SSEA3 and Sialyl Lewis a Glycan Expression Is Controlled by B3GALT5 LTR through Lamin A-NFYA and SIRT1-STAT3 Signaling in Human ES Cells

 ,
,  ,
,
Abstract
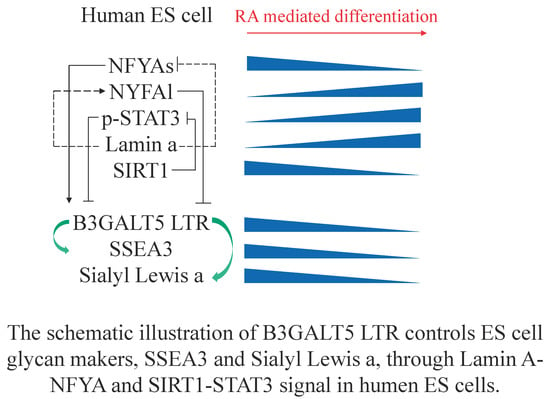
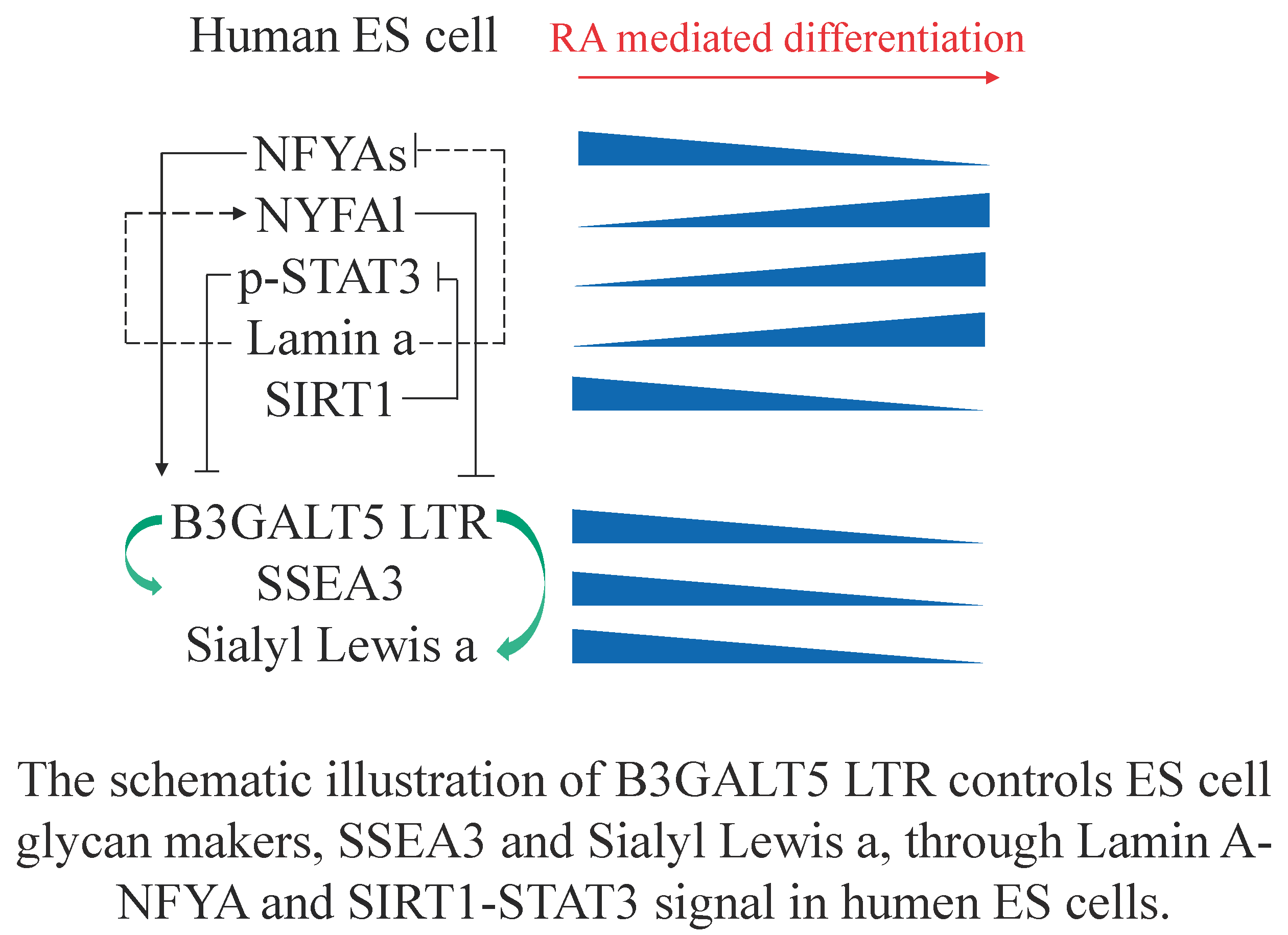
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Cell Culture, and Drug Treatment
2.2. Flow Cytometry
2.3. Plasmid Construction
2.4. Transfection of Cells with Plasmids or Short Interfering RNAs (siRNAs)
2.5. Luciferase Assay
2.6. Western Blotting
2.7. ChIP Assay
2.8. Real-Time Quantitative PCR and Primers
2.9. Statistical Analysis
3. Results
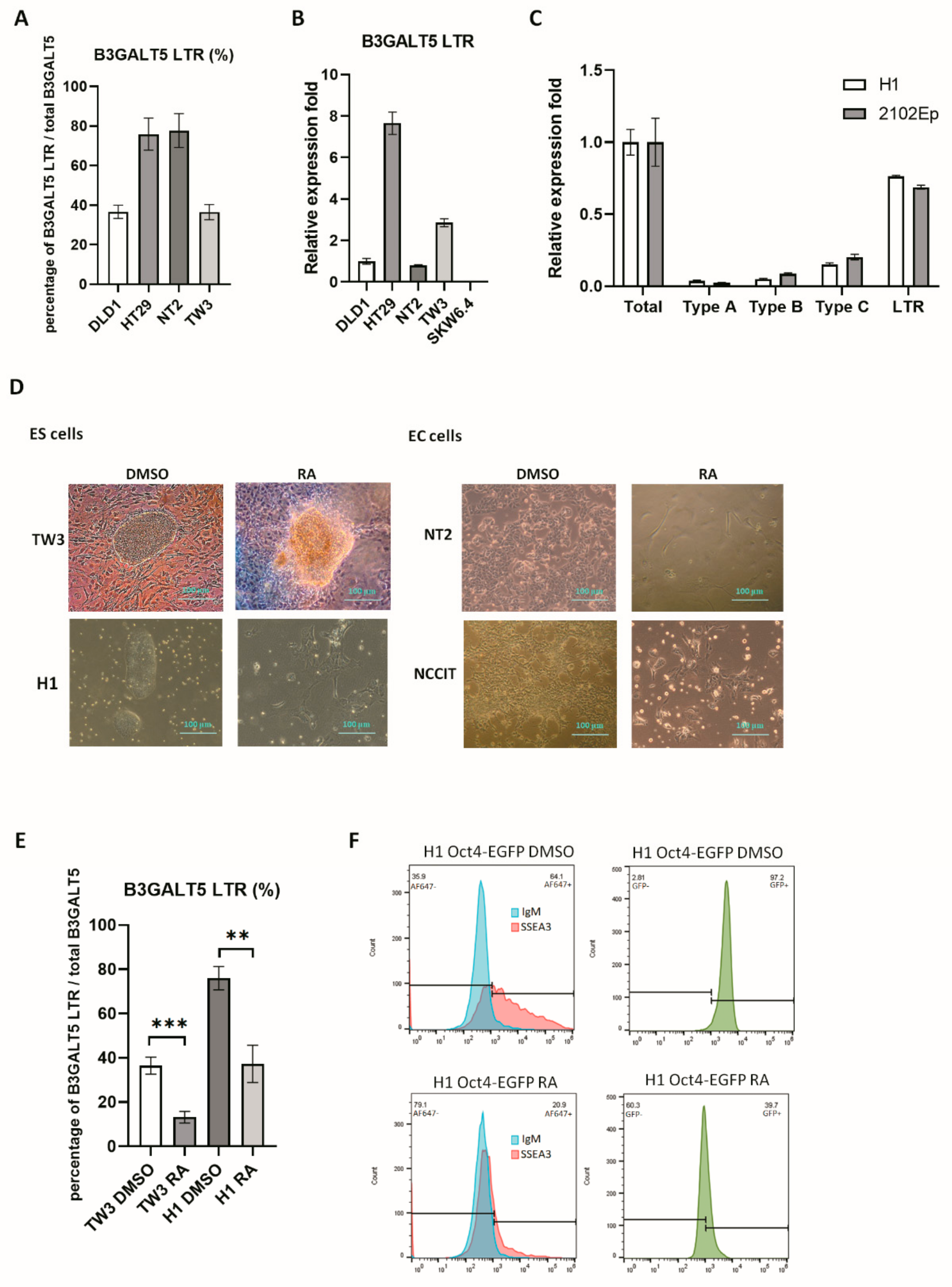
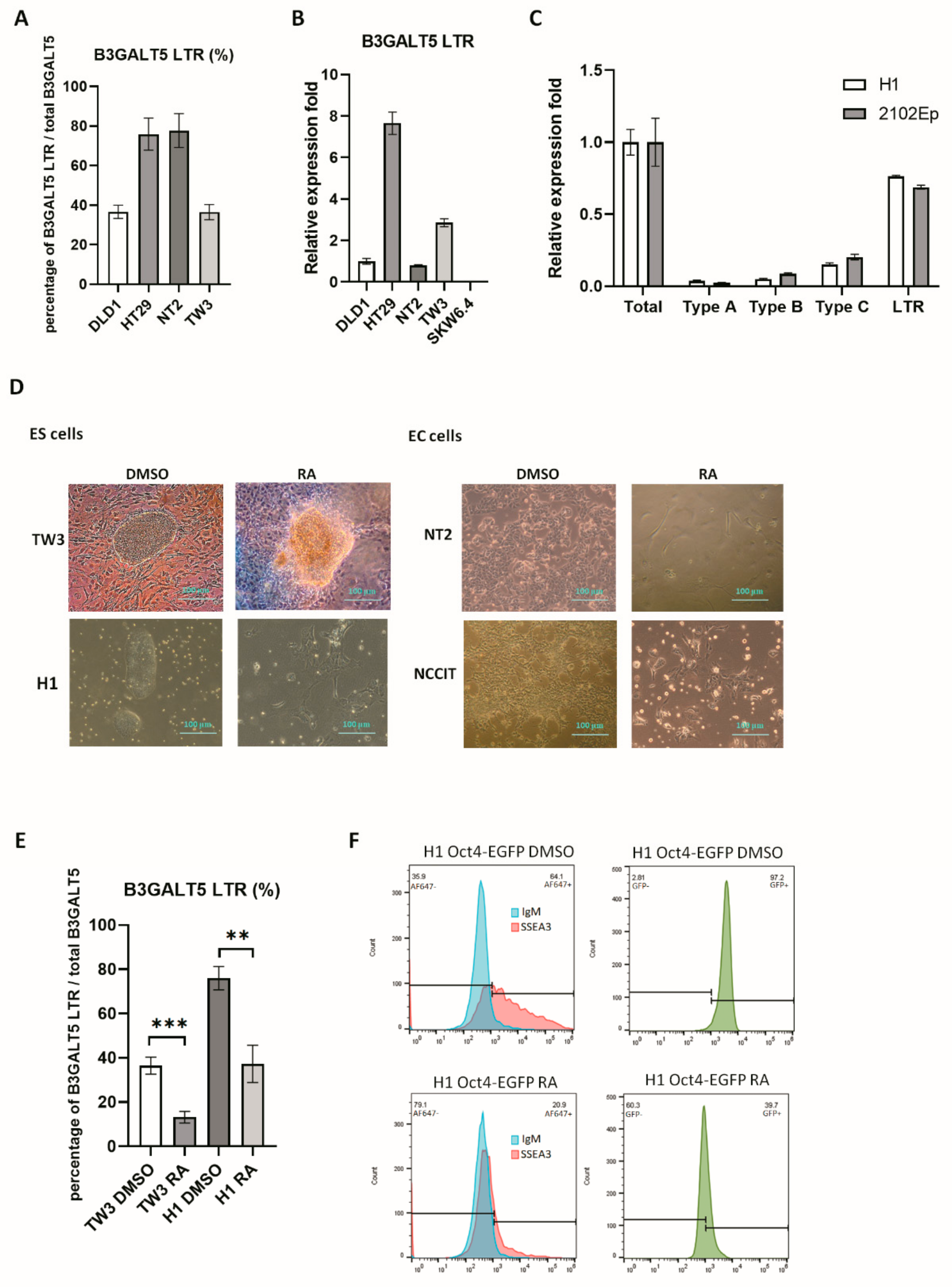
3.1. B3GALT5-LTR is Highly Expressed in ES and EC Cells
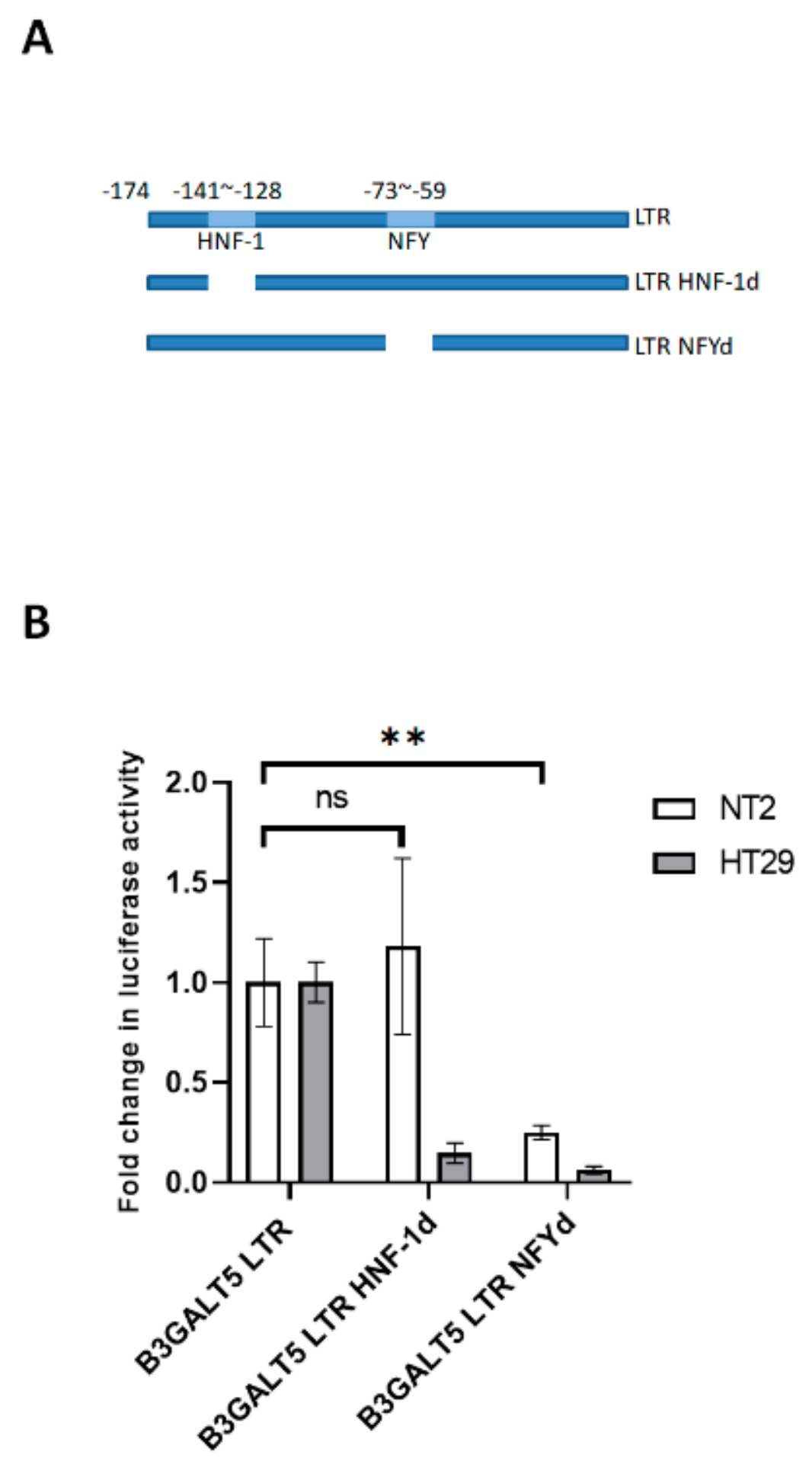
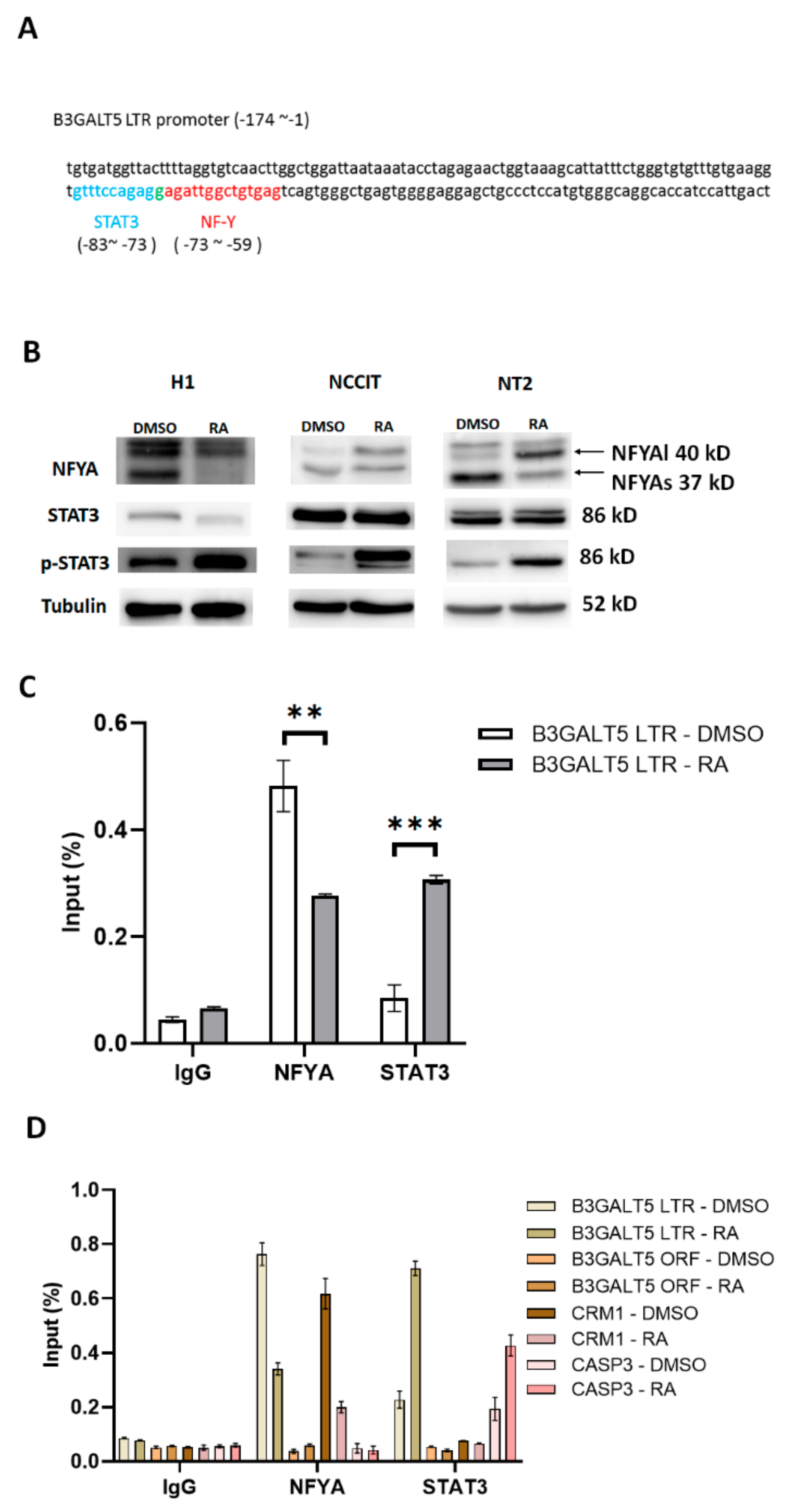
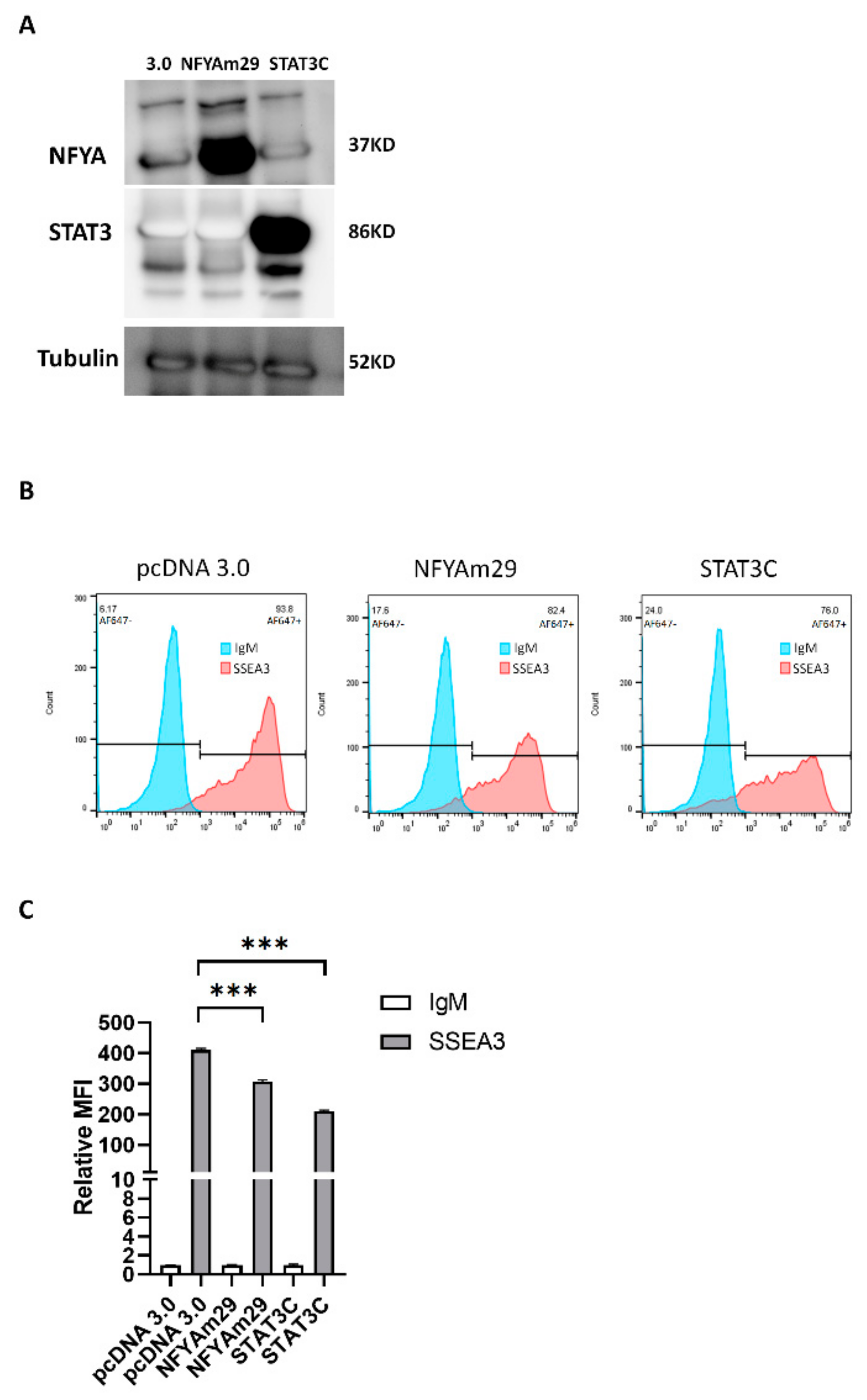
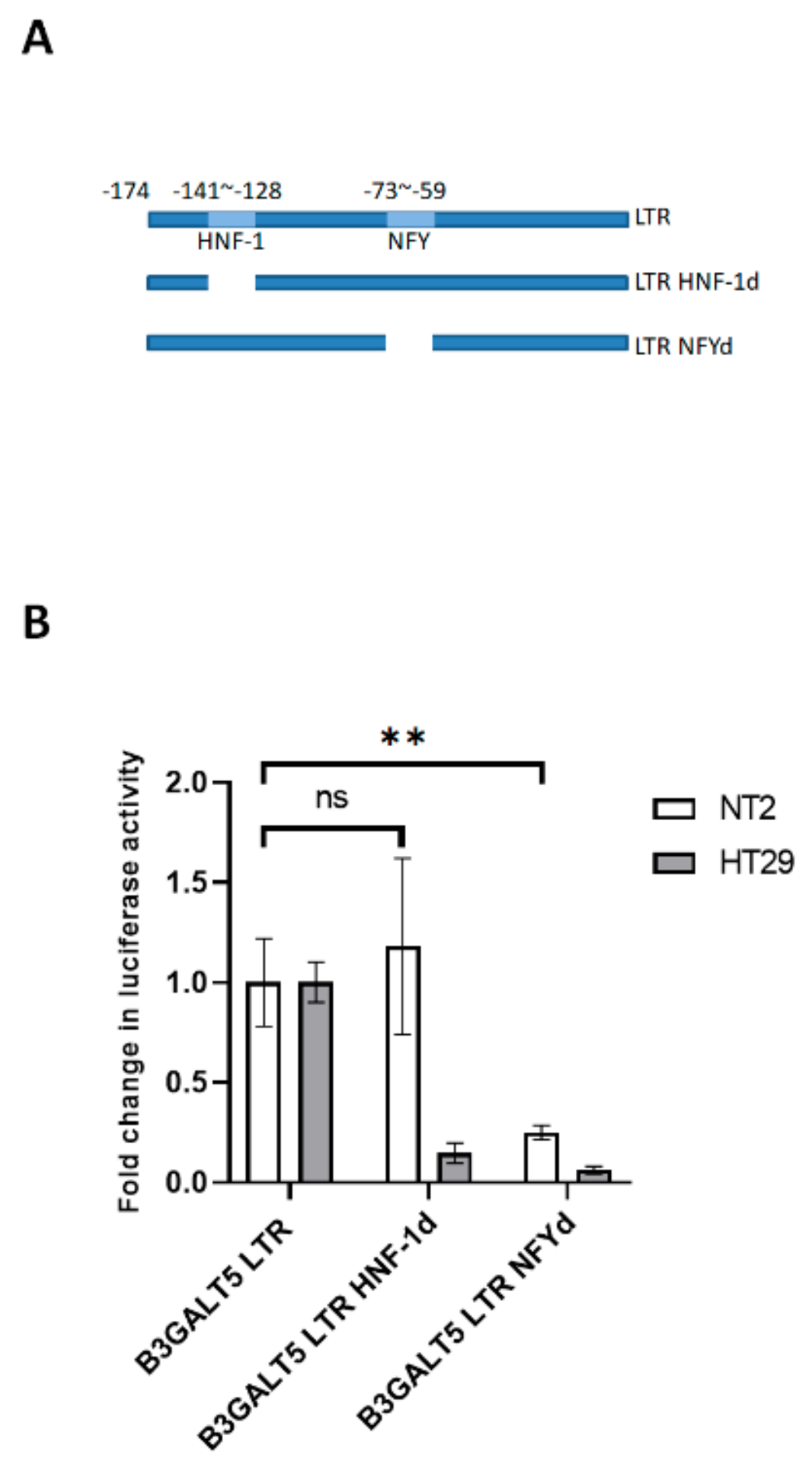
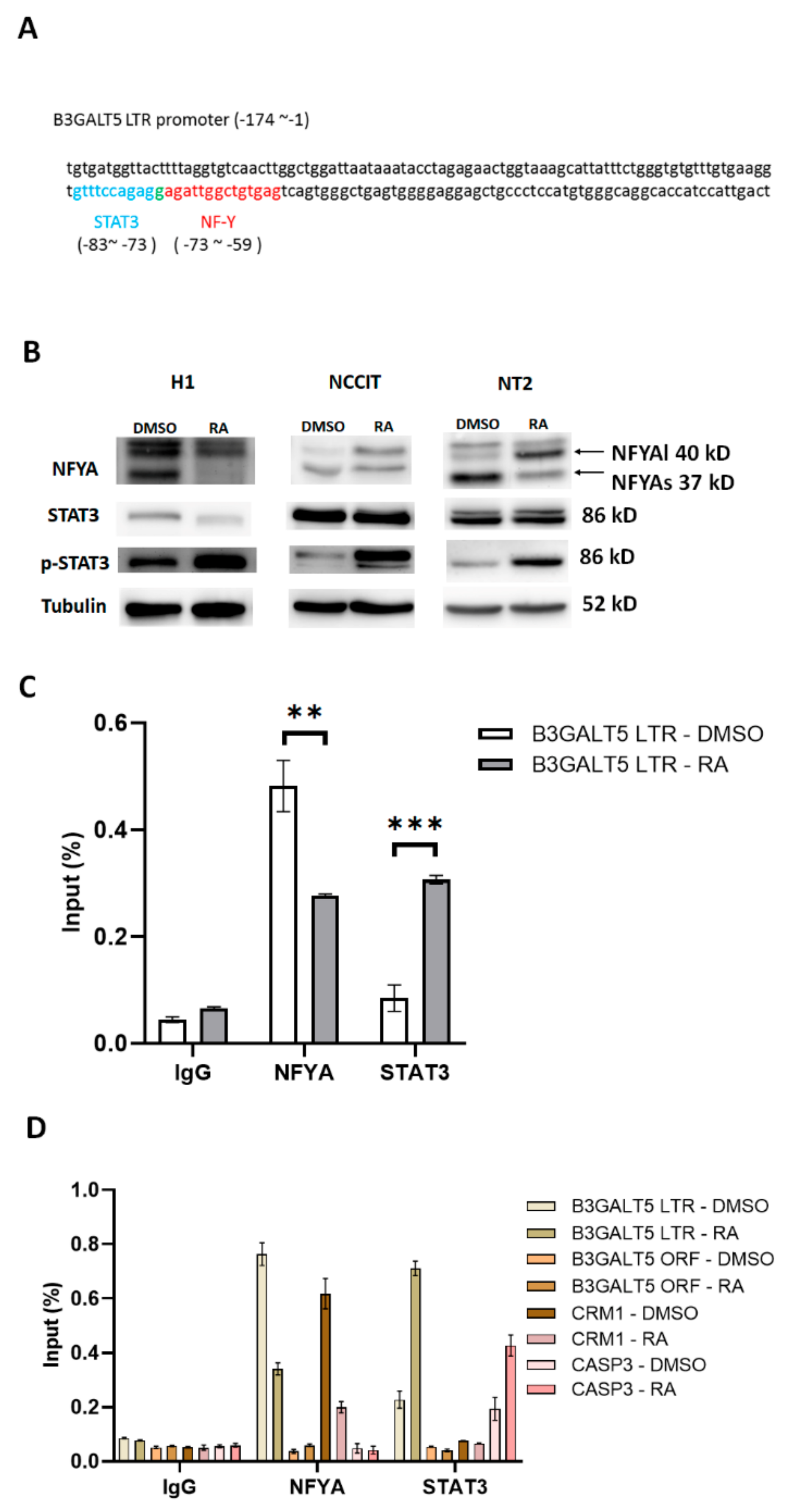
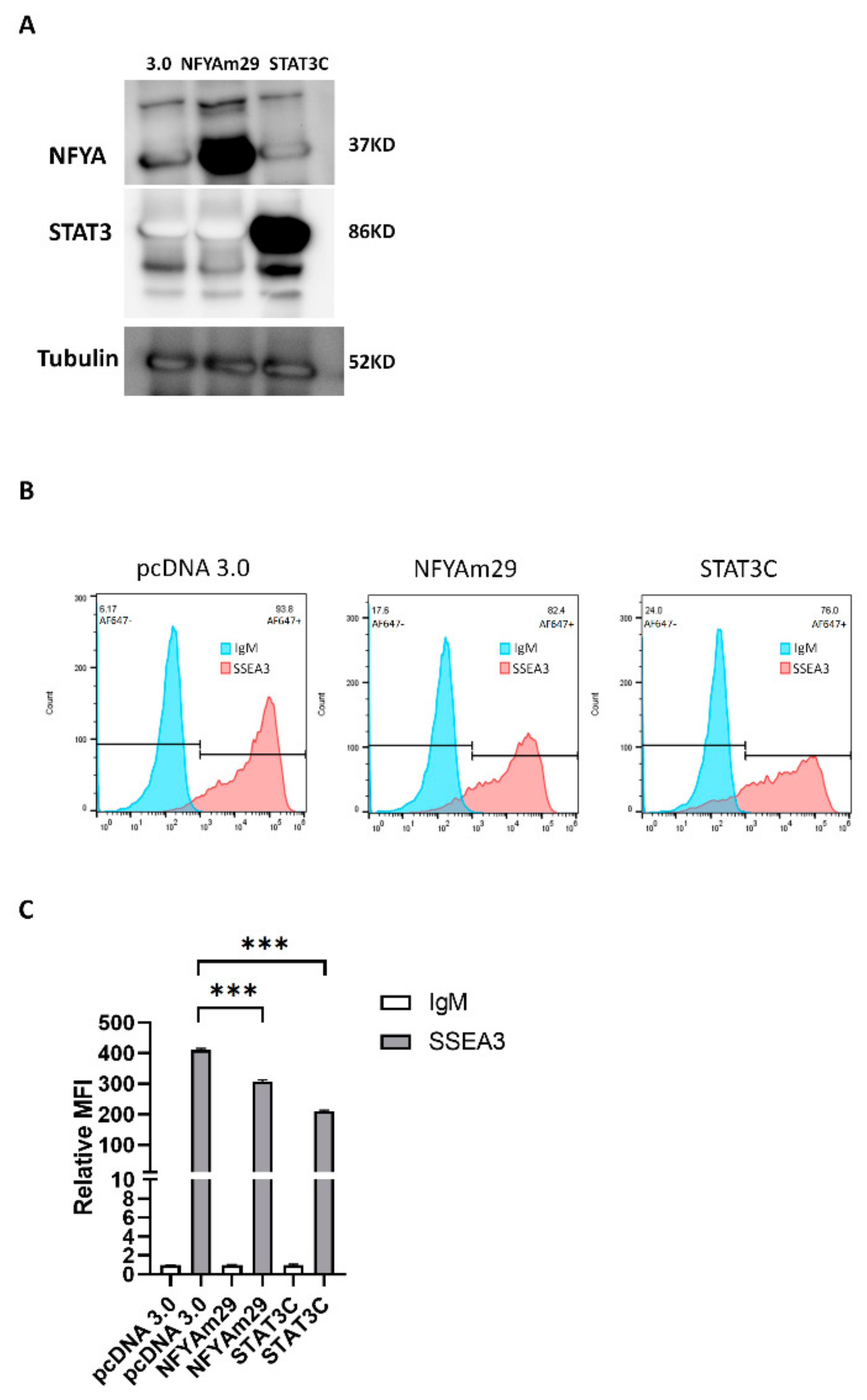
3.2. NFYA and STAT3, but Not HNF-1, Are the Transcription Factors that Regulate B3GALT5-LTR Expression in ES and EC Cells
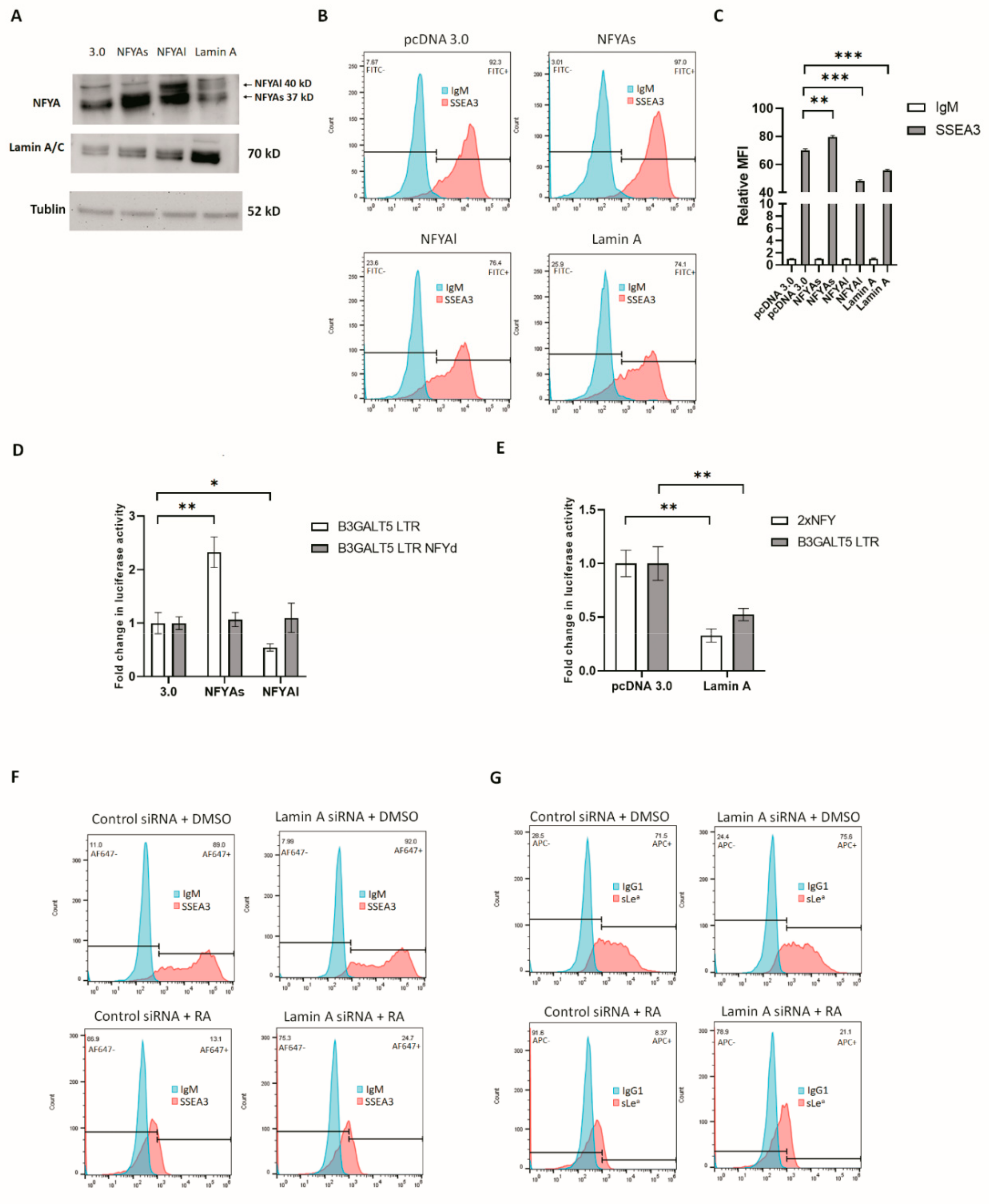
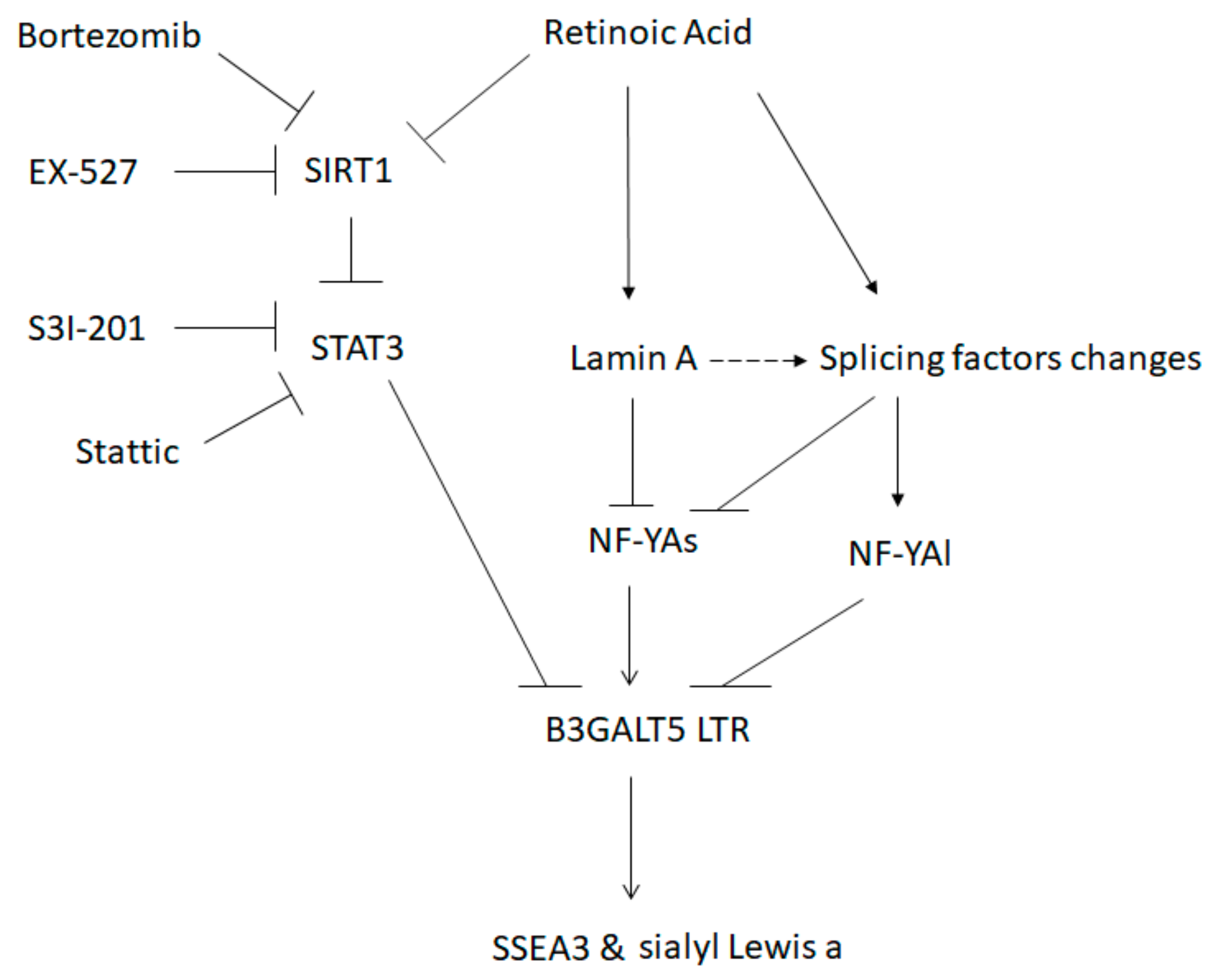
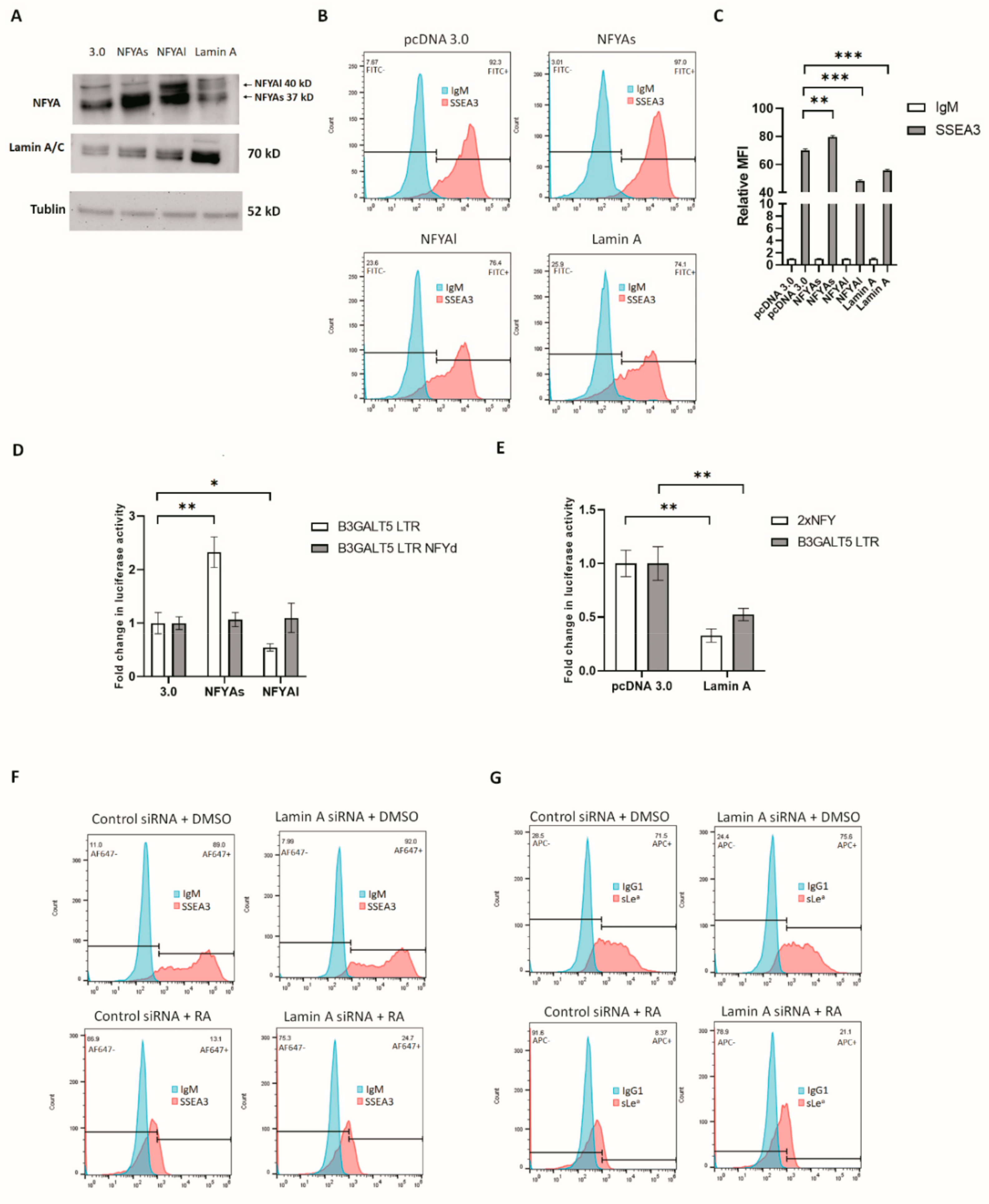
3.3. RA-Mediated Lamin A-NFYA Pathway Regulates B3GALT5-LTR Promoter and Represses Production of SSEA3 and Sialyl Lewis a
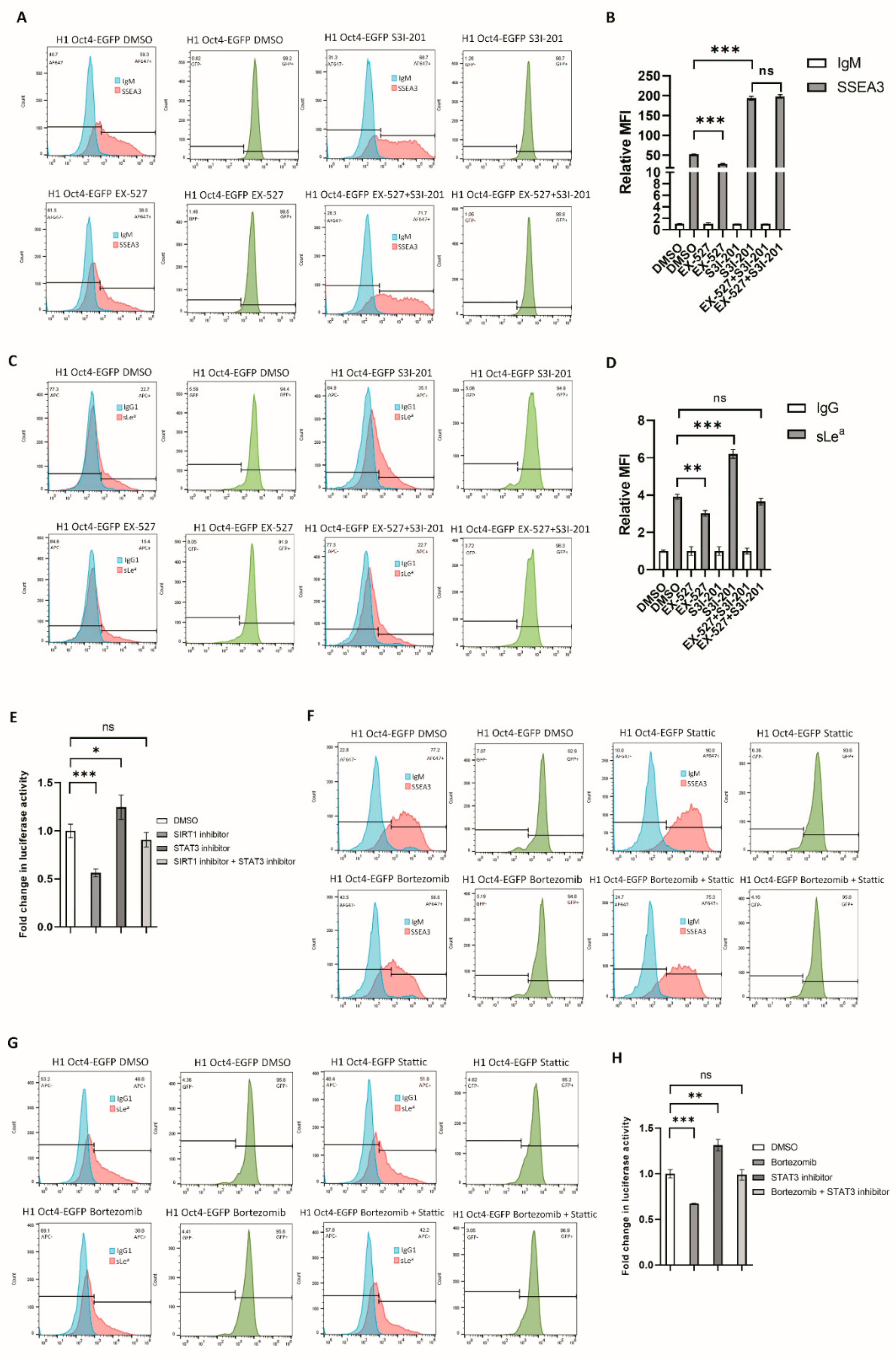
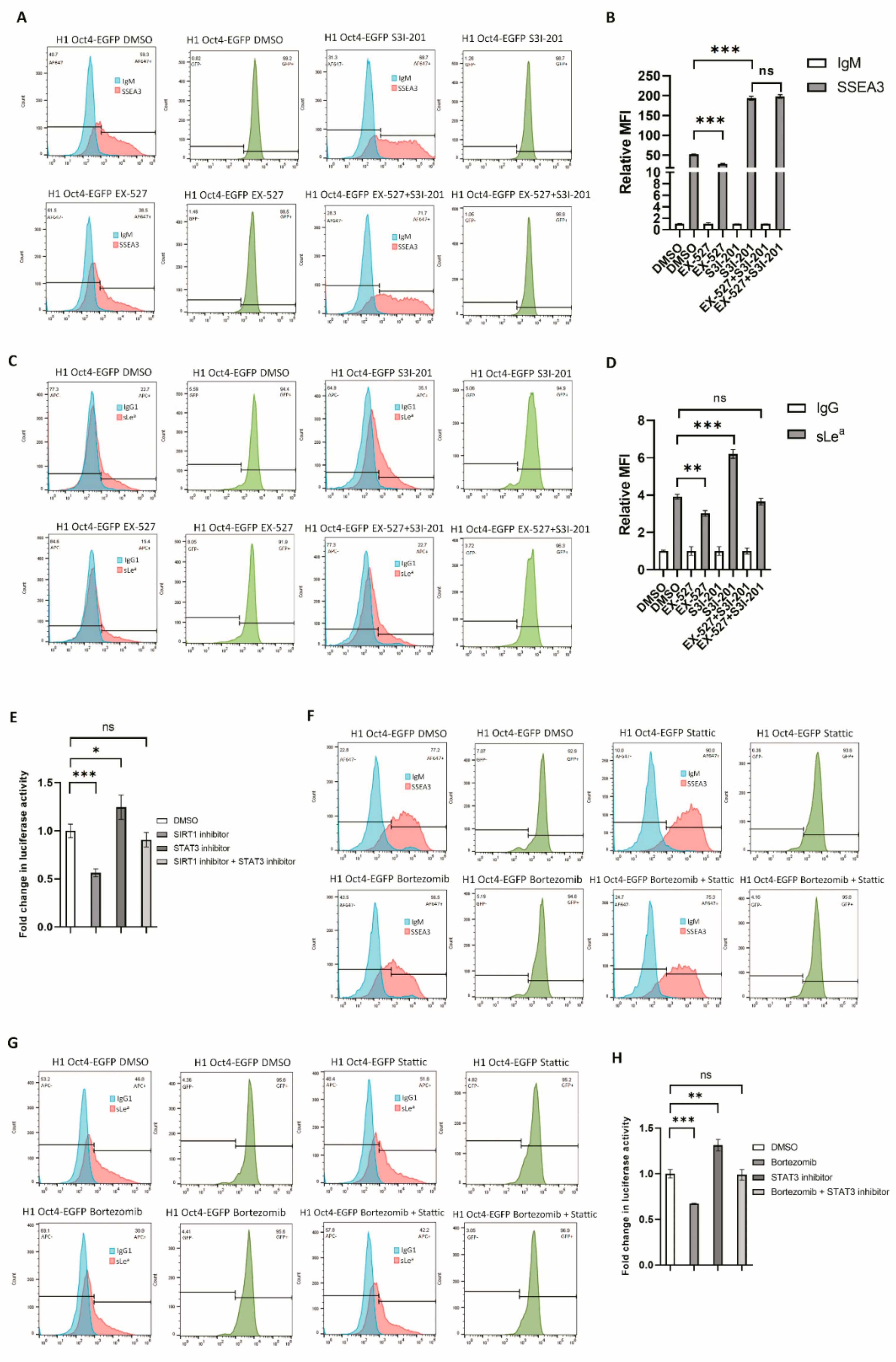
3.4. RA-Mediated SIRT1-STAT3 Pathway Regulates B3GALT5-LTR Promoter Activity and Thereby Represses SSEA3 and Sialyl Lewis a Synthesis
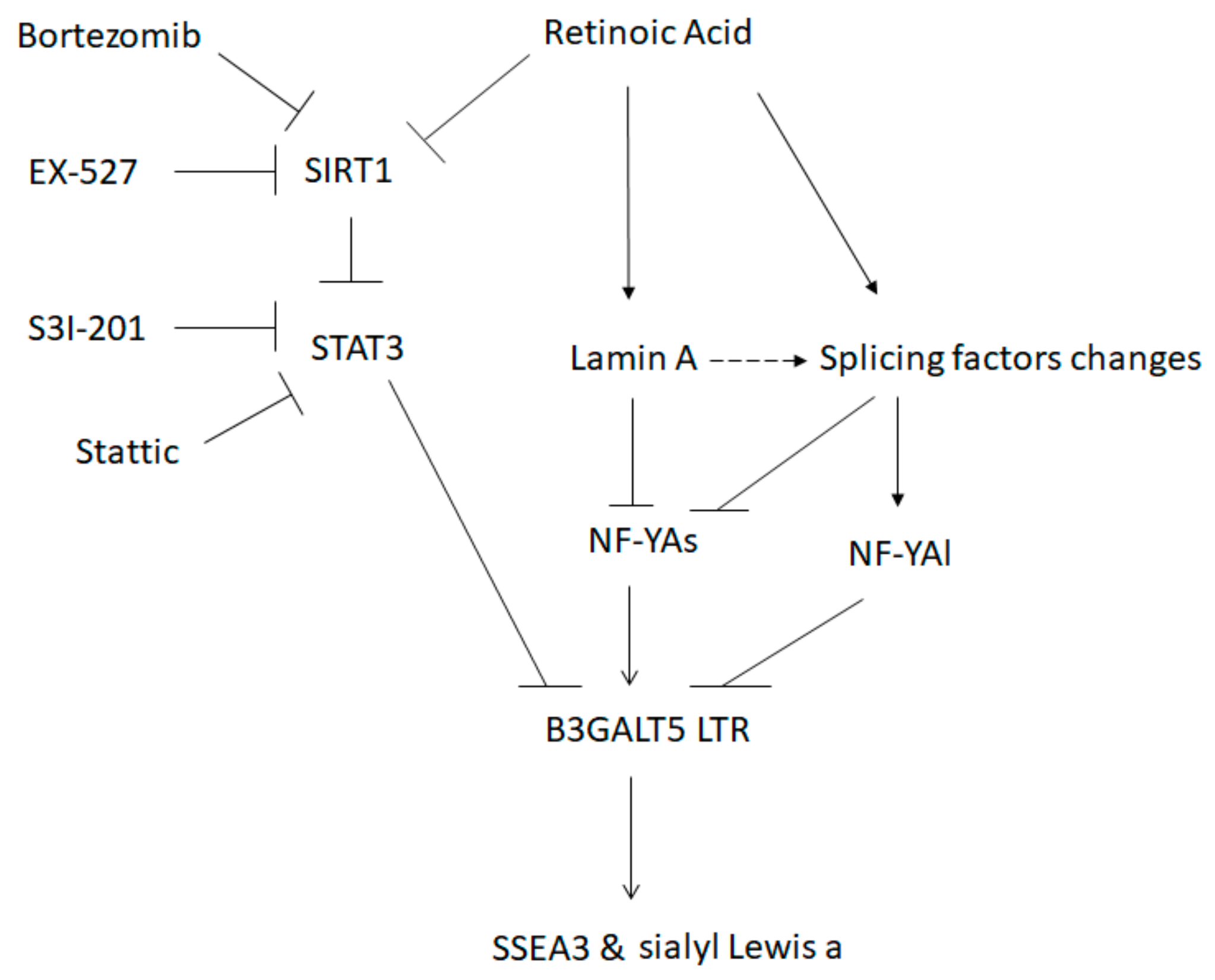
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kannagi, R.; Cochran, N.A.; Ishigami, F.; Hakomori, S.; Andrews, P.W.; Knowles, B.B.; Solter, D. Stage-specific embryonic antigens (SSEA-3 and -4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J. 1983, 2, 2355–2361. [Google Scholar] [CrossRef]
- Zhou, D.; Henion, T.R.; Jungalwala, F.B.; Berger, E.G.; Hennet, T. The beta 1,3-galactosyltransferase beta 3GalT-V is a stage-specific embryonic antigen-3 (SSEA-3) synthase. J. Biol. Chem. 2000, 275, 22631–22634. