Sonic Hedgehog Ligand: A Role in Formation of a Mesenchymal Niche in Human Pancreatic Ductal Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Pancreatic Tissues
2.2. Immunohistochemistry and Immunocytochemistry
2.3. In Vitro Cell Cultures and Stem Cell Niche Models
2.4. Stem Cell Niche In Vitro Models
2.5. Three-Dimensional Tumour Growth Assay (3D-TGA)
2.6. RNA Extraction, cDNA Synthesis, and Gene Expression Analysis
2.7. Statistical Analysis
3. Results
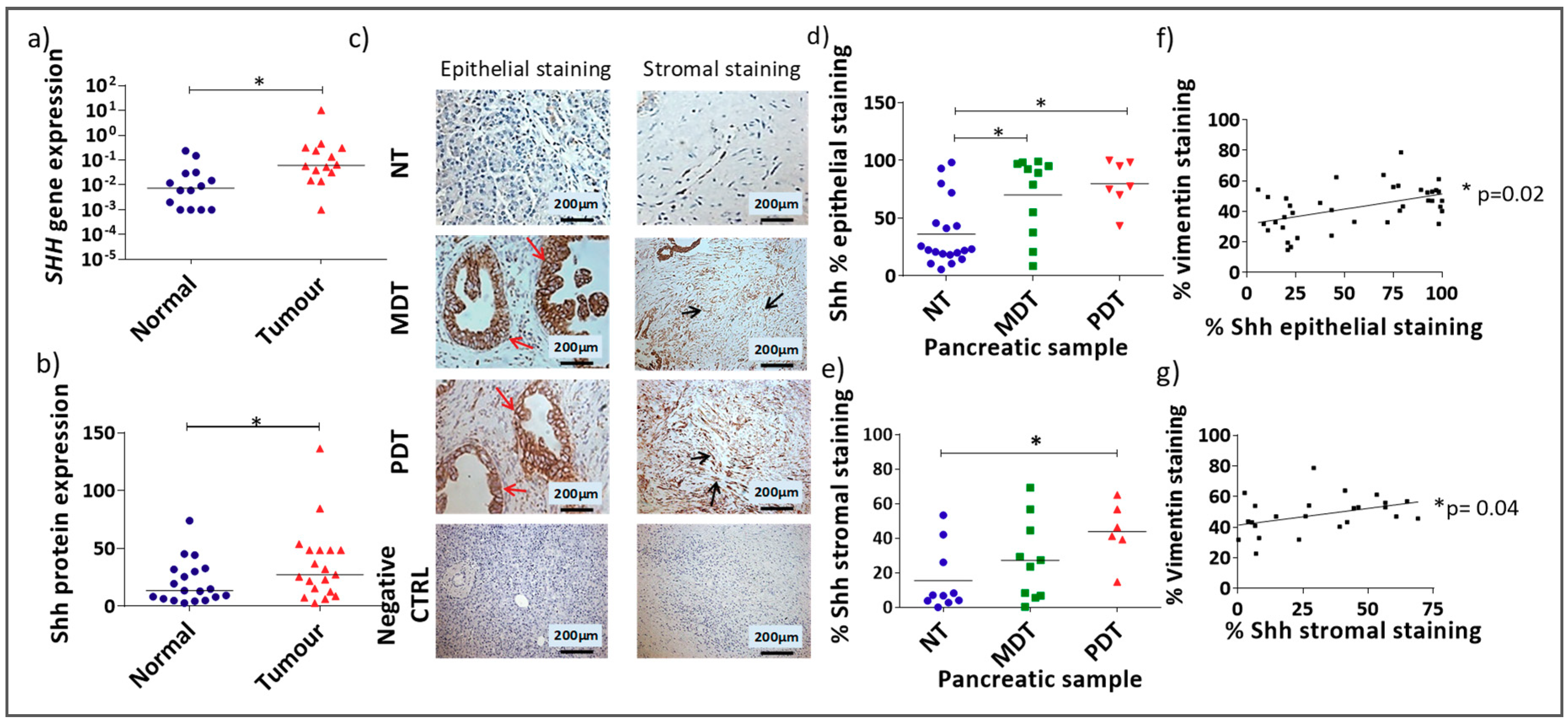
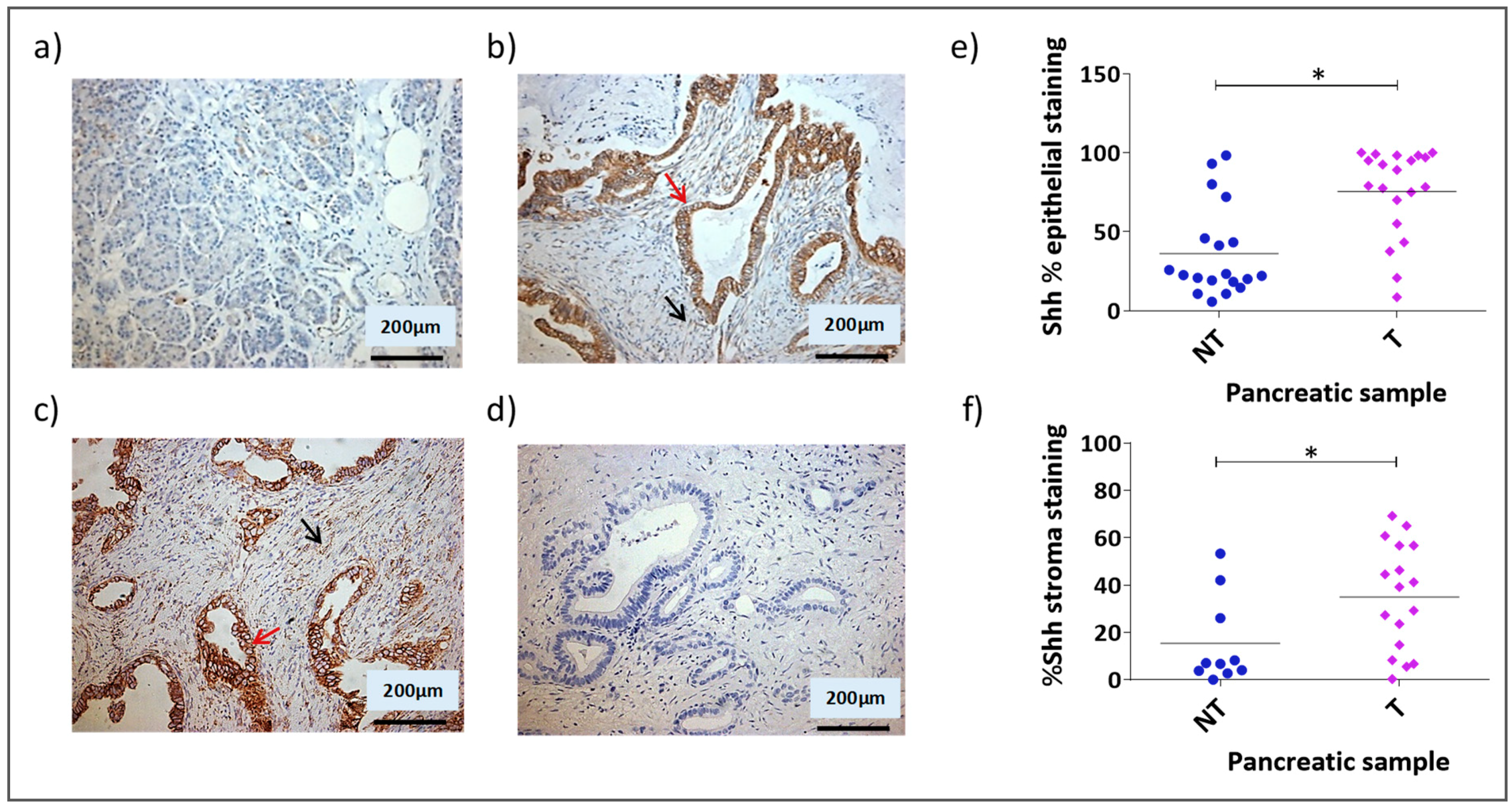
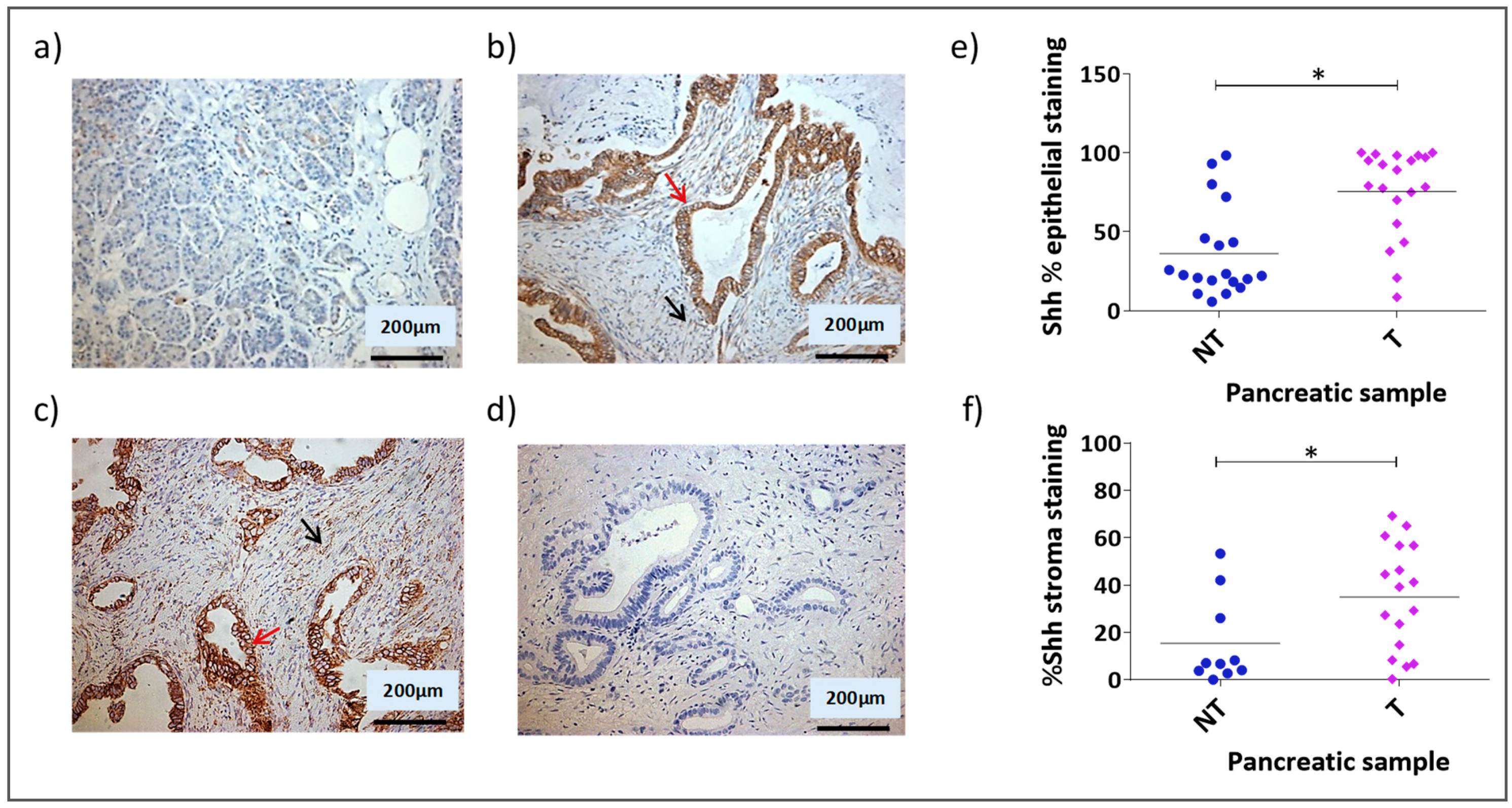
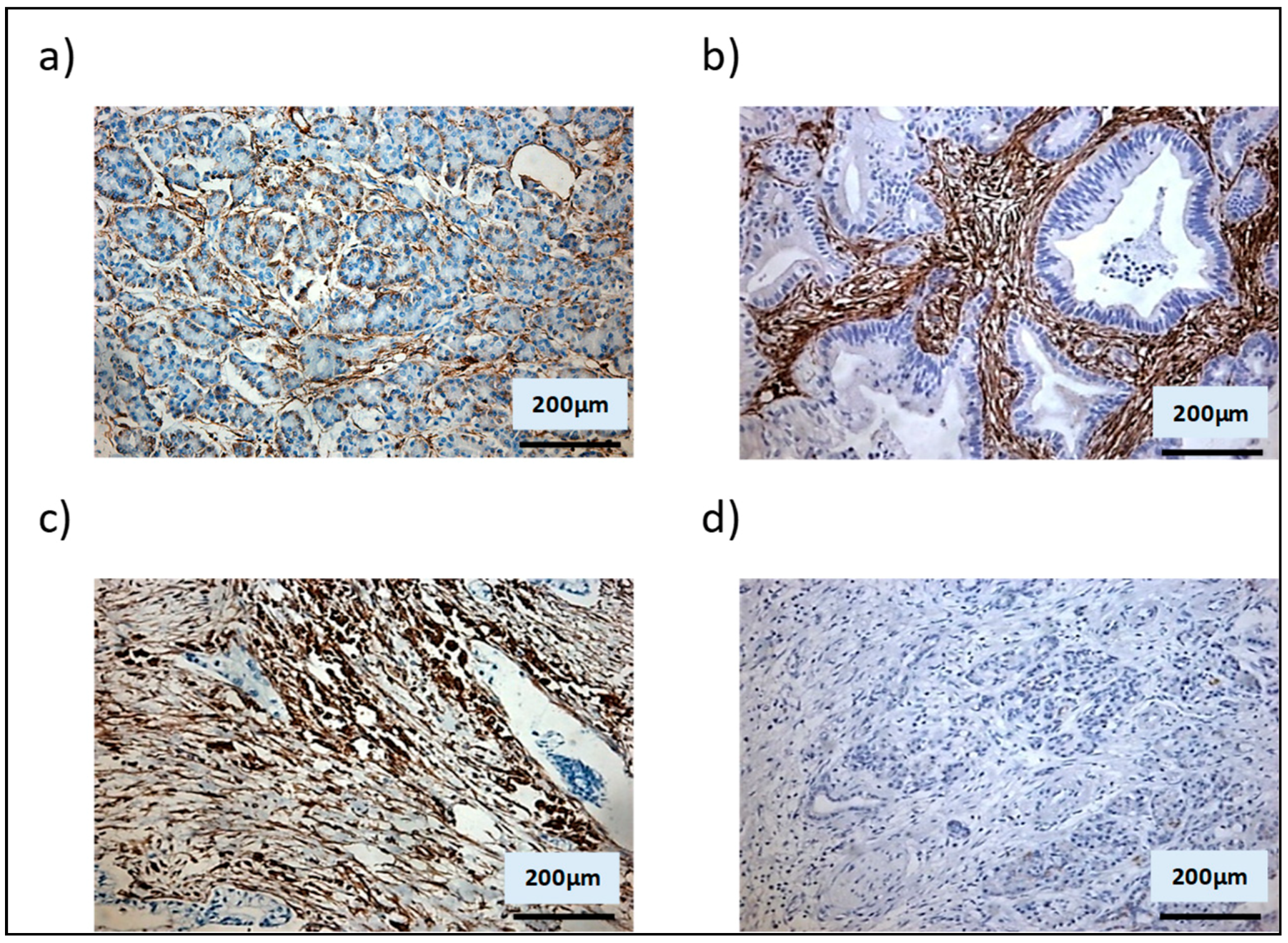
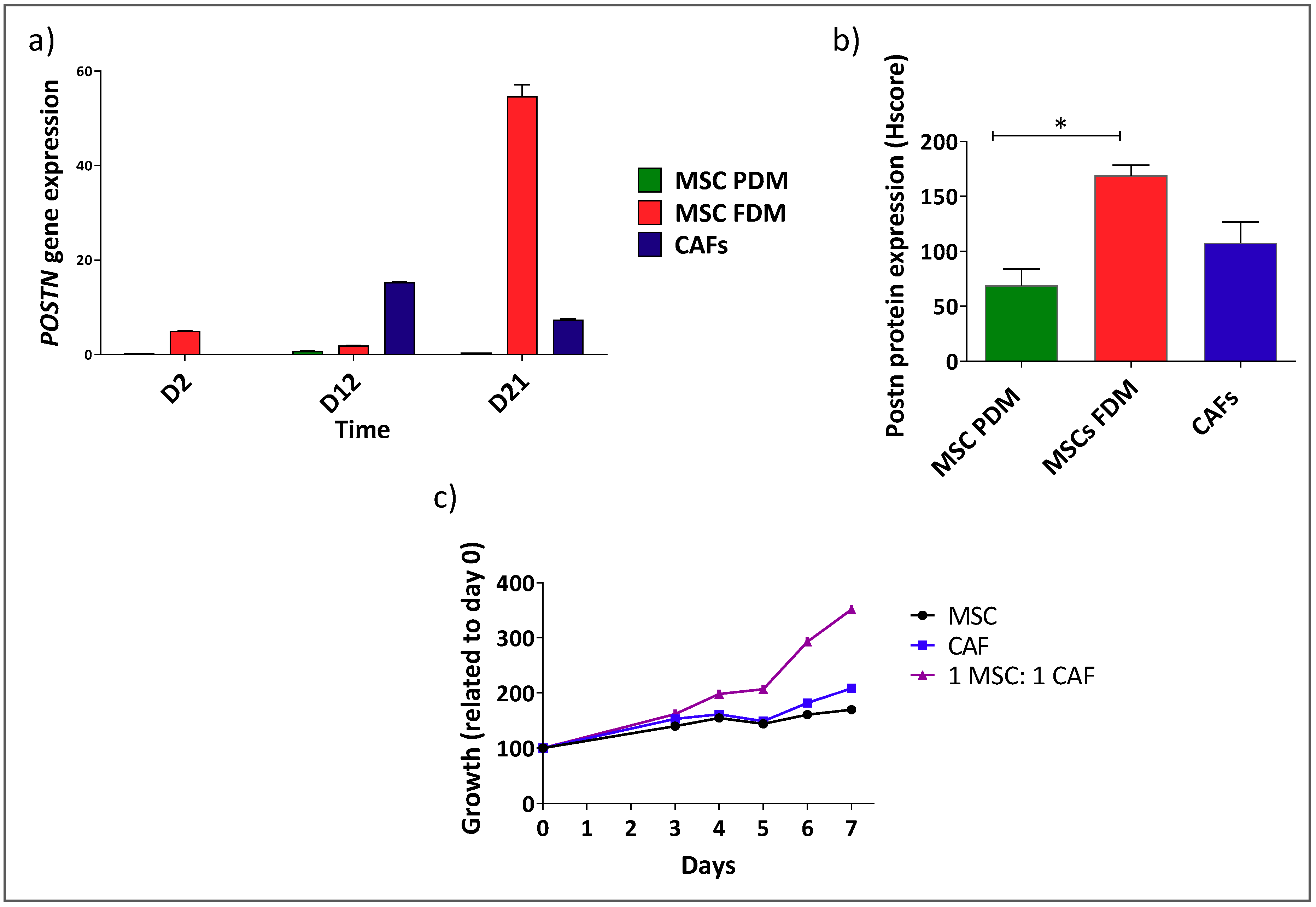
3.1. Shh Is Overexpressed in Both the Stromal and Epithelial Compartments of Advanced Pancreatic Tumours
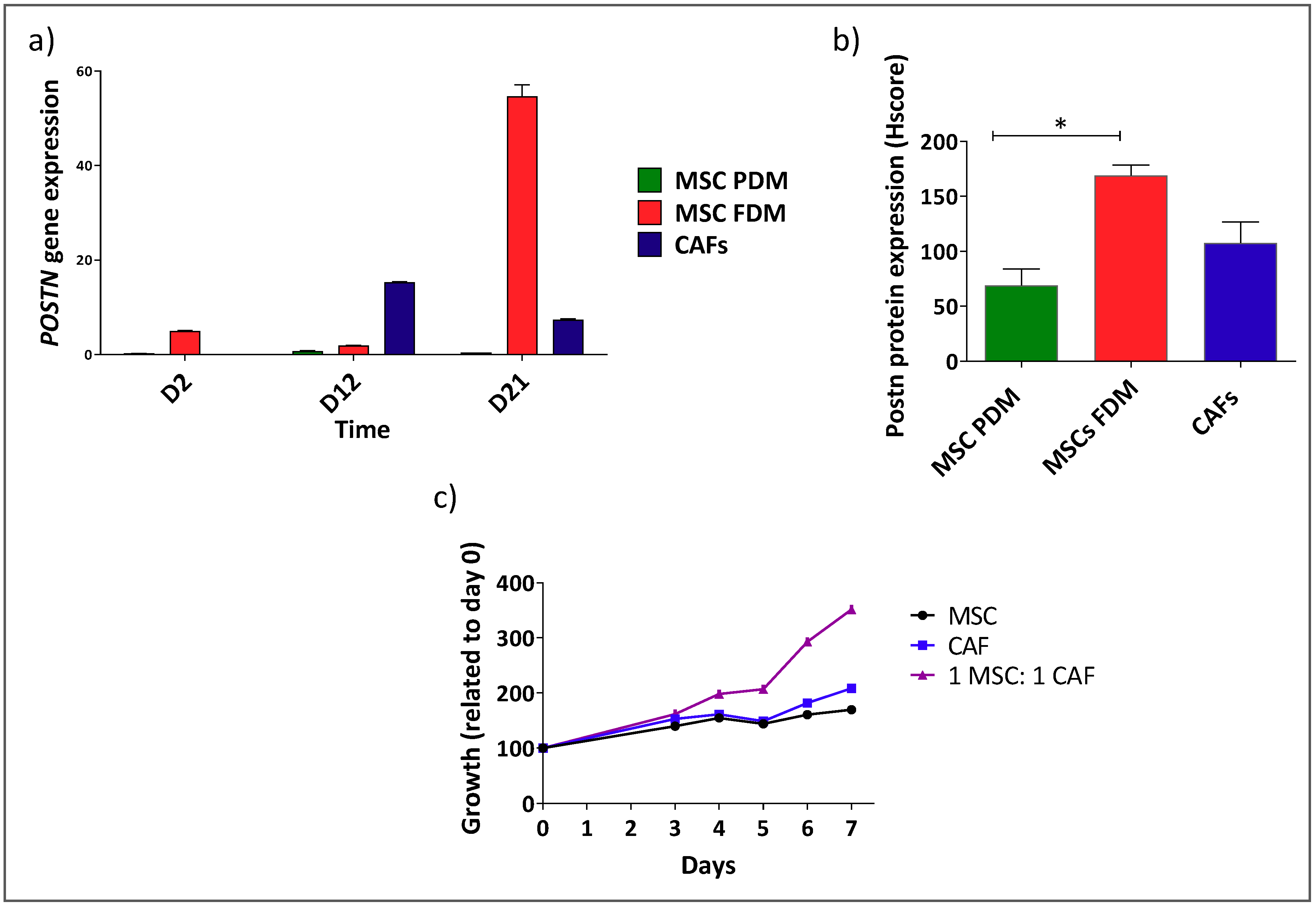
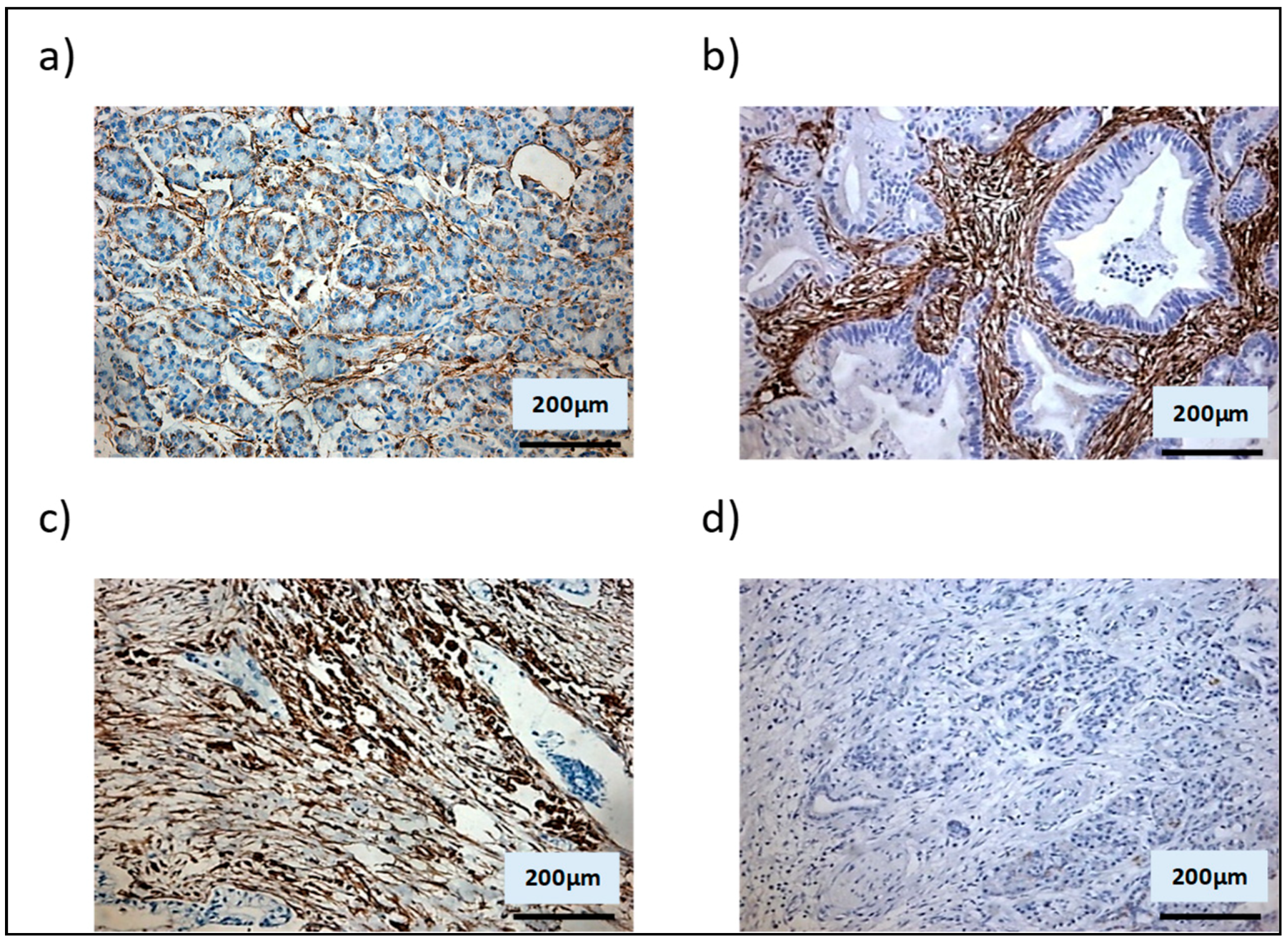
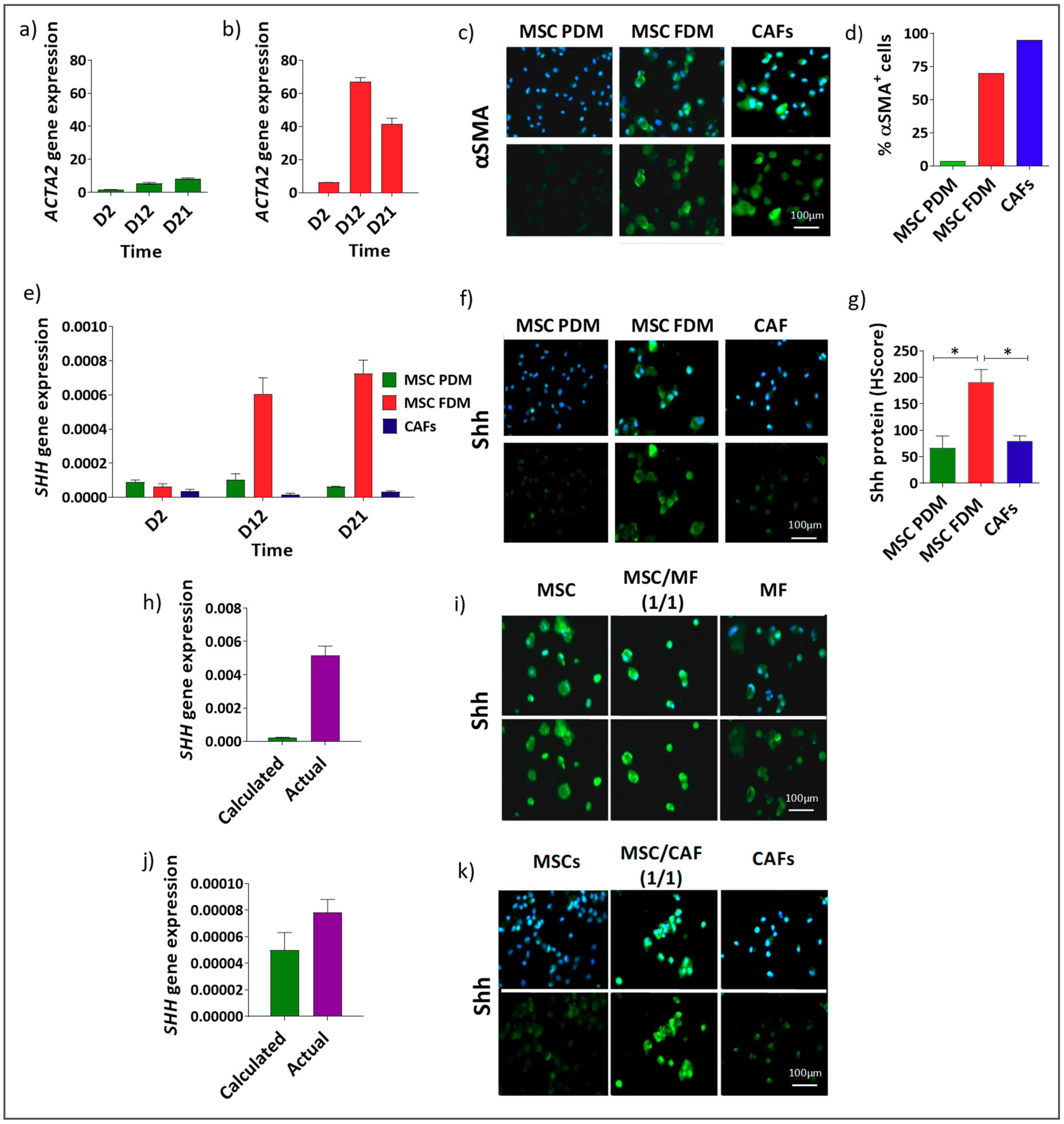
3.2. Shh Is Upregulated in a Mixed Population of αSMA− and αSMA+ Cells
3.3. αSMA+ and αSMA− Cells Interact Between Each Other in the Shh Expressing Mixed Population
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MSCs | mesenchymal stem cells |
| αSMA | alpha-smooth muscle actin-expressing |
| mSCN | mesenchymal stem cell niche |
| Shh | sonic hedgehog |
| PDAC | pancreatic adenocarcinoma |
| CAFs | cancer-associated fibroblasts |
| PSCs | pancreatic stellate cells |
| MMPs | matrix metalloproteinases |
| ECM | extracellular matrix |
| MDT | moderately differentiated tumour |
| PDT | poorly differentiated tumour |
| ROI | regions of interest |
| FDM | free differentiation medium |
