β3-Adrenoreceptor Activity Limits Apigenin Efficacy in Ewing Sarcoma Cells: A Dual Approach to Prevent Cell Survival

 , ,
, ,  ,
,  ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
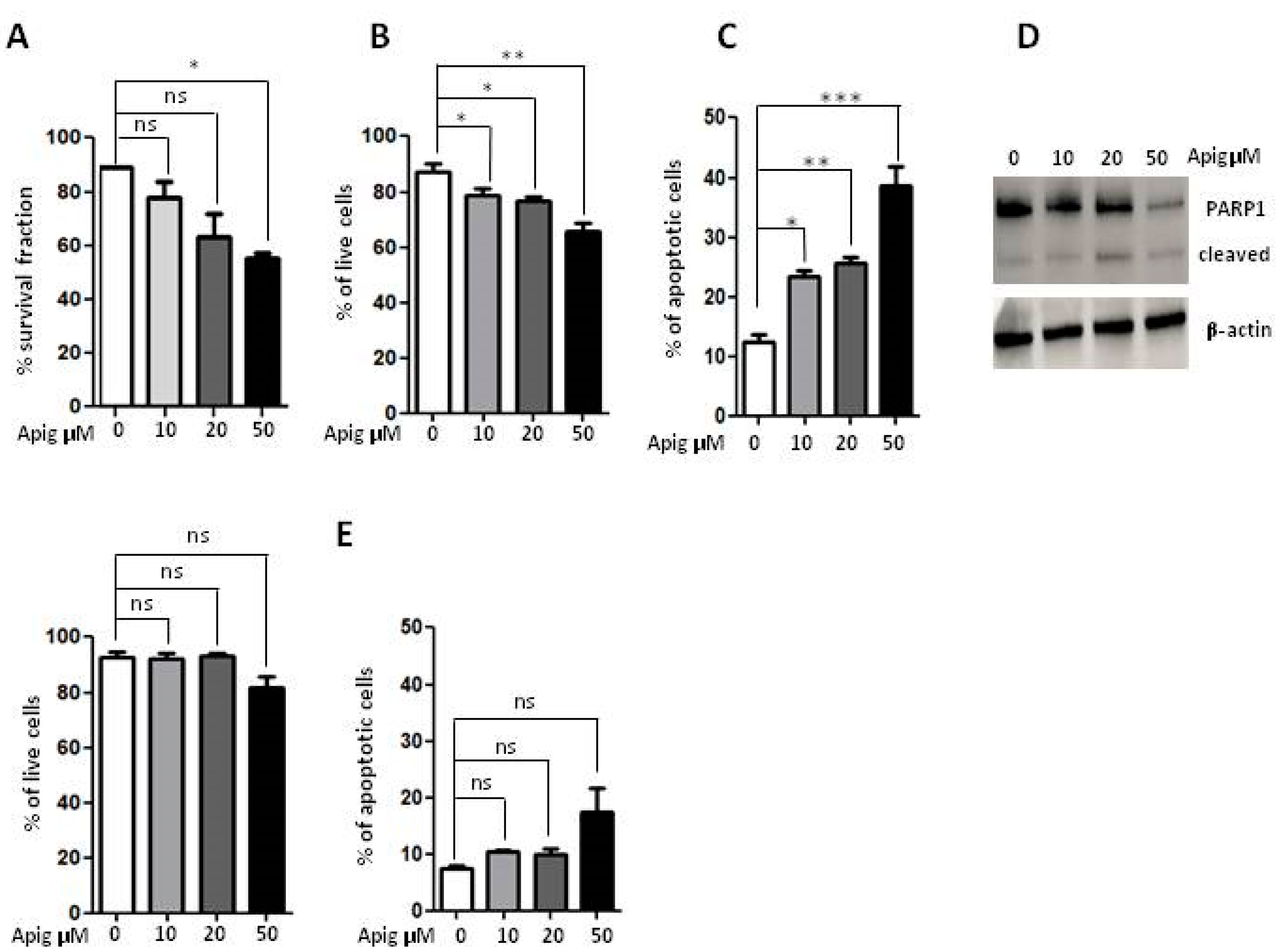
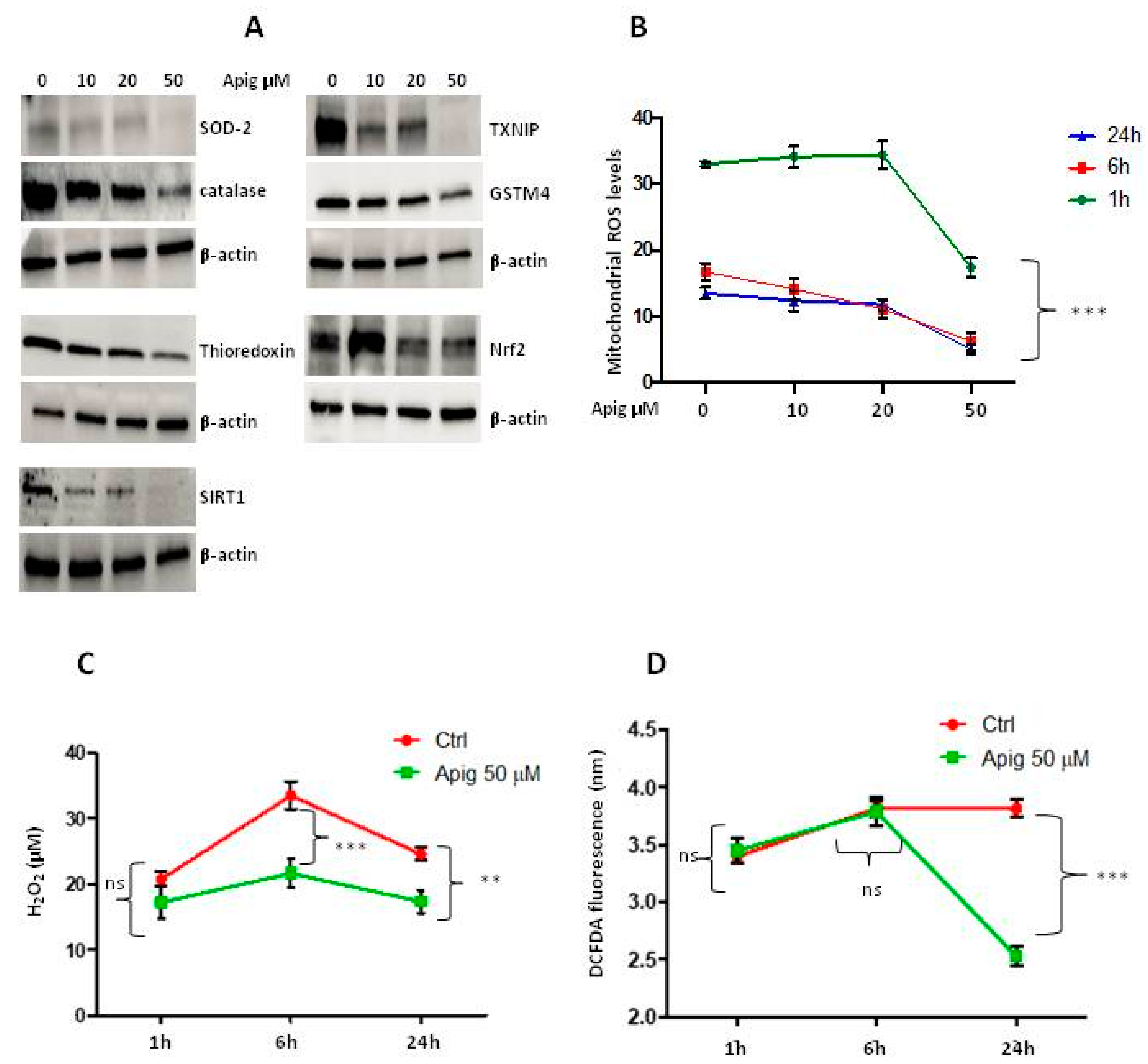
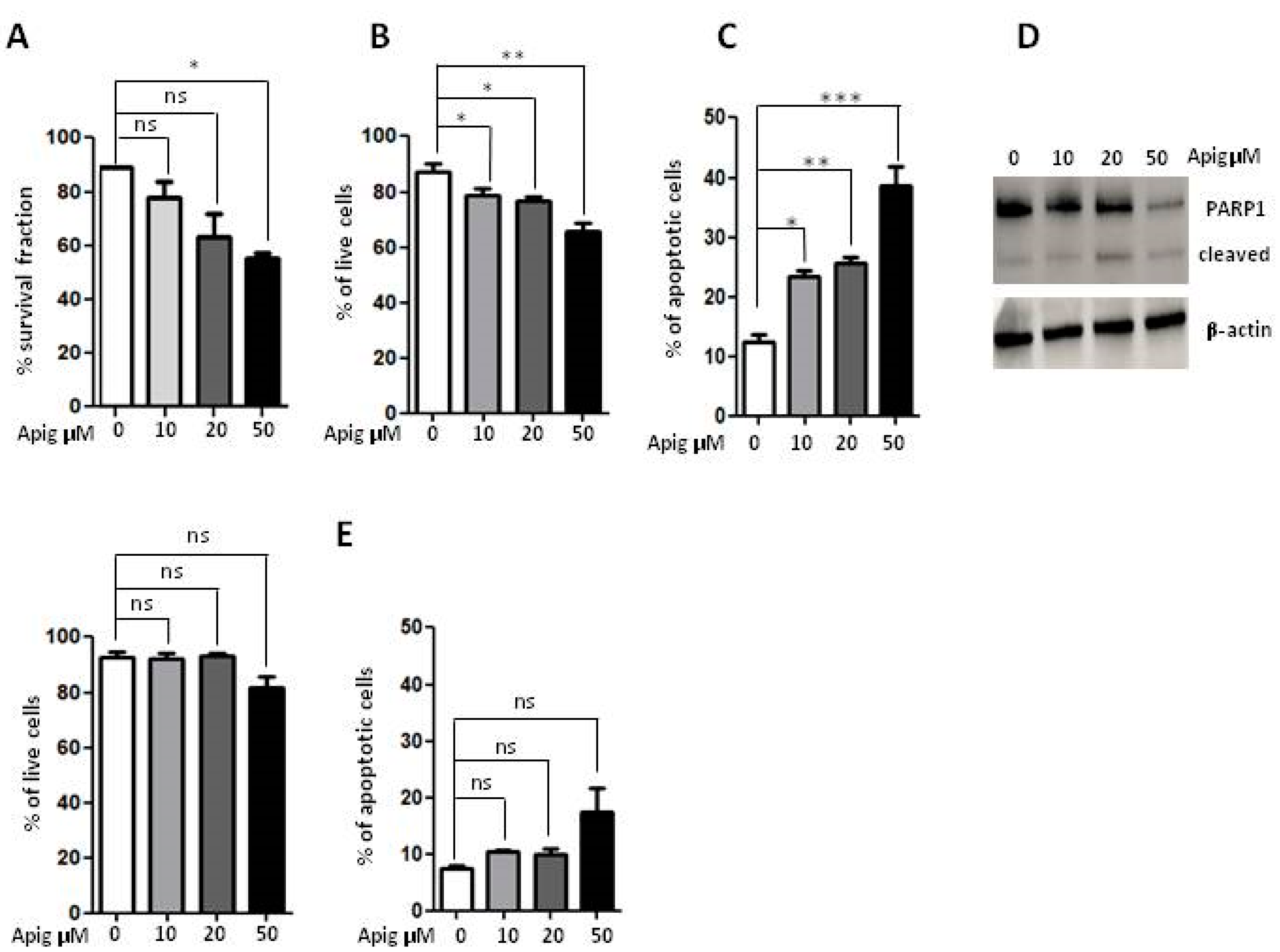
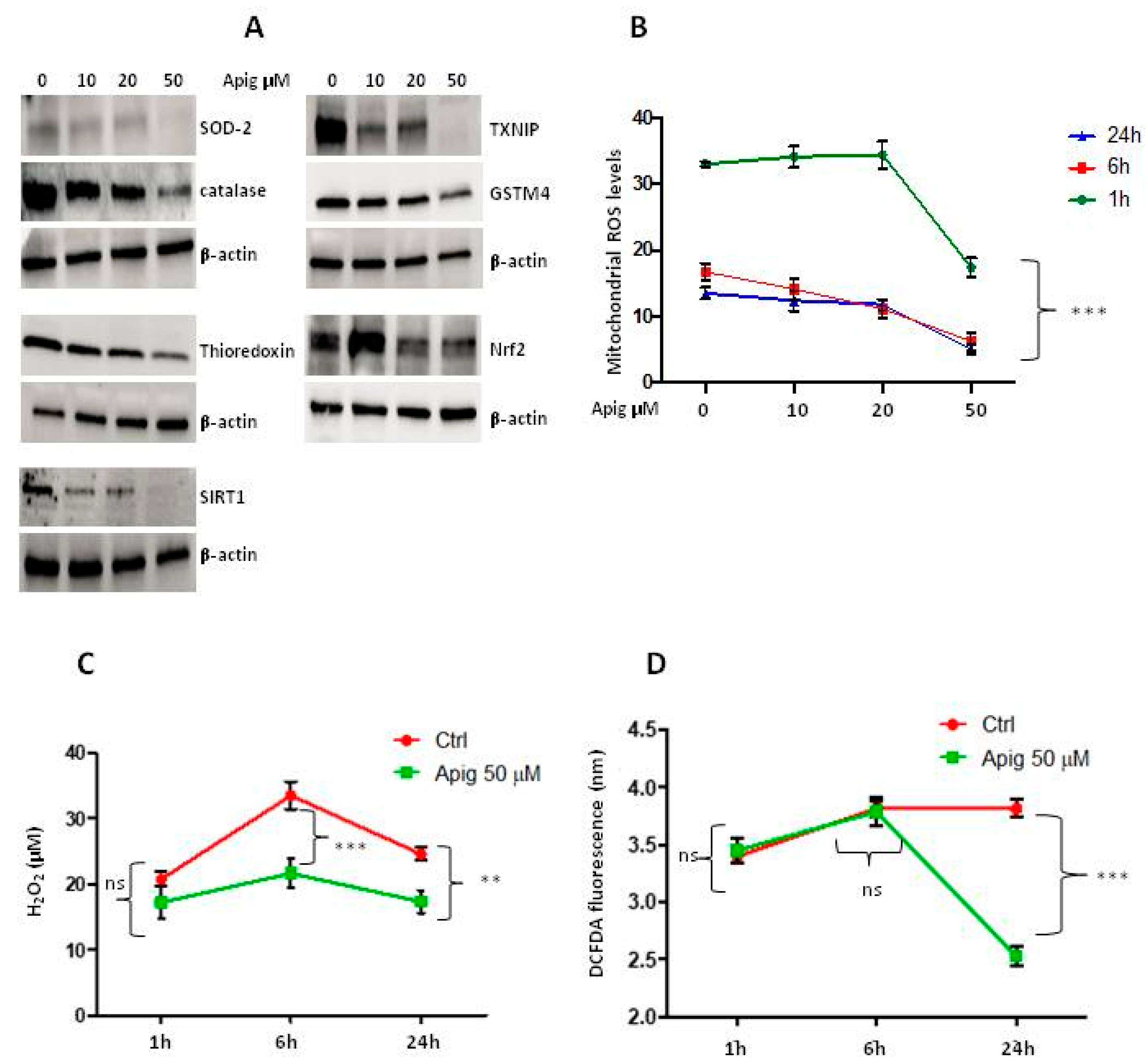
2.1. Apigenin Induces Cell Death and Inhibition of ROS and Antioxidant Activity in ES Cells
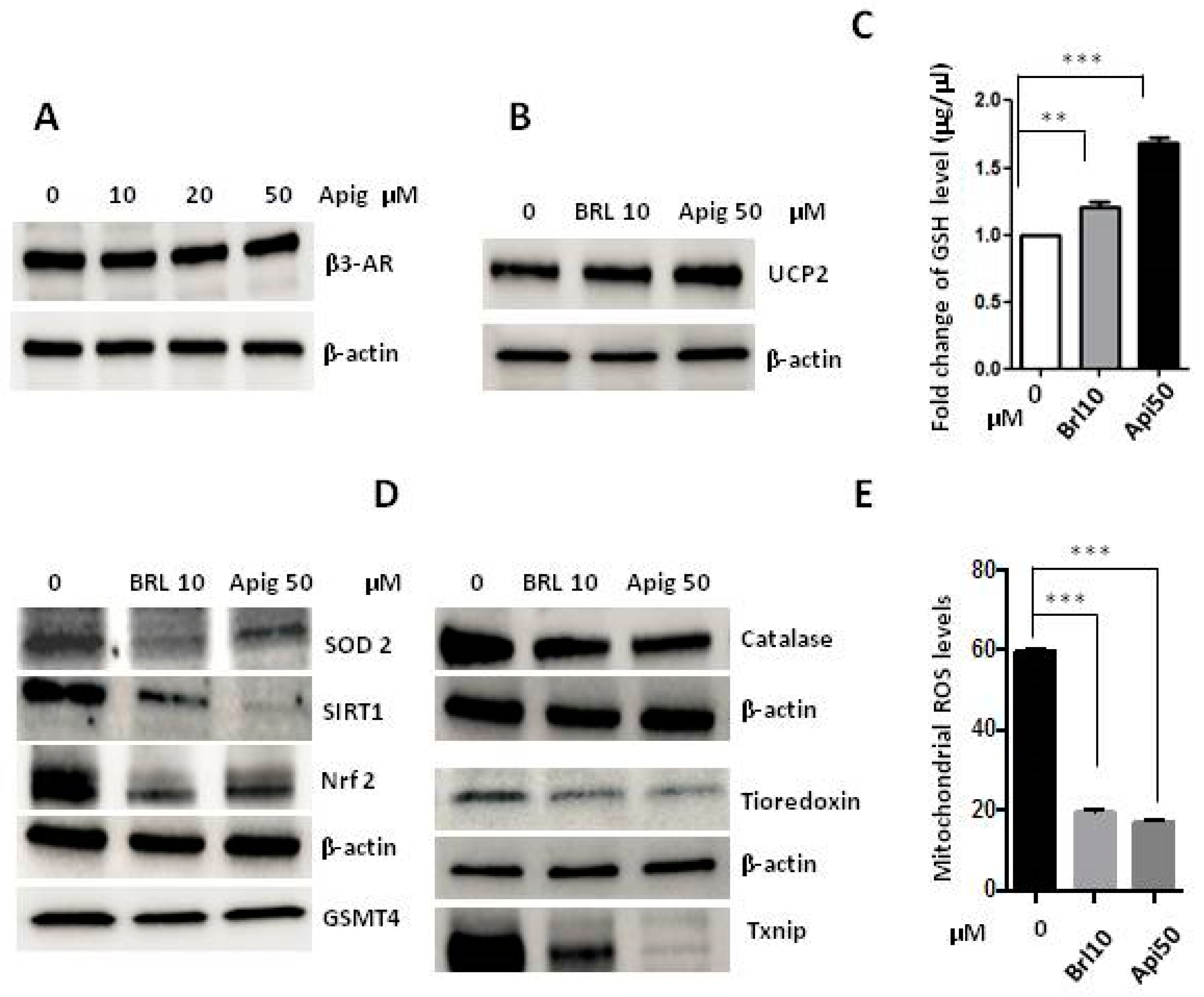
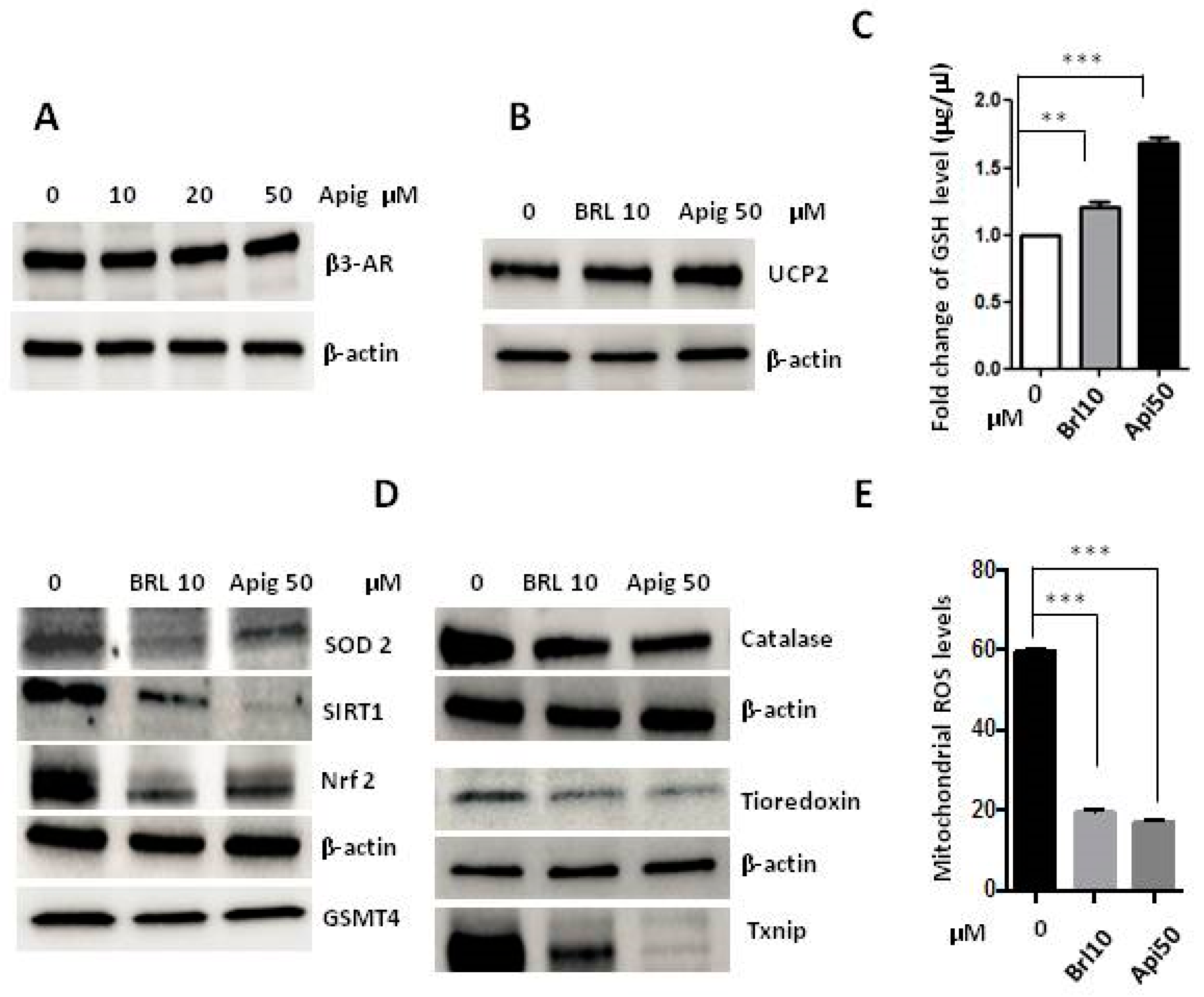
2.2. Apigenin Controls ROS Levels by Activation of UCP2 and GSH Accumulation
2.3. The Agonism of β3-AR Reproduces the Effect of Apigenin
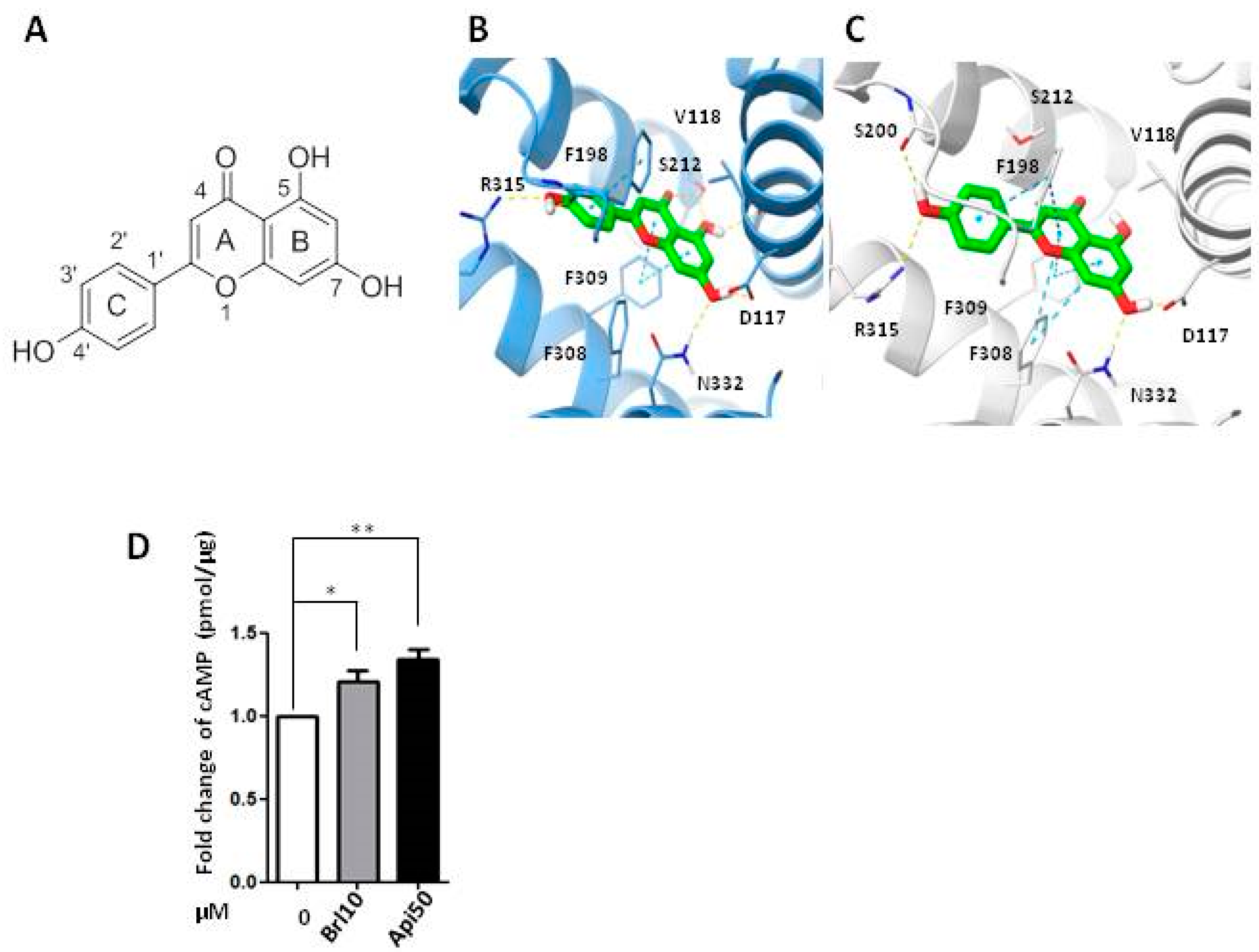
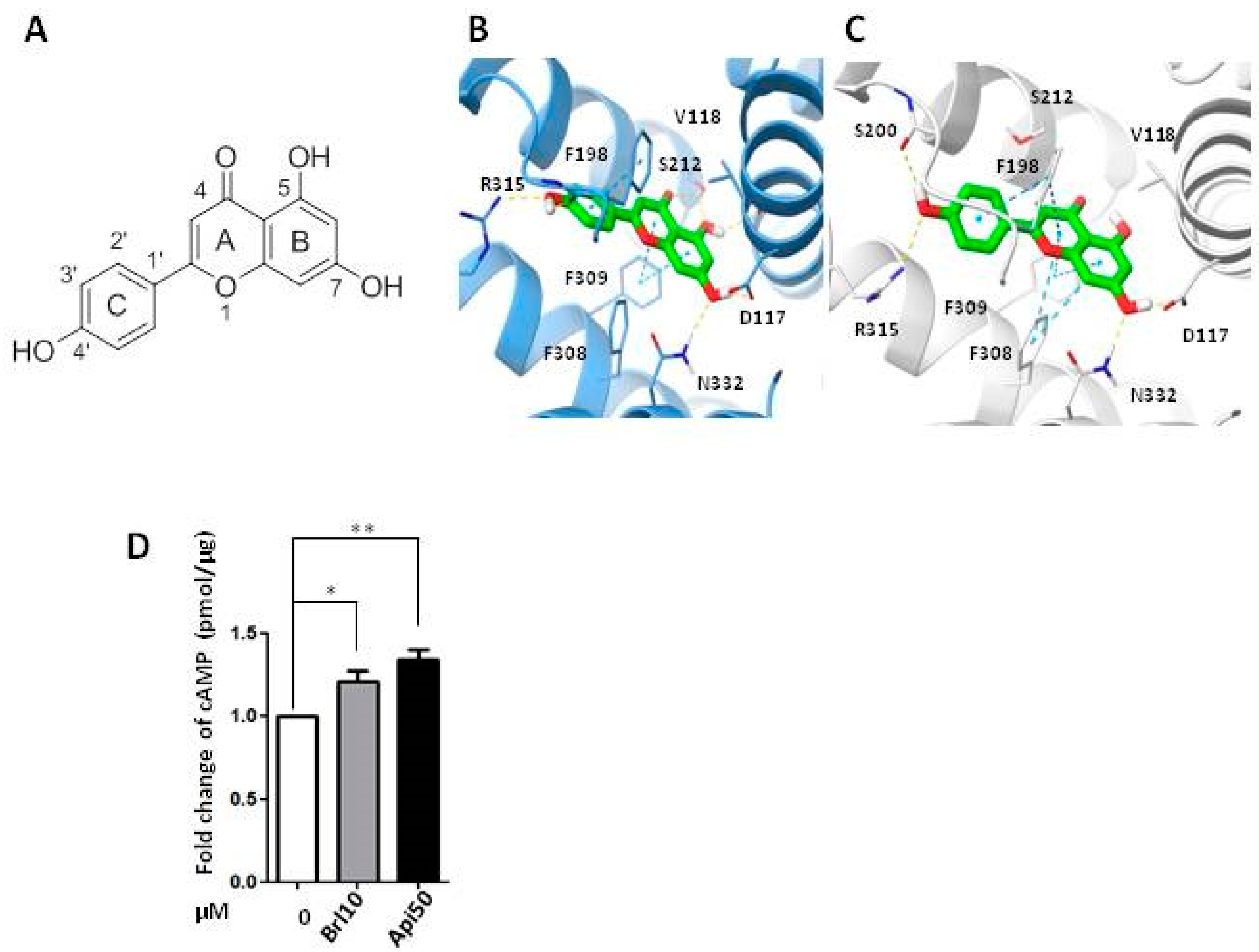
2.4. Apigenin Could Be a β3-AR Agonist
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures
4.3. Cell Treatments
4.4. MTT Assay
4.5. Western Blot Analysis
4.6. Glutathione Fluorometric Assay Kit
4.7. Cell Viability Analysis
4.8. cAMP Direct ImmunoAssay Kit
4.9. ROS Analysis
4.10. Mitochondria Isolation
4.11. In Silico Methods
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Kovar, H. Ewing’s Sarcoma and peripheral primitive neuroectodermal tumors after their genetic union. Curr. Opin. Oncol. 1998, 10, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Stamenkovic, I. The Biology of Ewing sarcoma. Cancer Lett. 2007, 254, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Balamuth, N.J.; Womer, R.B. Ewing’s sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef]
- Jain, S.; Kapoor, G. Chemotherapy in Ewing’s sarcoma. Indian J. Orthop. 2010, 44, 369–377. [Google Scholar] [PubMed]
