Oxidative Stress: A Key Modulator in Neurodegenerative Diseases



Abstract
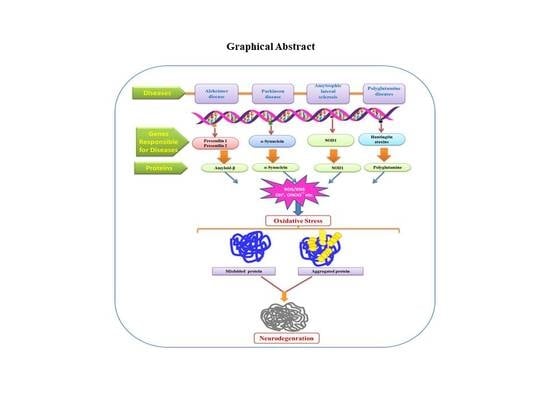
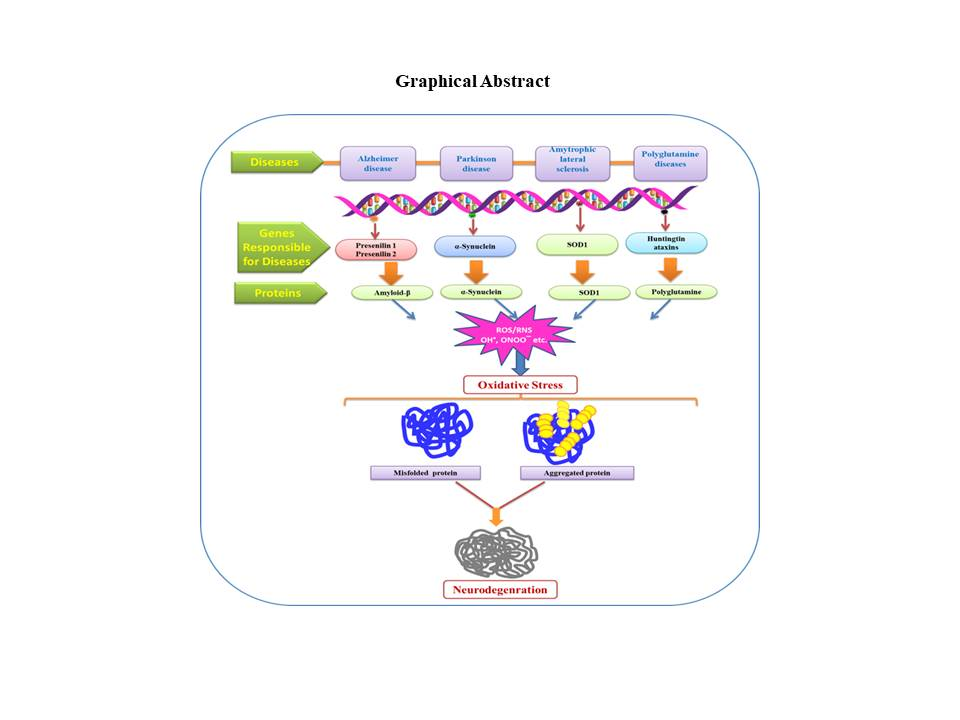{kind=link}
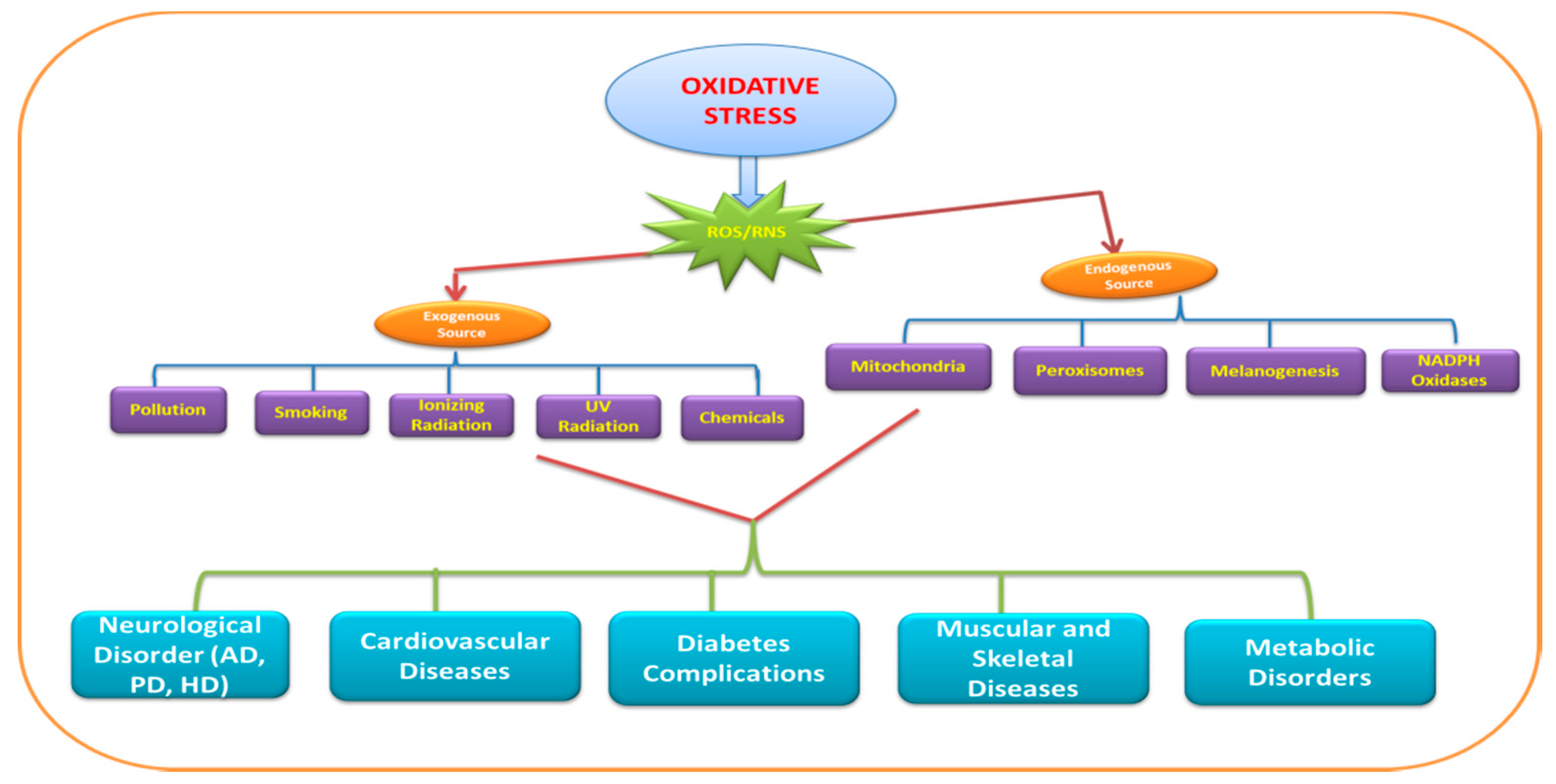{kind=link}
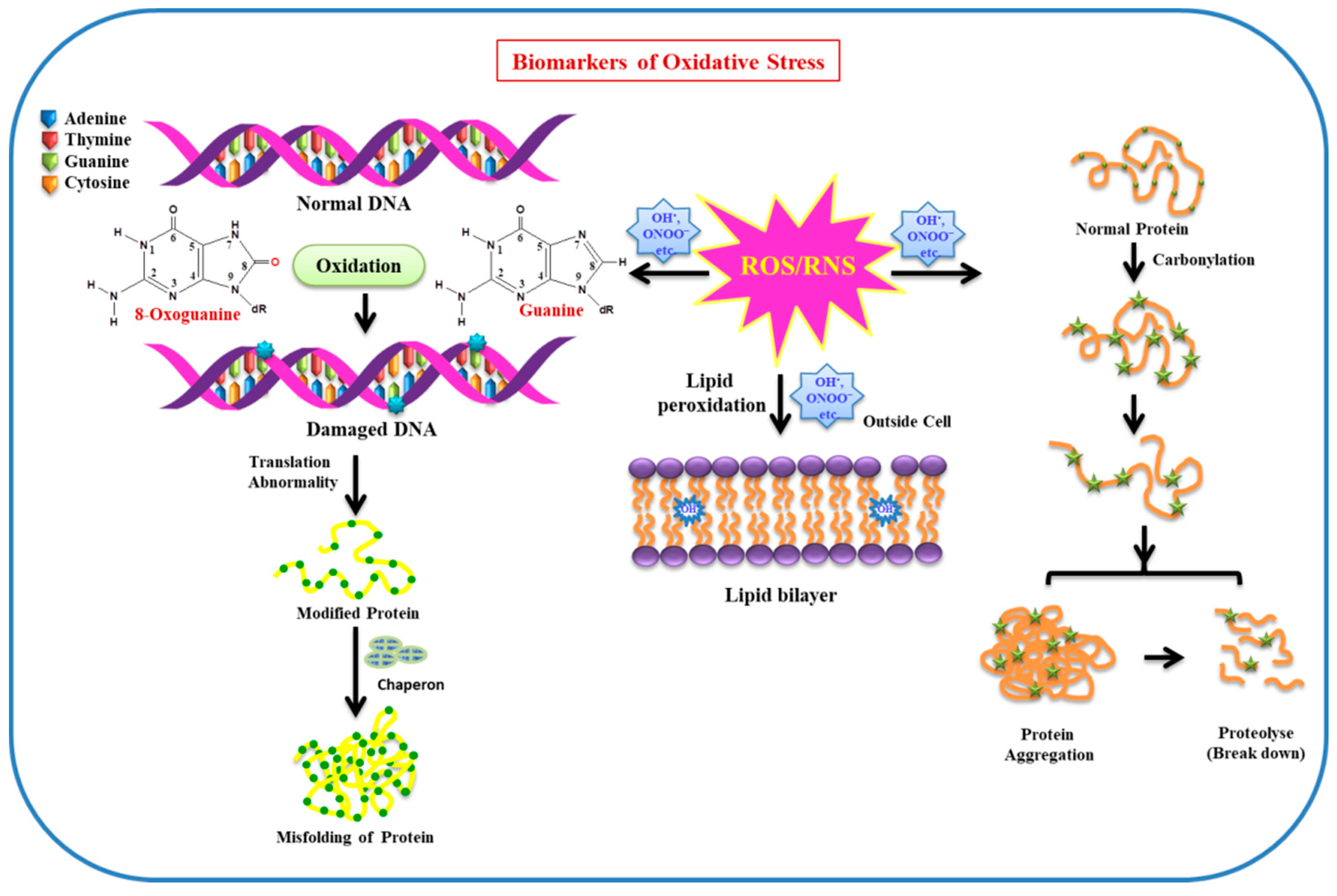{kind=link}
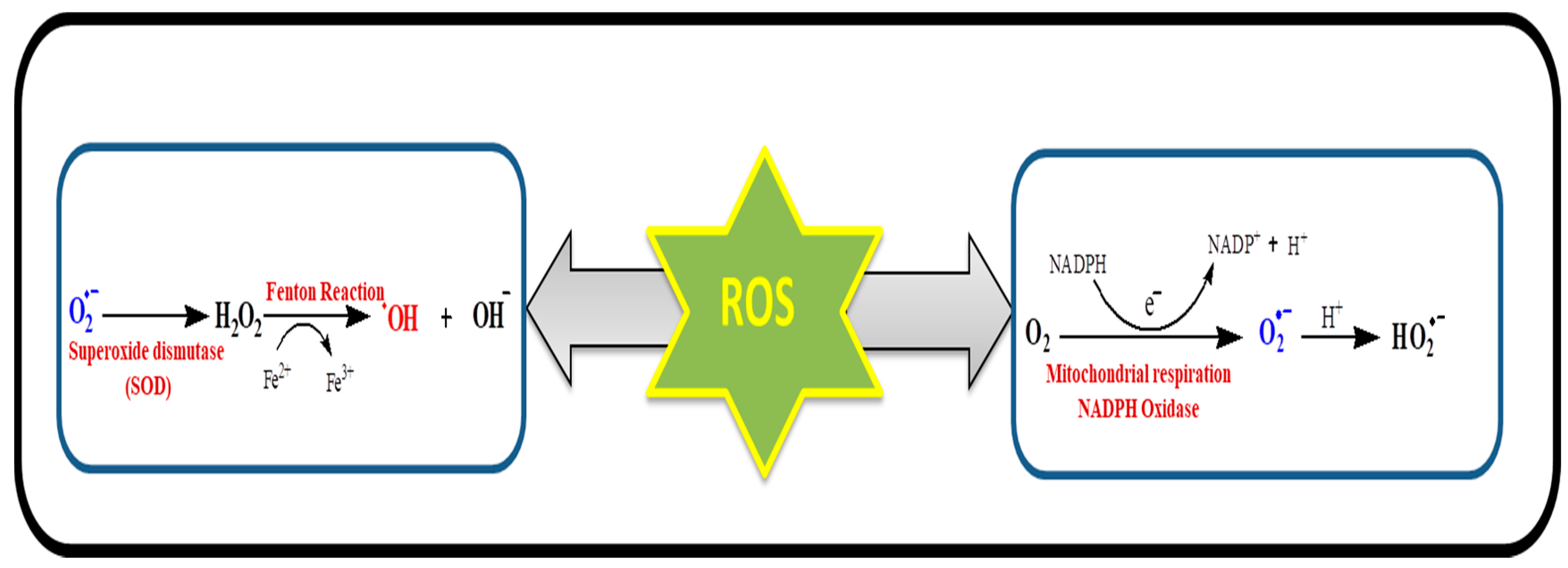{kind=link}
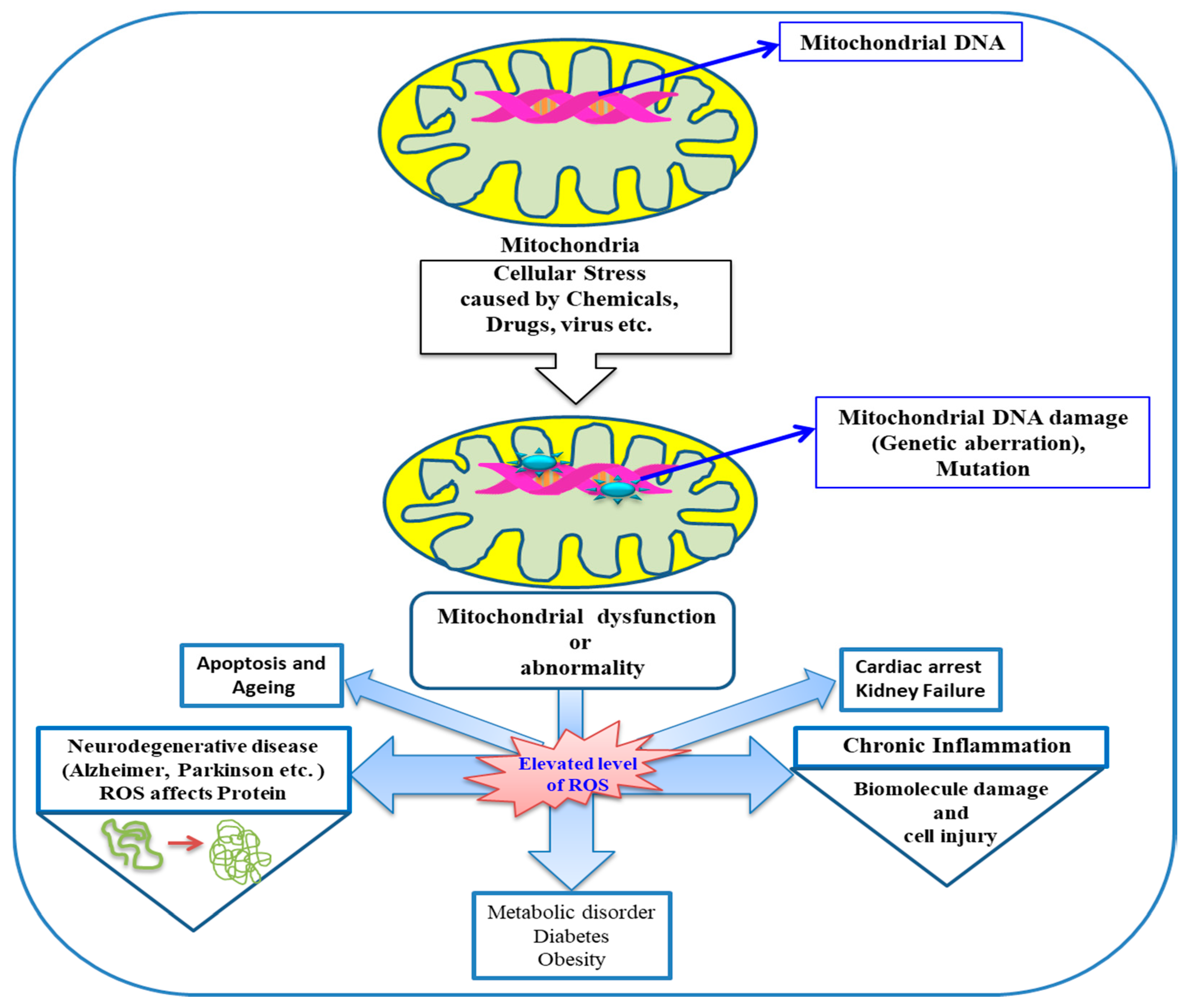{kind=link}
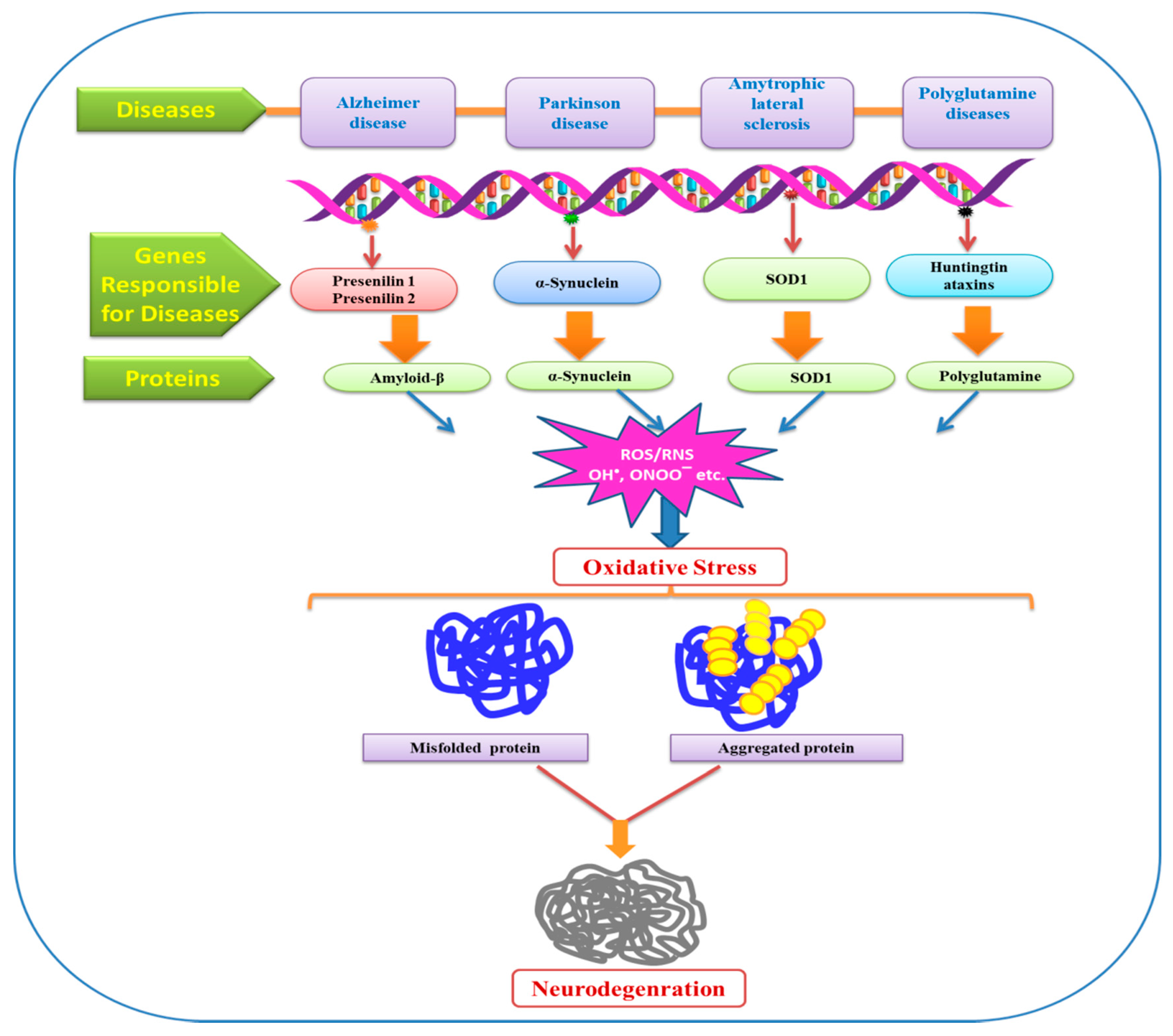{kind=link}
1. Introduction
2. Sensor and Biomarkers of Oxidative Stress
3. Mitochondria and Oxidative Stress
4. Role of Pro-Oxidants in Oxidative Stress
5. Role of Heavy Metals in Oxidative Stress
6. Concept of Ferroptosis and its Implications for Neurodegeneration
7. Role of Oxidative Stress Signaling in Senescence versus Apoptosis
8. Oxidative Stress: Modulator of Neurodegenerative Diseases
9. Alzheimer’s Disease (AD)
10. Parkinson’s Disease (PD)
11. Amyotrophic Lateral Sclerosis (ALS)
12. Huntington Disease (HD)
13. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Chiurchiù, V.; Orlacchio, A.; Maccarrone, M. Is Modulation of Oxidative Stress an Answer? The State of the Art of Redox Therapeutic Actions in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Storz, G. Redox sensing by prokaryotic transcription factors. Biochem. Pharmacol. 2000, 59, 1–6. [Google Scholar] [CrossRef]
- Aikens, J.; Dix, T.A. Perhydroxyl radical (HOO) initiated lipid peroxidation: The role of fatty acid hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098. [Google Scholar] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; MacCarrone, M. Chronic inflammatory disordersandtheirredox control from molecular mechanisms totherapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 2605–2641. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Harvey, L.; Arnold, B.; Lawrence, Z.; Paul, M.; David, B. Molecular Cell Biology, 4th ed.; Publisher W.H. Freeman & Co. Ltd.: San Francisco, CA, USA, 1999; pp. 197–433. [Google Scholar]
- Guido, K.; John, C.R. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar]
- Uday, B.; Dipak, D.; Ranajit, B.K. Reactive oxygen species: Oxidative damage and pathogenesis. Curr. Sci. 1990, 77, 658–666. [Google Scholar]
- Poulson, H.E.; Prieme, H.; Loft, S. Role of oxidative DNA damage in cancer initiation and promotion. Eur. J. Cancer Prev. 1998, 7, 9–16. [Google Scholar]
