Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Mutations in Patients
2.3. Patients
2.4. Cell Culture
2.5. Metabolite Extraction and Profiling by Targeted LC-MS
2.6. Proteomics Sample Preparation with Label-Free Quantification (LFQ)
2.7. LC-MS Instrument Settings for Shotgun Proteome Profiling and Data Analysis
2.8. Experimental Design, Statistical Rationale, Pathway, and Data Analyses
2.9. Simulations of Electron Transfer between the Iron–Sulfur Clusters of NDUFS1 and Prediction of Protein Stability of p.Phe124Leu in ND5
2.10. Measurement of Respiratory Chain Enzyme Activities
2.11. Live Cell Respiration Assay by Seahorse XFe96
2.12. Blue Native PAGE, Western Blot, and In-Gel Activity Assay of CI
2.13. Protein Sequence Alignment
3. Results
3.1. Substituted Amino Acids in ND5 and NDUFS1 are Highly Conserved from Bacteria to Human
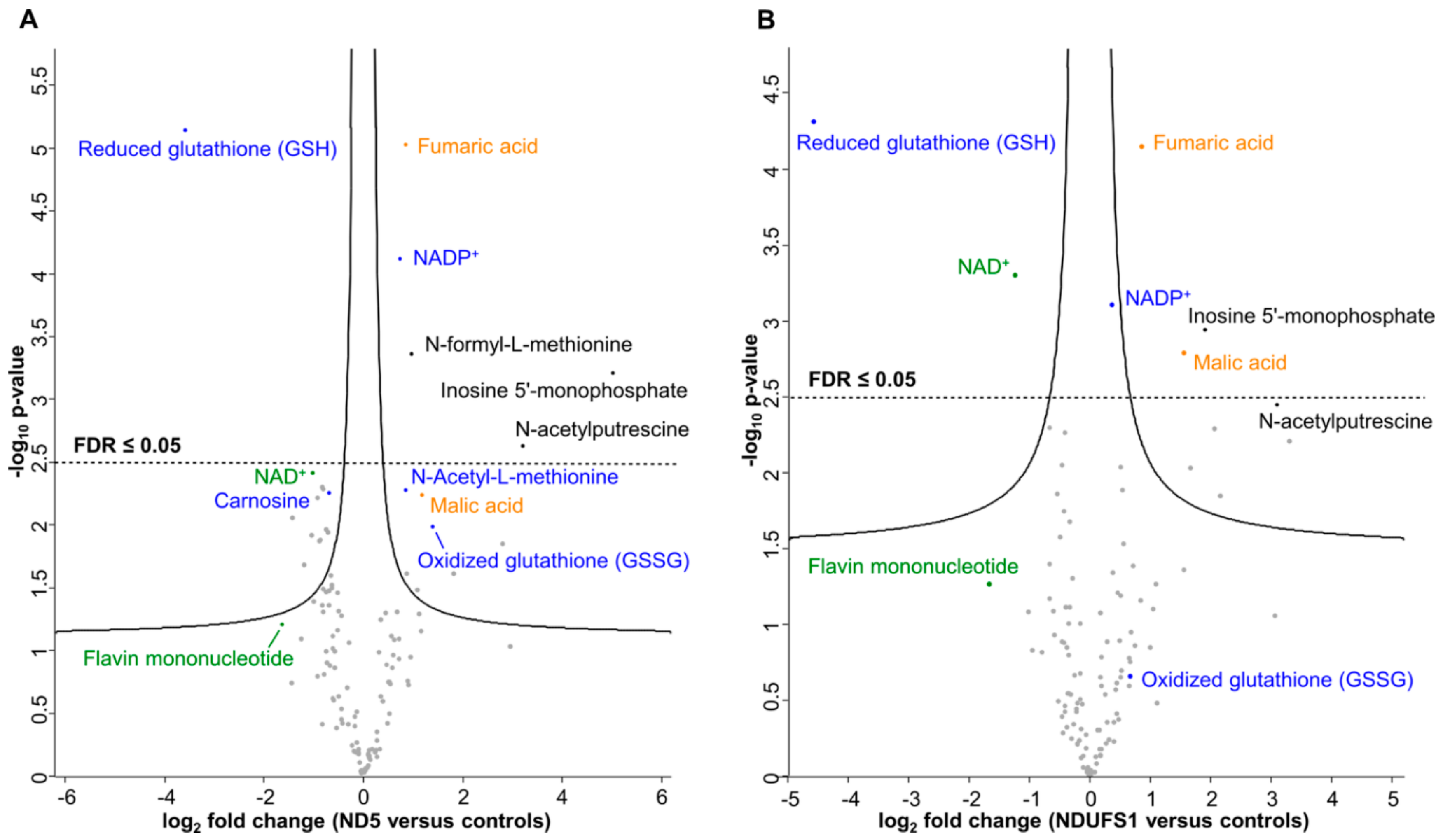
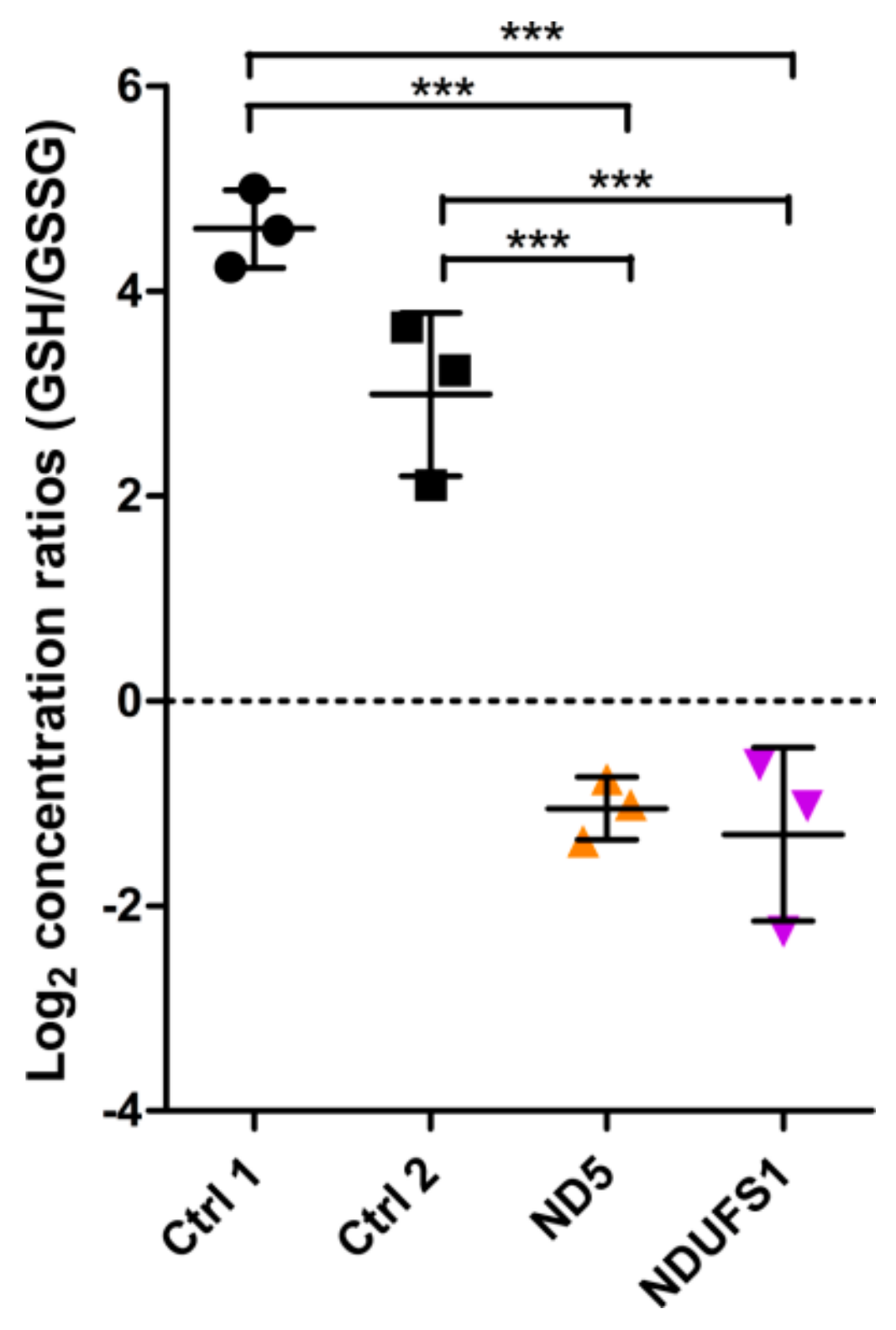
3.2. Metabolome Profiling Revealed a Decrease of the GSH/GSSG Ratio in Both Patients
3.3. The TCA Cycle Metabolites—Fumaric and Malic Acid—Significantly Increased in Both Patients
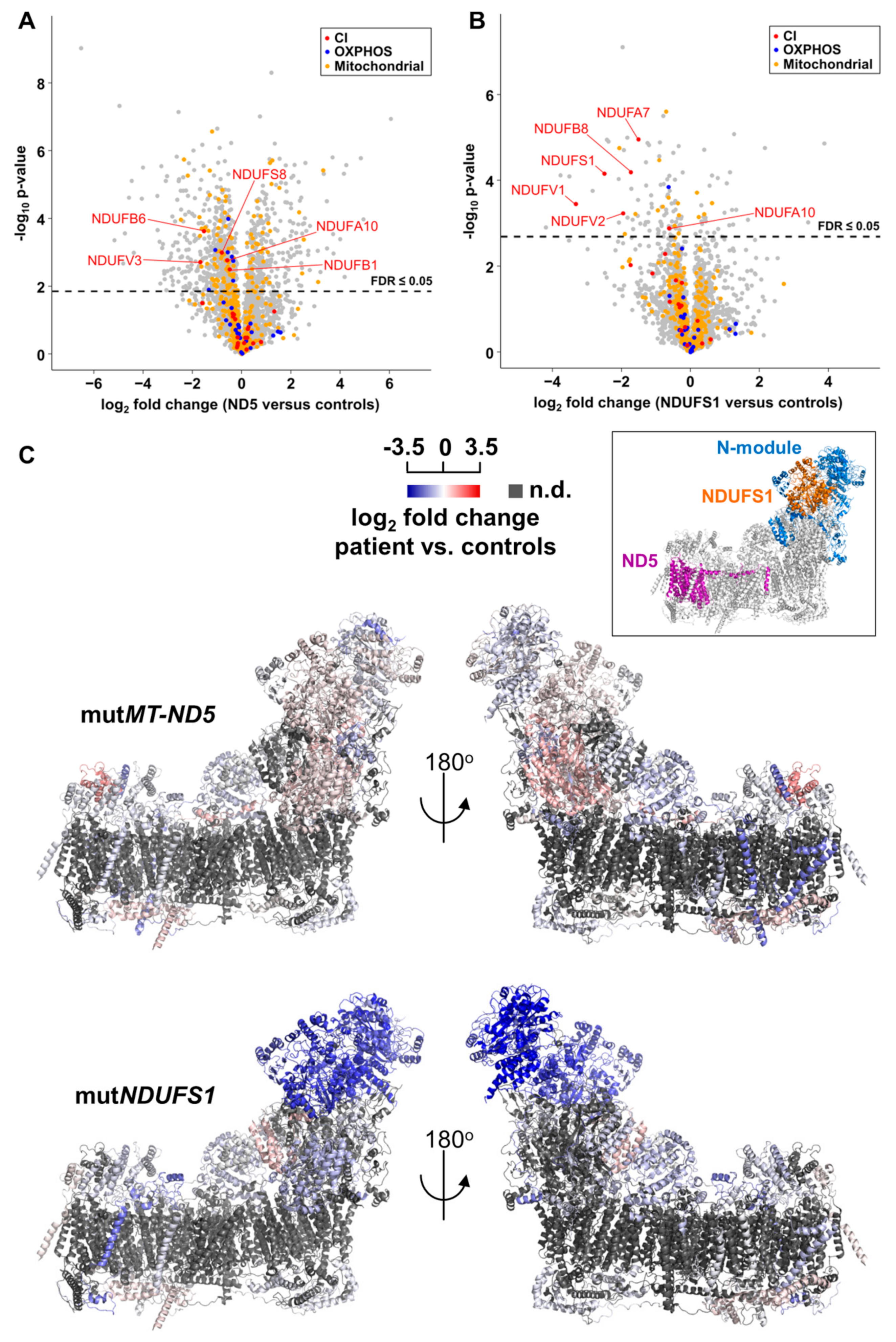
3.4. Proteome Profiling
3.5. Gene Set Enrichment Analyses Reveal Glycolysis is Upregulated in the MT-ND5 Mutation and the Respiratory Chain is Down-Regulated in the NDUFS1 Mutations
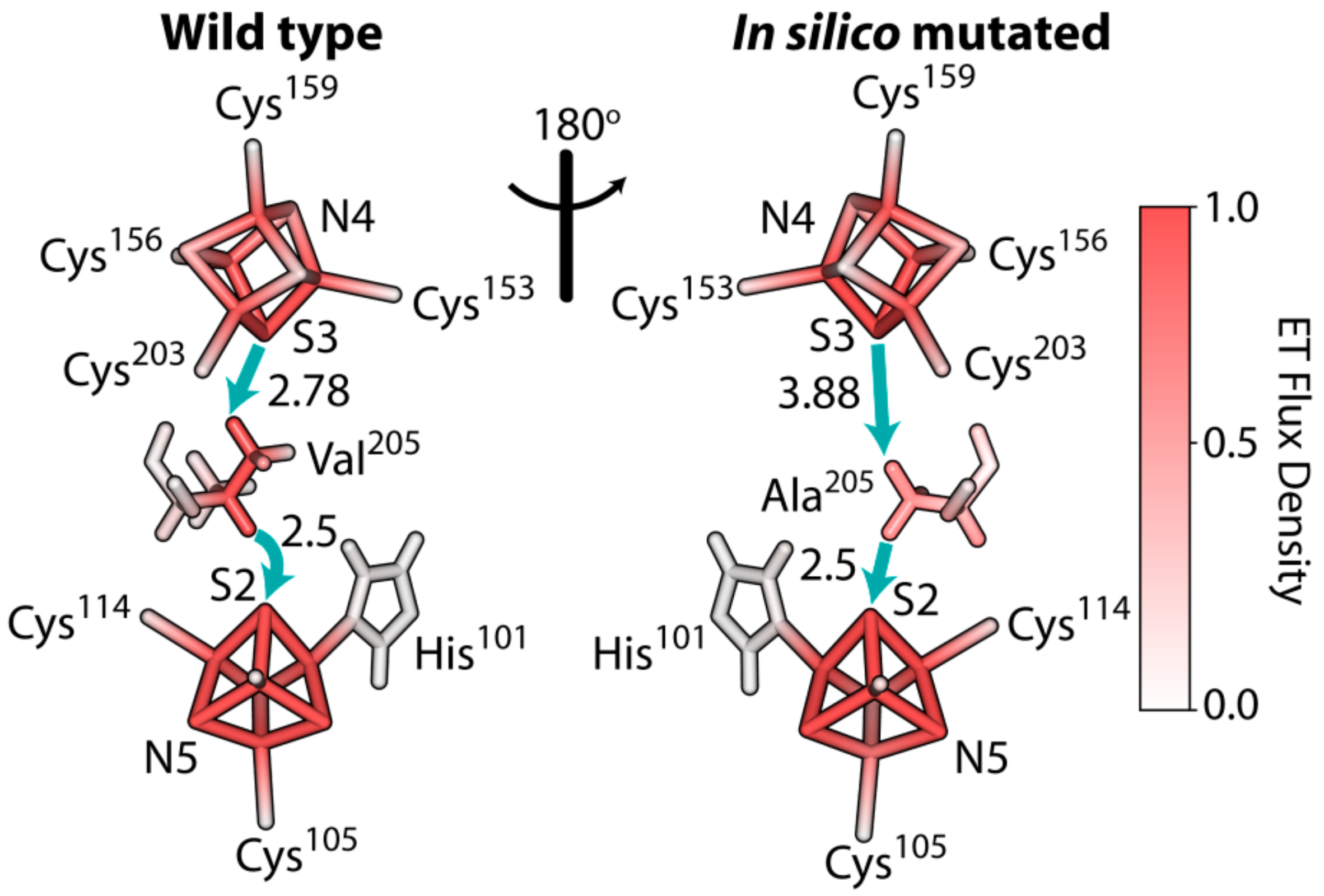
3.6. The Rate of Electron Tunneling between the Iron–Sulfur Clusters N4 and N5 of NDUFS1 Was Predicted to Decrease Dramatically in a V228A Mutant
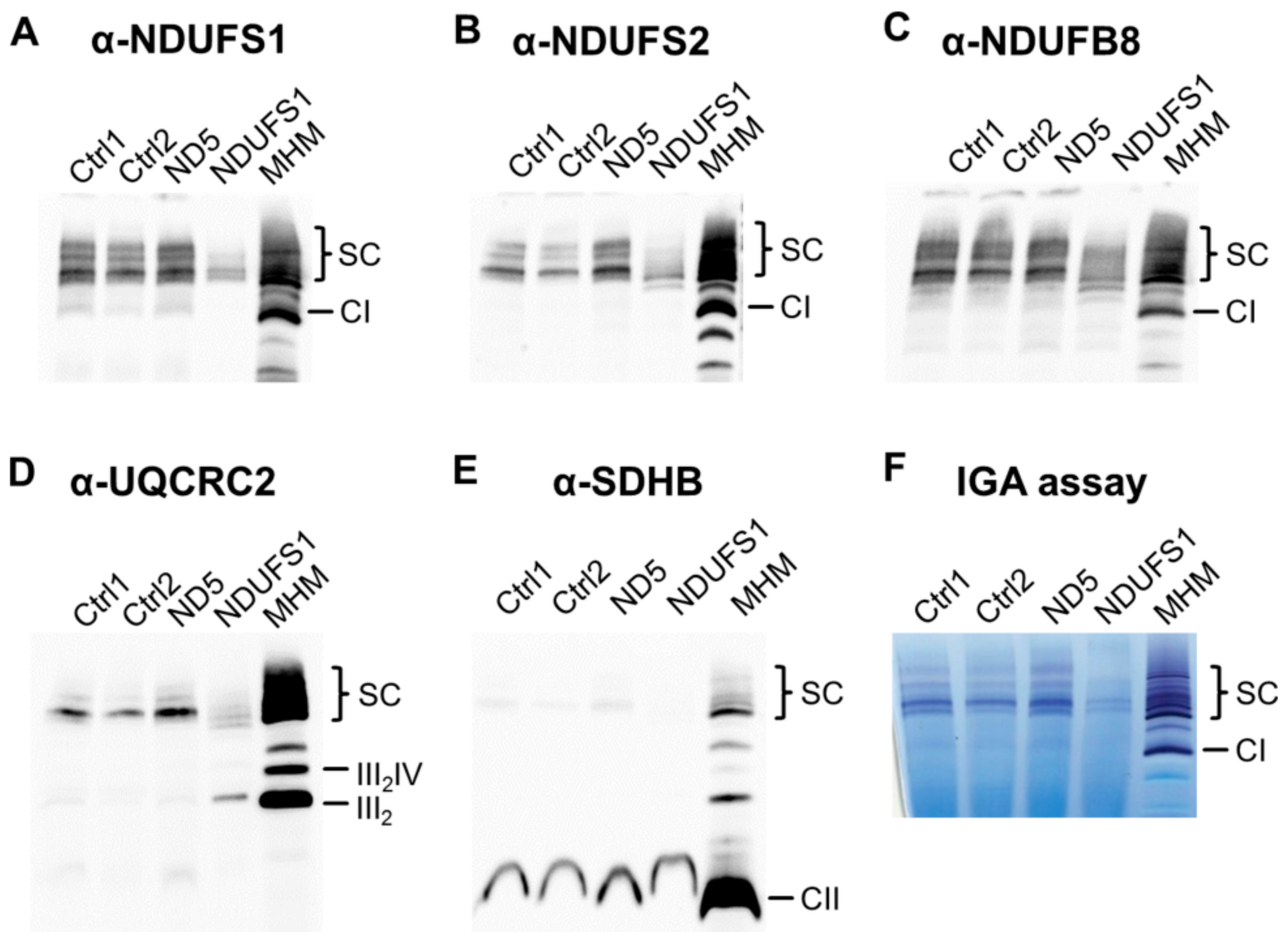
3.7. Decreased Stability of CI in Mutated NDUFS1 Prevents the Formation of Supercomplexes
3.8. Isolated CI Deficiency in Both Patients
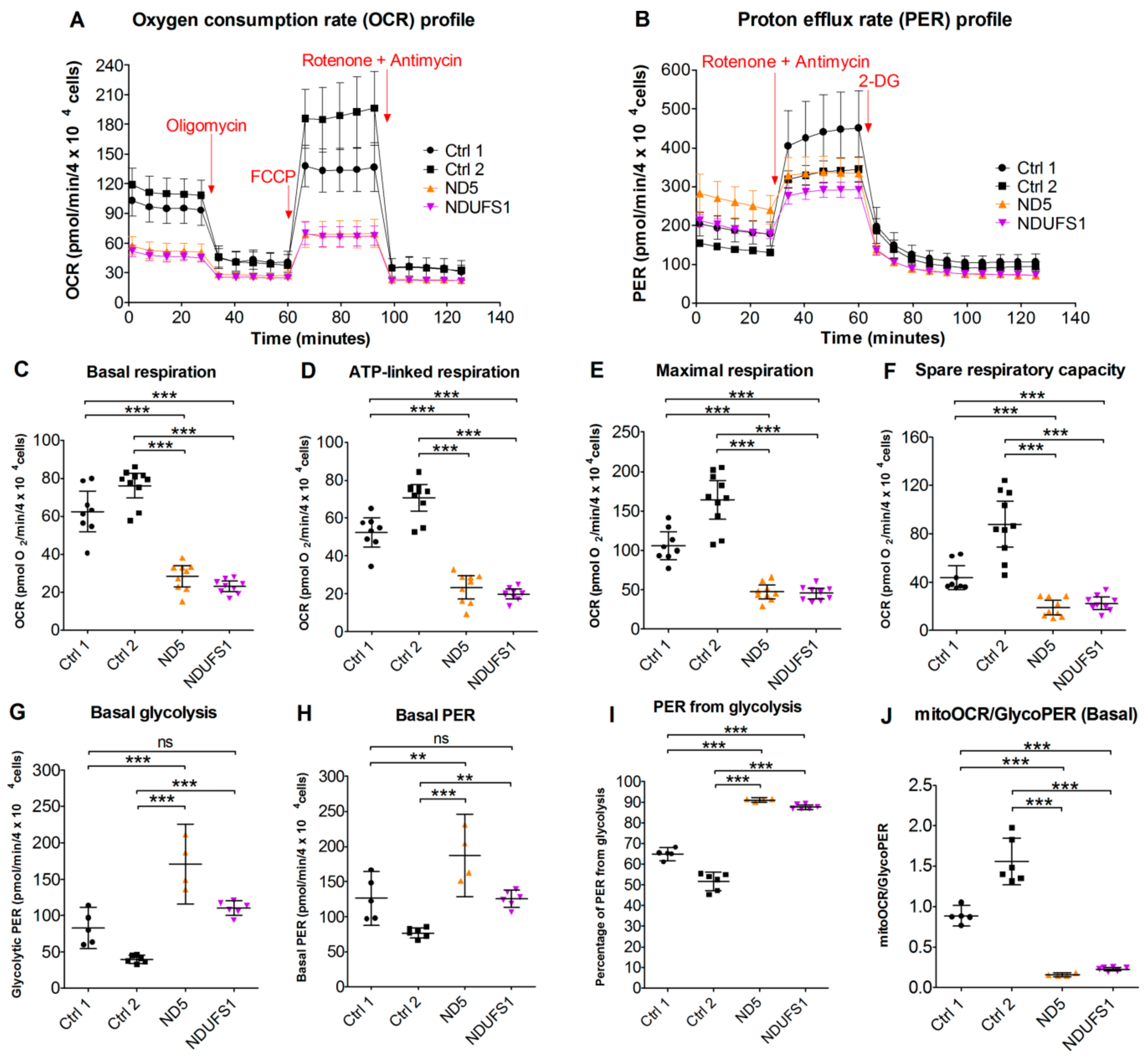
3.9. Live Cell Respiration Assays Revealed a Low Oxygen Consumption Rate in Both Patients
4. Discussion
4.1. Specific Disassembly of the N-Module and the Entire Respirasome in Mutated NDUFS1
4.2. Disruption of The Electron Flow in Mutated NDUFS1
4.3. The Stalling of Proton Translocation in Mutated ND5 Is Assumed to Stop Electron Flow Without Any Consequences for Respirasome Formation
