Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
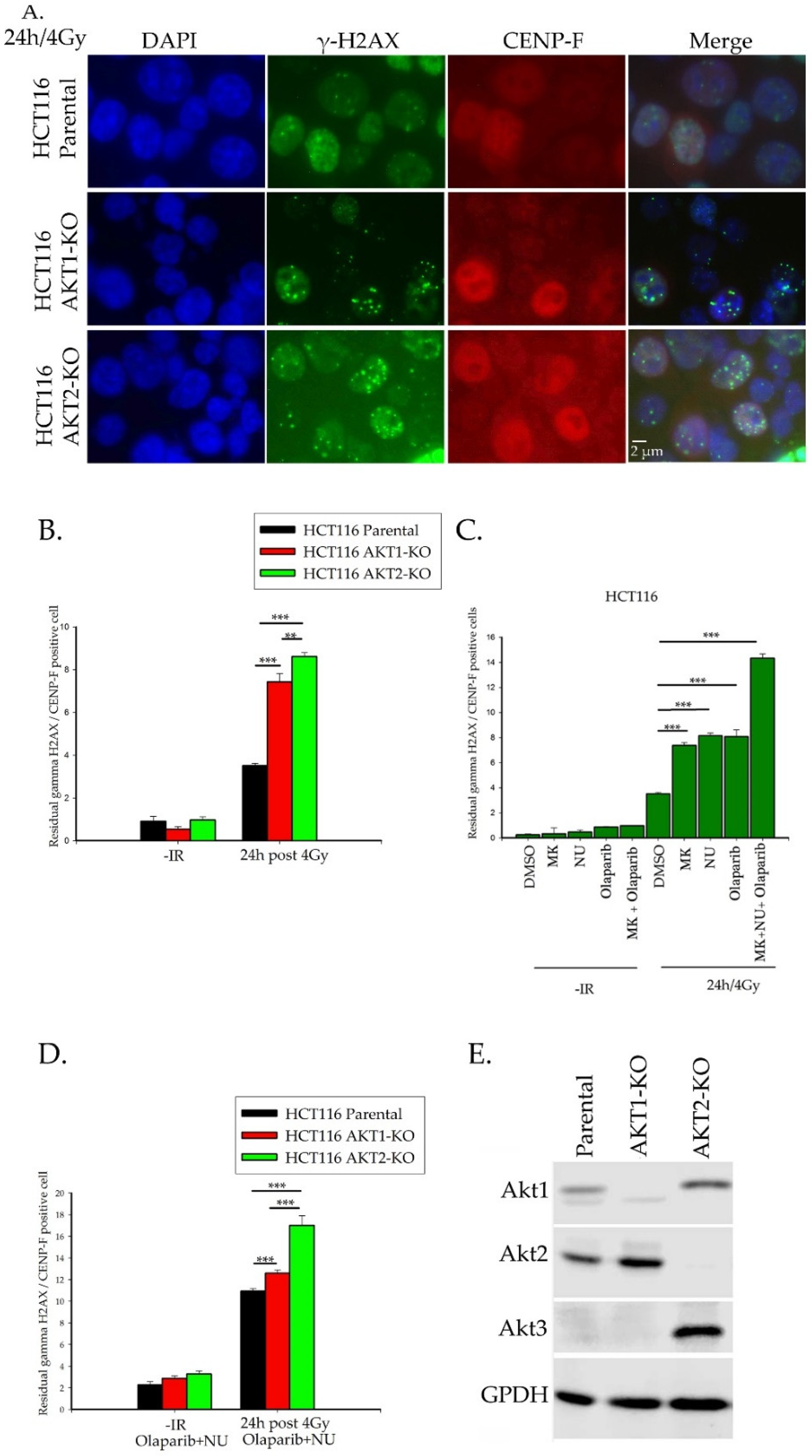
2.1. Importance of Akt1 and Akt2 for HR Repair of DSBs
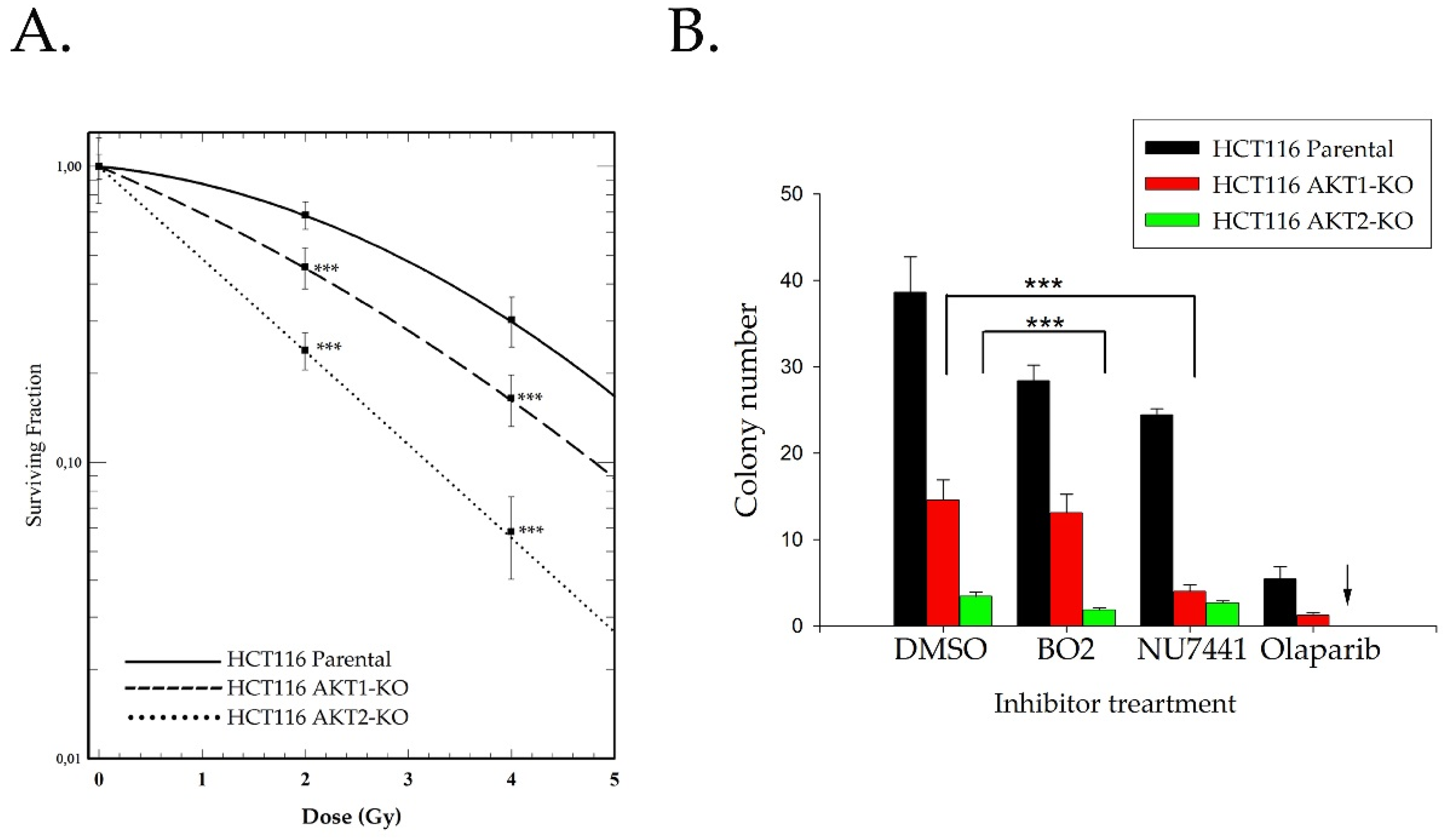
2.2. Akt Mediates Radioresistance Through Stimulated Homologous Recombination
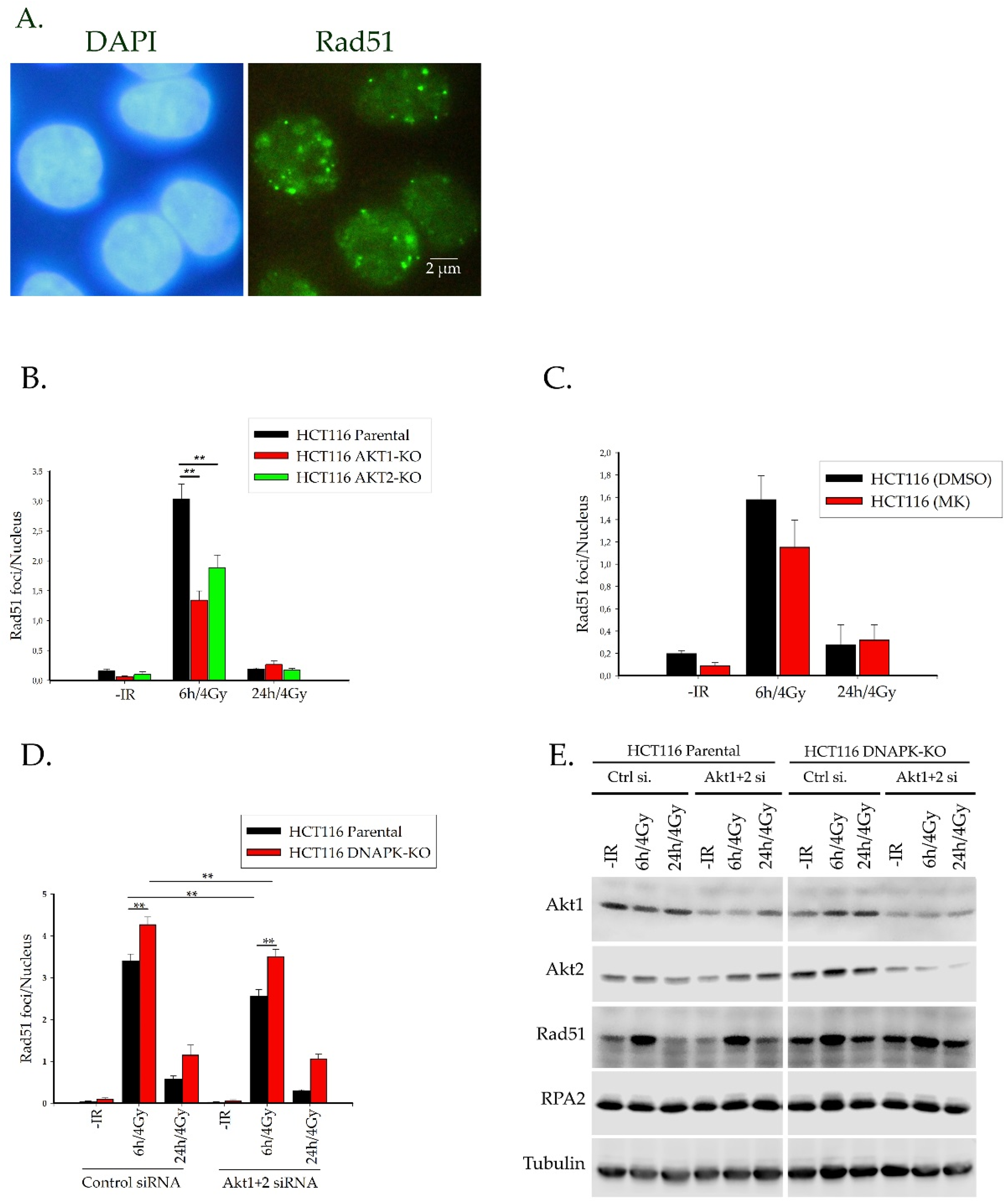
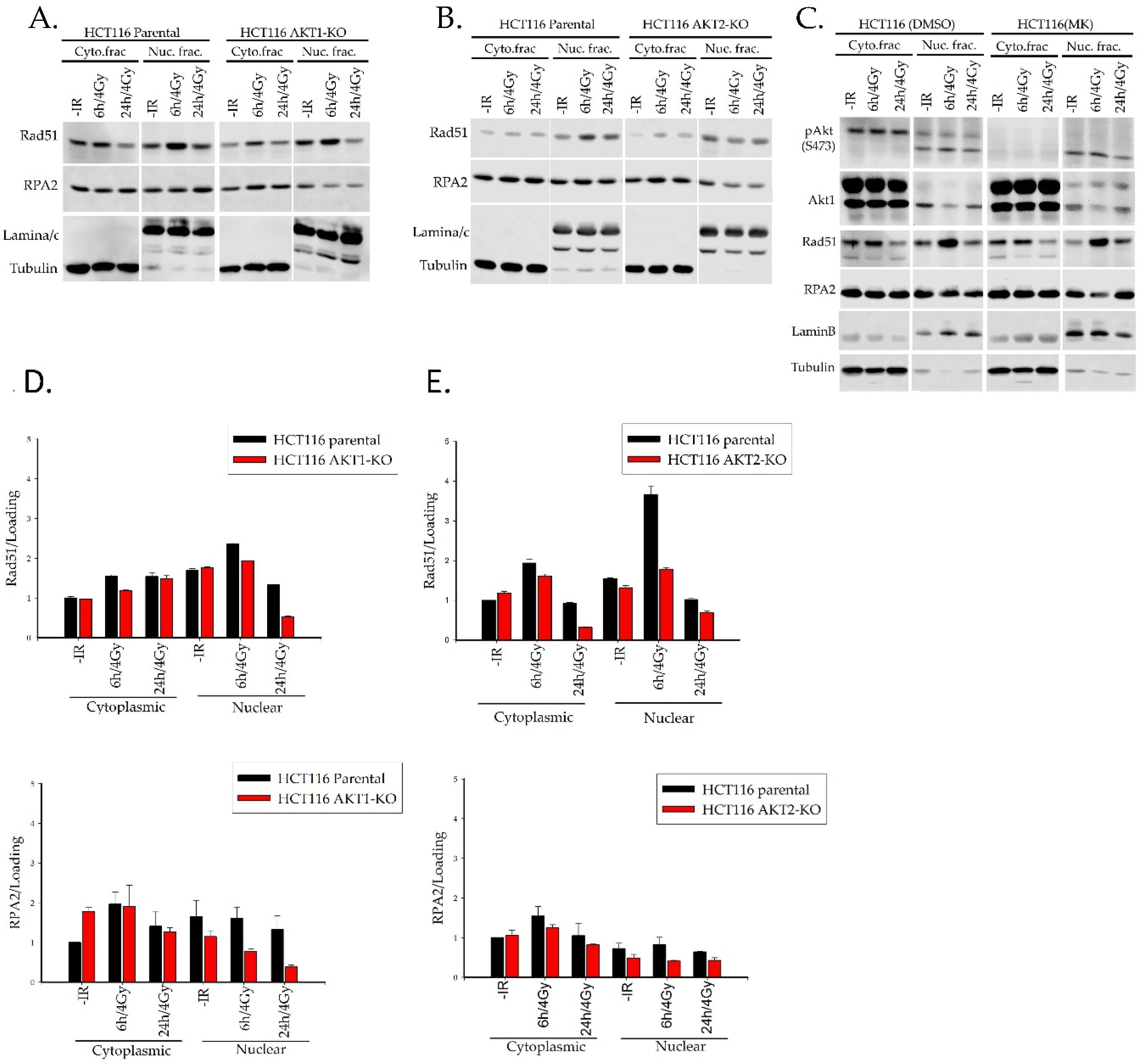
2.3. Akt1 and Akt2 Affect the Regulation of Rad51 Translocation and its Loading to the Site of Damage
2.3.1. Rad51 Foci Formation and Nuclear Translocation
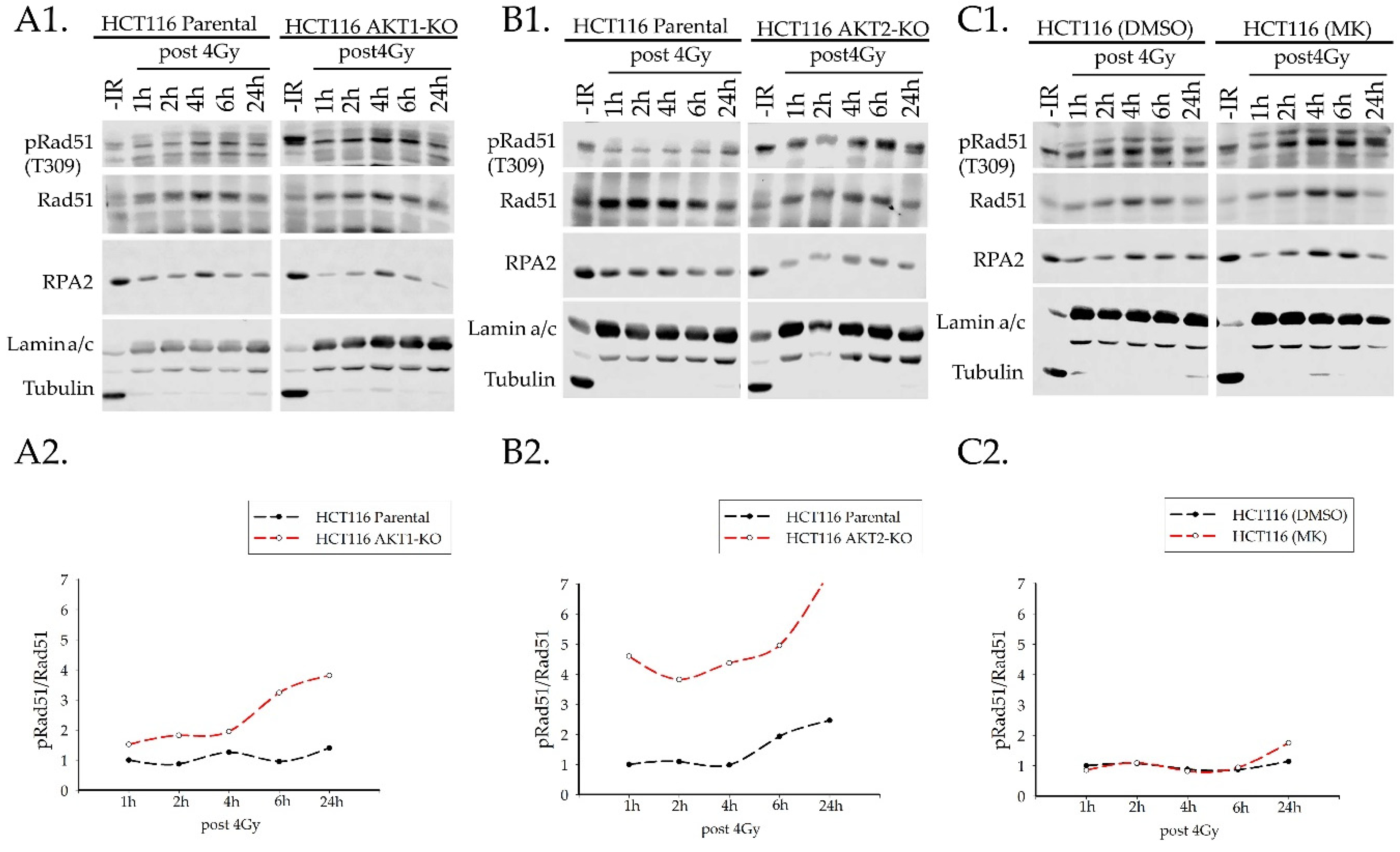
2.3.2. Rad51 Phosphorylation
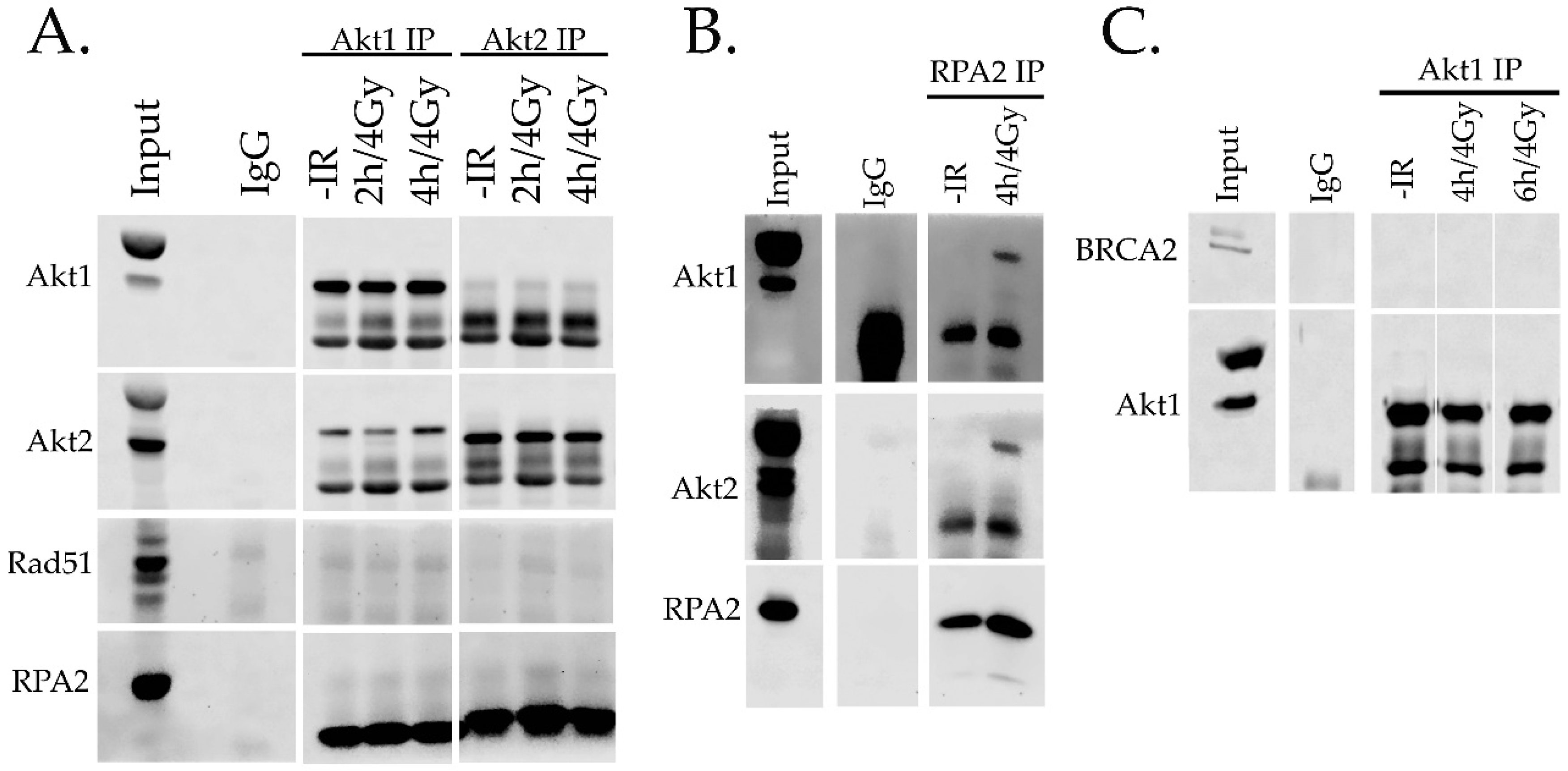
2.4. Interaction of Akt with HR Proteins
3. Discussion
4. Materials and Methods
4.1. Cell lines, Antibodies, and Reagents
4.2. Subcellular Fractionation
4.3. Irradiation
4.4. siRNA Transfection
4.5. Immunofluorescence Analysis
4.6. Immunoprecipitation
4.7. Colony Formation Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DSBs | double strand breaks |
| HRR | homologous recombination repair |
| NHEJ | non homologous end joining |
| A-NHEJ | alternative non homologous recombination |