- Yun-Zhong, F.; Sheng, Y.; Guoyao, W. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar]
- von Arnim, C.A.F.; Gola, U.; Biesalski, H.K. More than thesumofitsparts? Nutritionin Alzheimer’s disease. Nutrition 2010, 26, 694–700. [Google Scholar] [CrossRef]
- El-Bach’a, R.S.; De-Lima-Filho, J.L.; Guedes, R.C.A. Dietary antioxidant deficiency facilitates cortical spreading depression induced by photoactivated riboflavin. Nutr. Neurosci. 1998, 1, 205–212. [Google Scholar] [CrossRef]
- Mandel, S.; Grünblatt, E.; Riederer, P.; Gerlach, M.; Levites, Y.; Youdim, M.B.H. Neuroprotective strategies in Parkinson’s disease: An update on progress. CNS Drugs 2003, 17, 729–762. [Google Scholar] [CrossRef]
- Yu, Y.C.; Kuo, C.L.; Cheng, W.L.; Liu, C.S.; Hsieh, M. Decreased antioxidant enzyme activity and increased mitochondrial DNA damage in cellular models of Machado Joseph disease. J. Neurosci. Res. 2009, 87, 1884–1891. [Google Scholar] [CrossRef]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef]
- Brown, H.A.; Murphy, R.C. Working towards an exegesis for lipids in biology. Nat. Chem. Biol. 2009, 5, 602–606. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Munshi, S.; Banerjee, K.; Thakurta, I.G.; Sinha, M.; Bagh, M.B. Mitochondrial Dysfunction during Brain Aging: Role of Oxidative Stress and Modulation by Antioxidant Supplementation. Aging Dis. 2011, 2, 242–256. [Google Scholar]
- Federico, A.; Cardaioli, E.; da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Junhwan, K.; Paul, E.M.; Robert, G.S.; Vernon, E.A.; Charles, L.H. Cardiolipin: Characterization of distinct oxidized molecular species. J. Lipid Res. 2011, 52, 125–135. [Google Scholar]
- Sen, T.; Sen, N.; Jana, S.; Khan, F.H.; Chatterjee, U.; Chakrabarti, S. Depolarization and cardiolipin depletion in aged rat brain mitochondria: Relationship with oxidative stress and electron transport chain activity. Neurochem. Int. 2007, 50, 719–725. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive Oxygen Species and the Central Nervous System. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s Disease. Front. Aging Neurosci. 2010, 2, 1–11. [Google Scholar] [CrossRef]