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.J.; Kuo, H.H.; Lin, C.H.; Chen, Y.Y.; Yang, B.C.; Cheng, Y.Y.; Yu, A.L.; Khoo, K.H.; Yu, J. Switching of the core structures of glycosphingolipids from globo- and lacto- to ganglio-series upon human embryonic stem cell differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 22564–22569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.H.; Lin, R.J.; Hung, J.T.; Hsieh, C.B.; Hung, T.H.; Lo, F.Y.; Ho, M.Y.; Yeh, C.T.; Huang, Y.L.; Yu, J.; et al. High expression FUT1 and B3GALT5 is an independent predictor of postoperative recurrence and survival in hepatocellular carcinoma. Sci. Rep. 2017, 7, 10750. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, S.; Togayachi, A.; Kudo, T.; Nishihara, S.; Watanabe, M.; Kubota, T.; Kitajima, M.; Shiraishi, N.; Sasaki, K.; Andoh, T.; et al. Cloning, expression, and characterization of a novel UDP-galactose:beta-N-acetylglucosamine beta1,3-galactosyltransferase (beta3Gal-T5) responsible for synthesis of type 1 chain in colorectal and pancreatic epithelia and tumor cells derived therefrom. J. Biol. Chem. 1999, 274, 12499–12507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, K.; Aoki, M.; Kannagi, R. Transcription factors c-Myc and CDX2 mediate E-selectin ligand expression in colon cancer cells undergoing EGF/bFGF-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2012, 109, 7776–7781. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Lee, A.S.; Volkmer, J.P.; Sahoo, D.; Nag, D.; Mosley, A.R.; Inlay, M.A.; Ardehali, R.; Chavez, S.L.; Pera, R.R.; et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011, 29, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Chuang, P.K.; Hsiao, M.; Hsu, T.L.; Chang, C.F.; Wu, C.Y.; Chen, B.R.; Huang, H.W.; Liao, K.S.; Chen, C.C.; Chen, C.L.; et al. Signaling pathway of globo-series glycosphingolipids and β1,3-galactosyltransferase V (β3GalT5) in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3518–3523. [Google Scholar] [CrossRef] [Green Version]
- Engle, D.D.; Tiriac, H.; Rivera, K.D.; Pommier, A.; Whalen, S.; Oni, T.E.; Alagesan, B.; Lee, E.J.; Yao, M.A.; Lucito, M.S.; et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019, 364, 1156–1162. [Google Scholar] [CrossRef]
