The Complex Interplay between Metabolic Reprogramming and Epigenetic Alterations in Renal Cell Carcinoma




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
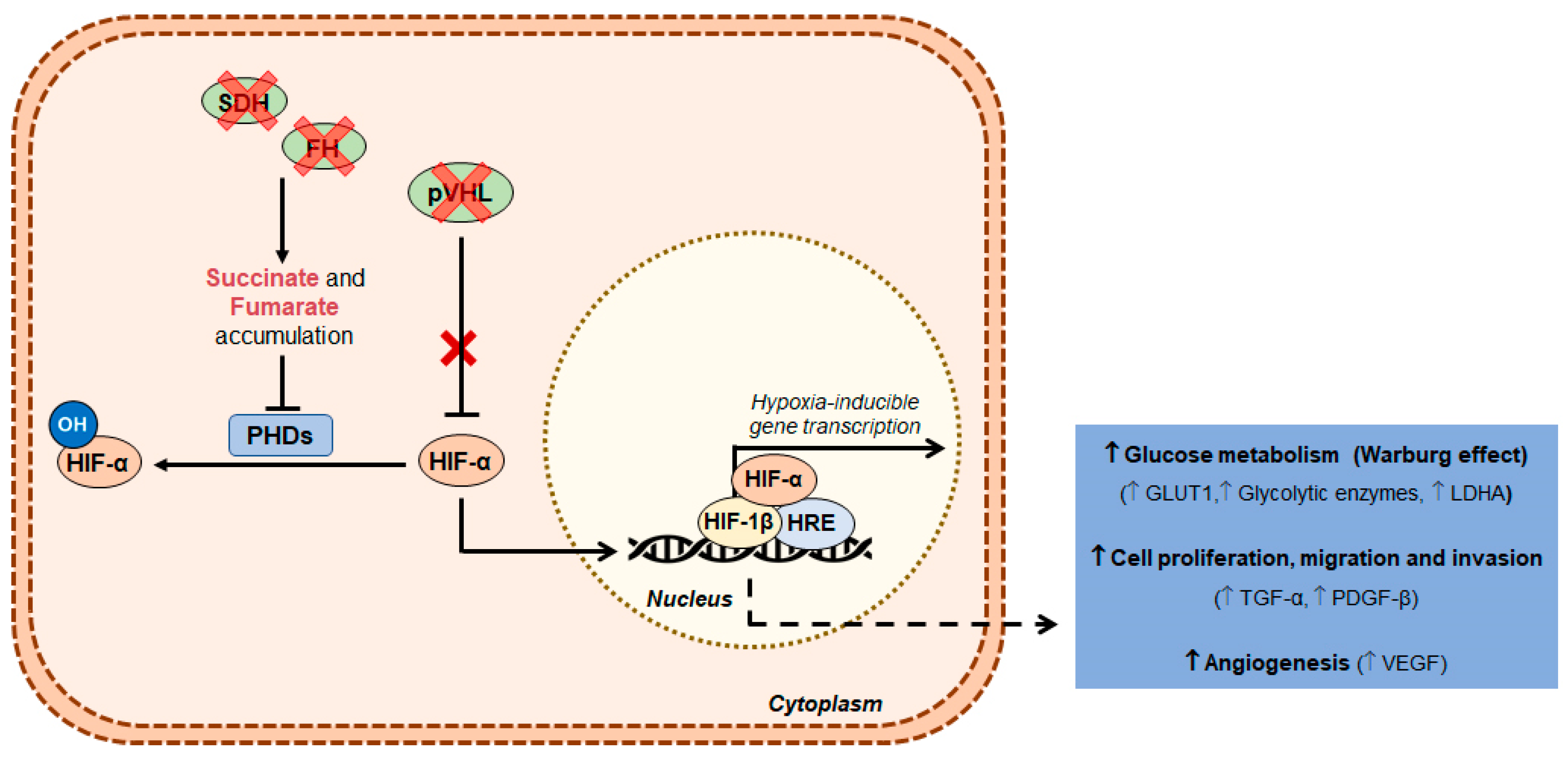
2. Clear Cell Renal Cell Carcinoma
3. Papillary Renal Cell Carcinoma
4. Tumor Metabolism
4.1. Warburg Effect
4.2. Metabolic Players in Renal Cell Carcinoma
4.3. Role of Lactate in Tumor Microenvironment
