Preparation of Methacrylate-Based Polymers Modified with Chiral Resorcinarenes and Their Evaluation as Sorbents in Norepinephrine Microextraction


Abstract
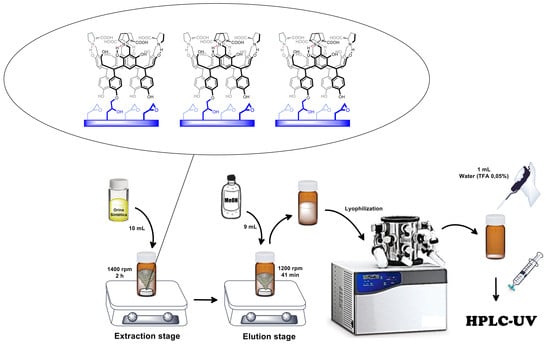
:
1. Introduction
2. Materials and Methods
2.1. General Experimental Information
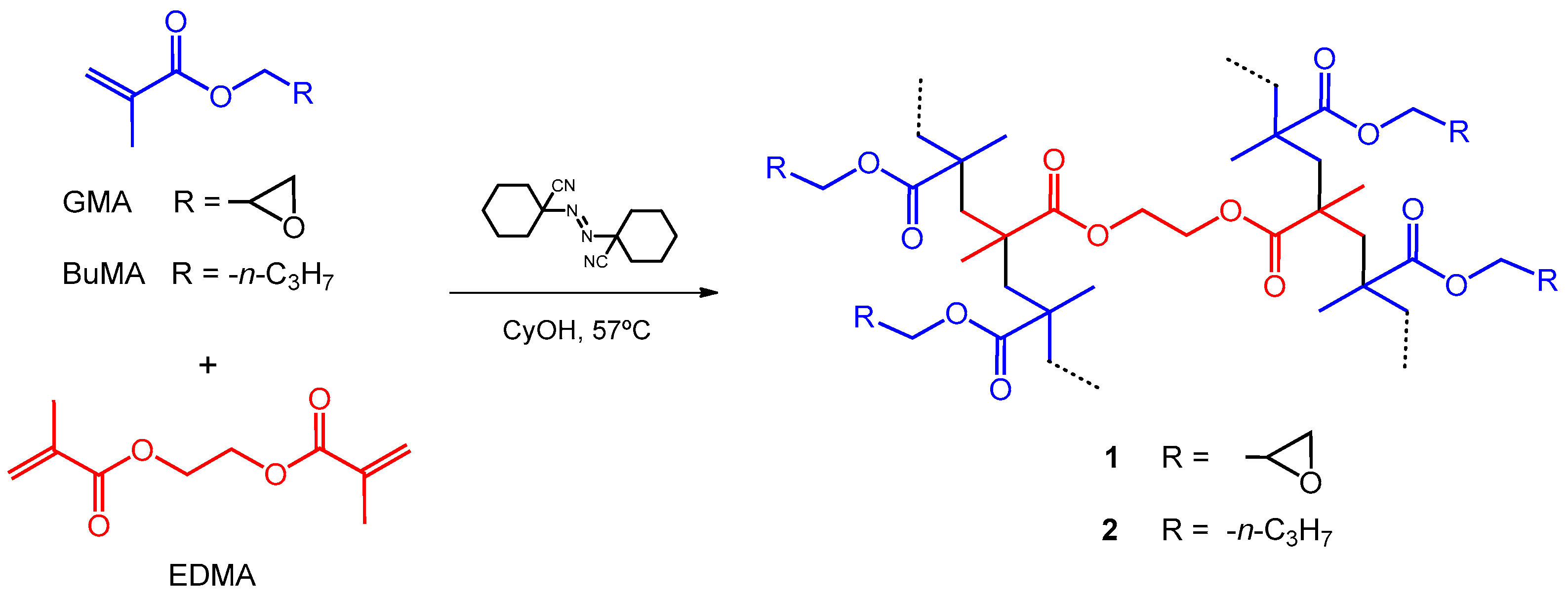
2.2. Preparation of Polymers
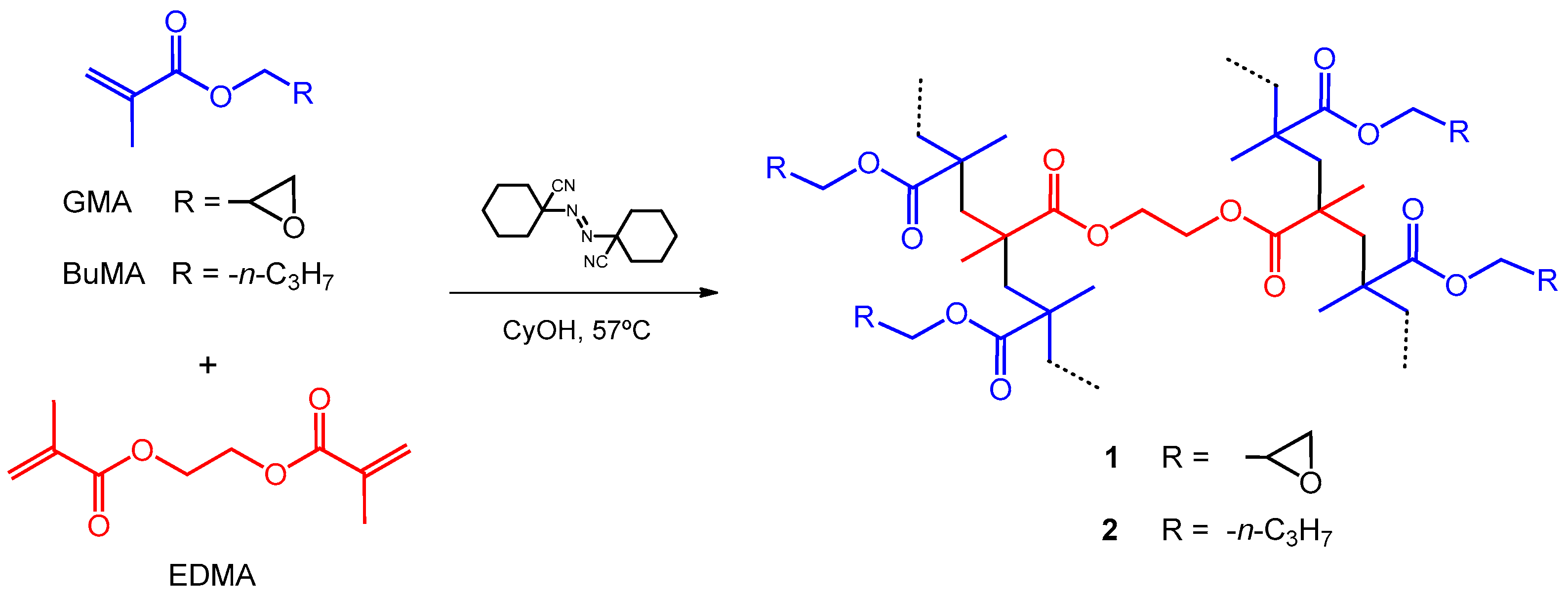
2.2.1. Poly(GMA–co–EDMA) (1)
2.2.2. Poly(BuMA–co–EDMA) (2)
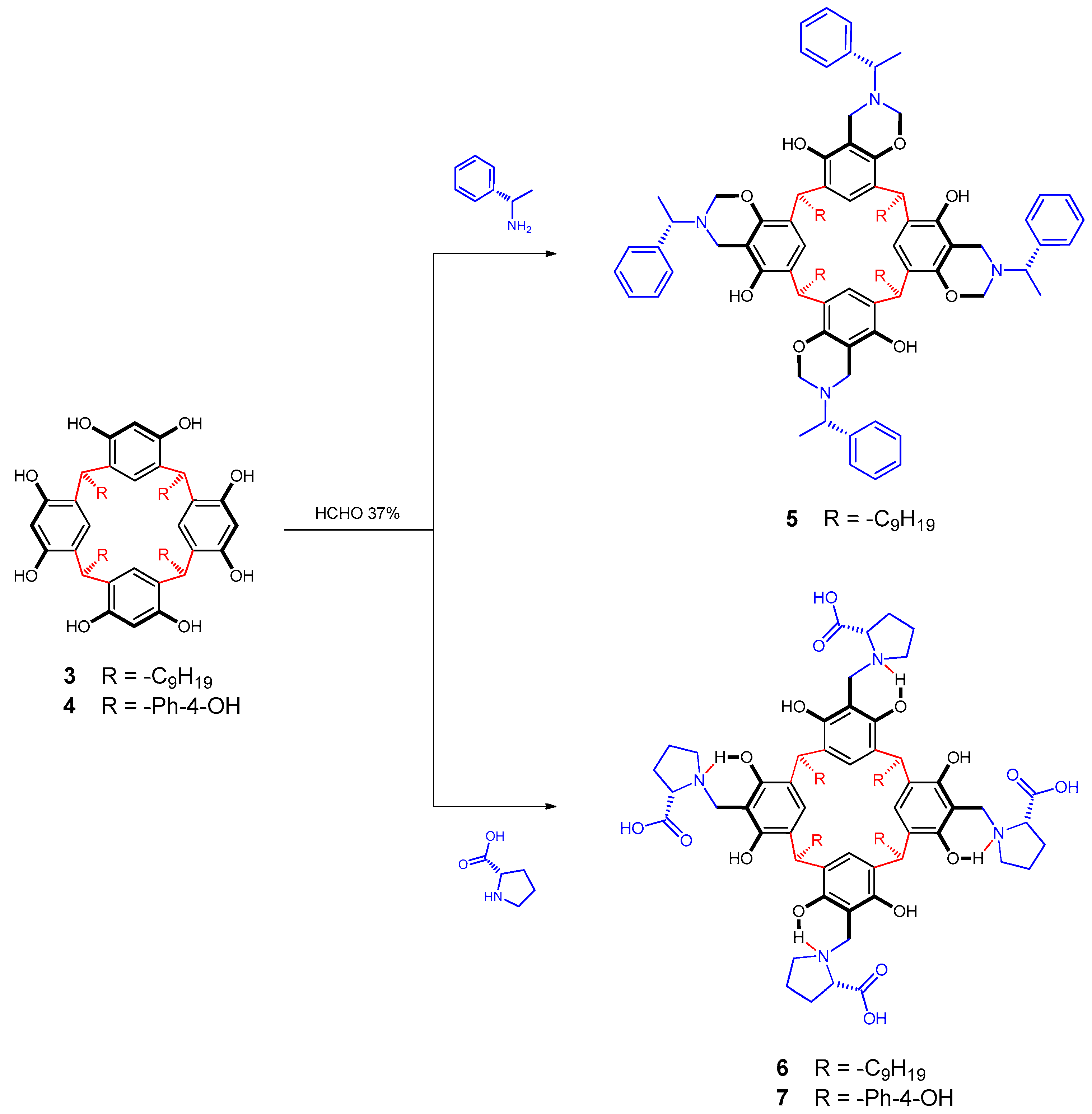
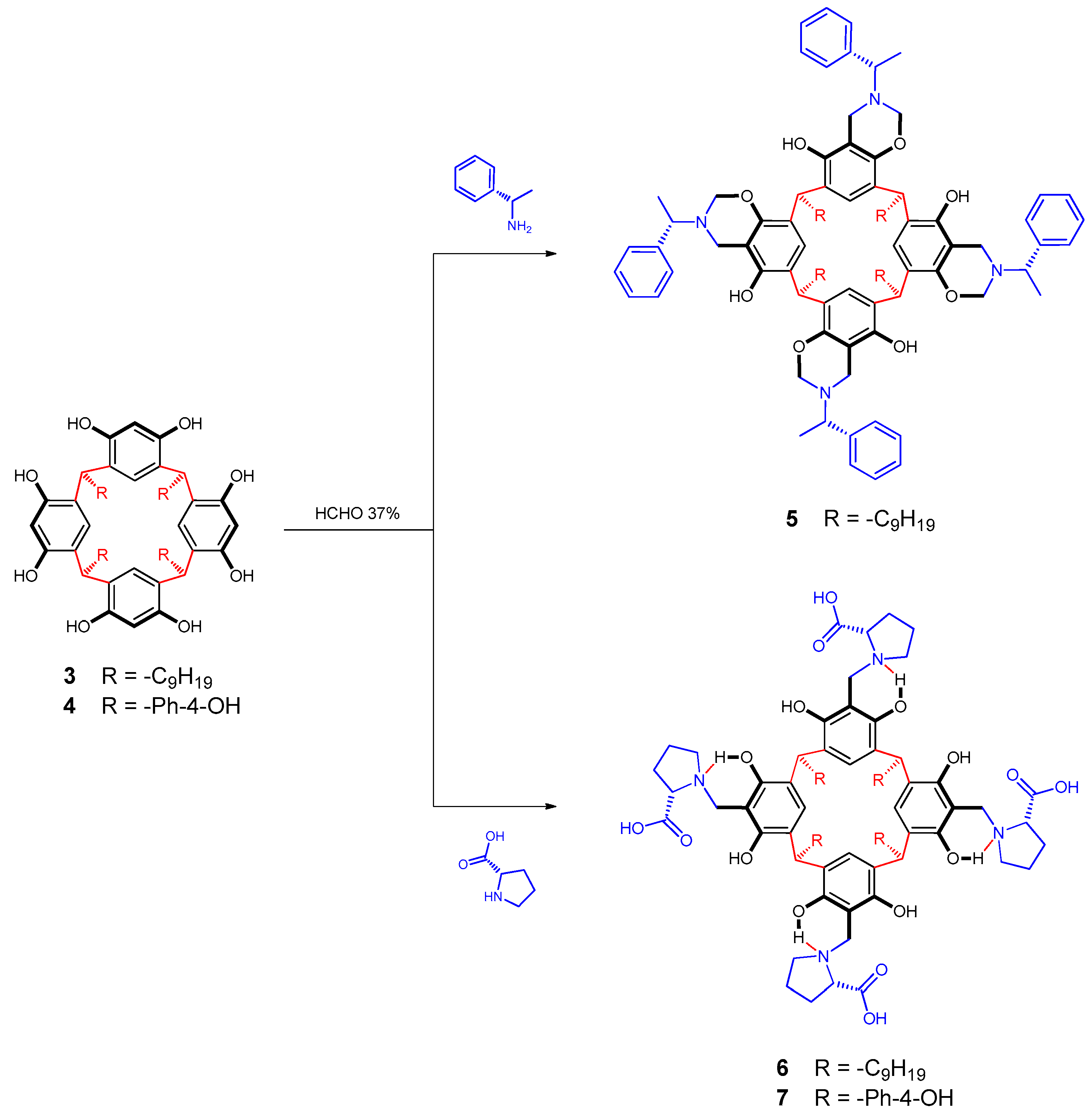
2.3. Synthesis of Chiral Resorcinarenes
2.3.1. Functionalization of Resorcinarene 3 with S-(-)-1-phenylethylamine
2.3.2. Functionalization of Resorcinarenes 3 and 4 with l-proline
2.4. Polymeric Modifications of 1 and 2 with Chiral Resorcinarenes 5–7
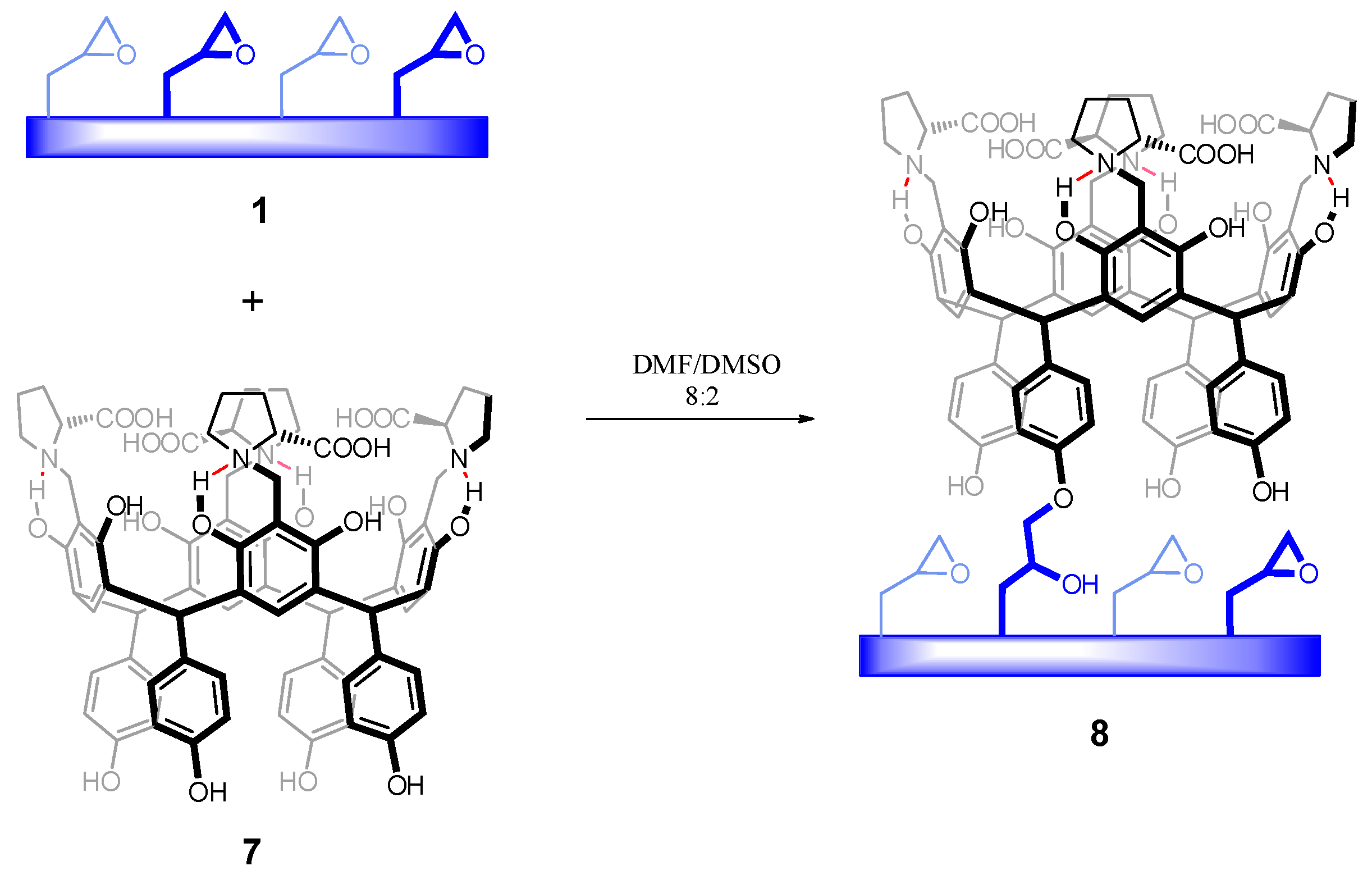
2.4.1. Chemical Modification of Polymer 1 with Chiral Resorcinarene 7
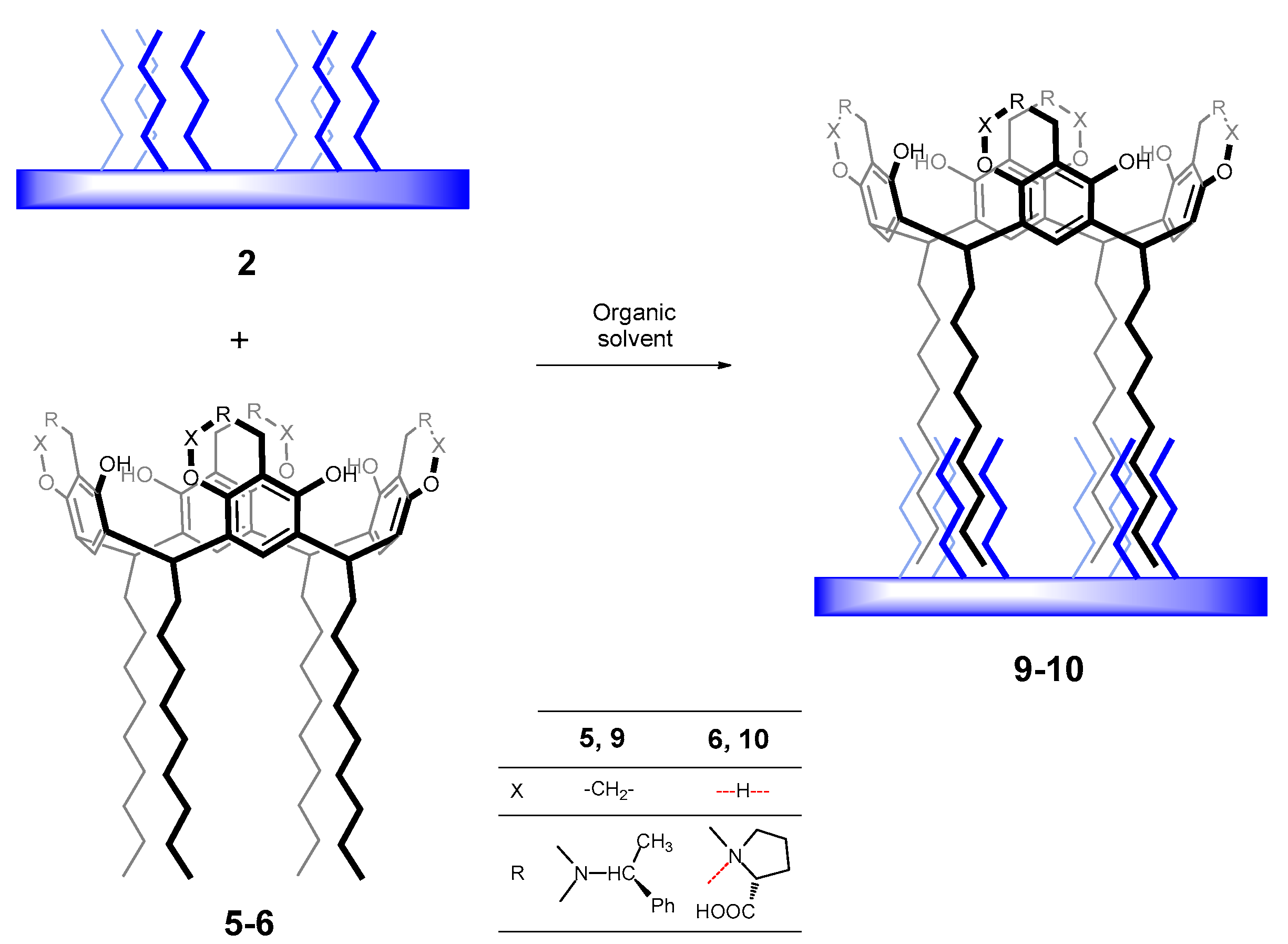
2.4.2. Physical Modification of Polymer 2 with Chiral Resorcinarenes 5 and 6
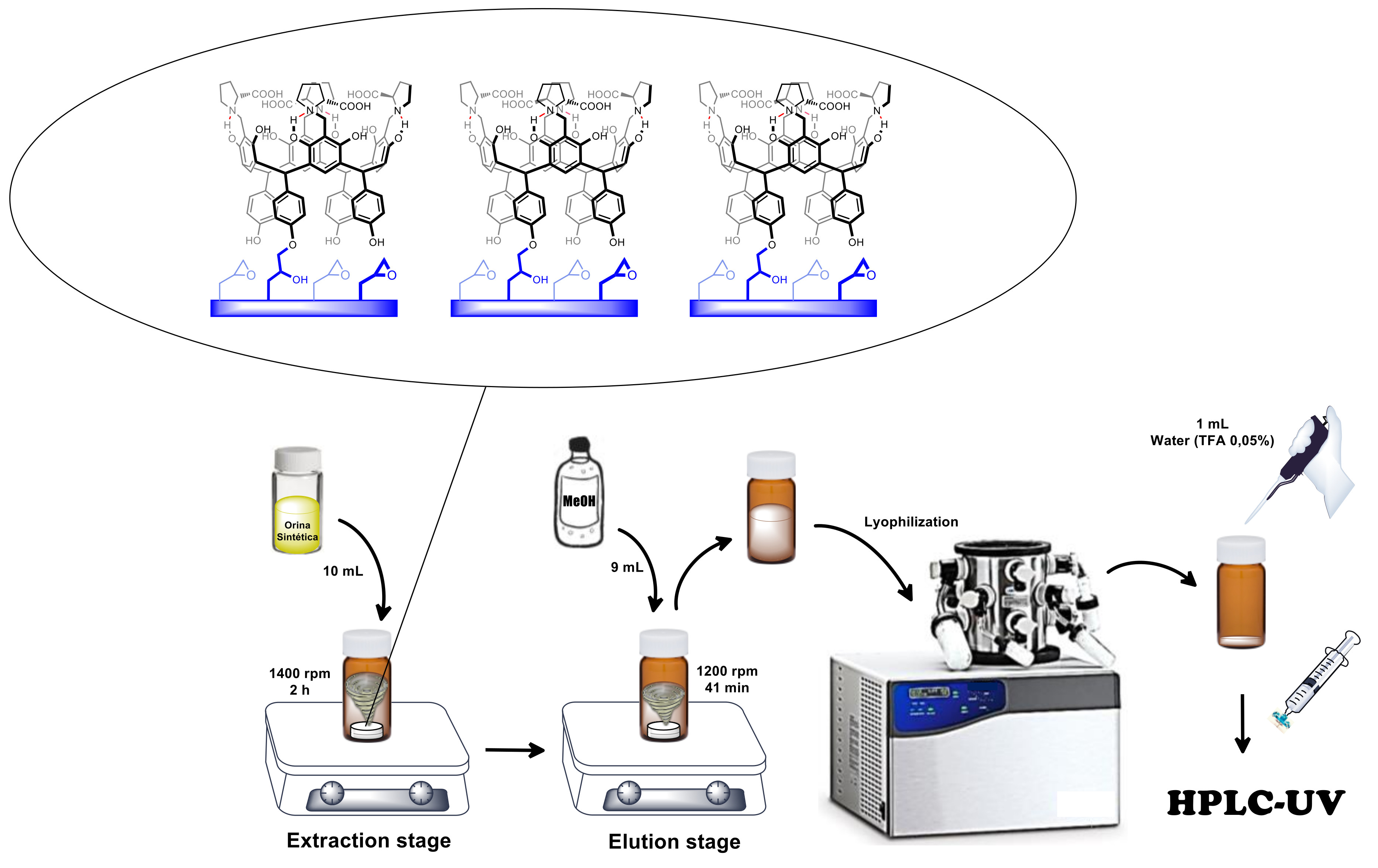
2.5. Microextraction and Quantification of Norepinephrine
2.5.1. RDSE Protocol
2.5.2. Detection and Quantification via HILIC-HPLC-UV
3. Results
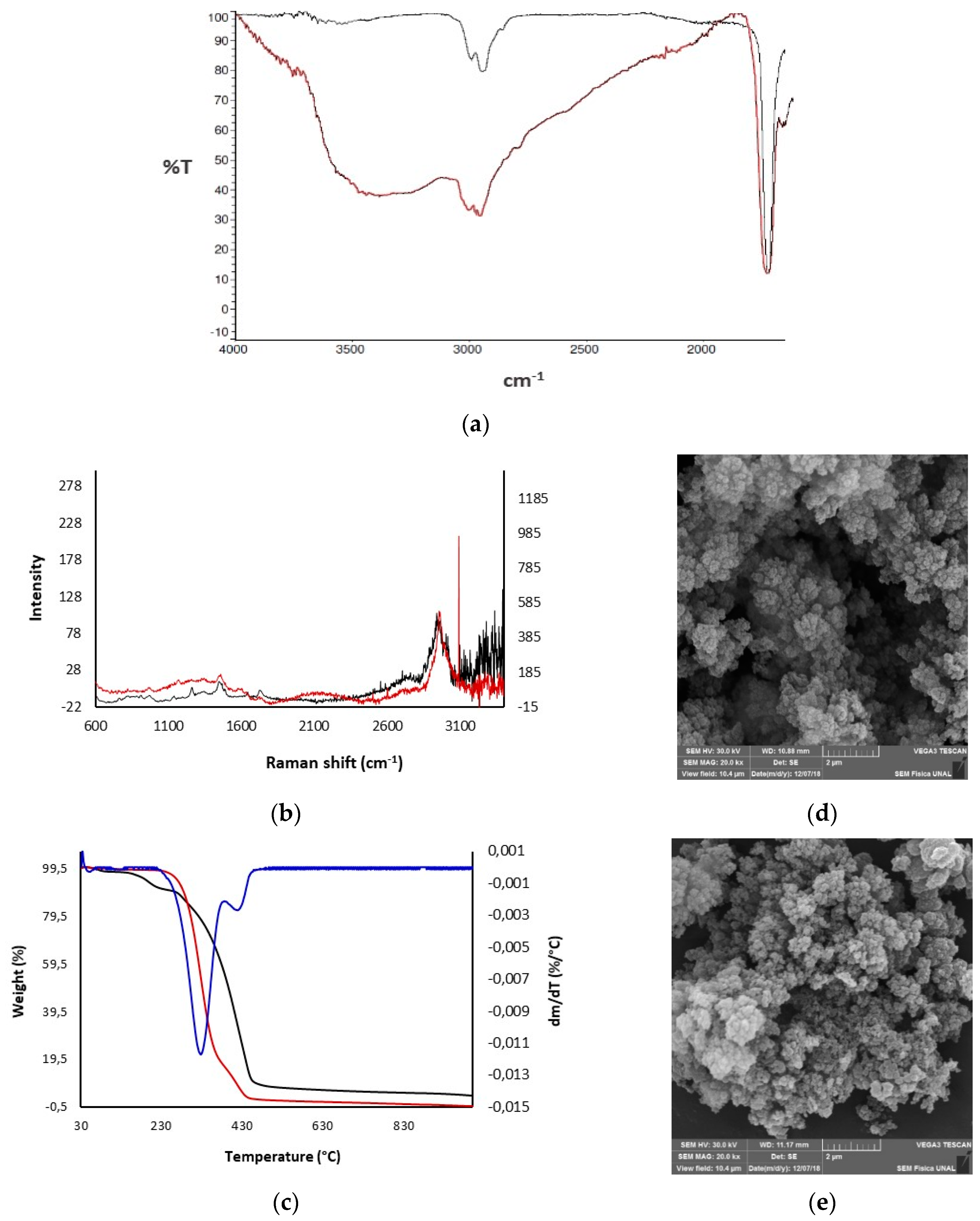
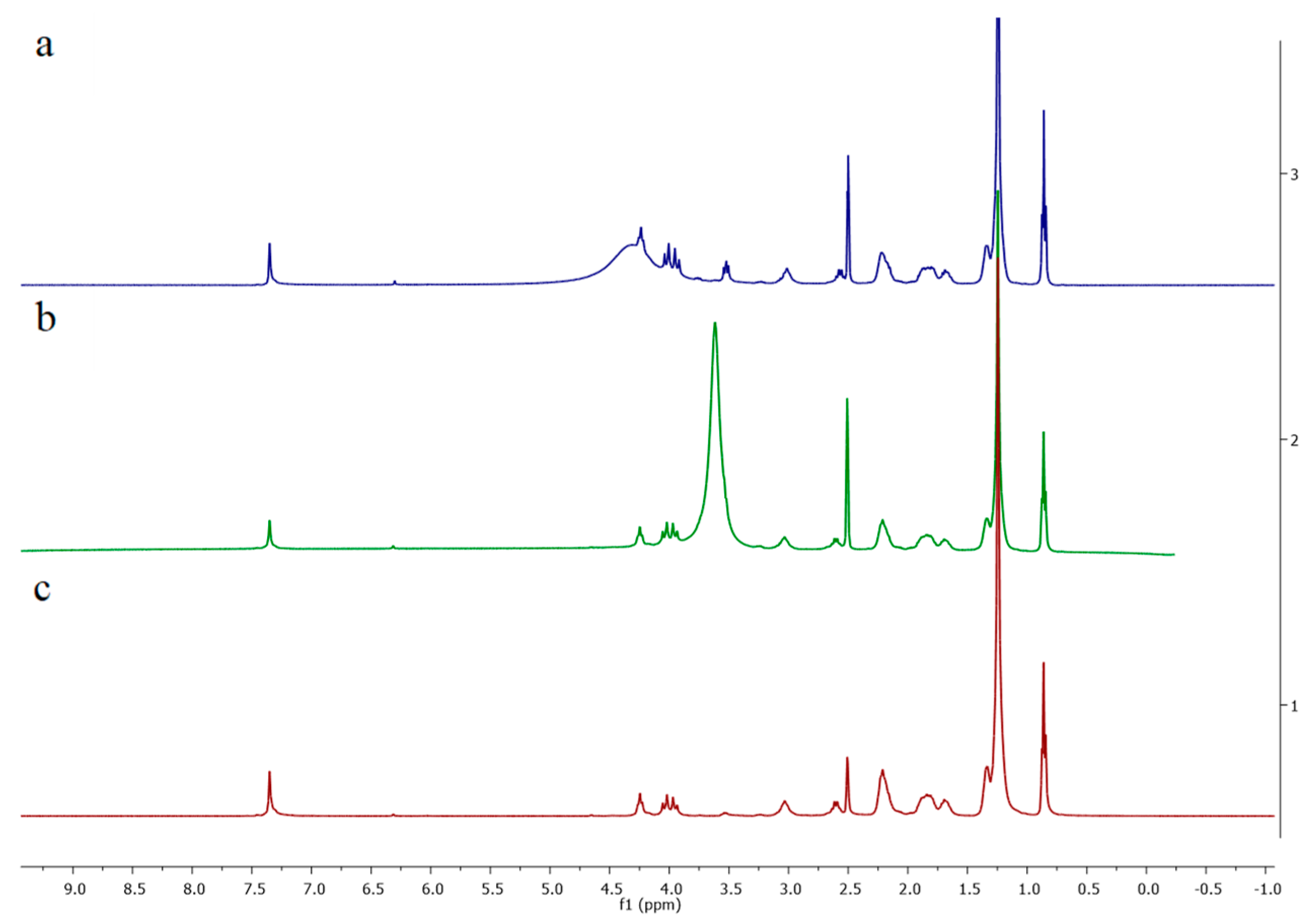
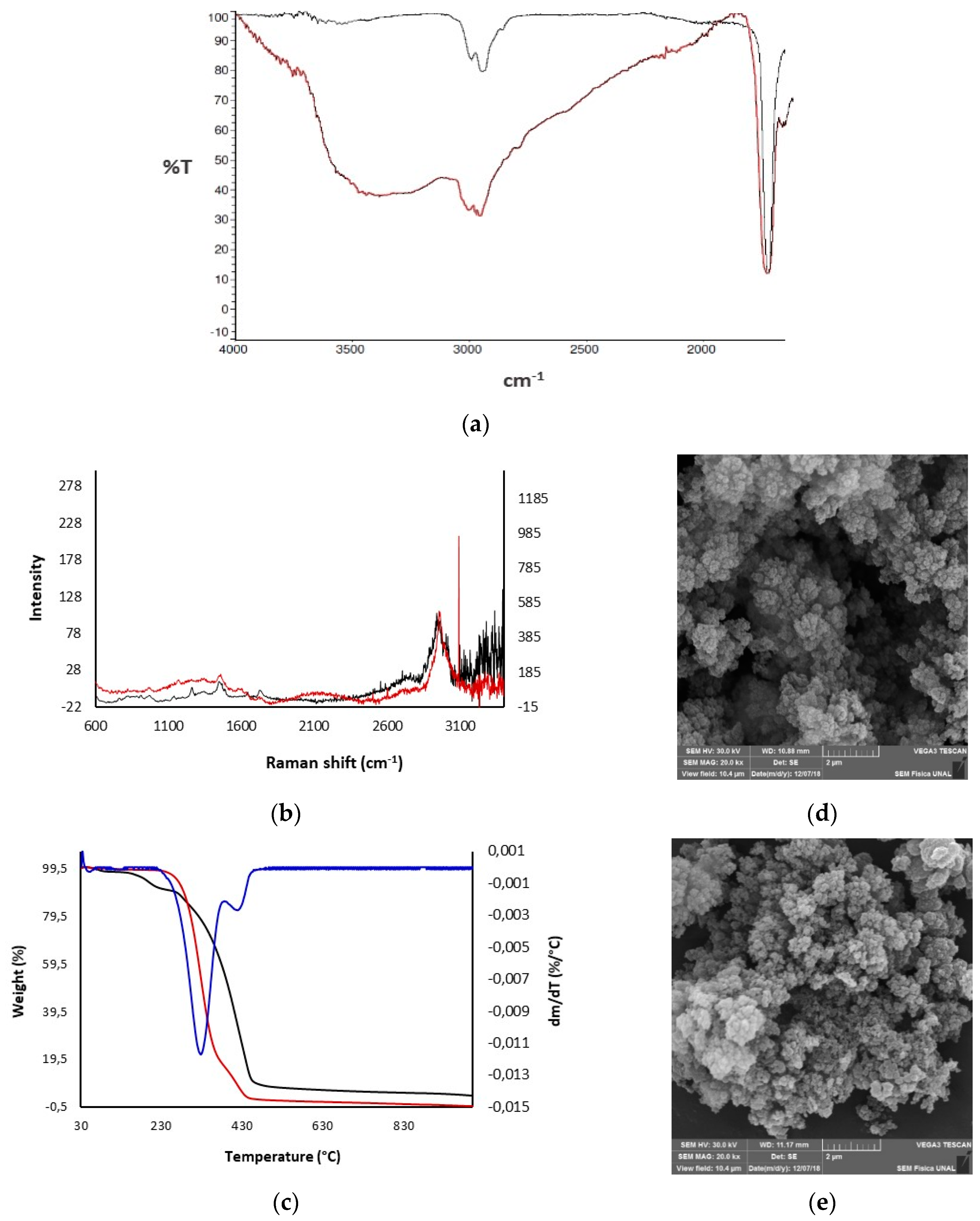
3.1. Preparation of Polymers
3.2. Synthesis of Chiral Resorcinarenes
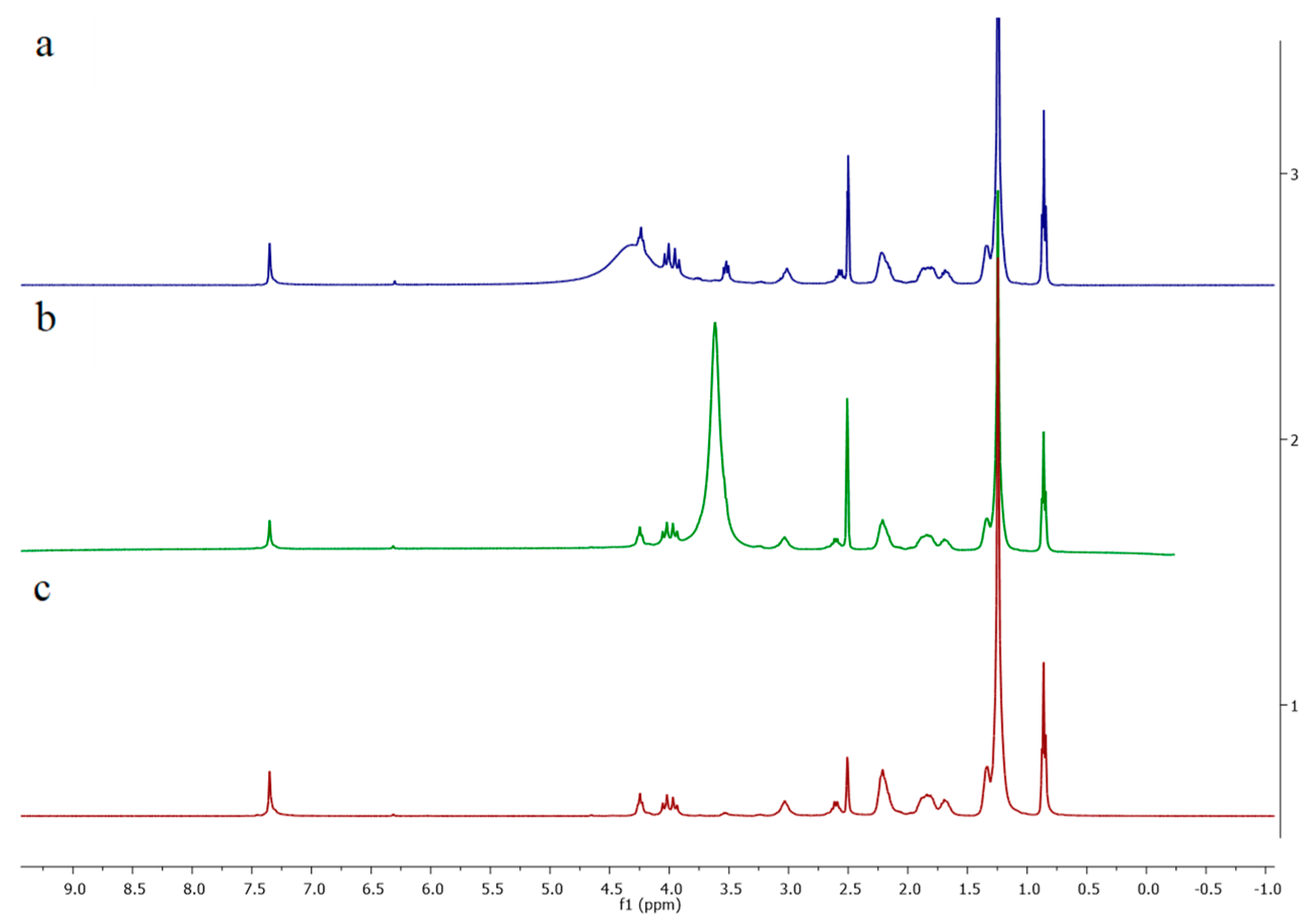
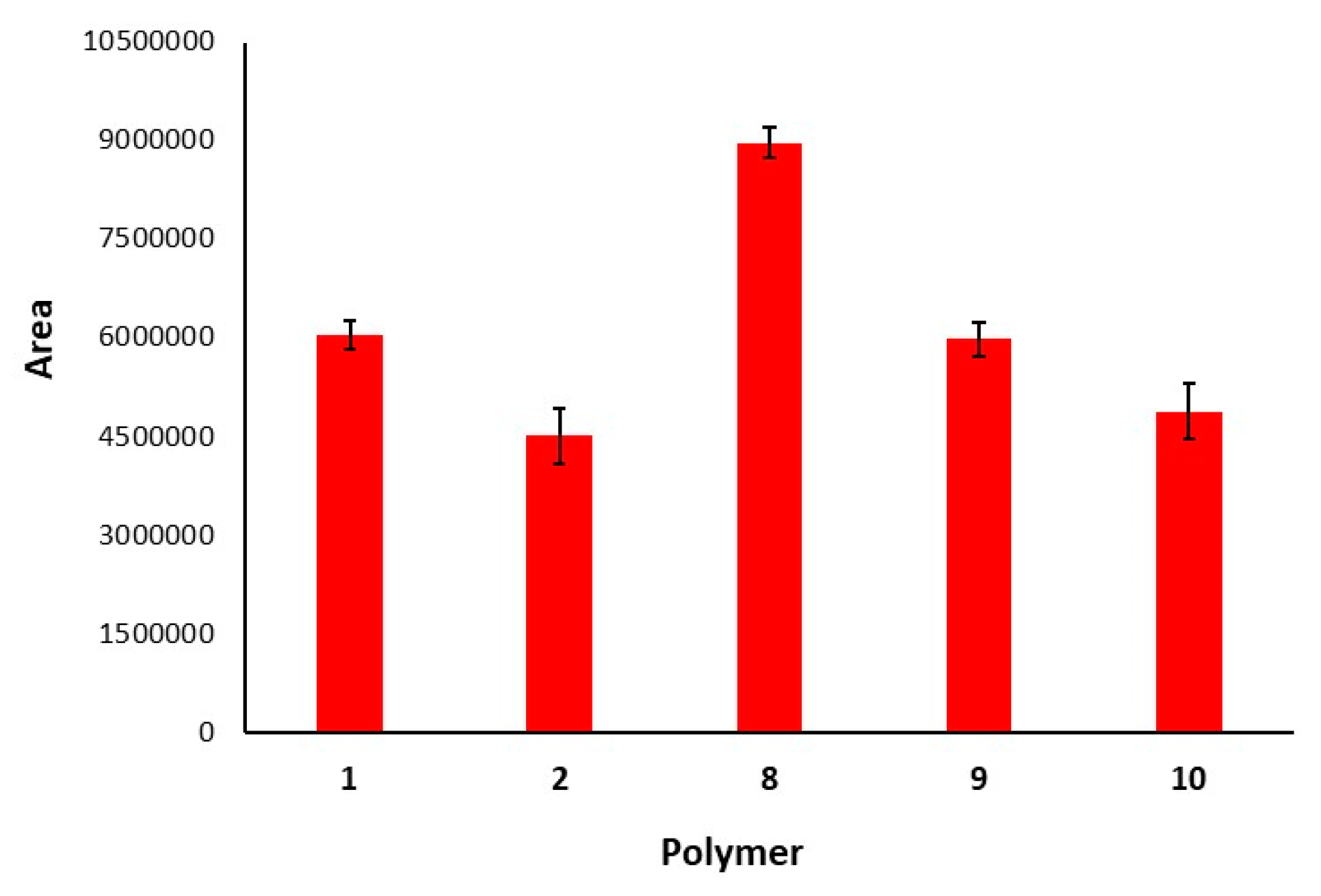
3.3. Polymeric Modifications of 1 and 2 with Chiral Resorcinarenes 5–7
3.3.1. Chemical Modification of Polymer 1 with Chiral Resorcinarene 7
3.3.2. Physical Modification of Polymer 2 with Chiral Resorcinarenes 5 and 6
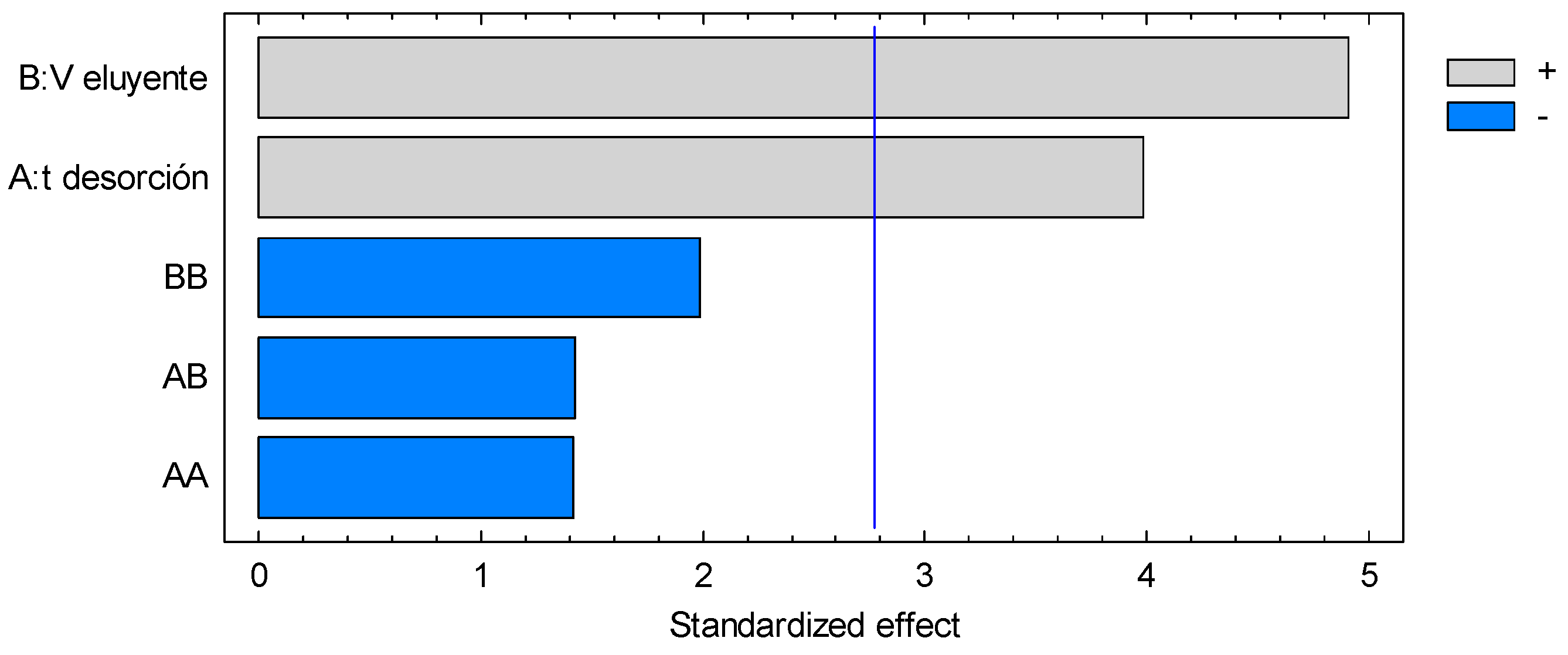
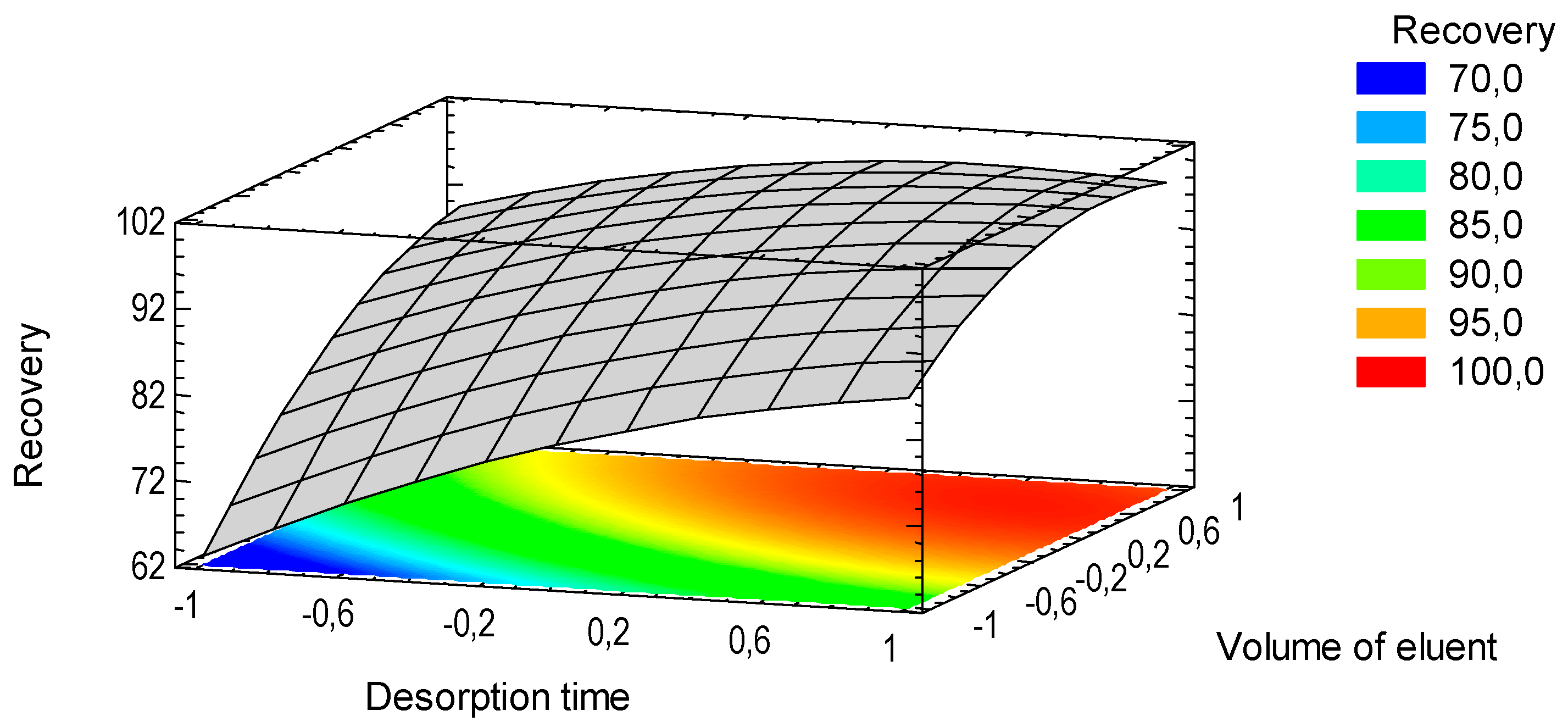
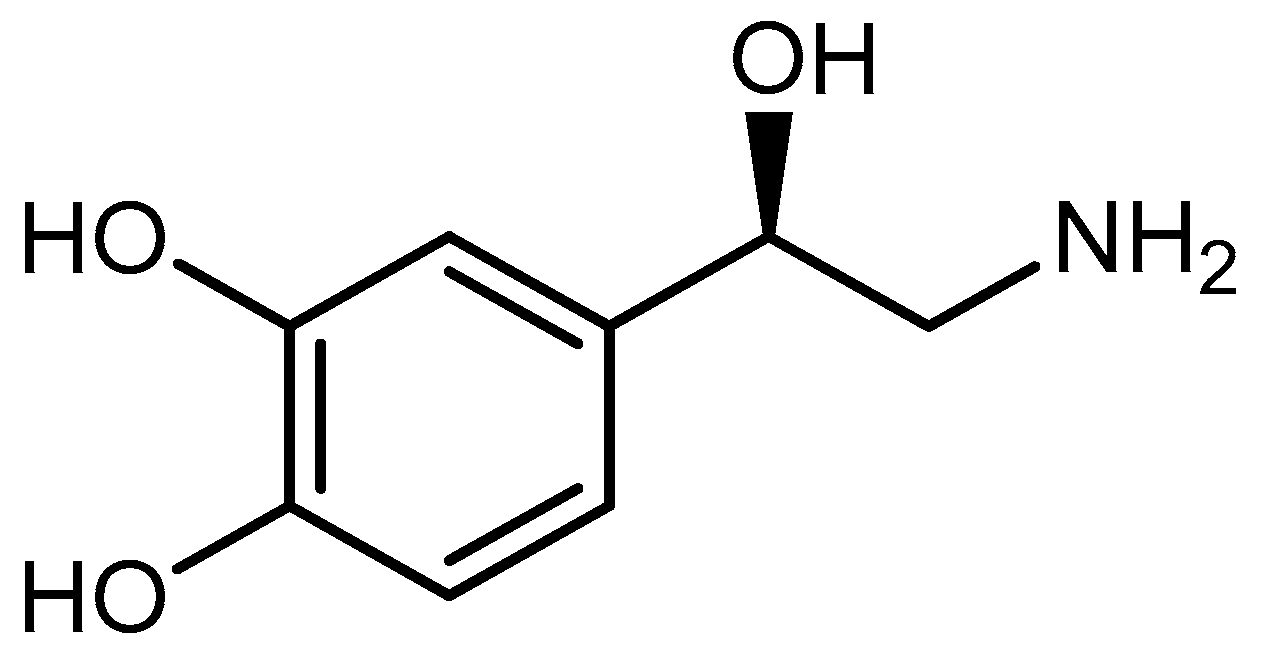
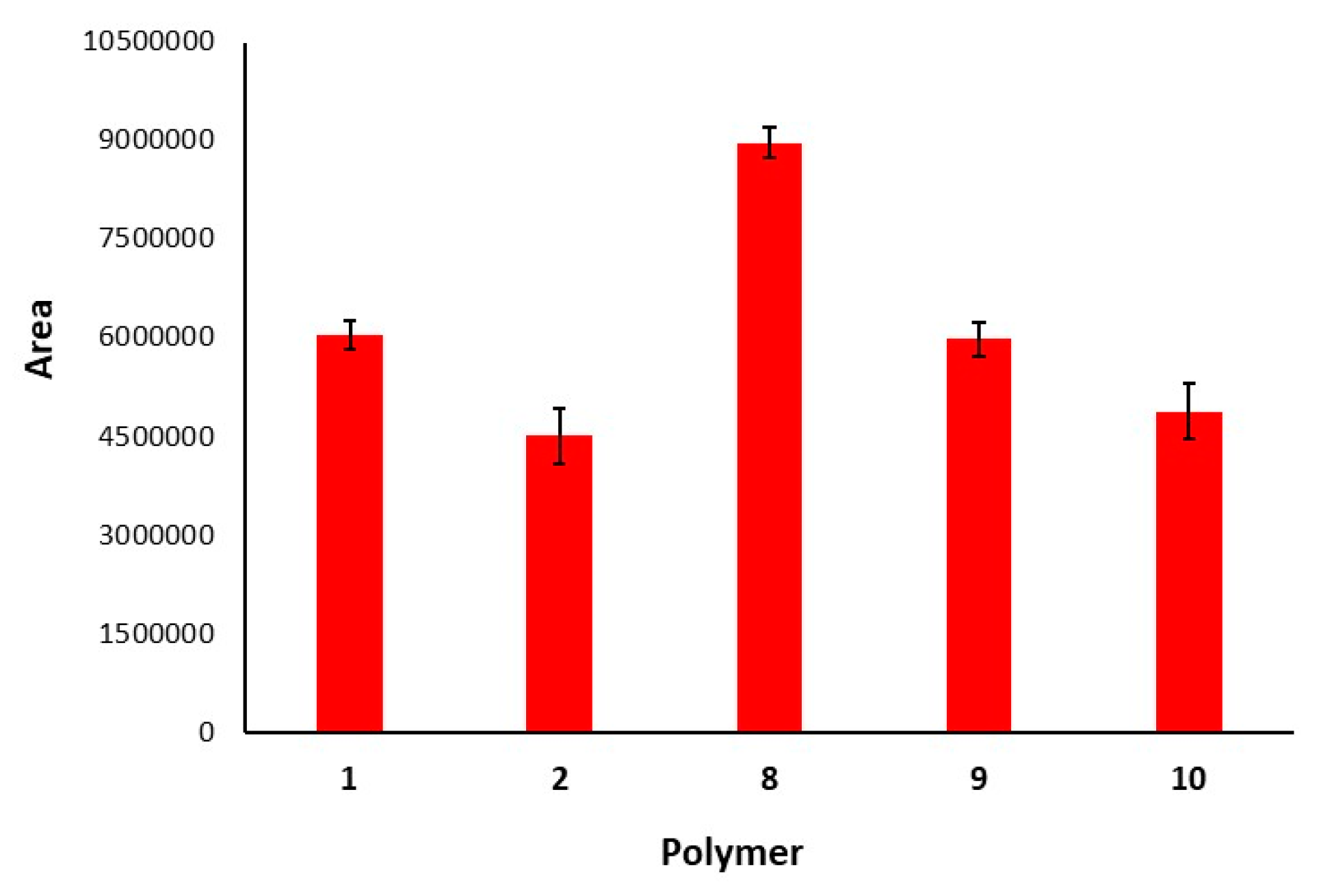
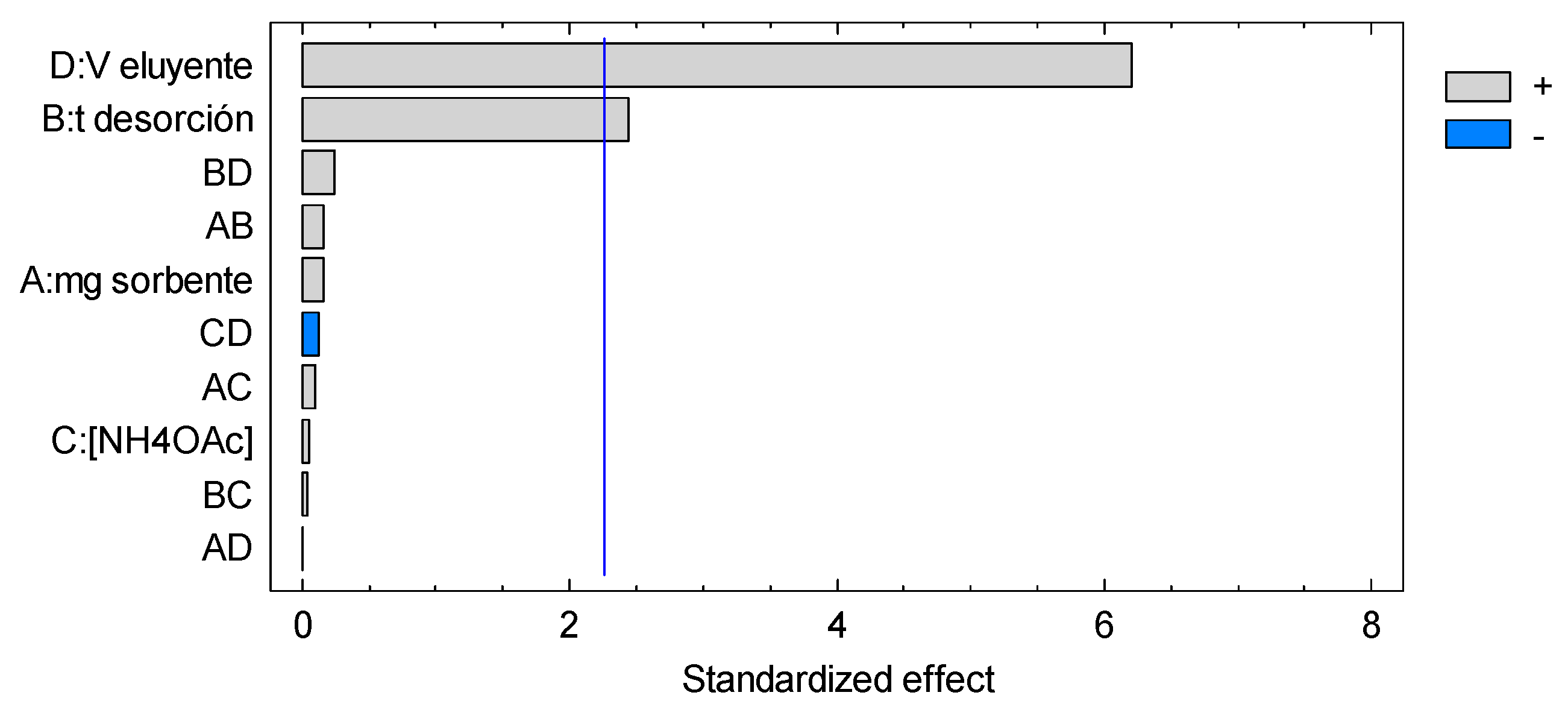
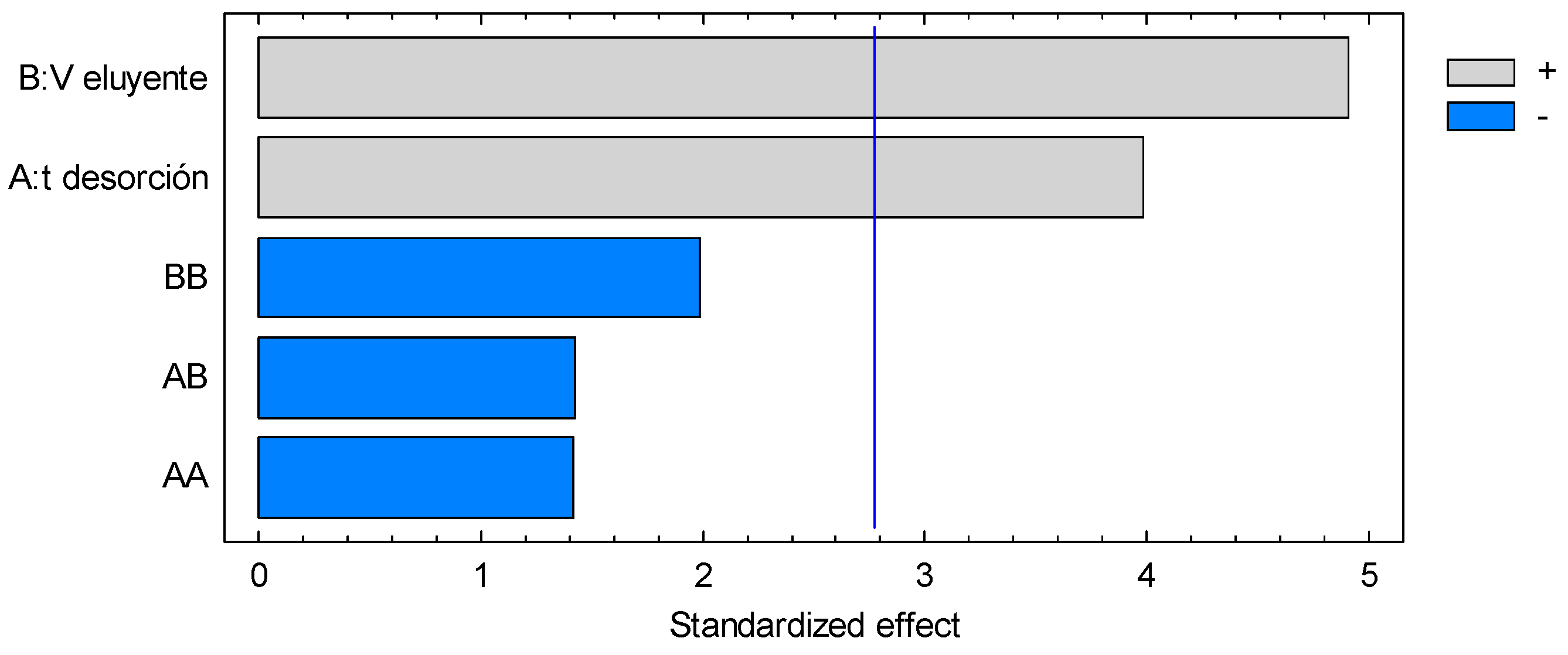
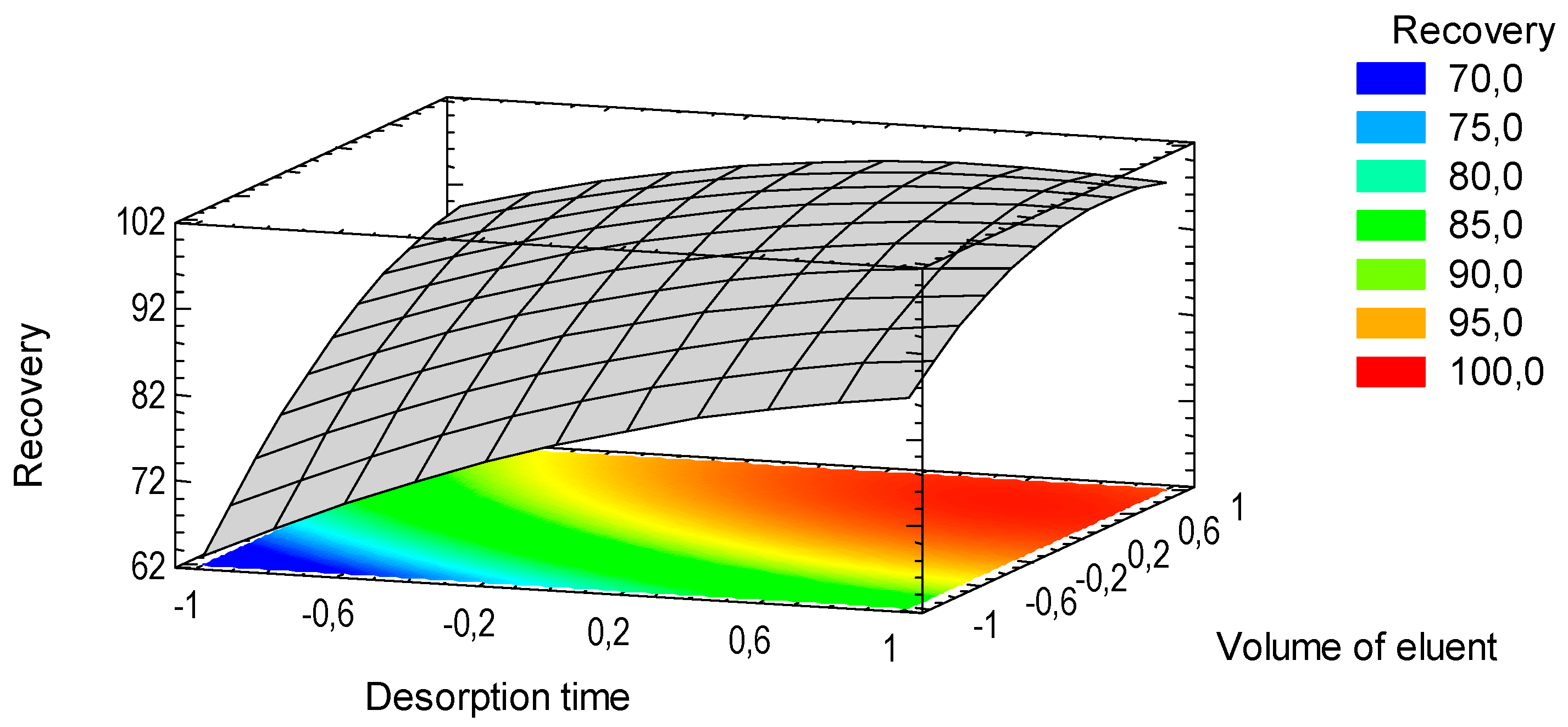
3.4. Microextraction and Quantification of Norepinephrine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sofuoglu, M.; Sewell, R.A. Norepinephrine and stimulant addiction. Addict. Biol. 2008, 14, 119–129. [Google Scholar] [CrossRef]
- Thomas, J.; Khanam, R.; Vohora, D. A validated HPLC-UV method and optimization of sample preparation technique for norepinephrine and serotonin in mouse brain. Pharm. Biol. 2015, 53, 1539–1544. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-Q.; Wang, Y.; Qu, J.-S.; Deng, J.-J.; Kang, X.-J. Selective extraction of catecholamines by packed fiber solid-phase using composite nanofibers composing of polymeric crown ether with polystyrene. Biomed. Chromatogr. 2014, 29, 103–109. [Google Scholar] [CrossRef]
