Dissecting Aging and Senescence—Current Concepts and Open Lessons

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
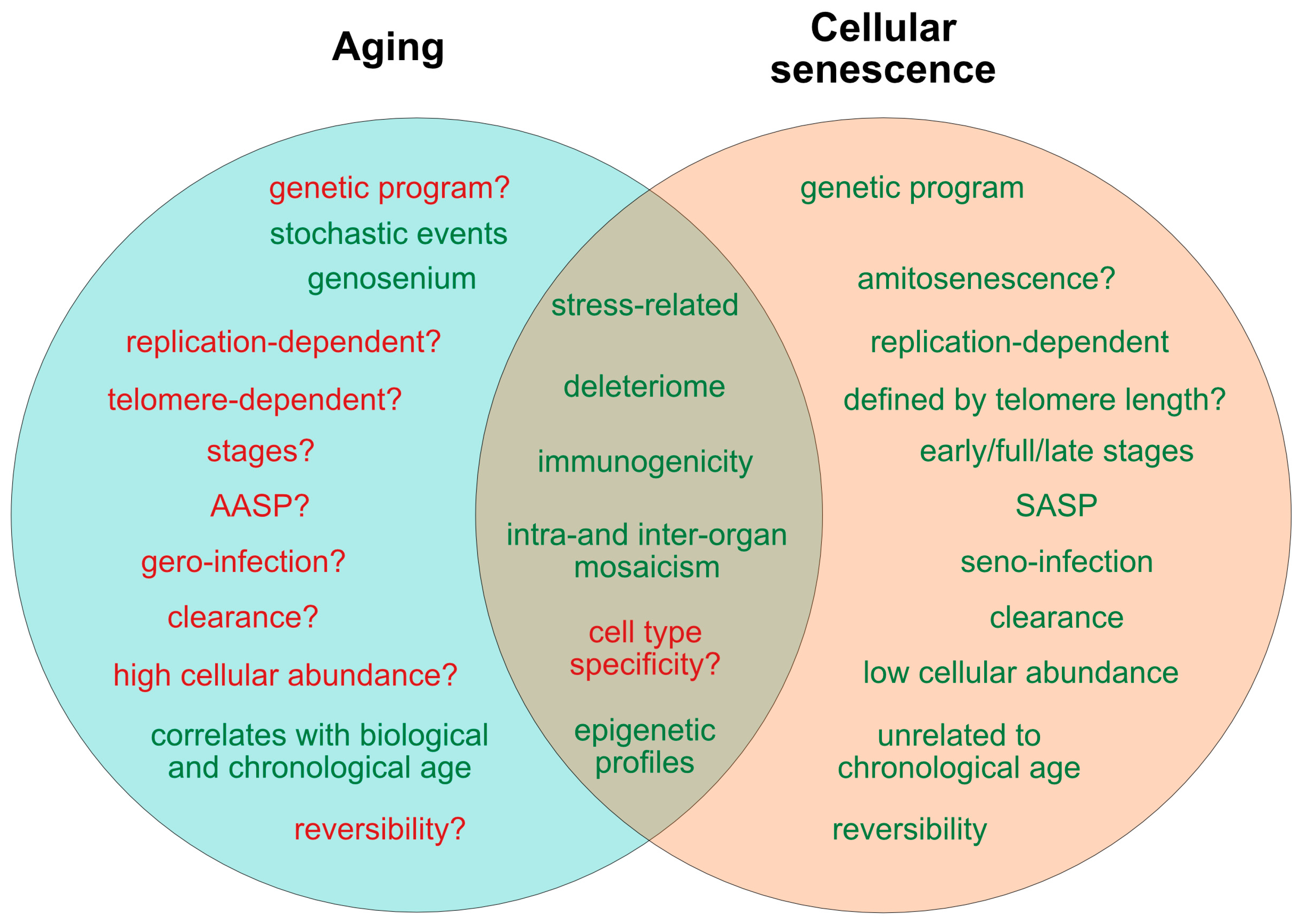
2. Current Concepts of Aging and Senescence
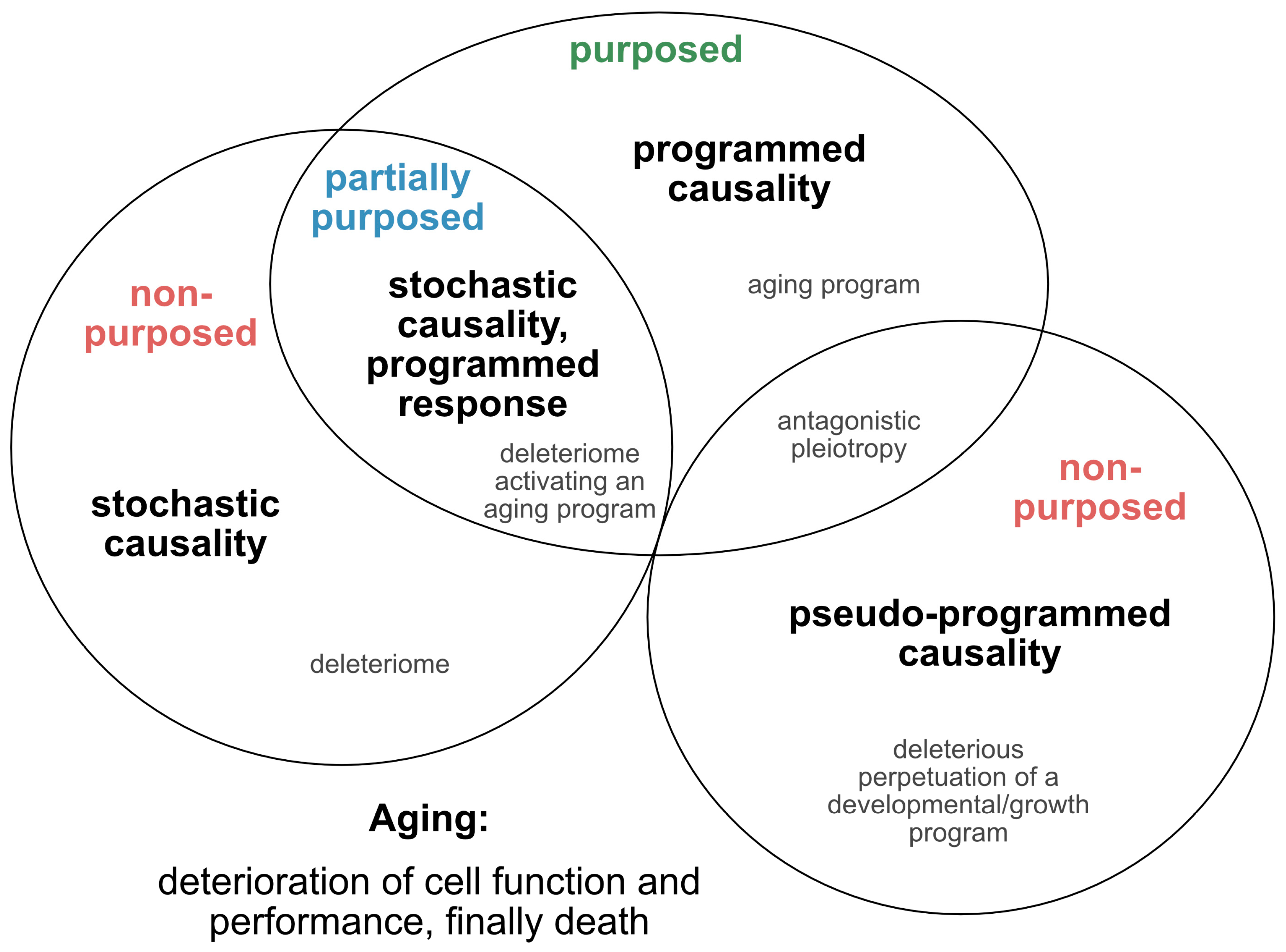
2.1. Is Aging Programmed?
2.2. Single Nucleotide Variants as Genetic Markers Supporting Programmed Aging?
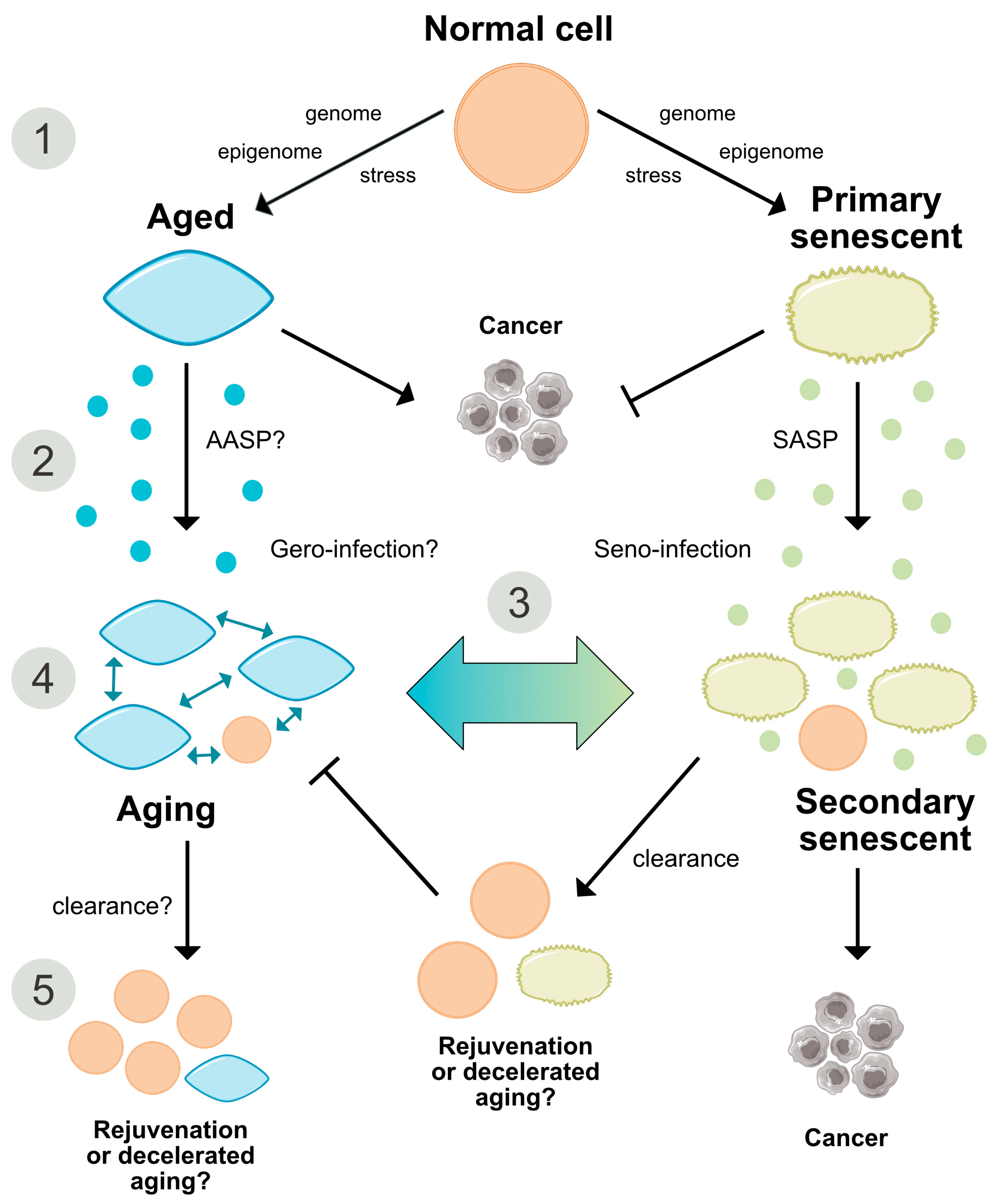
2.3. Senescence—A Programmed Process
2.4. Role of Cell Replication, Telomere Shortening and DNA Integrity for Aging and Senescence
2.5. Role of the Immune System in Cellular Senescence and Aging
2.6. Are Both Senescence- and Aging-Associated Alterations Mediated by a Cell-Specific Secretome?
2.7. States of Aging and Senescence
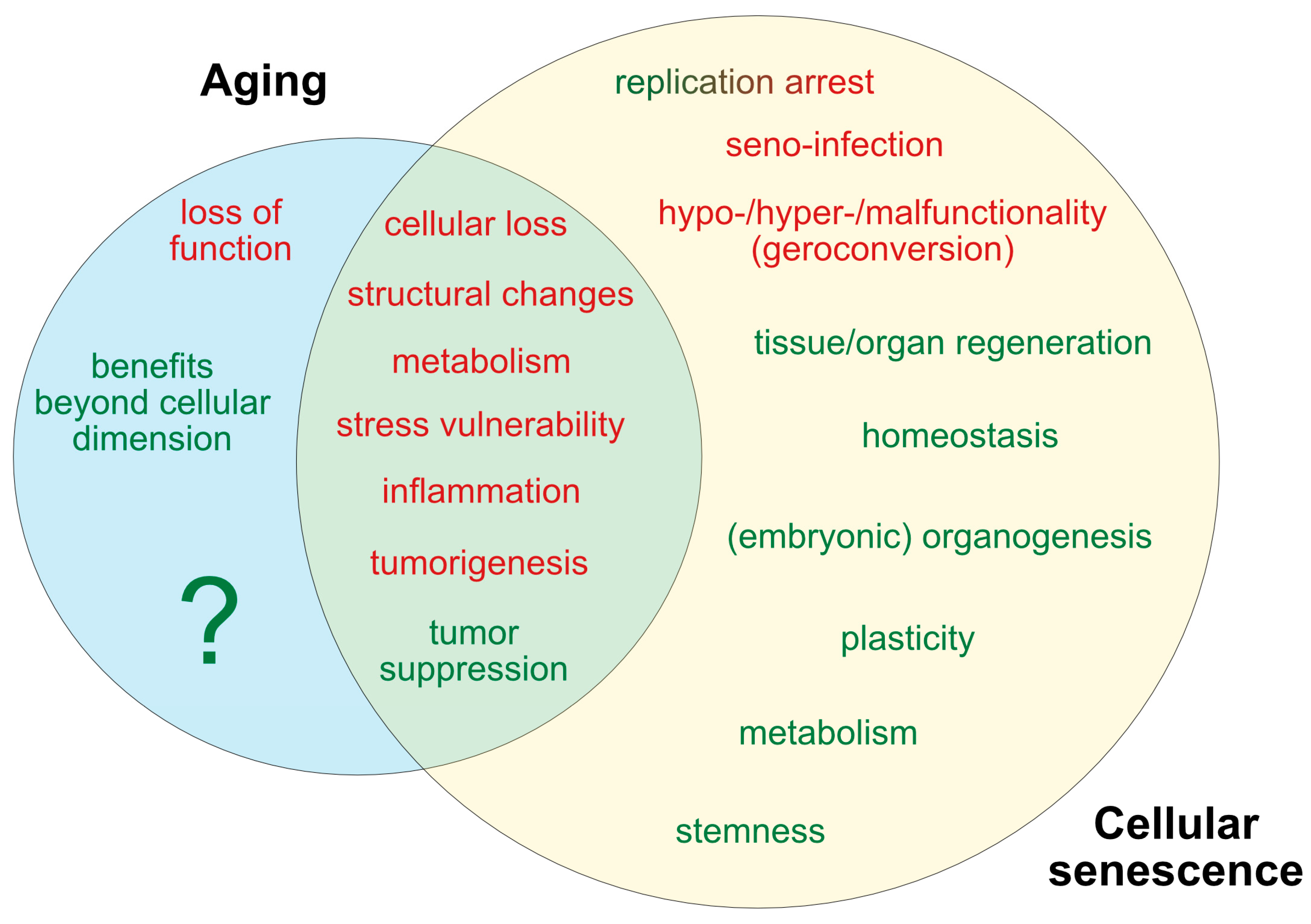
3. Markers of Aging and Senescence
Aging of Senescent Cells
4. Beneficial Functions of Senescence and Aging
4.1. Embryonic Patterning, Tissue Regeneration and Repair
4.2. The Beneficial Effects of Aging
5. Epigenetic Mechanisms in Cellular Senescence and Aging
6. Reversibility of Aging and Senescence
7. Conclusions and Perspectives
7.1. Conclusions
7.2. Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AASP | Aging-associated secretory phenotype |
| ALS | Amyotrophic lateral sclerosis |
| CCL-2 | C-C motif chemokine ligand 2 |
| Cdk/CdkI | Cyclin-dependent kinase/Cyclin-dependent kinase inhibitor |
| cGAS | Cyclic guanosine monophosphate – adenosine monophosphate synthase |
| CSF | Colony-stimulating factor |
| CXCL | Chemokine C-X-C motif ligand |
| DDR | DNA damage response |
| DNA-SCARS | DNA segments with chromatin alterations reinforcing senescence |
| DPP4 | Dipeptidyl peptidase-4 |
| FoxO | Forkhead box protein O |
| GH | Growth hormone |
| HLA | Human leukocyte antigen |
| HR | Homologous recombination |
| IGF-1 | Insulin-like growth factor-1 |
| iPSCs | Inducible pluripotent stem cells |
| LINE-1 | Long interspersed nuclear element-1 |
| LTR | Long terminal repeats |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix-metalloprotease |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor κB |
| NK cell | Natural killer cell |
| OIS | Oncogene-induced senescence |
| PI3K | Phosphoinositid-3-kinase |
| PML | Promyelocytic leukemia protein |
| ROS | Reactive oxygen species |
| RS | Replicative senescence |
| SA-β-Gal | Senescence-associated-β-galactosidase |
| SAHF | Senescence-associated heterochromatic foci |
| SASP | Senescence associated secretory phenotype |
| SIPS | Stress-induced premature senescence |
| STH | Somatotropin |
| STING | Stimulator of interferon genes |
| TC-NER | Transcription-coupled nucleotide excision repair |
References
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Boil. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Aramillo Irizar, P.; Schauble, S.; Esser, D.; Groth, M.; Frahm, C.; Priebe, S.; Baumgart, M.; Hartmann, N.; Marthandan, S.; Menzel, U.; et al. Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nat. Commun. 2018, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Penndorf, D.; Witte, O.W.; Kretz, A. DNA plasticity and damage in amyotrophic lateral sclerosis. Neural Regen. Res. 2018, 13, 173–180. [Google Scholar] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Hayflick, L. The establishment of a line (WISH) of human amnion cells in continuous cultivation. Exp. Cell Res. 1961, 23, 14–20. [Google Scholar] [CrossRef]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef]