- Mecocci, P.; Beal, M.F.; Cecchetti, R.; Polidori, M.C.; Cherubini, A.; Chionne, F.; Avellini, L.; Romano, G.; Senin, U. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Mol. Chem. Neuropathol. 1997, 31, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Lin, M.T.; Zheng, L.; Liu, G.J.; Ahn, C.H.; Kim, L.M.; Mauck, W.M.; Twu, F.; Beal, M.F.; Johns, D.R. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiol. Aging 2004, 25, 71–81. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef]
- Tian, F.; Tong, T.J.; Zhang, Z.Y.; McNutt, M.A.; Liu, X.W. Age-dependent down-regulation of mitochondrial 8-oxoguanine DNA glycosylase in SAM-P/8 mouse brain and its effect on brain aging. Rejuvenation Res. 2009, 12, 209–215. [Google Scholar] [CrossRef]
- Imam, S.Z.; Karahalil, B.; Hogue, B.A.; Souza-Pinto, N.C.; Bohr, V.A. Mitochondrial and nuclear DNArepair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol. Aging 2006, 27, 1129–1136. [Google Scholar] [CrossRef]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch. Biochem. Biophys. 2008, 476, 107–112. [Google Scholar] [CrossRef]
- Chobot, V.; Hadacek, F. Exploration of pro-oxidant and antioxidant activities of the flavonoid myricetin. Redox Rep. 2011, 16, 242–247. [Google Scholar] [CrossRef]
- Ramasamy, C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets. Eur. J. Pharmacol. 2006, 545, 51–64. [Google Scholar] [CrossRef]
- Ono, K.; Hamaguchi, T.; Naiki, H.; Yamada, M. Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim. Biophys. Acta 2006, 1762, 575–586. [Google Scholar] [CrossRef]
- Seo, M.Y.; Lee, S.M. Protective effect to flow dose of ascorbic acid on hepatobiliary function in hepatic ischemia/repersfusioninrats. J. Hepatol. 2002, 36, 72–77. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative Stress, Prooxidants, and Antioxidants: The Interplay. BioMed Res. Int. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental Pollutants as risk factors for neurodegenerative disorder: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, N.; Pandey, R.; Gupta, V.K.; Sharma, B. Biochemical and molecular targets of heavy metals and their actions. In Biomedical Applications of Metals; Rai, M., Ingle, A., Medici, S., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Singh, N.; Gupta, V.K.; Kumar, A.; Sharma, B. Synergistic effects of heavy metals and pesticides in living systems. Front. Chem. 2017, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Risher, J.F.; Amler, S.N. Mercury exposure: Evaluation and intervention the inappropriate use of chelating agents in the diagnosis and treatment of putative mercury poisoning. Neurotoxicology 2005, 26, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Ayensu, W.K.; Ninashvili, N.; Sutton, D. Environmental exposure to mercury and its toxicopathologic implications for public health. Environ. Toxicol. 