- Rozet, E.; Morello, R.; Lecomte, F.; Martin, G.B.; Chiap, P.; Crommen, J.; Boos, K.S.; Hubert, P. Performances of a multidimensional on-line SPE-LC-ECD method for the determination of three major catecholamines in native human urine: Validation, risk and uncertainty assessments. J. Chromatogr. B 2006, 844, 251–260. [Google Scholar] [CrossRef]
- Tsunoda, M.; Aoyama, C.; Ota, S.; Tamura, T.; Funatsu, T. Extraction of catecholamines from urine using a monolithic silica disk-packed spin column and high-performance liquid chromatography-electrochemical detection. Anal. Methods 2011, 3, 582. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Ji, C.; McDonald, T.; Walton, J.; Groeber, E.A.; Steenwyk, R.C.; Lin, Z. Ultra sensitive measurement of endogenous epinephrine and norepinephrine in human plasma by semi-automated SPE-LC-MS/MS. J. Chromatogr. B 2012, 895–896, 186–190. [Google Scholar] [CrossRef]
- He, H.; Carballo-Jane, E.; Tong, X.; Cohen, L.H. Measurement of catecholamines in rat and mini-pig plasma and urine by liquid chromatography-tandem mass spectrometry coupled with solid phase extraction. J. Chromatogr. B 2015, 997, 154–161. [Google Scholar] [CrossRef]
- Fotopoulou, M.A.; Ioannou, P.C. Post-column terbium complexation and sensitized fluorescence detection for the determination of norepinephrine, epinephrine and dopamine using high-performance liquid chromatography. Anal. Chim. Acta 2002, 462, 179–185. [Google Scholar] [CrossRef]
- Lu, H.; Yu, J.; Wang, J.; Wu, L.; Xiao, H.; Gao, R. Simultaneous quantification of neuroactive dopamine serotonin and kynurenine pathway metabolites in gender-specific youth urine by ultra performance liquid chromatography tandem high resolution mass spectrometry. J. Pharm. Biomed. Anal. 2016, 122, 42–51. [Google Scholar] [CrossRef]
- Kumar, A.; Hart, J.P.; McCalley, D.V. Determination of catecholamines in urine using hydrophilic interaction chromatography with electrochemical detection. J. Chromatogr. A 2011, 1218, 3854–3861. [Google Scholar] [CrossRef]
- Li, X.; Li, S.; Kellermann, G. Pre-analytical and analytical validations and clinical applications of a miniaturized, simple and cost-effective solid phase extraction combined with LC-MS/MS for the simultaneous determination of catecholamines and metanephrines in spot urine samples. Talanta 2016, 159, 238–247. [Google Scholar] [CrossRef]
