
Advanced Glycation End Products of Bovine Serum Albumin Suppressed Th1/Th2 Cytokine but Enhanced Monocyte IL-6 Gene Expression via MAPK-ERK and MyD88 Transduced NF-κB p50 Signaling Pathways

 , ,
, ,
Abstract
:1. Introduction
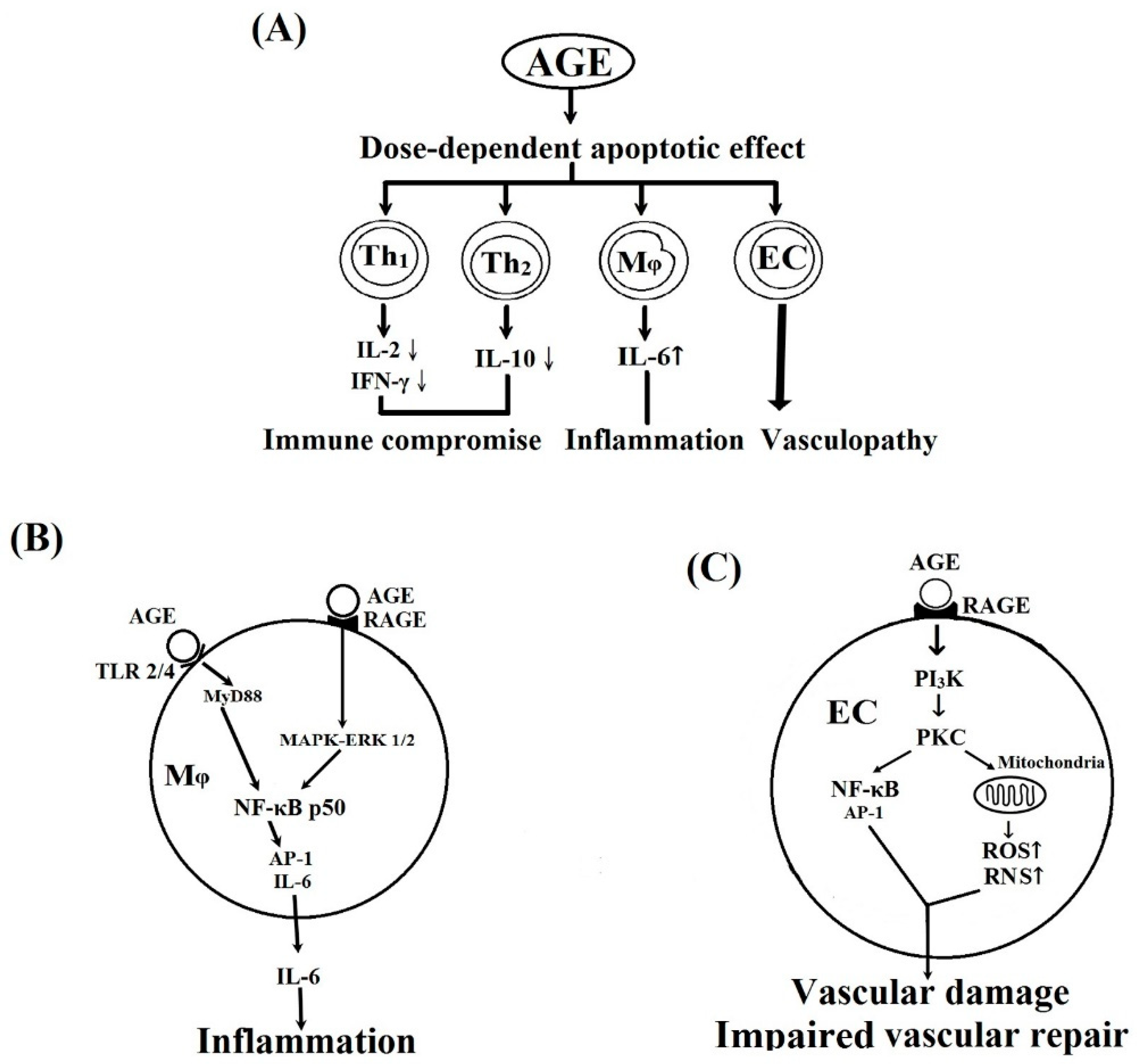
2. Results and Discussion
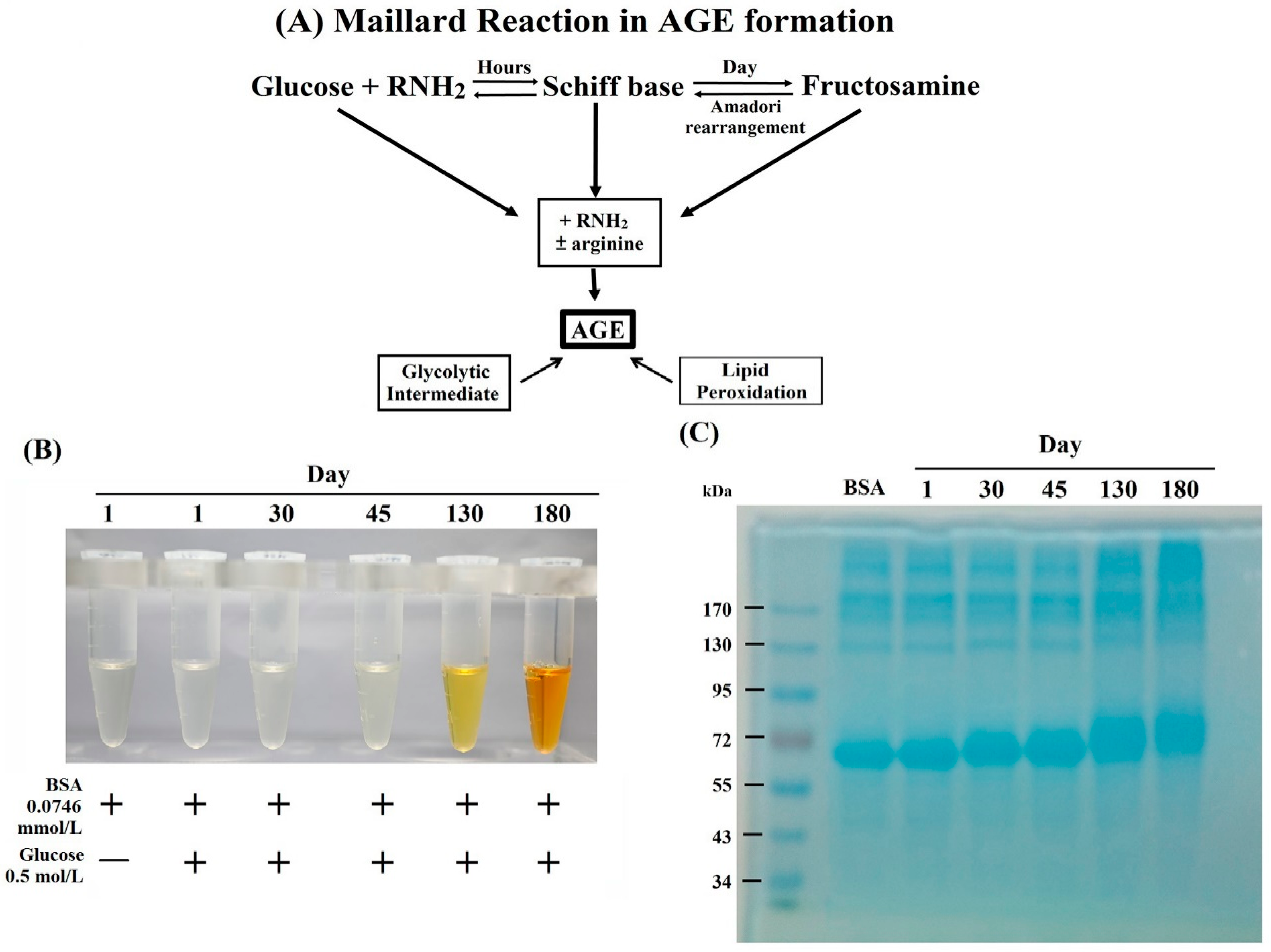
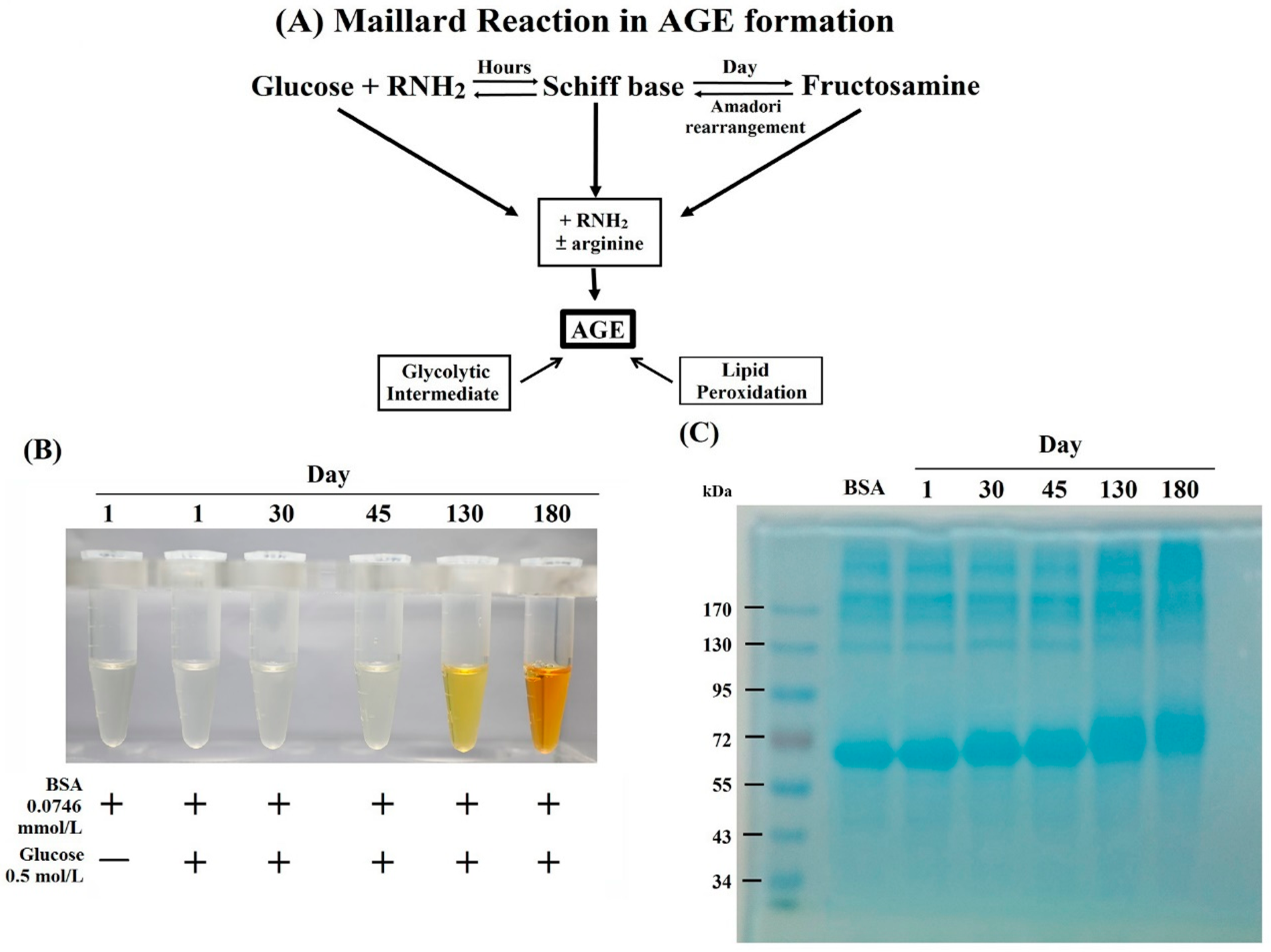
2.1. Formation, Changes of Color and Molecular Weight, and Amino Acid Residue Glycation in BSA–AGE
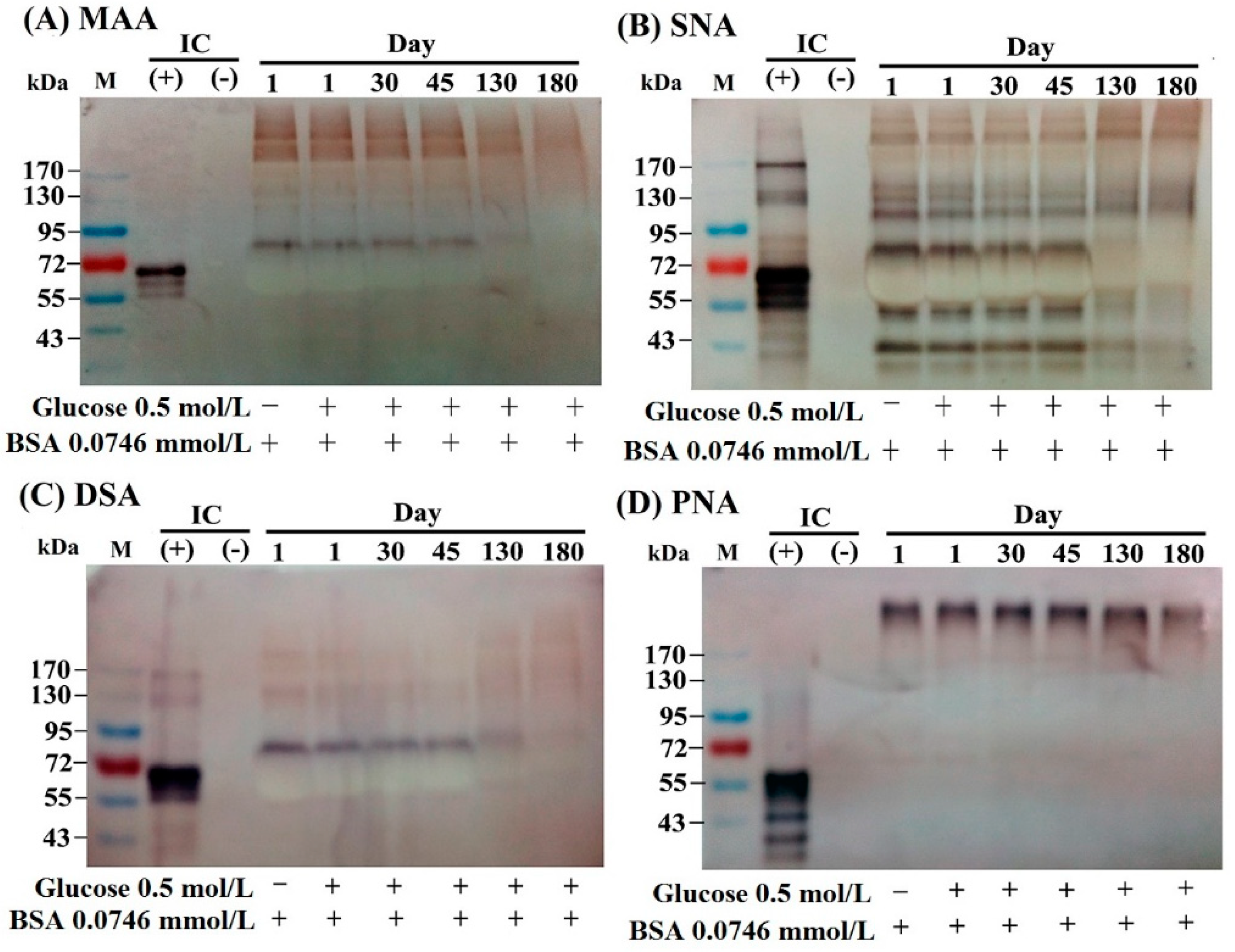
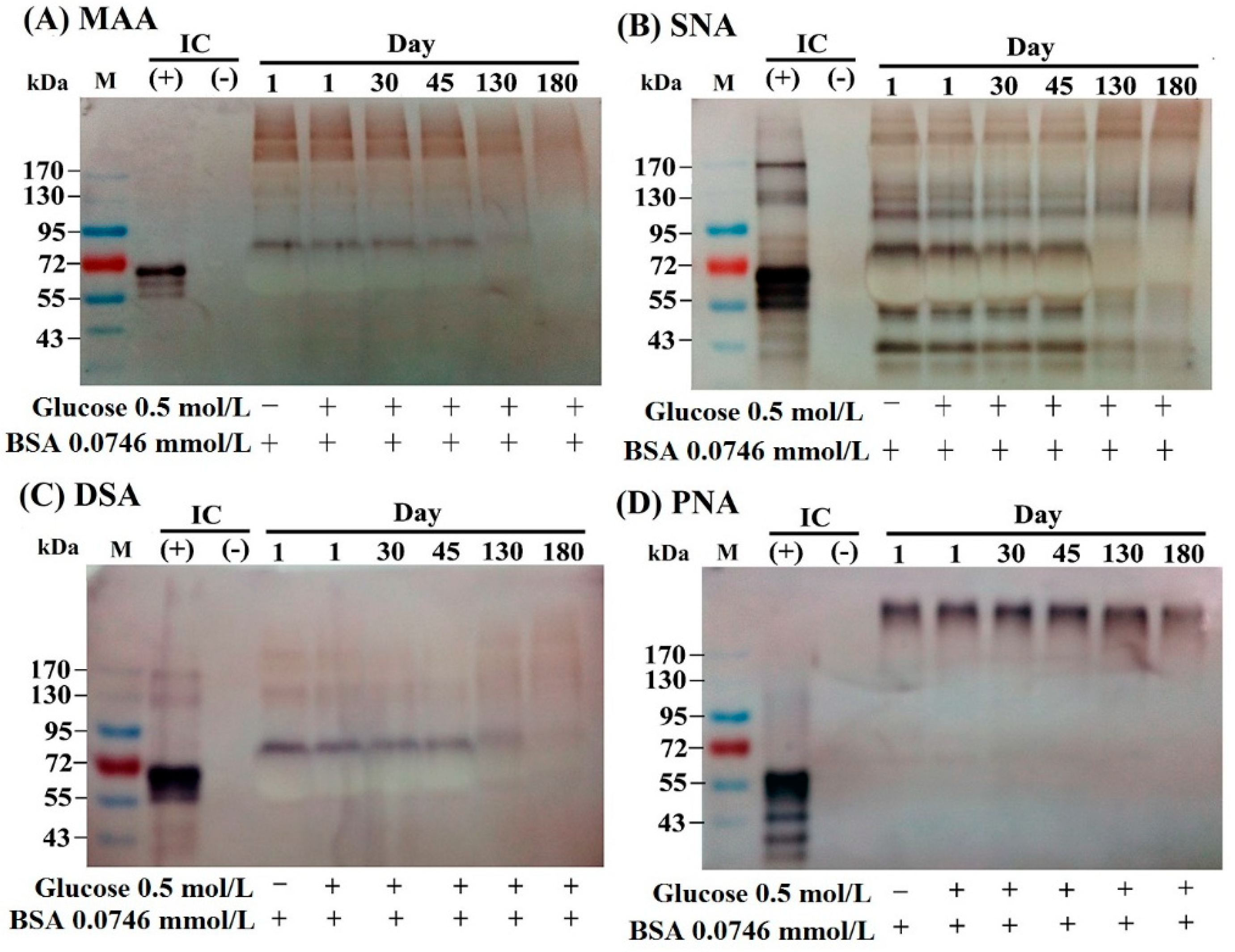
2.2. The Lectin-Binding Capacity of Different BSA–AGEs
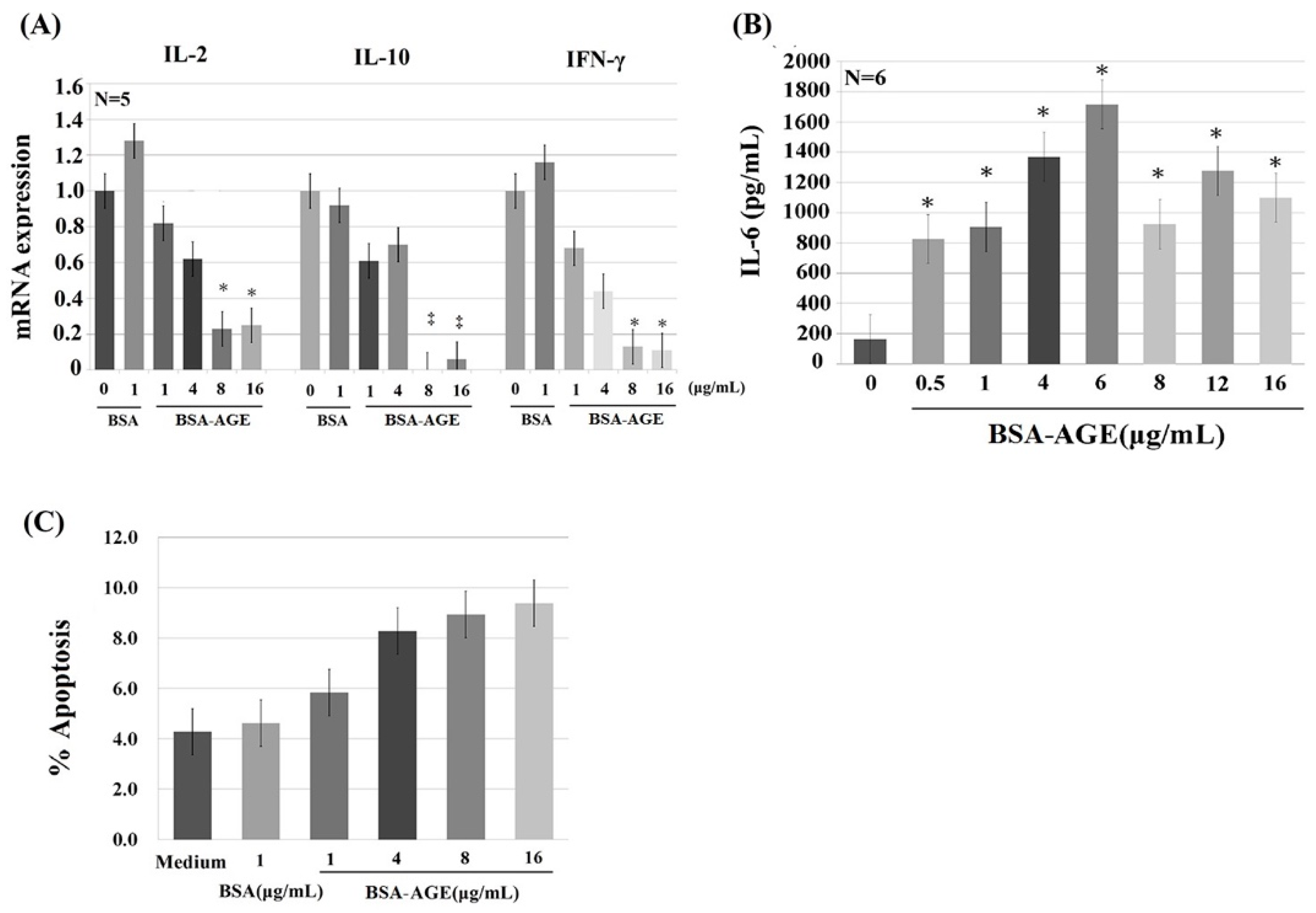
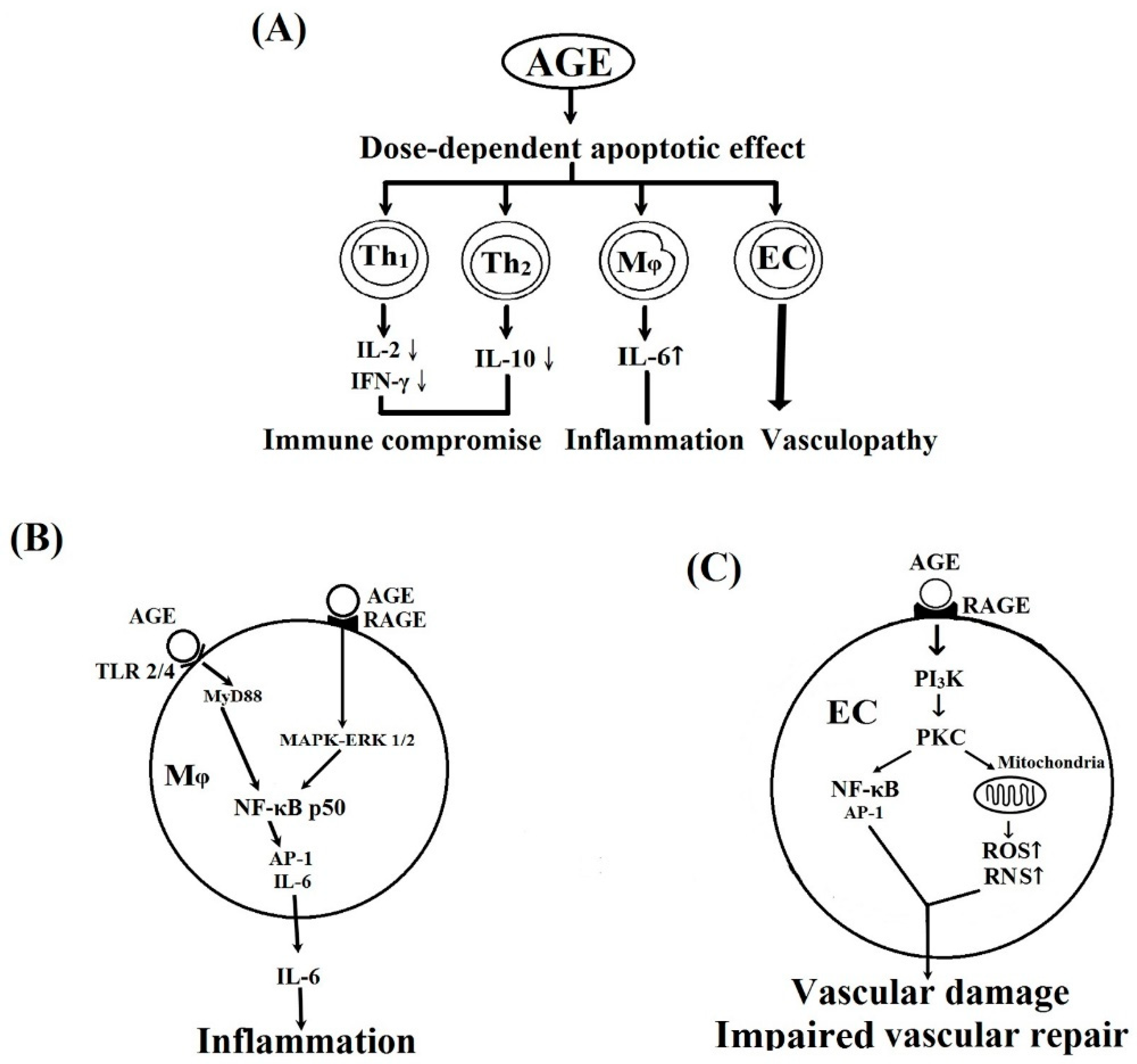
2.3. Dose-Dependent Effects of BSA–AGE on Th1 (IL-2 and IFN-γ) and Th2 (IL-10) Cytokine mRNA Expression, and Monocyte IL-6 Production by Anti-CD3 + Anti-CD28 Activated Normal Human Mononuclear Cells
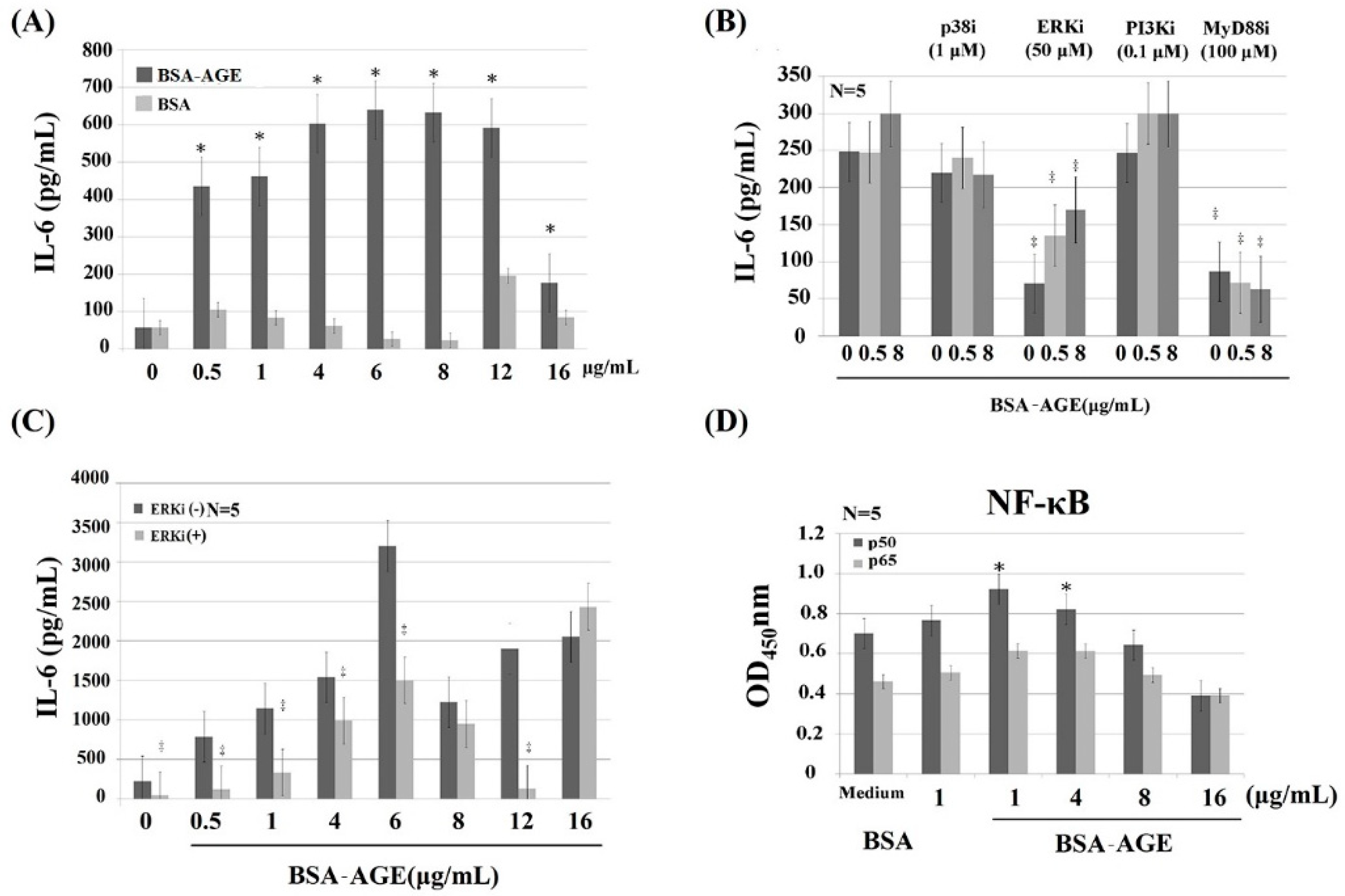
2.4. MAPK-ERK and MyD88 Transduced NF-κB Signaling Pathways are Involved in BSA–AGE-Enhanced IL-6 Production by Activated Normal MNCs
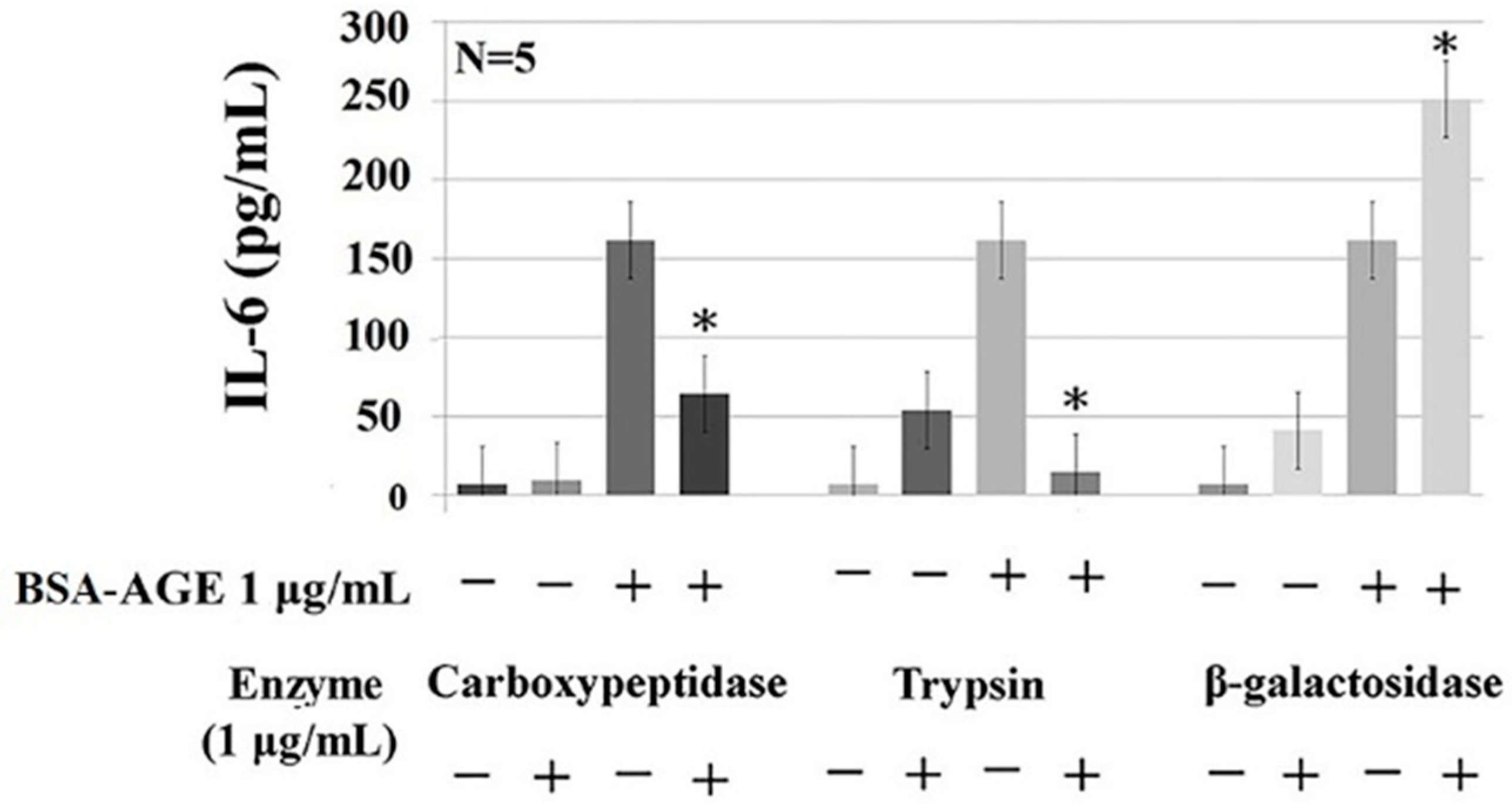
2.5. Protein- and Glycoprotein-Degrading Enzymes Decrease Whereas a Particular Glycome-Modifying Enzyme Enhances IL-6 Production by Activated MNCs
3. Materials and Methods
3.1. Preparation of BSA–AGE
3.2. Identification of AGE
3.2.1. Observation of Color Changes
3.2.2. Changes of Relative Molecular Weight of BSA–AGE Detected by 10% SDS-PAGE
3.2.3. Measurement of Nε-(Carboxymethyl)-lysine (CML) Content in BSA–AGE Molecule
3.2.4. Detection of the Number of Glycation Sites in Lysine, Arginine, and Cysteine in Different BSA–AGEs by LC/MS Proteomics Analysis
3.2.5. Detection of the Lectin-Binding Capacity of Different BSA–AGE Molecules
3.3. Isolation of Mononuclear Cells from Peripheral Blood of Normal Human Volunteers
3.4. Incubation of MNCs with Different Concentrations of Day-180 AGE
3.5. Detection of Th1/Th2 Cytokine mRNA Expression
3.6. Detection of IL-6 Concentration in the Culture Supernatants by ELISA
3.7. Detection of Cell Apoptosis by ELISA
3.8. Identification of Signaling Pathway(s) of BSA–AGE-Enhanced Monocyte IL-6 Production by Anti-CD3 + Anti-CD28 Antibodies-Activated Normal MNCs Using Different Protein Inhibitors
3.9. Detection of Intranuclear NF-κB Components, p65 and p50, by ELISA
3.10. Detection of the Effects of Carbohydrate-, Protein-, and Carbohydrate-and-Protein-Degrading-Enzyme-Digested BSA–AGE on IL-6 Production by Anti-CD3 + Anti-CD28 Activated Normal MNCs
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end products |
| BSA | bovine serum albumin |
| CML | Nε-(carboxymethyl)-lysine |
| CEL | Nε-(carboxyethyl)-lysine |