- Petralia, R.S.; Mattson, M.P.; Yao, P.J. Aging and longevity in the simplest animals and the quest for immortality. Ageing Res. Rev. 2014, 16, 66–82. [Google Scholar] [CrossRef]
- Ain, Q.; Schmeer, C.; Penndorf, D.; Fischer, M.; Bondeva, T.; Förster, M.; Haenold, R.; Witte, O.W.; Kretz, A. Cell cycle-dependent and -independent telomere shortening accompanies murine brain aging. Aging 2018, 10, 3397–3420. [Google Scholar] [CrossRef]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Boil. 2018, 28, 595–607. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, T.V.; Gladyshev, V.N. A Disease or not a Disease? Aging as a Pathology. Trends Mol. Med. 2016, 22, 995–996. [Google Scholar] [CrossRef] [PubMed]
- Galkin, F.; Zhang, B.; Dmitriev, S.E.; Gladyshev, V.N. Reversibility of irreversible aging. Ageing Res. Rev. 2019, 49, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. An Unsolved Problem of Biology; H. K. Lewis: London, UK, 1952; p. 24. [Google Scholar]
- Williams, G.C. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 1957, 11, 398. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mitteldorf, J.; Skulachev, V.P. Programmed and altruistic ageing. Nat. Rev. Genet. 2005, 6, 866–872. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Aging: ROS or TOR. Cell Cycle 2008, 7, 3344–3354. [Google Scholar] [CrossRef]
- Gladyshev, V.N. Aging: Progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 2016, 15, 594–602. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Aging is not programmed: Genetic pseudo-program is a shadow of developmental growth. Cell Cycle 2013, 12, 3736–3742. [Google Scholar] [CrossRef]
- Goldsmith, T.C. Evolution of aging theories: Why modern programmed aging concepts are transforming medical research. Biochemistry 2016, 81, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Kawahara, T.L.; Segal, E.; Chang, H.Y. Reversal of aging by NFκB blockade. Cell Cycle 2008, 7, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Simone, E.; Grimaldi, M.; Musto, V.; Mancini, F.P. Sirtuins as Mediator of the Anti-Ageing Effects of Calorie Restriction in Skeletal and Cardiac Muscle. Int. J. Mol. Sci. 2018, 19, 928. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 longevity factor suppresses NF-κB-driven immune responses: Regulation of aging via NF-κB acetylation? BioEssays 2008, 30, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Suuronen, T.; Huuskonen, J.; Kaarniranta, K. NEMO shuttle: A link between DNA damage and NF-κB activation in progeroid syndromes? Biochem. Biophys. Res. Commun. 2008, 367, 715–718. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Huuskonen, J.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. Interaction of aging-associated signaling cascades: Inhibition of NF-κB signaling by longevity factors FoxOs and SIRT1. Cell. Mol. Life Sci. 2008, 65, 1049–1058. [Google Scholar] [CrossRef]