- Diniz, M.E.R.; Vilhena, L.S.; Paulo, B.P.; Barbosaa, T.C.C.; Mateo, E.C. Simultaneous Determination of Catecholamines and Metanephrines in Urine by Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry: Successful Clinical Application. J. Braz. Chem. Soc. 2015, 26, 1684–1691. [Google Scholar] [CrossRef]
- Woo, H.I.; Yang, J.S.; Oh, H.J.; Cho, Y.Y.; Kim, J.H.; Park, H.D.; Lee, S.Y. A simple and rapid analytical method based on solid-phase extraction and liquid chromatography-tandem mass spectrometry for the simultaneous determination of free catecholamines and metanephrines in urine and its application to routine clinical analysis. Clin. Biochem. 2016, 49, 573–579. [Google Scholar] [CrossRef]
- Li, X.S.; Li, S.; Kellermann, G. Simultaneous extraction and determination of monoamine neurotransmitters in human urine for clinical routine testing based on a dual functional solid phase extraction assisted by phenylboronic acid coupled with liquid chromatography-tandem mass spectromet. Anal. Bioanal. Chem. 2017, 409, 2859–2871. [Google Scholar] [CrossRef]
- Shen, Y.; Cheng, L.; Guan, Q.; Lu, J.; Wang, X. Development and validation of a liquid chromatography tandem mass spectrometry method for the measurement of urinary catecholamines in diagnosis of pheochromocytoma. Biomed. Chromatogr. 2017, 31, 1–7. [Google Scholar] [CrossRef]
- Naccarato, A.; Gionfriddo, E.; Sindona, G.; Tagarelli, A. Development of a simple and rapid solid phase microextraction-gas chromatography-triple quadrupole mass spectrometry method for the analysis of dopamine, serotonin and norepinephrine in human urine. Anal. Chim. Acta 2014, 810, 17–24. [Google Scholar] [CrossRef]
- Sarafraz-Yazdi, A.; Razavi, N. Application of molecularly-imprinted polymers in solid-phase microextraction techniques. Trends Anal. Chem. 2015, 73, 81–90. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, S.; Lim, J.-M.; Lee, Y.-I. Molecularly imprinted solid phase microextraction fiber for trace analysis of catecholamines in urine and serum samples by capillary electrophoresis. Talanta 2012, 99, 270–276. [Google Scholar] [CrossRef]
- Saracino, M.A.; Santarcangelo, L.; Raggi, M.A.; Mercolini, L. Microextraction by packed sorbent (MEPS) to analyze catecholamines in innovative biological samples. J. Pharm. Biomed. Anal. 2015, 104, 122–129. [Google Scholar] [CrossRef]
- Konieczna, L.; Roszkowska, A.; Synakiewicz, A.; Stachowicz-Stencel, T.; Adamkiewicz-Drozynska, E.; Baczek, T. Analytical approach to determining human biogenic amines and their metabolites using eVol microextraction in packed syringe coupled to liquid chromatography mass spectrometry method with hydrophilic interaction chromatography column. Talanta 2016, 150, 331–339. [Google Scholar] [CrossRef]