| EC | endothelial cell |
| Gal | galactosamine |
| GlcNAc | N-acetyl-glucosamine |
| IL | interleukin |
| Inflamm-aging | inflammation in the aging group |
| IFN-γ | interferon-γ |
| MNC | mononuclear cell |
| Mφ | monocyte/macrophage |
| PSS | progressive systemic sclerosis |
| RA | rheumatoid arthritis |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SLE | systemic lupus erythematosus |
| Th | helper T cell |
| TNF-α | tumor necrosis factor-α |
References
- Suji, G.; Sivakani, S. Glucose, glycation and aging. Biogerontology 2004, 5, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. A. Biol. Sci. Med. Sci. 2010, 65, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-H.; Shin, D.-G.; Kim, J.-R.; Cho, K.-H. Senescence-related truncation and multimerization of apolipoprotein A-I in high-density lipoprotein with an elvated level of advanced glycated end products and cholesteryl ester transfer activity. J. Gerontol. Biol. Med. Sci. 2010, 65, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Taniguchi, N. Advanced glycation in diabetes, aging and age-related diseases: Editorial and dedication. Glycoconj. J. 2016, 33, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Gautieri, A.; Passini, F.S.; Silvan, U.; Quizar-Sicairos, M.; Carimati, G.; Volpi, P.; Moretti, M.; Schoenhuber, H.; Redaelli, A.; Berli, M.; et al. Advanced glycation end-products: Mechanics of aged collagen from molecule to tissue. Matrix Biol. 2017, 59, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.-S.; Park, S.; Kim, J. The role of glycation in the pathogenesis of aging and its prevention through herbal products and physical exercise. J. Exerc. Nutr. Biochem. 2017, 21, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, D.; Sun, L.; Lu, T.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and prospective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef]
- Qin, Q.; Nu, J.; Wang, Z.; Xu, W.; Qiao, Z.; Gu, Y. Heparanase induced by advanced glycation end products in various types of neurodegenerative diseases: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Rojas, A.; Mirales, M.A. Advanced glycation and endothelial functions: A link towards vascular complications in diabetes. Life Sci. 2004, 76, 715–730. [Google Scholar] [CrossRef]
- Tan, K.C.; Shiu, S.W.; Wong, Y.; Tam, X. Serum advanced glycation end products (AGEs) are associated with insulin resistance. Diabetes Metab. Res. Rev. 2011, 27, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Zhong, Z.; Mima, A.; King, G.L. Molecular mechanisms of diabetes vascular complications. Diabetes Investig. 2010, 1, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp. Gerontol. 