2003, 18, 149–175. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Kumar, A.; Gupta, V.K.; Sharma, B. Biochemical and molecular bases of lead-induced toxicity in mammalian systems and possible mitigations. Chem. Res. Toxicol. 2018, 31, 1009–1021. [Google Scholar] [CrossRef]
- Ahamed, M.; Siddiqui, M.K.J. Low level lead exposure andoxidative stress: Current opinions. Clin. Chim. Acta 2007, 383, 57–64. [Google Scholar] [CrossRef]
- Sun, H.J.; Rathinasabapathi, B.; Wu, B.; Luo, J.; Pu, L.P.; Ma, L.Q. Arsenic and selenium toxicity and their interactive effects in humans. Environ. Int. 2014, 69, 148–158. [Google Scholar] [CrossRef]
- Sharma, B.; Singh, S.; Siddiqi, N.J. Biomedical implications of heavy metals induced imbalances in redox systems. Biomed. Res. Int. 2014, 2014, 640754. [Google Scholar] [CrossRef]
- Salazar-Flores, J.; Torres-Jasso, J.H.; Rojas-Bravo, D.; Reyna-Villela, Z.M.; Torres-Sánchez, E.D. Effects of Mercury, Lead, Arsenic and Zinc to Human Renal Oxidative Stress and Functions: A Review. J. Heavy Met. Toxic. Dis. 2018, 4, 1–16. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 2018, 8, 574. [Google Scholar] [CrossRef]
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2019, 1–14. [Google Scholar] [CrossRef]
- Wu, Y.; Song, J.; Wang, Y.; Wang, X.; Culmsee, C.; Zhu, C. The Potential Role of Ferroptosis in Neonatal Brain Injury. Front. Neurosci. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Raychaudhuri, S. A minimal model of signaling network elucidates cell-to-cell stochastic variability in apoptosis. PLoS ONE 2010, 5, e11930. [Google Scholar] [CrossRef] [PubMed]
- Colavitti, R.; Finkel, T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, D.; Xiao, H. Methods of cellular senescence induction using oxidative stress. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.C.; Hutchison, E.R.; Eitan, E.; Mattson, M.P. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 2014, 15, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Choudhury, A.R. Reactive Oxygen Species (ROS) and responses of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 1–13. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.P.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Liu, J.; Wang, J.; Lee, S.; Wen, R. Copper-caused oxidative stress triggers the activation of antioxidant enzymes via ZmMPK3 in maize leaves. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Berg, D.; Youdim, M.B.; Riederer, P. Redox imbalance. Cell Tissue Res. 2004, 318, 201–213. [Google Scholar] [CrossRef]
- McQuillen, P.S.; Ferriero, D.M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Rougemont, M.; Do, K.Q.; Castagné, V. New model of glutathione deficit during development: Effect on lipid peroxidation in the rat brain. J. Neurosci. Res. 2002, 70, 774–783. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Hritcu, L.; Ciobica, A.; Artenie, V. Effects of right-unilateral 6-hydroxydopamine infusion-induced memory impairment and oxidative stress: Relevance for Parkinson’s disease. Cent. Eur. J. Biol. 2008, 3, 250–257. [Google Scholar] [CrossRef]
- Smith, M.A. Oxidative stress and iron imbalance in Alzheimer disease: How rust became the fuss! J. Alzheimers Dis. 2006, 9, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Piroddi, M.; Galli, F.; Butterfield, D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res. 2008, 33, 2540–2546. [Google Scholar] [CrossRef]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Pratico, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 316523. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Sinha, M.; Bhowmick, P.; Banerjee, A.; Chakrabarti, S. Antioxidant role of amyloid Î2 protein in cell-free and biological systems: Implication for the pathogenesis of Alzheimer disease. Free Radic. Biol. Med. 2013, 56, 184–192. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Craddock, T.J.; Tuszynski, J.A.; Chopra, D.; Casey, N.; Goldstein, L.E.; Hameroff, S.R.; Tanzi, R.E. The zinc dyshomeostasis hypothesis of Alzheimer’s disease. PLoS ONE 2012, 7, e33552. [Google Scholar] [CrossRef]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Fagét, K.Y. Zinc takes the center stage: Its paradoxical role in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2003, 41, 44–56. [Google Scholar] [CrossRef]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease (Review). Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Endrach, M.; Leuner, K.; Eckert, A.; Müller, W. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S265–S279. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [PubMed]