- Saraji, M.; Shahvar, A. Selective micro solid-phase extraction of epinephrine, norepinephrine and dopamine from human urine and plasma using aminophenylboronic acid covalently immobilized on magnetic nanoparticles followed by high-performance liquid chromatography-fluorescence detection. Anal. Methods 2016, 8, 830–839. [Google Scholar] [CrossRef]
- Vasconcelos, I.; Fernandes, C. Magnetic solid phase extraction for determination of drugs in biological matrices. Trends Anal. Chem. 2017, 89, 41–52. [Google Scholar] [CrossRef]
- Lee, M.; Oh, S.Y.; Pathak, T.S.; Paeng, I.R.; Cho, B.Y.; Paeng, K.J. Selective solid-phase extraction of catecholamines by the chemically modified polymeric adsorbents with crown ether. J. Chromatogr. A 2007, 1160, 340–344. [Google Scholar] [CrossRef]
- Chen, L.; Wang, H.; Xu, Z.; Zhang, Q.; Liu, J.; Shen, J.; Zhang, W. High-throughput and selective solid-phase extraction of urinary catecholamines by crown ether-modified resin composite fiber. J. Chromatogr. A 2018, 1561, 48–55. [Google Scholar] [CrossRef]
- Torvinen, M.; Kalenius, E.; Sansone, F.; Casnati, A.; Jänis, J. Noncovalent Complexation of Monoamine Neurotransmitters and Related Ammonium Ions by Tetramethoxy Tetraglucosylcalix[4]arene. J. Am. Soc. Mass Spectrom. 2012, 23, 359–365. [Google Scholar] [CrossRef]
- Fraschetti, C.; Letzel, M.C.; Paletta, M.; Mattay, J.; Speranza, M.; Filippi, A.; Aschi, M.; Rozhenko, A.B. Cyclochiral resorcin[4]arenes as effective enantioselectors in the gas phase. J. Mass Spectrom. 2012, 47, 72–79. [Google Scholar] [CrossRef]
- Bianchi, F.; Mattarozzi, M.; Betti, P.; Bisceglie, F.; Careri, M.; Mangia, A.; Sidisky, L.; Ongarato, S.; Dalcanale, E. Innovative Cavitand-Based Sol-Gel Coatings for the Environmental Monitoring of Benzene and Chlorobenzenes via Solid-Phase Microextraction. Anal. Chem. 2008, 80, 6423–6430. [Google Scholar] [CrossRef]
- Pinalli, R.; Barboza, T.; Bianchi, F.; Massera, C.; Ugozzoli, F.; Dalcanale, E. Detection of amphetamine precursors with quinoxaline-bridged cavitands. Supramol. Chem. 2013, 25, 682–687. [Google Scholar] [CrossRef]
- Bianchi, F.; Bedini, A.; Riboni, N.; Pinalli, R.; Gregori, A.; Sidisky, L.M.; Dalcanale, E.; Careri, M. Cavitand-based solid-phase microextraction coatings for the selective detection of nitroaromatic explosives in air and soil. Anal. Chem. 2014, 86, 10646–10652. [Google Scholar] [CrossRef]