| PDM | preventing differentiation medium |
| 3D-TGA | three-dimensional tumour growth assay |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| IHC | immunohistochemistry |
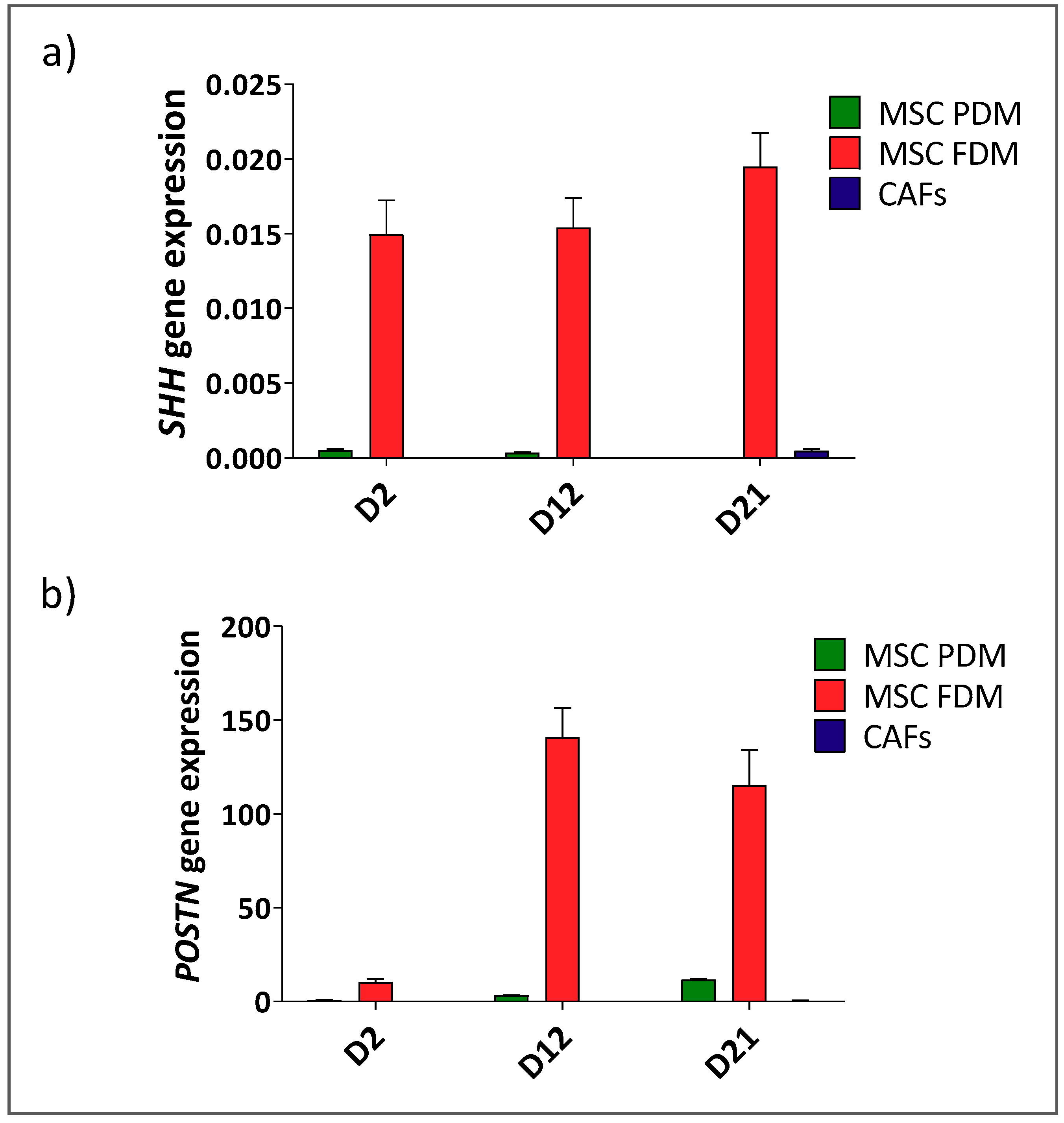
Appendix A

References
- Oliani, C.; Padovani, M.; Manno, P.; Barana, D.; Falconi, M.; Bassi, C.; Cavallini, G.; Pederzoli, P.; Cetto, G.L. Gemcitabine and continuous infusion of 5-fluorouracil in locally advanced and metastatic pancreatic cancer: A phase I-II study. Anticancer Res. 2004, 24, 2107–2112. [Google Scholar] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, Y.A. Desmoplasia: A response or a niche? Cancer Discov. 2012, 2, 772–774. [Google Scholar] [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, C.J.; Colvin, E.K.; Pinese, M.; Chang, D.K.; Morey, A.L.; Musgrove, E.A.; Pajic, M.; Apte, M.; Henshall, S.M.; Sutherland, R.L.; et al. Recruitment and Activation of Pancreatic Stellate Cells from the Bone Marrow in Pancreatic Cancer: A Model of Tumor-Host Interaction. PLoS ONE 2011, 6, e26088. [Google Scholar] [CrossRef]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Anderberg, C.; Pietras, K. On the origin of cancer-associated fibroblasts. Cell Cycle 2009, 8, 1461–1462. [Google Scholar] [CrossRef]
- Schofield, R. Relationship between spleen colony-forming cell and hematopoietic stem-cell hypothesis. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Lin, N.; Tang, Z.; Deng, M.; Zhong, Y.; Lin, J.; Yang, X.; Xiang, P.; Xu, R. Hedgehog-mediated paracrine interaction between hepatic stellate cells and marrow-derived mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2008, 372, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Wang, C.Y.; Yang, L.; Zhan, S.D.; Zhang, M.; Tian, K. Contribution of murine bone marrow mesenchymal stem cells to pancreas regeneration after partial pancreatectomy in mice. Cell Biol. Int. 2012, 36, 823–831. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef]
- El-Zaatari, M.; Tobias, A.; Grabowska, A.M.; Kumari, R.; Scotting, P.J.; Kaye, P.; Atherton, J.; Clarke, P.A.; Powe, D.G.; Watson, S.A. De-regulation of the sonic hedgehog pathway in the InsGas mouse model of gastric carcinogenesis. Br. J. Cancer 2007, 96, 1855–1861. [Google Scholar] [CrossRef]
- Bian, Y.-H.; Huang, S.H.; Yang, L.; Ma, X.L.; Xie, J.W.; Zhang, H.W. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J. Gastroenterol. 2007, 13, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.B.; Habbe, N.; Karikari, C.; Mullendore, M.; et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 2725–2735. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Kwon, J.; Ali, B.; Wang, E.; Patra, S.; Shridhar, V.; Mukherjee, P. Role of Hedgehog Signaling in Ovarian Cancer. Clin. Cancer Res. 2008, 14, 7659–7666. [Google Scholar] [CrossRef]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef]
- Chen, M.; Tanner, M.; Levine, A.C.; Levina, E.; Ohouo, P.; Buttyan, R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle 2009, 8, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Mao, J.; Zhang, Q.; Li, L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol. Lett. 2011, 2, 995–1001. [Google Scholar] [PubMed]
