejsova, L.; Kučera, T.; Prchal, L.; Muckova, L.; Rezacova, M.; et al. A Novel Class of Small Molecular Inhibitors with Radioprotective Properties. Eur. J. Med. Chem. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Dolezal, R.; Cabelova, P.; Horova, A.; Hruba, E.; Ricny, J.; Sedlacek, L.; Nepovimova, E.; Spilovska, K.; Andrs, M.; et al. 7-MEOTA–donepezil like compounds as cholinesterase inhibitors: Synthesis, pharmacological evaluation, molecular modeling and QSAR studies. Eur. J. Med. Chem. 2014, 82, 426–438. [Google Scholar] [CrossRef]
- PubChem (2R)-1-(4-Chloro-3-methylphenoxy)-3-[4-(2-hydroxyethyl)piperazin-1-yl]propan-2-ol. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/1539752 (accessed on 12 November 2019).
- Dains, F.B.; Brewster, R.Q.; Blair, J.S.; Thompson, W.C. THE SUBSTITUTED THIO-UREAS. III. THE SYNTHESIS OF THIAZOLIDINE AND THIAZANE DERIVATIVES. J. Am. Chem. Soc. 1922, 44, 2637–2643. [Google Scholar] [CrossRef]
- Hutchinson, T.H.; Bögi, C.; Winter, M.J.; Owens, J.W. Benefits of the maximum tolerated dose (MTD) and maximum tolerated concentration (MTC) concept in aquatic toxicology. Aquatic Toxic. 2009, 91, 197–202. [Google Scholar] [CrossRef]
- Thoolen, B.; Maronpot, R.R.; Harada, T.; Nyska, A.; Rousseaux, C.; Nolte, T.; Malarkey, D.E.; Kaufmann, W.; Küttler, K.; Deschl, U.; et al. Proliferative and Nonproliferative Lesions of the Rat and Mouse Hepatobiliary System. Toxicol. Pathol. 2010, 38, 5S–81S. [Google Scholar] [CrossRef]
- Ghosh, S.P.; Perkins, M.W.; Hieber, K.; Kulkarni, S.; Kao, T.-C.; Reddy, E.P.; Reddy, M.V.R.; Maniar, M.; Seed, T.; Kumar, K.S. Radiation protection by a new chemical entity, Ex-Rad: Efficacy and mechanisms. Radiat. Res. 2009, 171, 173–179. [Google Scholar] [CrossRef]
- Ghosh, S.P.; Kulkarni, S.; Perkins, M.W.; Hieber, K.; Pessu, R.L.; Gambles, K.; Maniar, M.; Kao, T.-C.; Seed, T.M.; Kumar, K.S. Amelioration of radiation-induced hematopoietic and gastrointestinal damage by Ex-RAD(R) in mice. J. Radia. Res. 2012, 53, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [Green Version]
- Hosseinimehr, S.J.; Shafiee, A.; Mozdarani, H.; Akhlagpour, S.; Froughizadeh, M. Radioprotective effects of 2-imino-3-[(chromone-2-yl)carbonyl] thiazolidines against gamma-irradiation in mice. J. Radiat. Res. 2002, 43, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun, G.-I.; Kim, S.; Choi, E.; Kim, C.S.; Lee, Y.