- Bertani, F.; Riboni, N.; Bianchi, F.; Brancatelli, G.; Sterner, E.S.; Pinalli, R.; Geremia, S.; Swager, T.M.; Dalcanale, E. Triptycene-Roofed Quinoxaline Cavitands for the Supramolecular Detection of BTEX in Air. Chem. Eur. J. 2016, 22, 3312–3319. [Google Scholar] [CrossRef]
- Riboni, N.; Trzcinski, J.W.; Bianchi, F.; Massera, C.; Pinalli, R.; Sidisky, L. Conformationally blocked quinoxaline cavitand as solid-phase microextraction coating for the selective detection of BTEX in air. Anal. Chim. Acta 2016, 905, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguirre, A.A.; Velásquez-Silva, B.A.; Palacio, C.; Baez, F.; Rivera-Monroy, Z.J.; Maldonado, M. Surface modification of poly(GMA-co-EDMA-co-MMA) with resorcarenes. J. Braz. Chem. Soc. 2018, 29, 1965–1972. [Google Scholar] [CrossRef]
- Castillo-Aguirre, A.A.; Velásquez-Silva, B.A.; Rivera-Monroy, Z.J.; Maldonado, M. Aminomethylated Calix[4]resorcinarenes as Modifying Agents for Glycidyl Methacrylate (GMA) Rigid Copolymers Surface. Polymers (Basel) 2019, 11, 1147. [Google Scholar] [Green Version]
- Castillo-Aguirre, A.; Rivera-Monroy, Z.; Maldonado, M. Selective O-Alkylation of the Crown Conformer of Tetra(4-hydroxyphenyl)calix[4]resorcinarene to the Corresponding Tetraalkyl Ether. Molecules 2017, 22, 1660. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguirre, A.A.; Rivera-Monroy, Z.J.; Maldonado, M. Analysis by RP-HPLC and Purification by RP-SPE of the C -Tetra(p-hydroxyphenyl)resorcinolarene Crown and Chair Stereoisomers. J. Anal. Methods Chem. 2019, 2019, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.; Sanabria, E.; Batanero, B.; Esteso, M. Apparent molal volume and viscosity values for a new synthesized diazoted resorcin[4]arene in DMSO at several temperatures. J. Mol. Liq. 2017, 231, 142–148. [Google Scholar] [CrossRef]
- Okanda, F.M.; El Rassi, Z. Affinity monolithic capillary columns for glycomics/proteomics: 1. Polymethacrylate monoliths with immobilized lectins for glycoprotein separation by affinity capillary electrochromatography and affinity nano-liquid chromatography in either a single colum. Electrophoresis 2006, 27, 1020–1030. [Google Scholar] [CrossRef]
- Wu, T.; Wang, H.-T.; Shen, B.; Du, Y.-P.; Wang, X.; Wang, Z.-P.; Zhang, C.-J.; Miu, W.-B. Determination of primary aromatic amines using immobilized nanoparticles based surface-enhanced Raman spectroscopy. Chin. Chem. Lett. 2016, 27, 745–748. [Google Scholar] [CrossRef]
- Manzo, V.; Honda, L.; Navarro, O.; Ascar, L.; Richter, P. Microextraction of non-steroidal anti-inflammatory drugs from waste water samples by rotating-disk sorptive extraction. Talanta 2014, 128, 486–492. [Google Scholar] [CrossRef]