- Mare, L.; Trinchera, M. Comparative analysis of retroviral and native promoters driving expression of beta1,3-galactosyltransferase beta3Gal-T5 in human and mouse tissues. J. Biol. Chem. 2007, 282, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Dunn, C.A.; Medstrand, P.; Mager, D.L. An endogenous retroviral long terminal repeat is the dominant promoter for human beta1,3-galactosyltransferase 5 in the colon. Proc. Natl. Acad. Sci. USA 2003, 100, 12841–12846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.A.; van de Lagemaat, L.N.; Baillie, G.J.; Mager, D.L. Endogenous retrovirus long terminal repeats as ready-to-use mobile promoters: The case of primate beta3GAL-T5. Gene 2005, 364, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Nguyen, H.N.; Reijo Pera, R.A. Enhanced generation of induced pluripotent stem cells from a subpopulation of human fibroblasts. PLoS ONE 2009, 4, e7118. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.O.; Moon, S.H.; Jeong, H.C.; Yi, J.Y.; Lee, T.H.; Shim, S.H.; Rhee, Y.H.; Lee, S.H.; Oh, S.J.; Lee, M.Y.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E3281–E3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, S.; Narimatsu, H.; Iwasaki, H.; Yazawa, S.; Akamatsu, S.; Ando, T.; Seno, T.; Narimatsu, I. Molecular genetic analysis of the human Lewis histo-blood group system. J. Biol. Chem. 1994, 269, 29271–29278. [Google Scholar] [PubMed]
- Henry, S.M.; Oriol, R.; Samuelsson, B.E. Detection and Characterization of Lewis Antigens in Plasma of Lewis-Negative Individuals Evidence of Chain Extension as a Result of Reduced Fucosyltransferase Competition. Vox Sang. 1994, 67, 387–396. [Google Scholar] [CrossRef]
- Lin, M.; Shieh, S.H. Postnatal development of red cell Le(a) and Le(b) antigens in Chinese infants. Vox Sang. 1994, 66, 137–140. [Google Scholar] [CrossRef]
- Yu, L.C.; Yang, Y.H.; Broadberry, R.E.; Chen, Y.H.; Chan, Y.S.; Lin, M. Correlation of a missense mutation in the human Secretor alpha 1,2-fucosyltransferase gene with the Lewis(a+b+) phenotype: A potential molecular basis for the weak Secretor allele (Sew). Biochem. J. 1995, 312, 329–332. [Google Scholar] [CrossRef]