2011, 46, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Miyata, T. Early- and advanced non-enzymatic glycation in diabetes vascular complications: The search for therapeutics. Amino Acids 2012, 42, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Chilelli, N.C.; Burlina, S.; Lappolla, A. AGEs rather than hyperglycemia, are responsible for microvascular complications in diabetes: A “glycoxidation-centric” point of view. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 913–919. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, G.E. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol. Metab. Clin. N. Am. 2013, 42, 697–719. [Google Scholar] [CrossRef] [PubMed]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2013, 3, 94–108. [Google Scholar] [CrossRef]
- Stefano, G.B.; Challenger, S.; Kream, R.M. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disoeders. Eur. J. Nutr. 2016, 55, 2339–2345. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef]
- Park, S.; Kang, H.-J.; Jeon, J.-H.; Kim, M.-J.; Lee, I.-K. Recent advances in the pathogenesis of microvascular complications in diabetes. Arch. Pharm. Res. 2019, 42, 251–261. [Google Scholar] [CrossRef]
- de Leeuw, K.; Graaff, R.; de Vries, R.; Dullaart, R.P.; Smit, A.J.; Kallenberg, C.G.; Bijl, M. Accumulation of advanced glycation endproducts in patients with systemic lupus erythematosus. Rheumatology 2007, 46, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaloudi, O.; Basta, G.; Perfetto, F.; Bartoli, F.; Del Rosso, A.; Miniatti, I.; Conforti, M.L.; Generini, S.; Guiducci, S.; Abbate, R.; et al. Circulating levels of Nε-(carboxymethyl)- lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007, 46, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Nienhuis, H.L.; Westra, J.; Smit, A.J.; Limburg, P.C.; Kallenberg, C.G.; Bijl, M. AGE and their receptor RAGE in systemic autoimmune diseases: An inflammation propagating factor contributing to accelerated atherosclerosis. Autoimmunity 2009, 42, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Danoniene, J.; Cypiene, A.; Ryliskyte, L.; Rugiene, R.; Ryliskiene, K.; Laucevicius, A. Skin autofluorescence in systemic sclerosis is related to the disease and vascular damage: A cross-sectional analytic study of comparative groups. Dis. Markers 2015, 2015, 837470. [Google Scholar]