References
- Noda, A. Radiation-induced unrepairable DSBs: Their role in the late effects of radiation and possible applications to biodosimetry. J. Radiat. Res. 2018, 59 (Suppl. 2), ii114–ii120. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, X. DNA double-strand break repair: A tale of pathway choices. Acta Biochim. Biophys. Sin. 2016, 48, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99. [Google Scholar] [CrossRef]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Srivastava, M.; Raghavan, S.C. DNA double-strand break repair inhibitors as cancer therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef]
- Liu, Q.; Turner, K.M.; Alfred Yung, W.K.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro Oncol. 2014, 16, 1313–1323. [Google Scholar] [CrossRef]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Lee, K.-J.; Fattah, K.R.; Lin, Y.-F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs–dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Haggblad Sahlberg, S.; Mortensen, A.C.; Haglof, J.; Engskog, M.K.; Arvidsson, T.; Pettersson, C.; Glimelius, B.; Stenerlow, B.; Nestor, M. Different functions of AKT1 and AKT2 in molecular pathways, cell migration and metabolism in colon cancer cells. Int. J. Oncol. 2017, 50, 5–14. [Google Scholar] [CrossRef]
- Toulany, M.; Maier, J.; Iida, M.; Rebholz, S.; Holler, M.; Grottke, A.; Juker, M.; Wheeler, D.L.; Rothbauer, U.; Rodemann, H.P. Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell Death Discov. 2017, 3, 17072. [Google Scholar] [CrossRef]
- Mueck, K.; Rebholz, S.; Harati, M.; Rodemann, H.; Toulany, M. AKT1 stimulates homologous recombination repair of DNA double-strand breaks in a Rad51-dependent manner. Int. J. Mol. Sci. 2017, 18, 2473. [Google Scholar] [CrossRef]
- Jia, Y.; Song, W.; Zhang, F.; Yan, J.; Yang, Q. Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway. Oncogene 2013, 32, 1943–1949. [Google Scholar] [CrossRef]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef]
- Ericson, K.; Gan, C.; Cheong, I.; Rago, C.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Huso, D.L.; Vogelstein, B.; Papadopoulos, N. Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Kötter, A.; Cornils, K.; Borgmann, K.; Dahm-Daphi, J.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Inhibition of PARP1-dependent end-joining contributes to Olaparib-mediated radiosensitization in tumor cells. Mol. Oncol. 2014, 8, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Philip, C.A.; Laskov, I.; Beauchamp, M.C.; Marques, M.; Amin, O.; Bitharas, J.; Kessous, R.; Kogan, L.; Baloch, T.; Gotlieb, W.H.; et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 2017, 17, 638. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, S.H.; Gustafsson, A.S.; Pendekanti, P.N.; Glimelius, B.; Stenerlow, B. The influence of AKT isoforms on radiation sensitivity and DNA repair in colon cancer cell lines. Tumour Biol. 2014, 35, 3525–3534. [Google Scholar] [CrossRef]
- Piscitello, D.; Varshney, D.; Lilla, S.; Vizioli, M.G.; Reid, C.; Gorbunova, V.; Seluanov, A.; Gillespie, D.A.; Adams, P.D. AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11. Oncogene 2018, 37, 427–438. [Google Scholar] [CrossRef]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.; Kearsley, J.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, e1437. [Google Scholar] [CrossRef]
- Landberg, G.; Erlanson, M.; Roos, G.; Tan, E.M.; Casiano, C.A. Nuclear autoantigen p330d/CENP-F: A marker for cell proliferation in human malignancies. Cytom. J. Int. Soc. Anal. Cytol. 1996, 25, 90–98. [Google Scholar] [CrossRef]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.-Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Dasgupta, Y.; Ren, S.-Y.; Gurdek, E.; Donlin, M.; Nieborowska-Skorska, M.; Fleury, F.; Skorski, T. Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia. Blood 2011, 118, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol. Cell 2019, 73, 224–237. [Google Scholar] [CrossRef]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Powell, C.; Mikropoulos, C.; Kaye, S.B.; Nutting, C.M.; Bhide, S.A.; Newbold, K.; Harrington, K.J. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treat. Rev. 2010, 36, 566–575. [Google Scholar] [CrossRef]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-dependent radiosensitization of human glioma