- Magwere, T.; Myatt, S.S.; Burchill, S.A. Manipulation of oxidative stress to induce cell death in Ewing’s sarcoma family of tumors. Eur. J. Cancer 2008, 44, 2276–2287. [Google Scholar] [CrossRef]
- Scotlandi, K.; Remondini, D.; Castellani, G.; Manara, M.C.; Nardi, F.; Cantiani, L.; Francesconi, M.; Mercuri, M.; Caccuri, A.M.; Serra, M.; et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J. Clin. Oncol. 2009, 27, 2209–2216. [Google Scholar] [CrossRef]
- Conklin, K. Dietary antioxidants during cancer chemotherapy: Impact on chemotherapeutic effectiveness and development of side effects. Nutr. Cancer 2000, 37, 1–18. [Google Scholar] [CrossRef]
- Ladas, E.; Kelly, K.M. The antioxidant debate. Explore (NY) 2010, 6, 75–85. [Google Scholar] [CrossRef]
- Peterson, J.; Dweyer, J. Flavonoids: Dietary occurrence and biochemical activity. Nutr. Res. 1998, 18, 1995–2018. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Apigenin: A promising molecule for cancer prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Afaq, F.; Mukhtar, H. Selective growth inhibitory, cell-cycle deregulatory and apoptotic response of apigenin in normal versus human prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2001, 287, 914–920. [Google Scholar] [CrossRef]
- Morrissey, C.; O’Neill, B.; Spengler, B.; Christoffel, V.; Fitzpatrick, J.M.; Watson, R.W. Apigenin drives the production of reactive species and initiates a mitochondrial mediated cell death pathway in prostate epithelial cells. Prostate 2005, 63, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Heideman, L.; Chung, C.S.; Pelling, K.J.; Koehler, D.F.; Birt, D.F. Cell cycle arrest at G2/M and growth inhibition by apigenin in human colon carcinoma cell lines. Mol. Carcinog. 2000, 28, 102–110. [Google Scholar] [CrossRef]