4.4. A Similar Pattern of Regulated Metabolites Was Identified in Both Patients, Mainly for ROS Defense and TCA Cycle Metabolites
4.5. CI Deficiency Leads to a Glycolytic Phenotype
4.6. Accumulation of Structural Proteins in Patients
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Vinothkumar, K.R.; Hirst, J. Structure of mammalian respiratory complex I. Nature 2016, 536, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Fearnley, I.M.; Skehel, J.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 2006, 281, 32724–32727. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Fearnley, I.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol. Cell. Proteomics MCP 2003, 2, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Degliesposti, G.; Fiedorczuk, K.; Skehel, M.; Sazanov, L.A. Purification of ovine respiratory complex I results in a highly active and stable preparation. J. Biol. Chem. 2016, 291, 24657–24675. [Google Scholar] [CrossRef]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef]
- Wallace, D.C.; Zheng, X.X.; Lott, M.T.; Shoffner, J.M.; Hodge, J.A.; Kelley, R.I.; Epstein, C.M.; Hopkins, L.C. Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 1988, 55, 601–610. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Prokisch, H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018, 62, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Haack, T.B.; Freisinger, P.; Karall, D.; Makowski, C.; Koch, J.; Feichtinger, R.G.; Zimmermann, F.A.; Rolinski, B.; Ahting, U.; et al. Spectrum of combined respiratory chain defects. J. Inherit. Metab. Dis. 2015, 38, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders—Past, present and future. Biochim. Biophys. Acta 2004, 1659, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain J. Neurol. 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Van Erven, P.M.; Gabreels, F.J.; Ruitenbeek, W.; Den Hartog, M.R.; Fischer, J.C.; Renier, W.O.; Trijbels, J.M.; Slooff, J.L.; Janssen, A.J. Subacute necrotizing encephalomyelopathy (Leigh syndrome) associated with disturbed oxidation of pyruvate, malate and 2-oxoglutarate in muscle and liver. Acta Neurol. Scand. 1985, 72, 36–42. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Idebenone: A review in Leber’s hereditary optic neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef]