- Yeung, F.; E Hoberg, J.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; A Frye, R.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef]
- Espín-Palazón, R.; Traver, D. The NF-κB family: Key players during embryonic development and HSC emergence. Exp. Hematol. 2016, 44, 519–527. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nature 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- E Drigo, R.A.; Lev-Ram, V.; Tyagi, S.; Ramachandra, R.; Deerinck, T.; Bushong, E.; Phan, S.; Orphan, V.; Lechene, C.; Ellisman, M.H.; et al. Age Mosaicism across Multiple Scales in Adult Tissues. Cell Metab. 2019, 30, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Arrojo, E.D.R.; Lev-Ram, V.; Ramachandra, R.; Deerinck, T.J.; Lechene, C.; Ellisman, M.H.; Hetzer, M.W. Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J. Cell Biol. 2019, 218, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Mansfeld, J.; Urban, N.; Priebe, S.; Groth, M.; Frahm, C.; Hartmann, N.; Gebauer, J.; Ravichandran, M.; Dommaschk, A.; Schmeisser, S.; et al. Branched-chain amino acid catabolism is a conserved regulator of physiological ageing. Nat. Commun. 2015, 6, 10043. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Salmonowicz, H.; Jurk, D.; Passos, J.F. Expansion and Cell-Cycle Arrest: Common Denominators of Cellular Senescence. Trends Biochem. Sci. 2019. [Google Scholar] [CrossRef]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef]
- Lee-Six, H.; Øbro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; DeMaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Nakano, M.; Nakashima, A.; Onishi, K.; Yamao, S.; Enari, M.; Kikkawa, U.; Kamada, S. Identification of cellular senescence-specific genes by comparative transcriptomics. Sci. Rep. 2016, 6, 31758. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.; Samarajiwa, S.A.; Cairns, J.M.; Menon, S.; Perez-Mancera, P.A.; Tomimatsu, K.; Bermejo-Rodriguez, C.; Ito, Y.; Chandra, T.; Narita, M.; et al. Phenotype specific analyses reveal distinct regulatory mechanism for chronically activated p53. PLoS Genet. 2015, 11, e1005053. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2019, 101275. [Google Scholar] [CrossRef] [PubMed]
- Westera, L.; Van Hoeven, V.; Drylewicz, J.; Spierenburg, G.; Van Velzen, J.F.; De Boer, R.J.; Tesselaar, K.; Borghans, J.A.M. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell 2015, 14, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef]
- Savas, J.N.; Toyama, B.H.; Xu, T.; Yates, J.R., 3rd; Hetzer, M.W. Extremely long-lived nuclear pore proteins in the rat brain. Science 2012, 335, 942. [Google Scholar] [CrossRef]
- Di Fagagna, F.D.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Whittemore, K.; Vera, E.; Martínez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Izumiyama-Shimomura, N.; Honma, N.; Sawabe, M.; Arai, T.; Kato, M.; Oshimura, M.; Nakamura, K.-I. Telomere lengths are characteristic in each human individual. Exp. Gerontol. 2002, 37, 523–531. [Google Scholar] [CrossRef]