- Chen, D.Y.; Chen, Y.M.; Lin, C.C.; Hsieh, C.; Wu, Y.C.; Hung, W.T.; Chen, H.H.; Lan, J.L. The potential role of advanced glycation end products (AGEs) and soluble receptors (sRAGE) in the pathogenesis of adult-onset Still’s disease. BMC Musculoskelet. Disord. 2015, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.R.; Moinuddin. Glycoxidation of histone proteins in autoimmune disorders. Clin. Chim. Acta 2015, 450, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Vytasek, R.; Sedova, L.; Vilim, V. Increased concentration of two different advanced glycation end-products detected by enzyme immunoassays with new monoclonal antibodies in sera of patients with rheumatoid arthritis. BMC Musculoskelet. Disord. 2010, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Kmani, I.; Bouzidi, H.; Zrour, S.; Bergaoui, N.; Hammami, M.; Kerkeni, M. Increased serum concentrations of Nε-carboxymethyl-lysine are related to the presence and severity of rheumatoid arthritis. Ann. Clin. Biochem. 2018, 55, 430–436. [Google Scholar]
- Kopec-Pyciarz, K.; Makulska, I.; Zwolinska, D.; Kaczmanski, K.; Baran, W. Skin autofluorescence, as a measure of AGE accumulation in individuals suffering from chronic plaque psoriasis. Mediat. Inflamm. 2018, 2018, 4016939. [Google Scholar] [CrossRef]
- Hmed, N.; Argirov, O.K.; Minhas, H.S.; Cordeiro, C.A.A.; Thronalley, P.J. Assay of advanced glycation endoproducts (AGEs): Surveying AGEs by chromatographic assay with derivatization of 6-aminoquinolyl-N- hydroxysuccinomidyl-carbamate and application to Nε-carboxymethyl-lysine and Nε-(1-caboxyethyl)lysine-modified albumin. Biochem. J. 2002, 364, 1–14. [Google Scholar]
- Nass, N.; Bartling, B.; Navarrete Santos, A.; Scheubel, R.J.; Borgermann, J.; Silber, R.E.; Simm, A. Advanced glycation end products in diabetes and ageing. Z. Gerontol. Geriat. 2007, 40, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Vlassopoulos, A.; Lean, M.E.J.; Combet, E. Oxidative stress, protein glycation and nutrition-interaction relevant to health and disease throughout the lifecycle. Proc. Nutr. Soc. 2014, 73, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, M.Y.; Rafi, Z.; Khan, H.; Siddiqui, Z.; Rehman, S.; Shahab, U.; Khan, M.S.; Saeed, M.; Alouffi, S.; et al. Oxidation, glycation and glycoxidation-the vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Mastrocola, R. Dietary sugars and endogenous formation of advanced glycation endproducts: Emerging mechanisms of disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Nedic, O.; Rattan, S.I.S.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signaling pathways in ageing and pathophysiology. Free Radic. Res. 2013, 47 (Suppl. 1), 28–38. [Google Scholar] [CrossRef]
- Cardenas-Leon, M.; Diaz-Diaz, E.; Anguellis-Medina, R.; Sanchez-Canales, P.; Diaz-Sanchez, V.; Larrea, F. Glycation and protein cross-linking in the diabetes and ageing pathogenesis. Rev. Investig. Clin. 2009, 61, 505–520. [Google Scholar] [PubMed]
- Simm, A. Protein glycation during aging in cardiovascular disease. J. Proteom. 2013, 92, 248–259. [Google Scholar] [CrossRef]