- Yan, R.; Peng, X.; Yuan, X.; Huang, D.; Chen, J.; Lu, Q.; Lv, N.; Luo, S. Suppression of growth and migration by blocking the hedgehog signaling pathway in gastric cancer cells. Cell. Oncol. 2013, 36, 421–435. [Google Scholar] [CrossRef]
- Xu, X.-F.; Guo, C.Y.; Liu, J.; Yang, W.J.; Xia, Y.J.; Xu, L.; Yu, Y.C.; Wang, X.P. Gli1 maintains cell survival by up-regulating IGFBP6 and Bcl-2 through promoter regions in parallel manner in pancreatic cancer cells. J. Carcinogen. 2009, 8, 13. [Google Scholar]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Nakamura, K.; Sasajima, J.; Mizukami, Y.; Sugiyama, Y.; Yamazaki, M.; Fujii, R.; Kawamoto, T.; Koizumi, K.; Sato, K.; Fujiya, M.; et al. Hedgehog Promotes Neovascularization in Pancreatic Cancers by Regulating Ang-1 and IGF-1 Expression in Bone-Marrow Derived Pro-Angiogenic Cells. PLoS ONE 2010, 5, e8824. [Google Scholar] [CrossRef]
- Yeung, T.M.; Chia, L.A.; Kosinski, C.M.; Kuo, C.J. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell. Mol. Life Sci. 2011, 68, 2513–2523. [Google Scholar] [CrossRef]
- Shaw, A.; Gipp, J.; Bushman, W. The Sonic Hedgehog pathway stimulates prostate tumor growth by paracrine signaling and recapitulates embryonic gene expression in tumor myofibroblasts. Oncogene 2009, 28, 4480–4490. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.X.; Jia, L.W.; Liu, W.M.; Miao, C.L.; Liu, S.; Cao, Y.J.; Duan, E.K. Role of sonic hedgehog in maintaining a pool of proliferating stem cells in the human fetal epidermis. Human Reprod. 2006, 21, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nat. Cell Boil. 2009, 458, 776–779. [Google Scholar]