- Karlseder, J. Senescence Induced by Altered Telomere State, Not Telomere Loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Santerre, M.; Tempera, I.; Martin, K.; Mukerjee, R.; Sawaya, B.E. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology 2017, 117, 364–375. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beauséjour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nature 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Rossiello, F.; Herbig, U.; Longhese, M.P.; Fumagalli, M.; Di Fagagna, F.D. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 2014, 26, 89–95. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and aging. Single Mol. Single Cell Seq. 2015, 847, 73–87. [Google Scholar]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.-A.; Bourdel, G.; Popovic, N.; A Rezende, F.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 2019, 18, e12841. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Hallmarks of human “immunosenescence”: Adaptation or dysregulation? Immun. Ageing 2012, 9, 15. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Olivieri, F.; Larbi, A. The integration of inflammaging in age-related diseases. Semin. Immunol. 2018, 40, 17–35. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Le, O.N.L.; Rodier, F.; Fontaine, F.; Coppé, J.-P.; Campisi, J.; DeGregori, J.; Laverdière, C.; Kokta, V.; Haddad, E.; Beauséjour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef]
- Lowe, D.; Horvath, S.; Raj, K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget 2016, 7, 8524–8531. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- West, M.D.; Shay, J.W.; Wright, W.E.; Linskens, M.H. Altered expression of plasminogen activator and plasminogen activator inhibitor during cellular senescence. Exp. Gerontol. 1996, 31, 175–193. [Google Scholar] [CrossRef]
- Kumazaki, T.; Kobayashi, M.; Mitsui, Y. Enhanced Expression of Fibronectin during in Vivo Cellular Aging of Human Vascular Endothelial Cells and Skin Fibroblasts. Exp. Cell Res. 1993, 205, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.-G.; Zhang, J.; Block, E.R.; Patel, J.M. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech. Ageing Dev. 2003, 124, 911–919. [Google Scholar] [CrossRef]
- Van Der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced Peroxynitrite Formation Is Associated with Vascular Aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Coppe, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Rane, A.; Demaria, M.; Campisi, J.; Andersen, J.K. Cellular senescence and the aging brain. Exp. Gerontol. 2015, 68, 3–7. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997–1007. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef] [PubMed]
- Leontieva, O.V.; Demidenko, Z.N.; Blagosklonny, M.V. Dual mTORC1/C2 inhibitors suppress cellular geroconversion (a senescence program). Oncotarget 2015, 6, 23238–23248. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef]
- Smith, L.K.; He, Y.; Park, J.-S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; E Plambeck, K.; Udeochu, J.; et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef]