- Bucala, R. Diabetes, aging and their tissue complications. J. Clin. Investig. 2014, 124, 1887–1888. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Folop, T.; Dupuis, G.; Witkowski, J.M.; Larbi, A. The role of immunosenescence in the development of age-related diseases. Rev. Investig. Clin. 2016, 68, 84–91. [Google Scholar]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, S.E.; Hoepelman, A.I. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol. Med. Microbiol. 1999, 26, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Frydrych, L.M.; Bian, G.; O’Lone, D.E.; Ward, P.A.; Delano, M.J. Obesity and type 2 diabetes mellitus drive immune dysfunction, infection development, and sepsis mortality. J. Leukoc. Biol. 2018, 104, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Garlick, R.L.; Mazer, J.S. The principal site of nonenzymatic glycosylation of human serum albumin in vivo. J. Biol. Chem. 1983, 258, 6142–6146. [Google Scholar] [PubMed]
- Shaklai, N.; Garlick, R.L.; Bunn, H.F. Nonenzymatic glycosylation of human serum albumin alters its conformation and function. J. Biol. Chem. 1984, 25, 3812–3817. [Google Scholar]
- Sattarahmady, N.; Moosavi-Movahedi, A.A.; Habibi-Rezaei, M.; Ahmadian, S.; Saboury, A.A.; Heli, H.; Sheibani, N. Detergency effects of nanofibrillar amyloid formation on glycation of human serum albumin. Carbohydr. Res. 2008, 343, 2229–2234. [Google Scholar] [CrossRef]
- Mendez, D.L.; Jensen, R.A.; McElroy, L.A.; Pena, J.M.; Esquerra, R.M. The effect of non-enzymatic glycation on the unfolding of human serum albumin. Arch. Biochem. Biophys. 2005, 444, 92–99. [Google Scholar] [CrossRef]
- Bouma, B.; Kroon-Batenburg, L.M.; Wu, Y.P.; Brunjes, B.; Posthuma, G.; Kranenburg, O.; de Groot, P.G.; Voest, E.E.; Gebbink, M.F. Glycation induces formation of amyloid cross-beta structure in albumin. J. Biol. Chem. 2003, 278, 41810–41819. [Google Scholar] [CrossRef]
- Chesne, S.; Rondeau, P.; Armenta, S.; Bourdon, E. Effects of oxidative modifications induced by the glycation of bovine serum albumin on its structure and on cultured adipose cells. Biochimie 2006, 88, 1467–1477. [Google Scholar] [CrossRef]
- Baraka-Vidot, J.; Guerin-Dubourg, A.; Dubois, F.; Payet, B.; Bourdon, E.; Rondeau, P. New insights into deleterious impacts of in vivo glycation on albumin antioxidant activities. Biochim. Biophys. Acta 2013, 1830, 3532–3541. [Google Scholar] [CrossRef] [PubMed]
- Bohney, J.P.; Feldhoff, R.C. Effects of nonenzymatic glycosylation and fatty acids on tryptophan binding to human serum albumin. Biochem. Pharmacol. 1992, 43, 1829–1834. [Google Scholar] [CrossRef]