- Claude, B.; Nehmé, R.; Morin, P. Analysis of urinary neurotransmitters by capillary electrophoresis: Sensitivity enhancement using field-amplified sample injection and molecular imprinted polymer solid phase extraction. Anal. Chim. Acta 2011, 699, 242–248. [Google Scholar] [CrossRef]
- Merhar, M.; Podgornik, A.; Barut, M.; Žigon, M.; Štrancar, A. Methacrylate monoliths prepared from various hydrophobic and hydrophilic monomers—Structural and chromatographic characteristics. J. Sep. Sci. 2003, 26, 322–330. [Google Scholar] [CrossRef]
- Carrasco-Correa, E.J.; Ramis-Ramos, G.; Herrero-Martínez, J.M. Methacrylate monolithic columns functionalized with epinephrine for capillary electrochromatography applications. J. Chromatogr. A 2013, 1298, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Kashapov, R.R.; Zakharova, L.Y.; Saifutdinova, M.N.; Kochergin, Y.S.; Gavrilova, E.L.; Sinyashin, O.G. Construction of a water-soluble form of amino acid C-methylcalix[4]resorcinarene. J. Mol. Liq. 2015, 208, 58–62. [Google Scholar] [CrossRef]
- El Gihani, M.T.E.; Heaney, H.; Slawin, A.M.Z. Highly Diastereoselective Functionalisation of Calix[4]resorcinarene Derivatives and Acid Catalysed Epimerisation Reactions. Tetrahedron Lett. 1995, 36, 4905–4908. [Google Scholar] [CrossRef]
- Iacono, M.; Connolly, D.; Heise, A. Fabrication of a GMA-co-EDMA monolith in a 2.0 mm i.d. polypropylene housing. Materials (Basel) 2016, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Vieira, C.M.S.; Mazurkievicz, M.; Lopez, A.M.; Debatin, V.; Micke, G.A.; Richter, P.; Rosero-Moreano, M.; Carasek, E. Exploiting green sorbents in rotating-disk sorptive extraction for the determination of parabens by high-performance liquid chromatography with tandem electrospray ionization triple quadrupole mass spectrometry. J. Sep. Sci. 2018, 41, 4047–4054. [Google Scholar] [CrossRef]
- Becerra-Herrera, M.; Miranda, V.; Arismendi, D.; Richter, P. Chemometric optimization of the extraction and derivatization of parabens for their determination in water samples by rotating-disk sorptive extraction and gas chromatography mass spectrometry. Talanta 2018, 176, 551–557. [Google Scholar] [CrossRef]
- Fiscal-Ladino, J.A.; Obando-Ceballos, M.; Rosero-Moreano, M.; Montaño, D.F.; Cardona, W.; Giraldo, L.F.; Richter, P. Ionic liquids intercalated in montmorillonite as the sorptive phase for the extraction of low-polarity organic compounds from water by rotating-disk sorptive extraction. Anal. Chim. Acta 2017, 953, 23–31. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Yang, X.; Yu, J. Central composite experimental design applied to the catalytic aromatization of isophorone to 3,5-xylenol. Chemom. Intell. Lab. Syst. 2007, 89, 45–50. [Google Scholar] [CrossRef]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef]