- A Villeda, S.; E Plambeck, K.; Middeldorp, J.; Castellano, J.M.; I Mosher, K.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Agba, O.B.; Lausser, L.; Huse, K.; Bergmeier, C.; Jahn, N.; Groth, M.; Bens, M.; Sahm, A.; Gall, M.; Witte, O.W.; et al. Tissue-, sex-, and age-specific DNA methylation of rat glucocorticoid receptor gene promoter and insulin-like growth factor 2 imprinting control region. Physiol. Genom. 2017, 49, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J. Insulin-like growth factor-I regulation of immune function: A potential therapeutic target in autoimmune diseases? Pharmacol. Rev. 2010, 62, 199–236. [Google Scholar] [CrossRef] [PubMed]
- Tia, N.; Singh, A.K.; Pandey, P.; Azad, C.S.; Chaudhary, P.; Gambhir, I.S. Role of Forkhead Box O (FOXO) transcription factor in aging and diseases. Gene 2018, 648, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Wolters, T.L.C.; Netea, M.G.; Hermus, A.R.M.M.; Smit, J.W.A.; Netea-Maier, R.T.; Hermus, A.R.; Smit, J.W. IGF1 potentiates the pro-inflammatory response in human peripheral blood mononuclear cells via MAPK. J. Mol. Endocrinol. 2017, 59, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Van Heemst, D.; Beekman, M.; Mooijaart, S.P.; Heijmans, B.T.; Brandt, B.W.; Zwaan, B.J.; Slagboom, P.E.; Westendorp, R.G.J.; Slagboom, P. Reduced insulin/IGF-1 signalling and human longevity. Aging Cell 2005, 4, 79–85. [Google Scholar] [CrossRef]
- Suh, Y.; Atzmon, G.; Cho, M.-O.; Hwang, D.; Liu, B.; Leahy, D.J.; Barzilai, N.; Cohen, P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. USA 2008, 105, 3438–3442. [Google Scholar] [CrossRef]
- Tazearslan, C.; Huang, J.; Barzilai, N.; Suh, Y. Impaired IGF1R signaling in cells expressing longevity-associated human IGF1R alleles. Aging Cell 2011, 10, 551–554. [Google Scholar] [CrossRef]
- Van Der Spoel, E.; Rozing, M.P.; Houwing-Duistermaat, J.J.; Slagboom, P.E.; Beekman, M.; De Craen, A.J.; Westendorp, R.G.; Van Heemst, D. Association analysis of insulin-like growth factor-1 axis parameters with survival and functional status in nonagenarians of the Leiden Longevity Study. Aging 2015, 7, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Milman, S.; Huffman, D.M.; Barzilai, N. The Somatotropic Axis in Human Aging: Framework for the Current State of Knowledge and Future Research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar] [CrossRef]
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M.; et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017, 13, e1006635. [Google Scholar] [CrossRef]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef]
- Bodea, G.O.; McKelvey, E.G.Z.; Faulkner, G.J. Retrotransposon-induced mosaicism in the neural genome. Open Biol. 2018, 8, 180074. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Macia, A.; Widmann, T.J.; Heras, S.R.; Ayllon, V.; Sanchez, L.; Benkaddour-Boumzaouad, M.; Munoz-Lopez, M.; Rubio, A.; Amador-Cubero, S.; Blanco-Jimenez, E.; et al. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017, 27, 335–348. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Cellular senescence in ageing, age-related disease and longevity. Curr. Vasc. Pharmacol. 2014, 12, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Yamakoshi, K.; Takahashi, A.; Hara, E. The p16INK4a-RB pathway: Molecular link between cellular senescence and tumor suppression. J. Med. Investig. 2004, 51, 146–153. [Google Scholar] [CrossRef]