- Way, T.D.; Kao, M.C.; Lin, J.K. Apigenin induces apoptosis through proteasomal degradation of HER2/neu in HER/neu-overexpressing breast cancer cells via the phosphatidylinositol 3-kinase/Akt-dependent pathway. J. Biol. Chem. 2004, 279, 4479–4489. [Google Scholar] [CrossRef]
- Wang, I.K.; Lin-Shiau, J.K.; Lin, J.K. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukemia HL-60 cells. Eur. J. Cancer 1999, 35, 1517–1525. [Google Scholar] [CrossRef]
- Zheng, P.W.; Chiang, L.C.; Lin, L.L. Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci. 2005, 75, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Chie, Y.S.; Tan, T.W.; Wu, S.H.; Ma, Y.S.; Ip, S.W.; Chung, J.W. Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax- and Bcl-2-triggered mitochondrial pathway. Int. J. Oncol. 2010, 36, 1477–1484. [Google Scholar]
- Chiang, L.C.; Ng, N.T.; Lin, I.C.; Kuo, P.I.; Lin, C.C. Anti-proliferative effect of apigenin and its apoptotic induction in human Hep G2 cells. Cancer Lett. 2006, 237, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Jeong, C.S.; Cho, S.Y.; Lee, Y.S. Mechanism of apoptosis induced by apigenin in HepG2 human hepatoma cells: Involvement of reactive oxygen species generated by NADPH oxidase. Arch. Pharm. Res. 2007, 30, 1328–1335. [Google Scholar] [CrossRef]
- Gupta, S.; Afaq, F.; Mukhtar, H. Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 2002, 21, 3727–3738. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Molecular mechanisms for apigenin-induced cell-cycle arrest and apoptosis of hormone refractory human prostate carcinoma DU145 cells. Mol. Carcinog. 2004, 39, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic. Biol. Med. 2008, 44, 1833–1845. [Google Scholar] [CrossRef] [PubMed]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; et al. Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 2014, 6, 4615–4632. [Google Scholar]