- Luo, G.; Guo, M.; Jin, K.; Liu, Z.; Liu, C.; Cheng, H.; Lu, Y.; Long, J.; Liu, L.; Xu, J.; et al. Optimize CA19-9 in detecting pancreatic cancer by Lewis and Secretor genotyping. Pancreatology 2016, 16, 1057–1062. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Luo, G.; Lu, R.; Shi, W.; Cheng, H.; Lu, Y.; Jin, K.; Yang, C.; Wang, Z.; Long, J.; et al. Distribution of Lewis and Secretor polymorphisms and corresponding CA19-9 antigen expression in a Chinese population. FEBS Open Bio 2017, 7, 1660–1671. [Google Scholar] [CrossRef]
- Narimatsu, H. Molecular biology of Lewis antigens--histo-blood type antigens and sialyl Lewis antigens as tumor associated antigens. Nihon Geka Gakkai Zasshi 1996, 97, 115–122. [Google Scholar]
- Cheng, E.H.; Chen, W.; Chang, S.Y.; Huang, J.J.; Huang, C.C.; Huang, L.S.; Liu, C.H.; Lee, M.S. Blastocoel volume is related to successful establishment of human embryonic stem cell lines. Reprod. Biomed. Online 2008, 17, 436–444. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Thomson, J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003, 21, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Chen, J.Y.; Lu, M.H.; Chang, L.T.; Lin, H.C.; Chang, Y.M.; Chao, C.F. Functional four-base A/T gap core sequence CATTAG of P53 response elements specifically bound tetrameric P53 differently than two-base A/T gap core sequence CATG bound both dimeric and tetrameric P53. Nucleic Acids Res. 2009, 37, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Brimble, S.N.; Sherrer, E.S.; Uhl, E.W.; Wang, E.; Kelly, S.; Merrill, A.H.; Robins, A.J.; Schulz, T.C. The cell surface glycosphingolipids SSEA-3 and SSEA-4 are not essential for human ESC pluripotency. Stem Cells 2007, 25, 54–62. [Google Scholar] [CrossRef]
- Zulueta, A.; Caretti, A.; Signorelli, P.; Dall’olio, F.; Trinchera, M. Transcriptional control of the B3GALT5 gene by a retroviral promoter and methylation of distant regulatory elements. FASEB J. 2014, 28, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Tronche, F.; Rollier, A.; Sourdive, D.; Cereghini, S.; Yaniv, M. NFY or a related CCAAT binding factor can be replaced by other transcriptional activators for co-operation with HNF1 in driving the rat albumin promoter in vivo. J. Mol. Biol. 1991, 222, 31–43. [Google Scholar] [CrossRef]
- Li, X.Y.; Hooft van Huijsduijnen, R.; Mantovani, R.; Benoist, C.; Mathis, D. Intron-exon organization of the NF-Y genes. Tissue-specific splicing modifies an activation domain. J. Biol. Chem. 1992, 267, 8984–8990. [Google Scholar]