- Martin, N.; Beach, D.; Gil, J. Ageing as developmental decay: Insights from p16INK4a. Trends Mol. Med. 2014, 20, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Micheli, L.; D’Andrea, G.; Ceccarelli, M.; Ferri, A.; Scardigli, R.; Tirone, F. p16Ink4a Prevents the Activation of Aged Quiescent Dentate Gyrus Stem Cells by Physical Exercise. Front. Cell. Neurosci. 2019, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef]
- Frescas, D.; Hall, B.M.; Strom, E.; Virtuoso, L.P.; Gupta, M.; Gleiberman, A.S.; Rydkina, E.; Balan, V.; Vujcic, S.; Chernova, O.B.; et al. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle 2017, 16, 1526–1533. [Google Scholar] [CrossRef]
- Stojiljkovic, M.R.; Ain, Q.; Bondeva, T.; Heller, R.; Schmeer, C.; Witte, O.W. Phenotypic and functional differences between senescent and aged murine microglia. Neurobiol. Aging 2019, 74, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Matheu, A.; Maraver, A.; Collado, M.; Garcia-Cao, I.; Cañamero, M.; Borras, C.; Flores, J.M.; Klatt, P.; Vina, J.; Serrano, M. Anti-aging activity of the Ink4/Arflocus. Aging Cell 2009, 8, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Matheu, A.; Pantoja, C.; Efeyan, A.; Criado, L.M.; Martín-Caballero, J.; Flores, J.M.; Klatt, P.; Serrano, M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genome Res. 2004, 18, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Andressoo, J.O.; De Wit, J.; Huijmans, J.; Beems, R.B.; Van Steeg, H.; Weeda, G.; Van Der Horst, G.T.J.; Van Leeuwen, W.; Themmen, A.P.N.; et al. Premature Aging in Mice Deficient in DNA Repair and Transcription. Science 2002, 296, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Bhanu, M.U.; Mandraju, R.; Bhaskar, C.; Kondapi, A.K. Cultured cerebellar granule neurons as an in vitro aging model: Topoisomerase IIβ as an additional biomarker in DNA repair and aging. Toxicol. In Vitro 2010, 24, 1935–1945. [Google Scholar] [CrossRef]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated β-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef]
- Walton, C.C.; Andersen, J.K. Unknown fates of (brain) oxidation or UFO: Close encounters with neuronal senescence. Free Radic. Biol. Med. 2019, 134, 695–701. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; López, G.G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.G.; Muller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; DiPietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Yang, X.; Indig, F.E.; Basu, S.K.; Ohnuma, K.; Morimoto, C.; Johnson, P.F.; et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017, 31, 1529–1534. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genome Res. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T.B.L. Can aging be programmed? A critical literature review. Aging Cell 2016, 15, 986–998. [Google Scholar] [CrossRef]
- Wang-Michelitsch, J.; Michelitsch, T. Traditional aging theories: Which ones are useful? arXiv 2015, arXiv:1503.07040. [Google Scholar]