- Yoshioka, Y.; Kadoi, H.; Yamamuro, A.; Ishimaru, Y.; Maeda, S. Noradrenaline increases intracellular glutathione in human astrocytoma U-251 MG cells by inducing glutamate-cysteine ligase protein via β3-adrenoceptor stimulation. Eur. J. Pharm. 2015, 772, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Calvani, M.; Cavallini, L.; Tondo, A.; Spinelli, V.; Ricci, L.; Pasha, A.; Bruno, G.; Buonvicino, D.; Bigagli, E.; Vignoli, M.; et al. β3-Adrenoreceptors Control Mitochondrial Dormancy in Melanoma and Embryionic Stem Cells. Oxid. Med. Cell Longev. 2018. [Google Scholar] [CrossRef]
- Warne, T.; Moukhametzianov, R.; Baker, J.G.; Nehmé, R.; Edwards, P.C.; Leslie, A.G.; Schertler, G.F.; Tate, C.G. The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature 2011, 469, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.; Choi, H.J.; Devree, B.T.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Zhou, S.; Starkov, A.; Froberg, M.K.; Leino, R.L.; Wallace, K.B. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001, 61, 771–777. [Google Scholar]
- Balentine, D.A.; Dwyer, J.T.; Erdman, J.W.; Ferruzzi, M.G.; Gaine, P.C.; Harnly, J.M.; Kwik Uribe, C.L. Recommendations on reporting requirements for flavonoids in research. Am. J. Clin. Nutr. 2015, 101, 1113–1125. [Google Scholar] [CrossRef]
- Jeyabal, P.V.; Syed, M.B.; Venkataraman, M.; Sambandham, J.K.; Sakthisekaran, D. Apigenin inhibits oxidative stress-induced macromolecular damage in N-nitrosodiethylamine (NDEA)-induced hepatocellular carcinogenesis in Wistar albino rats. Mol. Carcinog. 2005, 44, 11–20. [Google Scholar] [CrossRef]