- Nardini, M.; Gnesutta, N.; Donati, G.; Gatta, R.; Forni, C.; Fossati, A.; Vonrhein, C.; Moras, D.; Romier, C.; Bolognesi, M.; et al. Sequence-specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination. Cell 2013, 152, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Dolfini, D.; Minuzzo, M.; Pavesi, G.; Mantovani, R. The short isoform of NF-YA belongs to the embryonic stem cell transcription factor circuitry. Stem Cells 2012, 30, 2450–2459. [Google Scholar] [CrossRef]
- Sandhu, J.K.; Sikorska, M.; Walker, P.R. Characterization of astrocytes derived from human NTera-2/D1 embryonal carcinoma cells. J. Neurosci. Res. 2002, 68, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.Y.; Lin, Y.P.; Chen, Y.L.; Lee, Y.C.; Tai, C.C.; Wang, Y.T.; Chen, Y.J.; Kao, C.F.; Yu, J. Interplay between SIN3A and STAT3 mediates chromatin conformational changes and GFAP expression during cellular differentiation. PLoS ONE 2011, 6, e22018. [Google Scholar] [CrossRef]
- Kretzschmar, A.K.; Dinger, M.C.; Henze, C.; Brocke-Heidrich, K.; Horn, F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem. J. 2004, 377, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Moasser, M.M.; Khoo, K.S.; Maerz, W.J.; Zelenetz, A.; Dmitrovsky, E. Derivation and characterization of retinoid-resistant human embryonal carcinoma cells. Differentiation 1996, 60, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.R.; Matin, M.M.; Andrews, P.W. The CDK inhibitor p27 enhances neural differentiation in pluripotent NTERA2 human EC cells, but does not permit differentiation of 2102Ep nullipotent human EC cells. Mech. Dev. 2005, 122, 1034–1042. [Google Scholar] [CrossRef]
- Mantovani, R.; Li, X.Y.; Pessara, U.; Hooft van Huisjduijnen, R.; Benoist, C.; Mathis, D. Dominant negative analogs of NF-YA. J. Biol. Chem. 1994, 269, 20340–20346. [Google Scholar]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D1284. [Google Scholar] [CrossRef]
- Van der Watt, P.J.; Leaner, V.D. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim. Biophys. Acta 2011, 1809, 316–326. [Google Scholar] [CrossRef]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Grgurevic, S.; Faderl, S.; Ferrajoli, A.; Wierda, W.G.; Martinez, M.; et al. At High Levels, Constitutively Activated STAT3 Induces Apoptosis of Chronic Lymphocytic Leukemia Cells. J. Immunol. 2016, 196, 4400–4409. [Google Scholar] [CrossRef]