- Brink, J.; Hovmoller, S.; Ragan, C.I.; Cleeter, M.W.; Boekema, E.J.; van Bruggen, E.F. The structure of NADH:ubiquinone oxidoreductase from beef-heart mitochondria: Crystals containing an octameric arrangement of iron-sulphur protein fragments. Eur. J. Biochem. 1987, 166, 287–294. [Google Scholar] [CrossRef]
- Hirst, J.; Carroll, J.; Fearnley, I.M.; Shannon, R.J.; Walker, J.E. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim. Biophys. Acta 2003, 1604, 135–150. [Google Scholar] [CrossRef]
- Baranova, E.A.; Morgan, D.J.; Sazanov, L.A. Single particle analysis confirms distal location of subunits NuoL and NuoM in Escherichia coli complex I. J. Struct. Biol. 2007, 159, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Zickermann, V.; Kerscher, S.; Zwicker, K.; Tocilescu, M.A.; Radermacher, M.; Brandt, U. Architecture of complex I and its implications for electron transfer and proton pumping. Biochim. Biophys. Acta 2009, 1787, 574–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, R.W.; Morris, A.A.; Hutchinson, M.; Turnbull, D.M. Leigh disease associated with a novel mitochondrial DNA ND5 mutation. Eur. J. Hum. Genet. 2002, 10, 141–144. [Google Scholar] [CrossRef]
- Lebon, S.; Chol, M.; Benit, P.; Mugnier, C.; Chretien, D.; Giurgea, I.; Kern, I.; Girardin, E.; Hertz-Pannier, L.; de Lonlay, P.; et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J. Med. Genet. 2003, 40, 896–899. [Google Scholar] [CrossRef]
- Zhadanov, S.I.; Grechanina, E.Y.; Grechanina, Y.B.; Gusar, V.A.; Fedoseeva, N.P.; Lebon, S.; Munnich, A.; Schurr, T.G. Fatal manifestation of a de novo ND5 mutation: Insights into the pathogenetic mechanisms of mtDNA ND5 gene defects. Mitochondrion 2007, 7, 260–266. [Google Scholar] [CrossRef]
- Benit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rotig, A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001, 68, 1344–1352. [Google Scholar] [CrossRef]
- Pagniez-Mammeri, H.; Lombes, A.; Brivet, M.; Ogier-de Baulny, H.; Landrieu, P.; Legrand, A.; Slama, A. Rapid screening for nuclear genes mutations in isolated respiratory chain complex I defects. Mol. Genet. Metab. 2009, 96, 196–200. [Google Scholar] [CrossRef]