- Choi, S.S.; Omenetti, A.; Witek, R.P.; Moylan, C.A.; Syn, W.K.; Jung, Y.; Yang, L.; Sudan, D.L.; Sicklick, J.K.; Michelotti, G.A.; et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1093–G1106. [Google Scholar] [CrossRef]
- Onion, D.; Argent, R.H.; Reece-Smith, A.M.; Craze, M.L.; Pineda, R.G.; Clarke, P.A.; Ratan, H.L.; Parsons, S.L.; Lobo, D.N.; Duffy, J.P.; et al. 3-Dimensional Patient-Derived Lung Cancer Assays Reveal Resistance to Standards-of-Care Promoted by Stromal Cells but Sensitivity to Histone Deacetylase Inhibitors. Mol. Cancer Ther. 2016, 15, 753–763. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Büchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; Di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and angiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S.; et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef]
- Niyaz, M.; Khan, M.S.; Wani, R.A.; Shah, O.J.; Mudassar, S. Sonic Hedgehog Protein is Frequently Up-Regulated in Pancreatic Cancer Compared to Colorectal Cancer. Pathol. Oncol. 2018, 1–7. [Google Scholar] [CrossRef]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.M.; Chawla, A.; Houghton, J.; Zavros, Y. Sonic Hedgehog Mediates the Proliferation and Recruitment of Transformed Mesenchymal Stem Cells to the Stomach. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Rucki, A.A.; Foley, K.; Zhang, P.; Xiao, Q.; Kleponis, J.; Wu, A.A.; Sharma, R.; Mo, G.; Liu, A.; Van Eyk, J.; et al. Heterogeneous Stromal Signaling within the Tumor Microenvironment Controls the Metastasis of Pancreatic Cancer. Cancer Res. 2017, 77, 41–52. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz I Altaba, A. Melanomas require HEDGEHOG-GLI signaling reaulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef]
- Wei, R.; Lv, M.; Li, F.; Cheng, T.; Zhang, Z.; Jiang, G.; Zhou, Y.; Gao, R.; Wei, X.; Lou, J.; et al. Human CAFs promote lymphangiogenesis in ovarian cancer via the Hh-VEGF-C signaling axis. Oncotarget 2017, 8, 67315–67328. [Google Scholar] [CrossRef]
- Li, X.; Ma, Q.; Duan, W.; Liu, H.; Xu, H.; Wu, E. Paracrine sonic hedgehog signaling derived from tumor epithelial cells: A key regulator in the pancreatic tumor microenvironment. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 97–108. [Google Scholar] [CrossRef]
- Tsang, S.W.; Zhang, H.; Lin, C.; Xiao, H.; Wong, M.; Shang, H.; Yang, Z.-J.; Lu, A.; Yung, K.K.-L.; Bian, Z. Rhein, a Natural Anthraquinone Derivative, Attenuates the Activation of Pancreatic Stellate Cells and Ameliorates Pancreatic Fibrosis in Mice with Experimental Chronic Pancreatitis. PLoS ONE 2013, 8, e82201. [Google Scholar] [CrossRef]
- Chu, G.C.; Kimmelman, A.C.; Hezel, A.F.; DePinho, R.A. Stromal biology of pancreatic cancer. J. Cell. Biochem. 2007, 101, 887–907. [Google Scholar] [CrossRef]
- Korc, M. Pancreatic cancer-associated stroma production. Am. J. Surg. 2007, 194, S84–S86. [Google Scholar] [CrossRef]
- Merika, E.E.; Syrigos, K.N.; Saif, M.W. Desmoplasia in Pancreatic Cancer. Can We Fight It? Gastroenterol. Res. Practice 2012, 2012, 781765. [Google Scholar] [CrossRef]