- Lee, R.D. Rethinking the evolutionary theory of aging: Transfers, not births, shape senescence in social species. Proc. Natl. Acad. Sci. USA 2003, 100, 9637–9642. [Google Scholar] [CrossRef]
- Wagner, W. The Link between Epigenetic Clocks for Aging and Senescence. Front. Genet. 2019, 10, 303. [Google Scholar] [CrossRef]
- Wagner, W.; Fernandez-Rebollo, E.; Frobel, J. DNA-methylation changes in replicative senescence and aging: Two sides of the same coin? Epigenomics 2016, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; Van Eijk, K.; Berg, L.H.V.D.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef]
- Maegawa, S.; Hinkal, G.; Kim, H.S.; Shen, L.; Zhang, L.; Zhang, J.; Zhang, N.; Liang, S.; Donehower, L.A.; Issa, J.-P.J. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Raj, K. The Epigenetic Clock and Aging. In Epigenetics of Aging and Longevity; Academic Press: Cambridge, MA, USA, 2018; pp. 95–118. [Google Scholar] [CrossRef]
- Han, Y.; Eipel, M.; Franzen, J.; Sakk, V.; Dethmers-Ausema, B.; Yndriago, L.; Izeta, A.; de Haan, G.; Geiger, H.; Wagner, W. Epigenetic age-predictor for mice based on three CpG sites. eLife 2018, 7. [Google Scholar] [CrossRef]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Ferreira, M.R.P.; Deary, I.J.; Wagner, W. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging 2016, 8, 394–401. [Google Scholar] [CrossRef]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Maierhofer, A.; Flunkert, J.; Oshima, J.; Martin, G.M.; Haaf, T.; Horvath, S. Accelerated epigenetic aging in Werner syndrome. Aging 2017, 9, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.K.; Craig, J.M.; Theda, C.; Bækvad-Hansen, M.; Bybjerg-Grauholm, J.; Hansen, C.S.; Hollegaard, M.V.; Hougaard, D.M.; Mortensen, P.B.; Weinsheimer, S.M.; et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016, 17, 206. [Google Scholar] [CrossRef] [PubMed]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 2010, 9, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nature 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Pellegatta, S.; Favaro, R.; Pisati, F.; Roncaglia, P.; Testa, G.; Nicolis, S.K.; Finocchiaro, G.; Di Fagagna, F.D. DNA damage in mammalian neural stem cells leads to astrocytic differentiation mediated by BMP2 signaling through JAK-STAT. Stem Cell Rep. 2013, 1, 123–138. [Google Scholar] [CrossRef]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef]
- Prieur, A.; Besnard, E.; Babled, A.; Lemaitre, J.-M. p53 and p16INK4A independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat. Commun. 2011, 2, 473. [Google Scholar] [CrossRef]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef]
- Leikam, C.; Hufnagel, A.L.; Otto, C.; Murphy, D.J.; Mühling, B.; Kneitz, S.; Nanda, I.; Schmid, M.; Wagner, T.U.; Haferkamp, S.; et al. In vitro evidence for senescent multinucleated melanocytes as a source for tumor-initiating cells. Cell Death Dis. 2015, 6, e1711. [Google Scholar] [CrossRef] [PubMed]