- Kashani, A.; Thiffault, I.; Dilenge, M.E.; Saint-Martin, C.; Guerrero, K.; Tran, L.T.; Shoubridge, E.; van der Knaap, M.S.; Braverman, N.; Bernard, G. A homozygous mutation in the NDUFS1 gene presents with a mild cavitating leukoencephalopathy. Neurogenetics 2014, 15, 161–164. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Gielisch, I.; Meierhofer, D. Metabolome and proteome profiling of complex I deficiency induced by rotenone. J. Proteome Res. 2015, 14, 224–235. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Martens, L.; Hermjakob, H.; Jones, P.; Adamski, M.; Taylor, C.; States, D.; Gevaert, K.; Vandekerckhove, J.; Apweiler, R. PRIDE: The proteomics identifications database. Proteomics 2005, 5, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Stuchebrukhov, A.A. Electron tunneling in respiratory complex I. Proc. Natl. Acad. Sci. USA 2010, 107, 19157–19162. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 2016, 32, 2936–2946. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Zimmermann, F.; Mayr, J.A.; Neureiter, D.; Hauser-Kronberger, C.; Schilling, F.H.; Jones, N.; Sperl, W.; Kofler, B. Low aerobic mitochondrial energy metabolism in poorly- or undifferentiated neuroblastoma. BMC Cancer 2010, 10, 149. [Google Scholar] [CrossRef]
- Meierhofer, D.; Mayr, J.A.; Foetschl, U.; Berger, A.; Fink, K.; Schmeller, N.; Hacker, G.W.; Hauser-Kronberger, C.; Kofler, B.; Sperl, W. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 2004, 25, 1005–1010. [Google Scholar] [CrossRef]
- Berger, A.; Mayr, J.A.; Meierhofer, D.; Fotschl, U.; Bittner, R.; Budka, H.; Grethen, C.; Huemer, M.; Kofler, B.; Sperl, W. Severe depletion of mitochondrial DNA in spinal muscular atrophy. Acta Neuropathol. 2003, 105, 245–251. [Google Scholar] [PubMed]