- Miele, C.; Riboulet, A.; Maitan, M.A.; Oriente, F.; Romano, C.; Formisano, P.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J. Biol. Chem. 2003, 278, 47376–47387. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Bujo, H.; Yamagishi, S.; Takeuchi, M.; Imaizumi, T.; Saito, Y. Advanced glycation end products attenuate cellular insulin sensitivity by increasing the generation of intracellular reactive oxygen species in adipocytes. Diabet. Res. Clin. Pract. 2007, 76, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Taulescu, M.; Crisan, D.; Cosgarea, R.; Parvu, A.; Catoi, C.; Drugan, T. Expression of advanced glycation end-products on sun-exposed and non-exposed cutaneous sites during the ageing process in humans. PLoS ONE 2013, 8, e75003. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Niu, J.; Wang, Z.; Xu, W.; Qiao, Z.; Gu, Y. Heparanase induced by advanced glycation end products (AGEs) promotes macrophage migration involving RAGE and PI3K/AKT pathway. Cardiovasc. Diabetol. 2013, 12, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.; Babu, J.P. Glycated albumin produced in diabetic hyperglycemia promotes monocyte secretion of inflammatory cytokines and bacterial adherence to epithelial cells. J. Periodontol. Res. 2015, 50, 197–204. [Google Scholar] [CrossRef]
- Ehlermann, P.; Eggers, K.; Bierhaus, A.; Most, P.; Weichenhan, D.; Greten, J.; Nawroth, P.P.; Katus, H.A.; Remppis, A. Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc. Diabetol. 2006, 5, 6–14. [Google Scholar] [CrossRef]
- Tanaka, N.; Yonekura, H.; Yamagishi, S.; Fujimori, H.; Yamamoto, Y.; Yamamoto, H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17 beta-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 2000, 275, 25781–25790. [Google Scholar] [CrossRef]
- Nakamura, N.; Hasegawa, G.; Obayashi, H.; Yamazaki, M.; Ogata, M.; Nakano, K.; Yoshikawa, T.; Watanabe, A.; Kinoshita, S.; Fujinami, A.; et al. Increased concentration of pentosidine, an advanced glycation end product, and interleukin-6 in the vitreous of patients with proliferative diabetic retinopathy. Diabet. Res. Clin. Pract. 2003, 61, 93–101. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.J.; Lutgers, H.L. The clinical relevance of advanced glycation endproducts (AGE) and recent developments in pharmaceutics to reduce AGE accumulation. Curr. Med. Chem. 2004, 11, 2767–2784. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, F.; Guo, Z.; Shan, Y.; Zhang, X.; Liu, Z. Advanced glycation end products upregulate C-reactive protein synthesis by human hepatocytes through stimulation of monocyte IL-6 and IL-1 beta production. Scand. J. Immunol. 2007, 66, 555–562. [Google Scholar] [CrossRef]