- Lüth, H.; Münch, G.; Arendt, T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002, 953, 135–143. [Google Scholar] [CrossRef]
- Lüth, H.; Holzer, M.; Gärtner, U.; Staufenbiel, M.; Arendt, T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: Evidence for an induction by amyloid pathology. Brain Res. 2001, 913, 57–67. [Google Scholar] [CrossRef]
- Massaad, C.A. Neuronal and vascular oxidative stress in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef]
- Toda, N.; Ayajiki, K.; Okamura, T. Cerebral blood flow regulation by nitric oxide in neurological disorders. Can. J. Physiol. Pharmacol. 2009, 87, 581–594. [Google Scholar] [CrossRef]
- Liu, G.; Men, P.; Perry, G.; Smith, M.A. Nanoparticle and iron chelators as a potential novel Alzheimer therapy. Methods Mol. Biol. 2010, 610, 123–144. [Google Scholar] [PubMed]
- Albert, M.S.; Dekosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association work groups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Krüger, R. Converging environmental and genetic pathways in the pathogenesis of Parkinson’s disease. J. Neurol. Sci. 2011, 306, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P., Jr.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synucleinfibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Razmpour, R.; Bernard, L.P. Glutathione and Parkinson’s disease: Is this the elephant in the room? Biomed. Pharmacother. 2008, 62, 236–249. [Google Scholar] [CrossRef]
- Lesly, P.; Sun, Y.C.; Jae-won, S. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53–64. [Google Scholar]
- Hemmerle, A.M.; Herman, J.P.; Seroogy, K.B. Stress, depression and Parkinson’s disease. Exp. Neurol. 2012, 233, 79–86. [Google Scholar] [CrossRef]
- Zatta, P.; Lucchini, R.; Van Rensburg, S.J.; Taylor, A. The role of metals in neurodegenerative processes: Aluminum, manganese, and zinc. Brain Res. Bull. 2003, 62, 15–28. [Google Scholar] [CrossRef]
- Torres-Vega, A.; Pliego-Rivero, B.F.; Otero-Ojeda, G.A.; Gómez-Oliván, L.M.; VieyraReyes, P. Limbic system pathologies associated with deficiencies and excesses of the trace elements iron, zinc, copper, and selenium. Nutr. Rev. 2012, 70, 679–692. [Google Scholar] [CrossRef]