- Aretz, I.; Hardt, C.; Wittig, I.; Meierhofer, D. An Impaired Respiratory Electron Chain Triggers Down-regulation of the Energy Metabolism and De-ubiquitination of Solute Carrier Amino Acid Transporters. Mol. Cell. Proteomics MCP 2016, 15, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Heidler, J.; Strecker, V.; Csintalan, F.; Bleier, L.; Wittig, I. Quantification of protein complexes by blue native electrophoresis. Methods Mol. Biol. 2013, 1033, 363–379. [Google Scholar] [PubMed]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schagger, H. Functional assays in high-resolution clear native gels to quantify mitochondrial complexes in human biopsies and cell lines. Electrophoresis 2007, 28, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Moore, T.; Le, A.; Atkuri, K.; Shah, M.K.; Cusmano-Ozog, K.; Niemi, A.K.; Cowan, T.M. Degree of glutathione deficiency and redox imbalance depend on subtype of mitochondrial disease and clinical status. PLoS ONE 2014, 9, e100001. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Santa, T. Recent advances in analysis of glutathione in biological samples by high-performance liquid chromatography: A brief overview. Drug Discov. Ther. 2013, 7, 172–177. [Google Scholar] [CrossRef][Green Version]
- Das, K.C.; Misra, H.P. Hydroxyl radical scavenging and singlet oxygen quenching properties of polyamines. Mol. Cell. Biochem. 2004, 262, 127–133. [Google Scholar] [CrossRef]
- Fujisawa, S.; Kadoma, Y. Kinetic evaluation of polyamines as radical scavengers. Anticancer Res. 2005, 25, 965–969. [Google Scholar] [PubMed]
- Esteitie, N.; Hinttala, R.; Wibom, R.; Nilsson, H.; Hance, N.; Naess, K.; Tear-Fahnehjelm, K.; von Dobeln, U.; Majamaa, K.; Larsson, N.G. Secondary metabolic effects in complex I deficiency. Ann. Neurol. 2005, 58, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H. Lactic acidemia and mitochondrial disease. Mol. Genet. Metab. 2006, 89, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Weis, S.; Mayr, J.A.; Zimmermann, F.; Geilberger, R.; Sperl, W.; Kofler, B. Alterations of oxidative phosphorylation complexes in astrocytomas. Glia 2014, 62, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Hinchliffe, P.; Sazanov, L.A. Organization of iron-sulfur clusters in respiratory complex I. Science 2005, 309, 771–774. [Google Scholar] [CrossRef]