- Candore, G.; Di Lorenzo, G.; Melluso, M.; Cigna, D.; Colucci, A.T.; Modica, M.A.; Caruso, C. Gamma-interferon, interleukin-4 and interleukin-6 in vitro production in old subjects. Autoimmunity 1993, 16, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Candore, G.; Cigna, D.; Di Lorenzo, G.; Sireci, G.; Dieli, F.; Salerno, A. Cytokine production pathway in the elderly. Immunol. Res. 1996, 15, 84–90. [Google Scholar] [CrossRef]
- Huang, H.; Patel, D.D.; Manton, K.G. The immune system in aging: Roles of cytokines, T cells and NK cells. Front. Biosci. 2005, 10, 192–215. [Google Scholar] [CrossRef]
- Singh, T.; Newman, A.B. Inflammatory markers in population studies of aging. Ageing Res. Rev. 2011, 10, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Balardy, L.; Maulis, G.; Gaudin, C.; Payrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Starr, M.E.; Saito, M.; Mark Evers, B.; Saito, H. Age-associated increase in cytokine production during systemic inflammation-II: The role of IL-1b in age-dependent IL-6 upregulation in adipose tissue. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Liu, J.; Lau, C.W.; Huang, Y. IL-6 in diabetes and cardiovascular complications. Br. J. Pharmacol. 2014, 171, 3595–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigerlova, E.; Battaglia-Hsu, S.F. IL-6 signaling in diabetes nephropathy: From pathophysiology to therapeutic prospective. Cytokine Growth Factor Rev. 2017, 37, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Hassan-Zadeh, V. IL-6 signalling pathways and the development of type 2 diabetes. Immunopharmacology 2018, 26, 685–698. [Google Scholar] [CrossRef]
- Moshapa, F.T.; Riches-Suman, K.; Palmer, T.M. Therapeutic targeting of the proinflammatory IL-6-JAK/STAT signalling pathways responsible for vascular restenosis in type 2 diabetes mellitus. Cardiol. Res. Pract. 2019, 2019. [Google Scholar] [CrossRef]
- Ross-John, S.; Winthrop, K.; Calaboese, L. The role of IL-6 in host defence against infections: Immunobiology and clincal implications. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef] [PubMed]
- John, J.; O’Shea, M.G.; Siegel, R. Cytokines and Cytokine Receptors, 4th ed.; Elsiever: Cambridge, MA, USA, 2014; Volume 1, pp. 108–135. [Google Scholar]
- Takagi, M.; Kasayama, S.; Yamamoto, T.; Motomura, T.; Hashimoto, K.; Yamamoto, H.; Sato, B.; Okada, S.; Kishimoto, T. Advanced glycation endproducts stimulate interleukin-6 production by human bone-derived cells. J. Bone Miner. Res. 1997, 12, 439–446. [Google Scholar] [CrossRef]
- Chang, P.C.; Chien, L.Y.; Chong, L.Y.; Kuo, Y.P.; Hsiao, J.K. Glycated matrix up-regulates inflammatory signaling similarly to Porphyromonas gingivalis lipopolysaccharide. J. Periodontol. Res. 2013, 48, 184–193. [Google Scholar] [CrossRef]