- Mamat, S.; Ikeda, J.; Tian, T.; Wang, Y.; Luo, W.; Aozasa, K.; Morii, E. Transcriptional Regulation of Aldehyde Dehydrogenase 1A1 Gene by Alternative Spliced Forms of Nuclear Factor Y in Tumorigenic Population of Endometrial Adenocarcinoma. Genes Cancer 2011, 2, 979–984. [Google Scholar] [CrossRef]
- Basile, V.; Baruffaldi, F.; Dolfini, D.; Belluti, S.; Benatti, P.; Ricci, L.; Artusi, V.; Tagliafico, E.; Mantovani, R.; Molinari, S.; et al. NF-YA splice variants have different roles on muscle differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belluti, S.; Semeghini, V.; Basile, V.; Rigillo, G.; Salsi, V.; Genovese, F.; Dolfini, D.; Imbriano, C. An autoregulatory loop controls the expression of the transcription factor NF-Y. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Cicchillitti, L.; Manni, I.; Mancone, C.; Regazzo, G.; Spagnuolo, M.; Alonzi, T.; Carlomosti, F.; Dell’Anna, M.L.; Dell’Omo, G.; Picardo, M.; et al. The laminA/NF-Y protein complex reveals an unknown transcriptional mechanism on cell proliferation. Oncotarget 2017, 8, 2628–2646. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells 2006, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.; Huh, Y.J.; Cho, H.J.; Lee, B.; Park, J.; Hwang, D.Y.; Kim, D.W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem Cell Rep. 2017, 9, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Huang, Z.X.; Li, X.; Shen, K.F.; Liu, M.; Ouyang, H.D.; Zhang, S.B.; Ruan, Y.T.; Zhang, X.L.; Wu, S.L.; et al. Upregulation of NLRP3 via STAT3-dependent histone acetylation contributes to painful neuropathy induced by bortezomib. Exp. Neurol. 2018, 302, 104–111. [Google Scholar] [CrossRef]
- Chen, K.; Fan, J.; Luo, Z.F.; Yang, Y.; Xin, W.J.; Liu, C.C. Reduction of SIRT1 epigenetically upregulates NALP1 expression and contributes to neuropathic pain induced by chemotherapeutic drug bortezomib. J. Neuroinflamm. 2018, 15, 292. [Google Scholar] [CrossRef]