- Shao, H.; Jing, K.; Mahmoud, E.; Huang, H.; Fang, X.; Yu, C. Apigenin sensitizes colon cancer cells to antitumor activity of ABT-263. Mol. Cancer Ther. 2013, 12, 2640–2650. [Google Scholar] [CrossRef]
- Salmani, J.M.M.; Zhang, X.P.; Jacob, J.A.; Chen, B.A. Apigenin’s anticancer properties and molecular mechanisms of action: Recent advances and future prospectives. Chin. J. Nat. Med. 2017, 15, 321–329. [Google Scholar] [CrossRef]
- Ganai, S.A. Plant-derived flavone apigenin: The small-molecule with promising activity against therapeutically resistant prostate cancer. Biomed. Pharmacother 2017, 85, 47–56. [Google Scholar] [CrossRef]
- Eghbaliferiz, S.; Iranshahi, M. Prooxidant Activity of Polyphenols, Flavonoids, Anthocyanins and Carotenoids: Updated Review of Mechanisms and Catalyzing Metals. Phytother. Res. 2016, 30, 1379–1391. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, Y.; Chang, W. Kinetic deoxyribose degradation assay and its application in assessing the antioxidant activities of phenolic compounds in a Fenton-type reaction system. Anal. Chim. Acta 2003, 478, 129–137. [Google Scholar] [CrossRef]
- Fukumoto, L.; Mazza, G. Assessing antioxidant and prooxidant activities of phenolic compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef]
- Perron, N.R.; Brumaghim, J.L. A Review of the Antioxidant Mechanisms of Polyphenol Compounds Related to Iron Binding. Cell Biochem. Biophys. 2009, 53, 75–100. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pènicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Klaus, S.; Seivert, A.; Boeuf, S. Effect of the β3-adrenergic agonist Cl316,243 on functional differentiation of white and brown adipocytes in primary cell culture. Biochim. Byophis. Acta 2001, 1539, 85–92. [Google Scholar] [CrossRef]