- Rasheed, Z.; Akhtar, N.; Haqqi, T.M. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-kappa B in human osteoarthritis chondrocytes. Rheumatology 2011, 50, 838–851. [Google Scholar] [CrossRef]
- Cheng, A.; Dong, Y.; Zhu, F.; Liu, Y.; Hou, F.F.; Nie, J. AGE-LDL activates Toll like receptor 4 pathway and promotes inflammatory cytokines production in renal tubular epithelial cells. Int. J. Biol. Sci. 2013, 9, 94–107. [Google Scholar] [CrossRef]
- Parikh, A.A.; Salzman, A.L.; Kane, C.D.; Fischer, J.E.; Hasselgren, P.O. IL-6 production in human intestinal epithelial cells following stimulation with IL-1 beta is associated with activation of the transcription factor NF-kappa B. J. Surg. Res. 1998, 69, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cheng, K.; Mao, J.; Liu, F.; Liu, J.; Ye, M.; Zou, H. Quantitative proteomics reveals the kinetics of trypsin-catalyzed protein digestion. Anal. Bioanal. Chem. 2014, 406, 6247–6256. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Incubation Time of AGE | Nε(carboxymethyl)-Lysine (μg/mL) | Number(%) of Glycation in AGE | ||||
|---|---|---|---|---|---|---|
| Exp. 1 | Exp. 2 | Exp. 3 | Lysine | Arginine | Cysteine | |
| Day 1 | 0.056 | 0.049 | — | — | — | — |
| Day 30 | 0.136 | 0.269 | 0.458 | 43/56(77%) | 2/23(8%) | 0/35(0%) |
| Day 45 | 1.126 | 0.171 | 0.186 | — | — | — |
| Day 130 | 7.586 | 7.197 | 7.779 | 56/56(100%) | 1/23(4%) | 0/35(0%) |
| Day 180 | 10.916 | 10.664 | 9.509 | 56/56(100%) | 4/23(16%) | 0/35(0%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, C.-Y.; Wu, C.-H.; Lu, C.-H.; Kuo, Y.-M.; Li, K.-J.; Hsieh, S.-C.; Yu, C.-L. Advanced Glycation End Products of Bovine Serum Albumin Suppressed Th1/Th2 Cytokine but Enhanced Monocyte IL-6 Gene Expression via MAPK-ERK and MyD88 Transduced NF-κB p50 Signaling Pathways. Molecules 2019, 24, 2461. https://doi.org/10.3390/molecules24132461
Shen C-Y, Wu C-H, Lu C-H, Kuo Y-M, Li K-J, Hsieh S-C, Yu C-L. Advanced Glycation End Products of Bovine Serum Albumin Suppressed Th1/Th2 Cytokine but Enhanced Monocyte IL-6 Gene Expression via MAPK-ERK and MyD88 Transduced NF-κB p50 Signaling Pathways. Molecules. 2019; 24(13):2461. https://doi.org/10.3390/molecules24132461
Chicago/Turabian StyleShen, Chieh-Yu, Cheng-Han Wu, Cheng-Hsun Lu, Yu-Min Kuo, Ko-Jen Li, Song-Chou Hsieh, and Chia-Li Yu. 2019. "Advanced Glycation End Products of Bovine Serum Albumin Suppressed Th1/Th2 Cytokine but Enhanced Monocyte IL-6 Gene Expression via MAPK-ERK and MyD88 Transduced NF-κB p50 Signaling Pathways" Molecules 24, no. 13: 2461. https://doi.org/10.3390/molecules24132461