-S. Pharmacology of natural radioprotectors. Arch. Pharma. Res. 2018, 41, 1033–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, V.; Pendergrass, J.A.; Kumar, K.S.; Landauer, M.R.; Seed, T.M. Radioprotection, pharmacokinetic and behavioural studies in mouse implanted with biodegradable drug (amifostine) pellets. Int. J. Radiat. Biol. 2002, 78, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Cassatt, D.R.; Fazenbaker, C.A.; Kifle, G.; Bachy, C.M. Effects of dose and schedule on the efficacy of ethyol: Preclinical studies. Semin. Oncol. 2003, 30, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Tichy, A.; Marek, J.; Havelek, R.; Pejchal, J.; Seifrtova, M.; Zarybnicka, L.; Filipova, A.; Rezacova, M.; Sinkorova, Z. New Light on An Old Friend: Targeting PUMA in Radioprotection and Therapy of Cardiovascular and Neurodegenerative Diseases. Curr. Drug Targets 2018, 19, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muckova, L.; Pejchal, J.; Jost, P.; Vanova, N.; Herman, D.; Jun, D. Cytotoxicity of acetylcholinesterase reactivators evaluated in vitro and its relation to their structure. Drug Chem. Toxicol. 2019, 42, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Misik, J.; Nepovimova, E.; Pejchal, J.; Kassa, J.; Korabecny, J.; Soukup, O. Cholinesterase Inhibitor 6-Chlorotacrine - In vivo Toxicological Profile and Behavioural Effects. Curr. Alzheimer Res. 2018, 15, 552–560. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2, 3, 4, 5, 6, 7, 8, 9 and 10 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Yield (%) | Melting Point (°C) | *ClogP |
|---|---|---|---|
| 2 | 78 | 172–173 | 2.255 |
| 3 | 75 | 157–158 | 1.948 |
| 4 | 79 | 189–190 | 1.478 |
| 5 | 68 | 185–186 | 0.682 |
| 6 | 69 | 181–182 | 1.671 |
| 7 | 61 | 182–183 | 0.524 |
| 8 | 27 | oil | −2.107 |
| 9 | 25 | oil | 2.090 |
| 10 | 90 | 154–155 | −0.009 |
| (A) | ||||||||||
| Cell line | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | DOX |
| Jurkat | 31 ± 14 | 95 ± 9 | 105 ± 5 | 98 ± 4 | 101 ± 1 | 100 ± 5 | 103 ± 5 | 104 ± 5 | 108 ± 7 | 2 ± 0 |
| MOLT-4 | 0 ± 0 | 81 ± 13 | 88 ± 6 | 93 ± 9 | 90 ± 7 | 93 ± 3 | 106 ± 8 | 103 ± 8 | 112 ± 6 | 1 ± 0 |
| A549 | 27 ± 3 | 94 ± 15 | 100 ± 4 | 86 ± 7 | 103 ± 7 | 95 ± 5 | 97 ± 5 | 107 ± 11 | 105 ± 9 | 42 ± 11 |
| HT-29 | 28 ± 6 | 85 ± 11 | 100 ± 2 | 91 ± 2 | 86 ± 4 | 96 ± 3 | 101 ± 1 | 102 ± 5 | 104 ± 11 | 55 ± 18 |
| PANC-1 | 39 ± 2 | 78 ± 6 | 97 ± 7 | 86 ± 16 | 92 ± 11 | 99 ± 10 | 98 ± 9 | 93 ± 12 | 95 ± 7 | 78 ± 7 |
| A2780 | 44 ± 9 | 84 ± 7 | 98 ± 2 | 93 ± 5 | 74 ± 3 | 94 ± 3 | 104 ± 4 | 90 ± 3 | 103 ± 6 | 8 ± 4 |
| HeLa | 25 ± 1 | 104 ± 13 | 106 ± 2 | 100 ± 4 | 108 ± 6 | 96 ± 8 | 103 ± 4 | 99 ± 9 | 110 ± 7 | 76 ± 12 |
| MCF-7 | 15 ± 1 | 84 ± 9 | 105 ± 4 | 101 ± 6 | 102 ± 8 | 105 ± 6 | 101 ± 5 | 100 ± 4 | 98 ± 2 | 32 ± 3 |