- López-Lluch, G.; Navas, P. Calorie restriction as an intervention in ageing. J. Physiol. 2016, 594, 2043–2060. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins, Aging, and Metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Calorie restriction and sirtuins revisited. Genome Res. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [PubMed]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 2019, 18, e12877. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induced pluripotent stem cells in medicine and biology. Development 2013, 140, 2457–2461. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Wu, J.; Platero-Luengo, A.; Sakurai, M.; Sugawara, A.; Gil, M.A.; Yamauchi, T.; Suzuki, K.; Bogliotti, Y.S.; Cuello, C.; Valencia, M.M.; et al. Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell 2017, 168, 473–486. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadinanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef]
- Romaniuk, A.; Paszel-Jaworska, A.; Toton, E.; Lisiak, N.; Holysz, H.; Krolak, A.; Grodecka-Gazdecka, S.; Rubis, B. The non-canonical functions of telomerase: To turn off or not to turn off. Mol. Biol. Rep. 2019, 46, 1401–1411. [Google Scholar] [CrossRef]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.-M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.C.; Zhang, W.; Patiño-Parrado, I.; Barrio-Alonso, E.; Garrido, J.-J.; Frade, J.M. Primary neurons can enter M-phase. Sci. Rep. 2019, 9, 4594. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Nakajima, S.; Böhm, S.; Bernstein, K.A.; Shen, Z.; Tsang, M.; Levine, A.S.; Lan, L. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E3495–E3504. [Google Scholar] [CrossRef]
- Welty, S.; Teng, Y.; Liang, Z.; Zhao, W.; Sanders, L.H.; Greenamyre, J.T.; Rubio, M.E.; Thathiah, A.; Kodali, R.; Wetzel, R.; et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 2018, 293, 1353–1362. [Google Scholar] [CrossRef]
- Nacarelli, T.; Sell, C. Targeting metabolism in cellular senescence, a role for intervention. Mol. Cell. Endocrinol. 2017, 455, 83–92. [Google Scholar] [CrossRef]
- Schneider, A.; Martin-Villalba, A.; Weih, F.; Vogel, J.; Wirth, T.; Schwaninger, M. NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 1999, 5, 554–559. [Google Scholar] [CrossRef]
- Haenold, R.; Weih, F.; Herrmann, K.-H.; Schmidt, K.-F.; Krempler, K.; Engelmann, C.; Nave, K.-A.; Reichenbach, J.R.; Lowel, S.; Witte, O.W.; et al. NF-κB controls axonal regeneration and degeneration through cell-specific balance of RelA and p50 in the adult CNS. J. Cell Sci. 2014, 127, 3052–3065. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence—Current Concepts and Open Lessons. Cells 2019, 8, 1446. https://doi.org/10.3390/cells8111446
Schmeer C, Kretz A, Wengerodt D, Stojiljkovic M, Witte OW. Dissecting Aging and Senescence—Current Concepts and Open Lessons. Cells. 2019; 8(11):1446. https://doi.org/10.3390/cells8111446
Chicago/Turabian StyleSchmeer, Christian, Alexandra Kretz, Diane Wengerodt, Milan Stojiljkovic, and Otto W. Witte. 2019. "Dissecting Aging and Senescence—Current Concepts and Open Lessons" Cells 8, no. 11: 1446. https://doi.org/10.3390/cells8111446
APA StyleSchmeer, C., Kretz, A., Wengerodt, D., Stojiljkovic, M., & Witte, O. W. (2019). Dissecting Aging and Senescence—Current Concepts and Open Lessons. Cells, 8(11), 1446. https://doi.org/10.3390/cells8111446