- Tieu, K.; Ischiropoulos, H.; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar] [CrossRef]
- Hunot, S.; Boissiers, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Eve, D.J.N.A.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Di Fonzo, A.; Rohe, C.F.; Ferreira, J.; Chien, H.F.; Vacca, L.; Stocchi, F.; Fabrizio, E.; Manfredi, M.; Vanacore, N.; Goldwurm, S.; et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 2005, 365, 412–415. [Google Scholar] [CrossRef]
- Nichols, W.C.; Pankratz, N.; Hernandez, D.; Paisan-Ruiz, C.; Jain, S.; Halter, C.A.; Michaels, V.E.; Reed, T.; Rudolph, A.; Shults, C.W.; et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005, 365, 410–412. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin, and mitochondrial Fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Canet-Avilés Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R.R.M. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Gonçalves, S.; Tenreiro, S.; Rosado Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with α-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Shinpo, K.; Ogata, A.; Tsuji, S.; Takeuchi, M.; Makita, Z.; Tashiro, K. Detection of N epsilon–(carboxymethyl)lysine (CML) and non–CML advanced glycation end–products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotroph. Lateral Scler Other Motor Neuron Disord. 2002, 3, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Mendez, E.F.; Sattler, R. BiomarkerdevelopmentforC9orf72 repeat expansion in ALS. Brain Res. 2015, 1607, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.K.; Jha, S.K.; Kar, R.; Ambasta, R.K.; Kumar, P. Role of oxidative stress, ER stress and ubiquitin proteasome system in neurodegeneration. MOJ Cell Sci. Rep. 2014, 1, 38–44. [Google Scholar]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study GroupII. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (phase II study). Amyotrop. Later. Sclerosis Off. Public World Fed. Neurol. Res. Group Motor Neuron Dis. 2006, 7, 241–245. [Google Scholar] [CrossRef]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.M.; Meyjes, F.E.P.; De Jong, J.M.B.V. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neuro Biol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.A.; Saccon, R.A.; Fratta, P.; Fisher, E.M.C. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Bosch, L.V.D.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Weston, W. Unifying Mechanism for Multiple Sclerosis and Amyotrophic Lateral Sclerosis: Reactive Oxygen Species, Oxidative Stress, and Antioxidants. J. Biopharm. Ther. Chal. 2018, 2, 1–8. [Google Scholar]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef]