- Crnogorac-Jurcevic, T.; Efthimiou, E.; Capelli, P.; Blaveri, E.; Baron, A.; Terris, B.; Jones, M.; Tyson, K.; Bassi, C.; Scarpa, A.; et al. Gene expression profiles of pancreatic cancer and stromal desmoplasia. Oncogene 2001, 20, 7437–7446. [Google Scholar] [CrossRef]
- Shinozaki, S.; Ohnishi, H.; Hama, K.; Kita, H.; Yamamoto, H.; Osawa, H.; Sato, K.; Tamada, K.; Mashima, H.; Sugano, K. Indian hedgehog promotes the migration of rat activated pancreatic stellate cells by increasing membrane type-1 matrix metalloproteinase on the plasma membrane. J. Cell Physiol. 2008, 216, 38–46. [Google Scholar] [CrossRef]
- Klieser, E.; Swierczynski, S.; Mayr, C.; Jäger, T.; Schmidt, J.; Neureiter, D.; Kiesslich, T.; Illig, R. Differential role of Hedgehog signaling in human pancreatic (patho-) physiology: An up to date review. World J. Gastrointest Pathophysiol. 2016, 7, 199–210. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef]
- Wang, W.Q.; Liu, L.; Xu, H.X.; Luo, G.P.; Chen, T.; Wu, C.T.; Xu, Y.F.; Xu, J.; Liu, C.; Zhang, B.; et al. Intratumoral alpha-SMA enhances the prognostic potency of CD34 associated with maintenance of microvessel integrity in hepatocellular carcinoma and pancreatic cancer. PLoS ONE 2013, 8, e71189. [Google Scholar]
- Anonymous. Infinity Reports Update from Phase 2 Study of Saridegib Plus Gemcitabine in Patients with Metastatic Pancreatic Cancer. Available online: www.businesswire.com/news/home/20120127005146/en/Infinity-ReportsUpdate-Phase-2-Study-Saridegib#.UxAvFfRdVxV (accessed on 8 May 2019).