| SAOS-2 | 28 ± 2 | 80 ± 4 | 99 ± 6 | 96 ± 4 | 94 ± 6 | 102 ± 7 | 102 ± 6 | 106 ± 2 | 97 ± 6 | 22 ± 4 |
| MRC-5 | 23 ± 2 | 84 ± 2 | 97 ± 8 | 98 ± 7 | 91 ± 4 | 100 ± 3 | 95 ± 7 | 102 ± 8 | 102 ± 3 | 22 ± 2 |
| (B) | ||||||||||
| Cell line | 2 - not tested | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | DOX |
| Jurkat | x | 12 ± 5 | 89 ± 1 | 98 ± 3 | 93 ± 2 | 96 ± 0 | 96 ± 13 | 79 ± 11 | 95 ± 4 | 2 ± 0 |
| MOLT-4 | x | 2 ± 0 | 92 ± 6 | 104 ± 7 | 112 ± 4 | 99 ± 3 | 81 ± 6 | 71 ± 4 | 100 ± 4 | 1 ± 0 |
| A549 | x | 13 ± 2 | 99 ± 0 | 104 ± 8 | 93 ± 3 | 115 ± 0 | 111 ± 11 | 98 ± 13 | 105 ± 2 | 42 ± 11 |
| HT-29 | x | 14 ± 5 | 83 ± 4 | 109 ± 2 | 97 ± 4 | 118 ± 2 | 154 ± 11 | 148 ± 2 | 113 ± 2 | 55 ± 18 |
| PANC-1 | x | 30 ± 4 | 111 ± 24 | 115 ± 7 | 111 ± 5 | 116 ± 3 | 117 ± 11 | 118 ± 5 | 111 ± 6 | 78 ± 7 |
| A2780 | x | 20 ± 5 | 89 ± 1 | 105 ± 8 | 95 ± 4 | 91 ± 2 | 76 ± 10 | 64 ± 5 | 96 ± 2 | 8 ± 4 |
| HeLa | x | 11 ± 3 | 137 ± 5 | 116 ± 10 | 104 ± 7 | 132 ± 3 | 100 ± 18 | 90 ± 14 | 121 ± 3 | 76 ± 12 |
| MCF-7 | x | 10 ± 2 | 90 ± 3 | 106 ± 1 | 94 ± 9 | 104 ± 6 | 96 ± 7 | 98 ± 11 | 96 ± 6 | 32 ± 3 |
| SAOS-2 | x | 10 ± 2 | 88 ± 4 | 111 ± 6 | 111 ± 4 | 109 ± 2 | 87 ± 7 | 90 ± 4 | 102 ± 2 | 22 ± 4 |
| MRC-5 | x | 9 ± 6 | 93 ± 5 | 102 ± 8 | 94 ± 2 | 104 ± 3 | 119 ± 16 | 120 ± 16 | 102 ± 8 | 22 ± 2 |
| Compounds | IC50 ± SEM (mM) | MTC (mM) |
|---|---|---|
| 2 | 0.04 ± 0.002 | 0.002 |
| 3 | 0.14 ± 0.01 | 0.01 |
| 4 | 0.64 ± 0.03 | 0.05 |
| 5 | 1.35 ± 0.12 | 0.05 |
| 6 | 0.45 ± 0.01 | 0.02 |
| 7 | 2.55 ± 0.21 | 0.39 |
| 8 | > 25 | 6.25 |
| 9 | 0.24 ± 0.01 | 0.09 |
| 10 | 5.06 ± 0.23 | 1.56 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipova, A.; Marek, J.; Havelek, R.; Pejchal, J.; Jelicova, M.; Cizkova, J.; Majorosova, M.; Muckova, L.; Kucera, T.; Prchal, L.; et al. Substituted Piperazines as Novel Potential Radioprotective Agents. Molecules 2020, 25, 532. https://doi.org/10.3390/molecules25030532
Filipova A, Marek J, Havelek R, Pejchal J, Jelicova M, Cizkova J, Majorosova M, Muckova L, Kucera T, Prchal L, et al. Substituted Piperazines as Novel Potential Radioprotective Agents. Molecules. 2020; 25(3):532. https://doi.org/10.3390/molecules25030532
Chicago/Turabian StyleFilipova, Alzbeta, Jan Marek, Radim Havelek, Jaroslav Pejchal, Marcela Jelicova, Jana Cizkova, Martina Majorosova, Lubica Muckova, Tomas Kucera, Lukas Prchal, and et al. 2020. "Substituted Piperazines as Novel Potential Radioprotective Agents" Molecules 25, no. 3: 532. https://doi.org/10.3390/molecules25030532