- Susulic, V.C.; Frederich, R.C.; Lawitts, J.; Tozzo, E.; Kahn, B.B.; Harper, M.E.; Himms-Hagen, J.; Flier, J.S.; Lowell, B.B. Targeted disruption of the beta 3-adrenergic receptor gene. J. Biol. Chem. 1995, 270, 29483–29492. [Google Scholar] [CrossRef] [PubMed]
- Atgiè, C.; D’Allaire, F.; Bukowiecki, L.J. Role of β1- and β3-adrenoceptors in the regulation of lipolysis and thermogenesis in rat brown adipocytes. Am. J. Physiol. 1997, 273, C1136–C1142. [Google Scholar] [CrossRef]
- Himms-Hagen, J.; Cui, J.; Danforth, E., Jr.; Taatjes, D.J.; Lang, S.S.; Waters, B.L.; Claus, T.H. Effect of CL-316,243, a thermogenic beta 3-agonist, on energy balance and brown and white adipose tissues in rats. Am. J. Physiol. 1994, 266, R1371–R1382. [Google Scholar]
- Ghorbani, M.; Claus, T.H.; Himms-Hagen, J. Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a β3-adrenoceptor agonist. Biochem. Pharmacol. 1997, 54, 121–131. [Google Scholar] [CrossRef]
- Hadi, T.; Douhard, R.; Dias, A.M.M.; Wendremaire, M.; Pezzè, M. Beta3 adrenergic receptor stimulation in human macrophages inhibits NADPHoxidase activity and induces catalase expression via PPARγ activation. Biochim. Biophys. Acta 2017, 1854, 1769–1784. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Andreeva, O.E. Apigenin Inhibits Growth of Breast Cancer Cells: The Role of ERα and HER2/neu. Acta Naturae 2015, 7, 133–139. [Google Scholar] [PubMed]
- Zhao, G.; Han, X.; Cheng, W.; Ni, J.; Zhang, Y.; Lin, J.; Song, Z. Apigenin inhibits proliferation and invasion, and induces apoptosis and cell cycle arrest in human melanoma cells. Oncol. Rep. 2017, 37, 2277–2285. [Google Scholar] [CrossRef]