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Hamada, S.; Shimosegawa, T. Pancreatic cancer stem cell and mesenchymal stem cell. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- De Jesus-Acosta, A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Maitra, A.; Rasheed, Z.; Zheng, L.; Rajeshkumar, N.V.; Le, D.T.; Hoering, A.; et al. A phase II study of vismodegib, a hedgehog (Hh) pathway inhibitor, combined with gemcitabine and nab-paclitaxel (nab-P) in patients (pts) with untreated metastatic pancreatic ductal adenocarcinoma (PDA). J. Clin. Oncol. 2014, 32, 257. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saini, F.; Argent, R.H.; Grabowska, A.M. Sonic Hedgehog Ligand: A Role in Formation of a Mesenchymal Niche in Human Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 424. https://doi.org/10.3390/cells8050424
Saini F, Argent RH, Grabowska AM. Sonic Hedgehog Ligand: A Role in Formation of a Mesenchymal Niche in Human Pancreatic Ductal Adenocarcinoma. Cells. 2019; 8(5):424. https://doi.org/10.3390/cells8050424
Chicago/Turabian StyleSaini, Francesca, Richard H. Argent, and Anna M. Grabowska. 2019. "Sonic Hedgehog Ligand: A Role in Formation of a Mesenchymal Niche in Human Pancreatic Ductal Adenocarcinoma" Cells 8, no. 5: 424. https://doi.org/10.3390/cells8050424
APA StyleSaini, F., Argent, R. H., & Grabowska, A. M. (2019). Sonic Hedgehog Ligand: A Role in Formation of a Mesenchymal Niche in Human Pancreatic Ductal Adenocarcinoma. Cells, 8(5), 424. https://doi.org/10.3390/cells8050424