- Qin, Q.; Li, H.; Shi, X.; Xu, G. Facile synthesis of Fe3O4@polyethyleneimine modified with 4-formylphenylboronic acid for the highly selective extraction of major catecholamines from human urine. J. Sep. Sci. 2015, 38, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Miekus, N.; Oledzka, I.; Plenis, A.; Kowalski, P.; Bien, E.; Miekus, A.; Krawczyk, M.A.; Adamkiewicz-Drozynska, E.; Baczek, T. Determination of urinary biogenic amines’ biomarker profile in neuroblastoma and pheochromocytoma patients by MEKC method with preceding dispersive liquid-liquid microextraction. J. Chromatogr. B 2016, 1036–1037, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, Y.; Chen, Y.; Ma, M.; Tan, Y.; Tang, H.; Chen, B. Determination of monoamine neurotransmitters in human urine by carrier-mediated liquid-phase microextraction based on solidification of stripping phase. Talanta 2015, 144, 356–362. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Proton | δ (ppm) | Correlation |
|---|---|---|---|
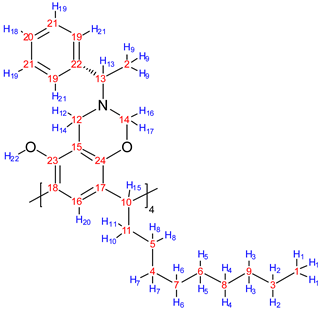 | 1 | 0.89 | 2 (1.28) |
| 9 | 1.37 | 13 (3.80) | |
| 10 | 2.15 | 8 (1.28) | |
| 10 | 2.15 | 15 (4.19) | |
| 11 | 2.20 | 8 (1.28) | |
| 11 | 2.20 | 15 (4.19) | |
| 12 | 3.73 | 14 (3.96) | |
| 12 | 3.73 | 16 (4.92) | |
| 14 | 3.96 | 16 (4.92) | |
| 16 | 4.92 | 17 (5.13) | |
| 18 | 6.95 | 19 (7.04) | |
| 19 | 7.04 | 21 (7.18) |
| Carbon | δ (ppm) | Correlation | |
|---|---|---|---|
| HMQC | HMBC | ||
| 1 | 14.1 | 1 (0.89) | 2, 3 (1.28) |
| 2 | 21.4 | 9 (1.37) | 13 (3.80) |
| 3 | 22.7 | 2 (1.28) | 1 (0.89), 3, 4 (1.28) |
| 4 | 28.1 | 7 (1.28) | 5, 6, 8 (1.28), 10 (2.15), 11 (2.20) |
| 5 | 29.4 | 8 (1.28) | 6, 7 (1.28), 10 (2.15), 11 (2.20), 15 (4.19) |
| 6 | 29.7 | 5 (1.28) | 3, 4, 6, 7 (1.28) |
| 7 | 29.8 | 6 (1.28) | 4, 5, 7, 8 (1.28) |
| 8 | 31.9 | 4 (1.28) | 2, 3, 5, 6 (1.28) |
| 9 | 32.0 | 3 (1.28) | 1 (0.89), 2, 4, 5 (1.28) |
| 10 | 32.7 | 15 (4.19) | 8 (1.28), 10 (2.15), 11 (2.20), 20 (7.11) |
| 11 | 33.7 | 10 (2.15), 11 (2.20) | 7, 8 (1.28), 15 (4.19) |
| 12 | 44.6 | 12 (3.73), 14 (3.96) | 13 (3.80), 16 (4.92), 17 (5.13) |
| 13 | 58.0 | 13 (3.80) | 9 (1.37), 12 (3.73), 14 (3.96), 16 (4.92), 17 (5.13), 21 (7.18) |
| 14 | 80.9 | 16 (4.92), 17 (5.13) | 12 (3.73), 13 (3.80), 14 (3.96) |