cells by inhibition of poly (ADP-Ribose) polymerase: Mechanisms and therapeutic potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287. [Google Scholar] [CrossRef]
- Rehman, F.L.; Lord, C.J.; Ashworth, A. The promise of combining inhibition of PI3K and PARP as cancer therapy. Cancer Discov. 2012, 2, 982–984. [Google Scholar] [CrossRef]
- Rychahou, P.G.; Kang, J.; Gulhati, P.; Doan, H.Q.; Chen, L.A.; Xiao, S.Y.; Chung, D.H.; Evers, B.M. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 20315–20320. [Google Scholar] [CrossRef] [PubMed]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Bouzakri, K.; Zachrisson, A.; Al-Khalili, L.; Zhang, B.B.; Koistinen, H.A.; Krook, A.; Zierath, J.R. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 2006, 4, 89–96. [Google Scholar] [CrossRef]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A. Nuclear acetyl-CoA production by ACLY promotes homologous recombination. Mol. Cell 2017, 67, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Ampferl, R.; Rodemann, H.P.; Mayer, C.; Höfling, T.T.A.; Dittmann, K. Glucose starvation impairs DNA repair in tumour cells selectively by blocking histone acetylation. Radiother. Oncol. 2018, 126, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavaré, J.M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammadian Gol, T.; Rodemann, H.P.; Dittmann, K. Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination. Int. J. Mol. Sci. 2019, 20, 6316. https://doi.org/10.3390/ijms20246316
Mohammadian Gol T, Rodemann HP, Dittmann K. Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination. International Journal of Molecular Sciences. 2019; 20(24):6316. https://doi.org/10.3390/ijms20246316
Chicago/Turabian StyleMohammadian Gol, Tahereh, H. Peter Rodemann, and Klaus Dittmann. 2019. "Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination" International Journal of Molecular Sciences 20, no. 24: 6316. https://doi.org/10.3390/ijms20246316
APA StyleMohammadian Gol, T., Rodemann, H. P., & Dittmann, K. (2019). Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination. International Journal of Molecular Sciences, 20(24), 6316. https://doi.org/10.3390/ijms20246316