- Nakamaru-Ogiso, E. Iron–Sulfur Clusters in Complex I. In A Structural Perspective on Respiratory Complex I: Structure and Function of NADH:ubiquinone Oxidoreductase; Sazanov, L., Ed.; Springer: Dordrecht, The Netherland, 2012; pp. 61–79. [Google Scholar]
- Lazarou, M.; McKenzie, M.; Ohtake, A.; Thorburn, D.R.; Ryan, M.T. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 2007, 27, 4228–4237. [Google Scholar] [CrossRef]
- Garcia, C.J.; Khajeh, J.; Coulanges, E.; Chen, E.I.; Owusu-Ansah, E. Regulation of Mitochondrial Complex I Biogenesis in Drosophila Flight Muscles. Cell Rep. 2017, 20, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef]
- Eubel, H.; Heinemeyer, J.; Braun, H.P. Identification and characterization of respirasomes in potato mitochondria. Plant Physiol. 2004, 134, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257. [Google Scholar] [CrossRef]
- Schägger, H.; Pfeiffer, K. The Ratio of Oxidative Phosphorylation Complexes I–V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar]
- Vartak, R.; Porras, C.A.; Bai, Y. Respiratory supercomplexes: Structure, function and assembly. Protein Cell 2013, 4, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef]
- Fedor, J.G.; Hirst, J. Mitochondrial Supercomplexes Do Not Enhance Catalysis by Quinone Channeling. Cell Metab. 2018, 28, 525–531. [Google Scholar] [CrossRef]
- Hirst, J.; Roessler, M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Esterhazy, D.; King, M.S.; Yakovlev, G.; Hirst, J. Production of reactive oxygen species by complex I (NADH:Ubiquinone oxidoreductase) from Escherichia coli and comparison to the enzyme from mitochondria. Biochemistry 2008, 47, 3964–3971. [Google Scholar] [CrossRef] [PubMed]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- King, M.S.; Sharpley, M.S.; Hirst, J. Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH:Ubiquinone oxidoreductase (Complex I) and production of reactive oxygen species. Biochemistry 2009, 48, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Leman, G.; Gueguen, N.; Desquiret-Dumas, V.; Kane, M.S.; Wettervald, C.; Chupin, S.; Chevrollier, A.; Lebre, A.S.; Bonnefont, J.P.; Barth, M.; et al. Assembly defects induce oxidative stress in inherited mitochondrial complex I deficiency. Int. J. Biochem. Cell