| 15 | 108.9 | - | 12 (3.73), 14 (3.96), 22 (7.66) |
| 16 | 121.1 | 20 (7.11) | 15 (4.19) |
| 17 | 123.5 | - | 10 (2.15), 11 (2.20), 15 (4.19), 20 (7.11) |
| 18 | 124.3 | - | 20 (7.11), 22 (7.66) |
| 19 | 127.0 | 21 (7.18) | 13 (3.80), 18 (6.95), 19 (7.04) |
| 20 | 127.1 | 18 (6.95) | 19 (7.04), 21 (7.18) |
| 21 | 128.2 | 19 (7.04) | 18 (6.95), 19 (7.04) |
| 22 | 144.5 | - | 9 (1.37), 13 (3.80) |
| 23 | 148.7 | - | 12 (3.73), 14 (3.96), 20 (7.11), 22 (7.66) |
| 24 | 149.6 | - | 12 (3.73), 14 (3.96), 15 (4.19), 16 (4.92), 17 (5.13), 20 (7.11) |
| Parameter | Concentration Level (μgL−1) | n | Value |
|---|---|---|---|
| Slope (standards) | 50–500 | 3 | 28,627 |
| Slope (fortifieds) | 50–500 | 3 | 9877 |
| R2 (standards) | 50–500 | 3 | 0.999 |
| R2 (fortifieds) | 50–500 | 3 | 0.998 |
| LOD (µgL−1) | 50 | 8 | 11.3 |
| LLOQ (µgL−1) | 50 | 8 | 34.0 |
| Recovery (%) | 50 | 6 | 96.2 |
| 500 | 6 | 101.7 | |
| RSD (%) | 50 | 6 | 7.0 |
| 500 | 6 | 5.6 |
| Extraction Technique | Quantification Technique | Quantification Mobile Phase | Recovery (%) | RSD (%) | LOD (µgL−1) | Linear Range | Ref. |
|---|---|---|---|---|---|---|---|
| SPE | HPLC-ECD | Buffer MeOH MeCN EDTA Octanesulfonic acid H3PO4 | 83.5 | 9.0 | 9.8 | 38–567 | [23] |
| DLLME | MEKC | NaOH 0.1 M Water Sodium tetraborate Sodium dodecyl sulfonate MeOH α-Cyclodextrin | 96.3 | 1.5 | 150 | 500–10,000 | [52] |
| CM-LPME-SSP | HPLC-ECD | Buffer MeOH Octanesulfonic acid | 97.5 | 13.0 | 8.2 | 15–2000 | [53] |
| MEPS (microextraction by packed sorbent) | HPLC-MS | Buffer Water MeCN | 96.1 | 3.8 | 5 | 20–200 | [20] |
| SPE | HPLC-MS/MS | Water MeOH Formic acid 0.25% | 101.2 | 4.1 | 1.9 | 10–210 | [12] |
| RDSE | HPLC-UV | Water MeOH Formic acid 0.05% | 99.0 | 6.3 | 11.3 | 50–500 | This work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo-Aguirre, A.; Maldonado, M. Preparation of Methacrylate-Based Polymers Modified with Chiral Resorcinarenes and Their Evaluation as Sorbents in Norepinephrine Microextraction. Polymers 2019, 11, 1428. https://doi.org/10.3390/polym11091428
Castillo-Aguirre A, Maldonado M. Preparation of Methacrylate-Based Polymers Modified with Chiral Resorcinarenes and Their Evaluation as Sorbents in Norepinephrine Microextraction. Polymers. 2019; 11(9):1428. https://doi.org/10.3390/polym11091428
Chicago/Turabian StyleCastillo-Aguirre, Alver, and Mauricio Maldonado. 2019. "Preparation of Methacrylate-Based Polymers Modified with Chiral Resorcinarenes and Their Evaluation as Sorbents in Norepinephrine Microextraction" Polymers 11, no. 9: 1428. https://doi.org/10.3390/polym11091428