- Trinchera, M.; Zulueta, A.; Caretti, A.; Dall’Olio, F. Control of Glycosylation-Related Genes by DNA Methylation: The Intriguing Case of the B3GALT5 Gene and Its Distinct Promoters. Biology 2014, 3, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Shimazaki, T.; Sobue, G.; Okano, H. Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Dev. Biol. 2004, 275, 124–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.J.; Yang, B.C.; Chen, J.M.; Lin, Y.H.; Huang, C.L.; Cheng, Y.Y.; Hsu, C.Y.; Khoo, K.H.; Shen, C.N.; Yu, J. Changes in glycosphingolipid composition during differentiation of human embryonic stem cells to ectodermal or endodermal lineages. Stem Cells 2011, 29, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M.A.; Bergmann, J.H.; Lazar, Z.; Spector, D.L. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 2013, 4, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.; Nigg, E.A. Ectopic expression of an A-type lamin does not interfere with differentiation of lamin A-negative embryonal carcinoma cells. J. Cell Sci. 1991, 100, 589–598. [Google Scholar]
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2017, 8, 7935–7945. [Google Scholar] [CrossRef] [Green Version]
- Cieply, B.; Park, J.W.; Nakauka-Ddamba, A.; Bebee, T.W.; Guo, Y.; Shang, X.; Lengner, C.J.; Xing, Y.; Carstens, R.P. Multiphasic and Dynamic Changes in Alternative Splicing during Induction of Pluripotency Are Coordinated by Numerous RNA-Binding Proteins. Cell Rep. 2016, 15, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Park, J.W.; Bebee, T.W.; Warzecha, C.C.; Guo, Y.; Shang, X.; Xing, Y.; Carstens, R.P. Determination of a Comprehensive Alternative Splicing Regulatory Network and Combinatorial Regulation by Key Factors during the Epithelial-to-Mesenchymal Transition. Mol. Cell Biol. 2016, 36, 1704–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, J.; Xia, Z.; Li, W.; Cooper, T.A. Neonatal cardiac dysfunction and transcriptome changes caused by the absence of Celf1. Sci. Rep. 2016, 6, 35550. [Google Scholar] [CrossRef] [Green Version]
- Pillman, K.A.; Phillips, C.A.; Roslan, S.; Toubia, J.; Dredge, B.K.; Bert, A.G.; Lumb, R.; Neumann, D.P.; Li, X.; Conn, S.J.; et al. miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA-binding protein Quaking. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Raz, R.; Lee, C.K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahéron, L.; Opitz, S.L.; Zaehres, H.; Lensch, M.W.; Lensch, W.M.; Andrews, P.W.; Itskovitz-Eldor, J.; Daley, G.Q. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells 2004, 22, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Fujimoto, Y.; Nakatsuji, N.; Suemori, H. STAT3 is dispensable for maintenance of self-renewal in nonhuman primate embryonic stem cells. Stem Cells 2004, 22, 861–872. [Google Scholar] [CrossRef]
- Aronica, A.; Avagliano, L.; Caretti, A.; Tosi, D.; Bulfamante, G.P.; Trinchera, M. Unexpected distribution of CA19.9 and other type 1 chain Lewis antigens in normal and cancer tissues of colon and pancreas: Importance of the detection method and role of glycosyltransferase regulation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3210–3220. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Goto, M.; Magnuson, B.; Abraham, S.; Ramanathan, N.; Hotaling, E.; Alaniz, A.A.; Kumar-Sinha, C.; Dziubinski, M.L.; Urs, S.; et al. HNF1A is a novel oncogene that regulates human pancreatic cancer stem cell properties. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, S.; Zhou, Y.; Li, S.; Li, Y.; Liao, W. HNF1A/CASC2 regulates pancreatic cancer cell proliferation through PTEN/Akt signaling. J. Cell Biochem. 2019, 120, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N.; et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, B.-H.; Lee, H.-Y.; Chou, C.-K.; Wu, P.-H.; Huang, H.-C.; Chao, C.-C.; Chung, H.-Y.; Kannagi, R. SSEA3 and Sialyl Lewis a Glycan Expression Is Controlled by B3GALT5 LTR through Lamin A-NFYA and SIRT1-STAT3 Signaling in Human ES Cells. Cells 2020, 9, 177. https://doi.org/10.3390/cells9010177
Cai B-H, Lee H-Y, Chou C-K, Wu P-H, Huang H-C, Chao C-C, Chung H-Y, Kannagi R. SSEA3 and Sialyl Lewis a Glycan Expression Is Controlled by B3GALT5 LTR through Lamin A-NFYA and SIRT1-STAT3 Signaling in Human ES Cells. Cells. 2020; 9(1):177. https://doi.org/10.3390/cells9010177
Chicago/Turabian StyleCai, Bi-He, Hsueh-Yi Lee, Chi-Kan Chou, Po-Han Wu, Hsiang-Chi Huang, Chia-Chun Chao, Hsiao-Yu Chung, and Reiji Kannagi. 2020. "SSEA3 and Sialyl Lewis a Glycan Expression Is Controlled by B3GALT5 LTR through Lamin A-NFYA and SIRT1-STAT3 Signaling in Human ES Cells" Cells 9, no. 1: 177. https://doi.org/10.3390/cells9010177