- Ujiki, M.B.; Ding, X.Z.; Salabat, M.R.; Bentrem, D.J.; Golkar, L.; Milam, B.; Talamonti, M.S.; Bell, R.H.; Iwamura, T.; Adrian, T.E. Apigenin inhibits pancreatic cancer cell proliferation trought G2/M cell cycle arrest. Mol. Cancer 2006, 5, 76. [Google Scholar] [CrossRef]
- Meyer, H.; Bolarinwa, A.; Wolfram, G.; Linseisen, J. Bioavailability of Apigenin from Apiin-Rich Parsley in Humans. Ann. Nutr. Metab. 2006, 50, 167–172. [Google Scholar] [CrossRef]
- Schrödinger Suite Release 2018-2 ((a) Maestro v.11.6; (b) Epik, v.4.4; (c) Impact, v.7.9; (d) Prime, v.5.2; (e) Macromodel v.12.0. (f) Glide, v.7.9; (g) Jaguar, v.10.0.); Schrödinger, L.L.C.: New York, NY, USA, 2018.
- Nocentini, A.; Bonardi, A.; Gratteri, P.; Cerra, B.; Gioiello, A.; Supuran, C.T. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J. Enzyme Inhib. Med. Chem. 2018, 33, 1453–1459. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Allam, H.A.; Mahmoud, W.R.; Bonardi, A.; Nocentini, A.; Gratteri, P.; Ibrahim, E.S.; Abdel-Aziz, H.A.; Supuran, C.T. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: Selective inhibitors for the tumor-associated hCA IX isoform. Eur. J. Med. Chem. 2018, 152, 1–9. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A673 Cells | % Dead Cells | |||
|---|---|---|---|---|
| Early | Late | Necrotic | Total | |
| 0 | 2.59 | 4.95 | 5.36 | 12.09 |
| APIG 10 μM | 6.51 | 8.36 | 8.56 | 23.43 |
| APIG 20 μM | 7.89 | 9.70 | 8.11 | 25.70 |
| APIG 50 μM | 19.00 | 16.3 | 3.68 | 38.68 |
| Lymphocytes | ||||
| 0 | 4.26 | 1.35 | 2.02 | 7.63 |
| APIG 10 μM | 5.66 | 2.42 | 2.28 | 10.36 |
| APIG 20 μM | 5.74 | 2.23 | 2.05 | 10.02 |
| APIG 50 μM | 7.04 | 3.22 | 7.01 | 17.27 |