- Folstein, S.; Abbott, M.H.; Chase, G.A.; Jensen, B.A.; Folstein, M.F. The association of affective disorder with Huntington’s disease in a case series and in families. Psychol. Med. 1983, 13, 537–542. [Google Scholar] [CrossRef]
- Li, S.H.; Li, X.J. Huntingtin and its role in neuronal degeneration. Neuroscientist 2004, 10, 467–475. [Google Scholar] [CrossRef]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Tunez, I.; Sanchez-Lopez, F.; Agüera, E.; Fernández-Bolaños, R.; Sanchez, F.M.; Tasset-Cuevas, I. Important Role of Oxidative Stress Biomarkers in Huntington’s disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Edamura, K.N.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s Disease. Biochim. Biophys. Acta 2012, 1822, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ratana, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Garg, V.K.; Singh, A.K.; Kumar, T. Role of free radicals and certain antioxidants in the management of huntington’s disease: A review. J. Anal. Pharm. Res. 2018, 7, 386–392. [Google Scholar] [CrossRef]
- Zheng, J.; Windericks, J.; Franssens, V.; Liu, B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s disease. Front. Mol. Neurosci. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Purushothuman, S. Oxidative stress in neurodegenerative conditions and the protective potential of a natural antioxidant, dietary saffron. Oxid. Antioxid. Med. Sci. 2015, 4, 1–7. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. https://doi.org/10.3390/molecules24081583
Singh A, Kukreti R, Saso L, Kukreti S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019; 24(8):1583. https://doi.org/10.3390/molecules24081583
Chicago/Turabian StyleSingh, Anju, Ritushree Kukreti, Luciano Saso, and Shrikant Kukreti. 2019. "Oxidative Stress: A Key Modulator in Neurodegenerative Diseases" Molecules 24, no. 8: 1583. https://doi.org/10.3390/molecules24081583
APA StyleSingh, A., Kukreti, R., Saso, L., & Kukreti, S. (2019). Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules, 24(8), 1583. https://doi.org/10.3390/molecules24081583