Biol. 2015, 65, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. A two-state stabilization-change mechanism for proton-pumping complex I. Biochim. Biophys. Acta 2011, 1807, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.; Blaza, J.N.; Varghese, F.; Hirst, J. Respiratory Complex I in Bos taurus and Paracoccus denitrificans Pumps Four Protons across the Membrane for Every NADH Oxidized. J. Biol. Chem. 2017, 292, 4987–4995. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, M.; Sakyiama, J.A.; Elguindy, M.M.; Nakamaru-Ogiso, E. Roles of subunit NuoL in the proton pumping coupling mechanism of NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli. J. Biochem. 2016, 160, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, L.A. Structure of Respiratory Complex I: “Minimal” Bacterial and “De luxe” Mammalian Versions. In Mechanisms of Primary Energy Transduction in Biology; The Royal Society of Chemistry: Cambridge, UK, 2018; pp. 25–59. [Google Scholar]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Esterhuizen, K.; van der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef]
- Williamson, J.R.; Cooper, R.H. Regulation of the citric acid cycle in mammalian systems. FEBS Lett. 1980, 117, K73–K85. [Google Scholar] [CrossRef]
- Varghese, F.; Atcheson, E.; Bridges, H.R.; Hirst, J. Characterization of clinically identified mutations in NDUFV1, the flavin-binding subunit of respiratory complex I, using a yeast model system. Hum. Mol. Genet. 2015, 24, 6350–6360. [Google Scholar] [CrossRef]
- Gostimskaya, I.S.; Grivennikova, V.G.; Cecchini, G.; Vinogradov, A.D. Reversible dissociation of flavin mononucleotide from the mammalian membrane-bound NADH:ubiquinone oxidoreductase (complex I). FEBS Lett. 2007, 581, 5803–5806. [Google Scholar] [CrossRef]
- Holt, P.J.; Efremov, R.G.; Nakamaru-Ogiso, E.; Sazanov, L.A. Reversible FMN dissociation from Escherichia coli respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 1777–1785. [Google Scholar] [CrossRef]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.; Brandt, U. Superoxide Radical Formation by Pure Complex I (NADH:Ubiquinone Oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 2005, 280, 30129–30135. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Benit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rotig, A.; et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Brecht, M.; Richardson, M.; Taranath, A.; Grist, S.; Thorburn, D.; Bratkovic, D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015, 19, 95–100. [Google Scholar] [PubMed]
- Ayalon, N.; Flore, L.A.; Christensen, T.G.; Sam, F. Mitochondrial encoded NADH dehydrogenase 5 (MT-ND5) gene point mutation presents as late onset cardiomyopathy. Int. J. Cardiol. 2013, 167, e143–e145. [Google Scholar] [CrossRef]
- Annunen-Rasila, J.; Ohlmeier, S.; Tuokko, H.; Veijola, J.; Majamaa, K. Proteome and cytoskeleton responses in osteosarcoma cells with reduced OXPHOS activity. Proteomics 2007, 7, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity Ratio Versus CS | CI | CI + III | CII | CII + III | CIII | CIV | F1Fo ATP Synthase |
|---|---|---|---|---|---|---|---|
| Patient MT-ND5 | 0.04 | 0.07 | 0.29 | 0.39 | 2.10 | 1.87 | 0.96 |
| Patient NDUFS1 | 0.05 | 0.14 | 0.29 | 0.36 | 2.20 | 0.97 | 1.16 |
| Reference range | 0.14–0.35 | 0.24–0.81 | 0.23–0.41 | 0.30–0.67 | 1.45–3.76 | 0.82–2.04 | 0.42–1.26 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, Y.; Hagras, M.A.; Konstantopoulou, V.; Mayr, J.A.; Stuchebrukhov, A.A.; Meierhofer, D. Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer. Cells 2019, 8, 1149. https://doi.org/10.3390/cells8101149
Ni Y, Hagras MA, Konstantopoulou V, Mayr JA, Stuchebrukhov AA, Meierhofer D. Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer. Cells. 2019; 8(10):1149. https://doi.org/10.3390/cells8101149
Chicago/Turabian StyleNi, Yang, Muhammad A. Hagras, Vassiliki Konstantopoulou, Johannes A. Mayr, Alexei A. Stuchebrukhov, and David Meierhofer. 2019. "Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer" Cells 8, no. 10: 1149. https://doi.org/10.3390/cells8101149
APA StyleNi, Y., Hagras, M. A., Konstantopoulou, V., Mayr, J. A., Stuchebrukhov, A. A., & Meierhofer, D. (2019). Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer. Cells, 8(10), 1149. https://doi.org/10.3390/cells8101149