| A673 Cells | % Live Cells |
|---|---|
| 0 | 87.20 |
| APIG 10 μM | 78.88 |
| APIG 20 μM | 76.90 |
| APIG 50 μM | 65.90 |
| Lymphocytes | |
| 0 | 92.70 |
| APIG 10 μM | 92.14 |
| APIG 20 μM | 93.41 |
| APIG 50 μM | 81.95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasha, A.; Vignoli, M.; Subbiani, A.; Nocentini, A.; Selleri, S.; Gratteri, P.; Dabraio, A.; Casini, T.; Filippi, L.; Fotzi, I.; et al. β3-Adrenoreceptor Activity Limits Apigenin Efficacy in Ewing Sarcoma Cells: A Dual Approach to Prevent Cell Survival. Int. J. Mol. Sci. 2019, 20, 2149. https://doi.org/10.3390/ijms20092149
Pasha A, Vignoli M, Subbiani A, Nocentini A, Selleri S, Gratteri P, Dabraio A, Casini T, Filippi L, Fotzi I, et al. β3-Adrenoreceptor Activity Limits Apigenin Efficacy in Ewing Sarcoma Cells: A Dual Approach to Prevent Cell Survival. International Journal of Molecular Sciences. 2019; 20(9):2149. https://doi.org/10.3390/ijms20092149
Chicago/Turabian StylePasha, Amada, Marina Vignoli, Angela Subbiani, Alessio Nocentini, Silvia Selleri, Paola Gratteri, Annalisa Dabraio, Tommaso Casini, Luca Filippi, Ilaria Fotzi, and et al. 2019. "β3-Adrenoreceptor Activity Limits Apigenin Efficacy in Ewing Sarcoma Cells: A Dual Approach to Prevent Cell Survival" International Journal of Molecular Sciences 20, no. 9: 2149. https://doi.org/10.3390/ijms20092149
APA StylePasha, A., Vignoli, M., Subbiani, A., Nocentini, A., Selleri, S., Gratteri, P., Dabraio, A., Casini, T., Filippi, L., Fotzi, I., Favre, C., & Calvani, M. (2019). β3-Adrenoreceptor Activity Limits Apigenin Efficacy in Ewing Sarcoma Cells: A Dual Approach to Prevent Cell Survival. International Journal of Molecular Sciences, 20(9), 2149. https://doi.org/10.3390/ijms20092149