5. Epigenetic Mechanisms
5.1. DNA Methylation
5.2. Histone Post-Translational Modifications
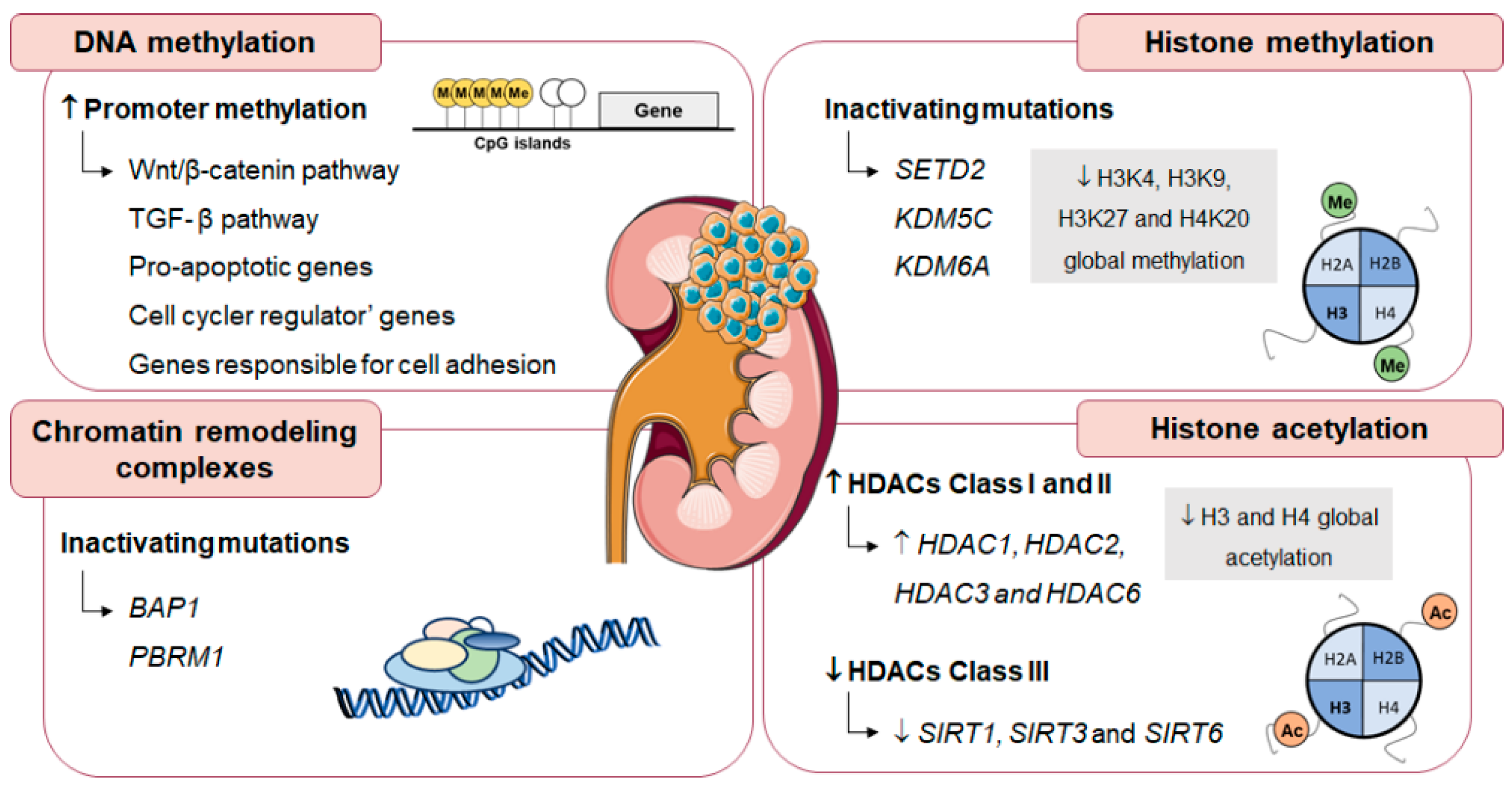
5.3. Epigenetic Alterations in Renal Cell Carcinoma
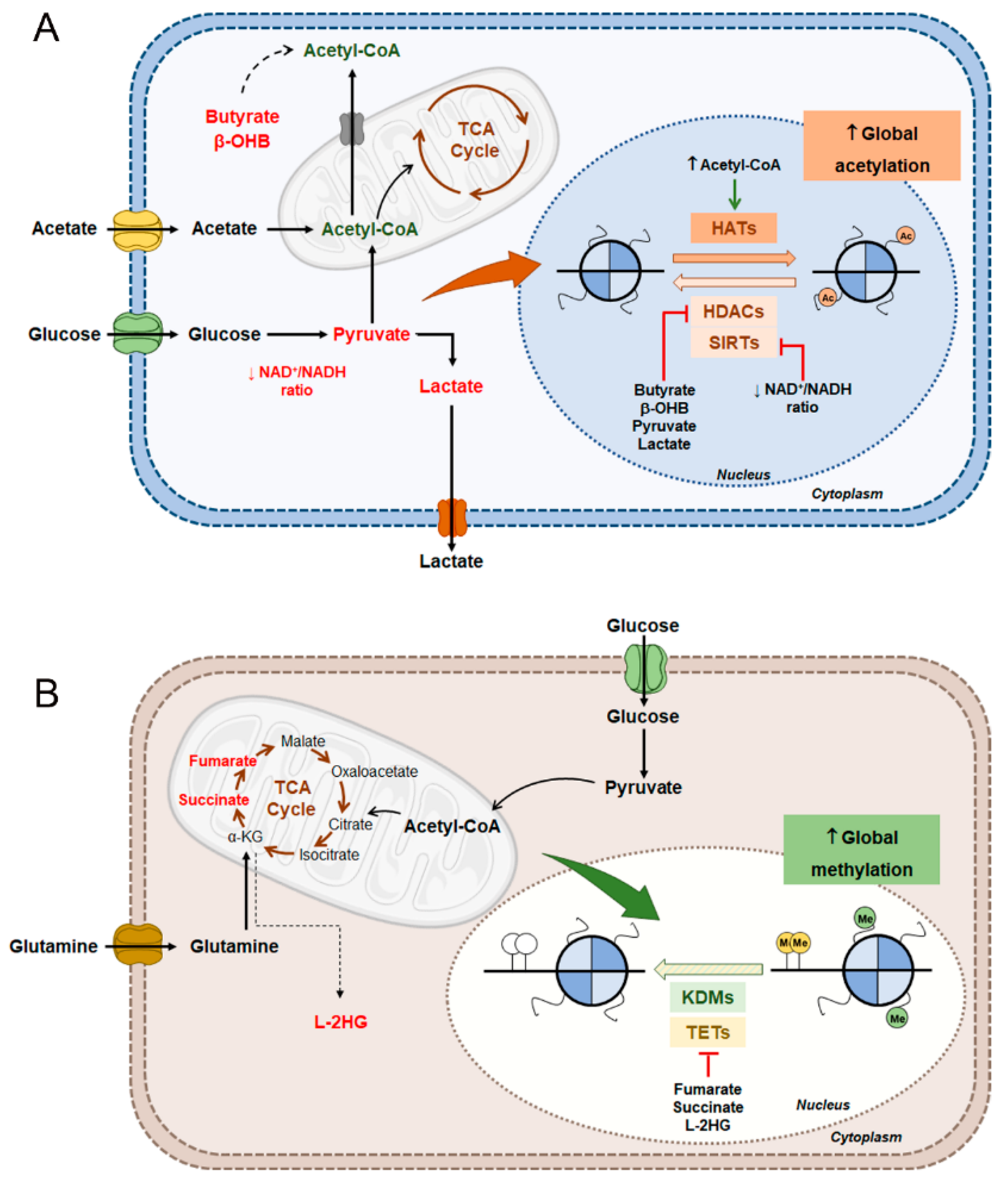
6. Interplay Between Metabolism and Epigenetic Mechanisms
6.1. Renal Cell Carcinoma Metabolism and Histone Acetylation Modulation
6.2. Renal Cell Carcinoma Metabolism in DNA and Histone Methylation Modulation
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef]
- Ljungberg, B.; Campbell, S.C.; Choi, H.Y.; Jacqmin, D.; Lee, J.E.; Weikert, S.; Kiemeney, L.A. The epidemiology of renal cell carcinoma. Eur. Urol. 2011, 60, 615–621. [Google Scholar] [CrossRef]
- Dhote, R.; Thiounn, N.; Debre, B.; Vidal-Trecan, G. Risk factors for adult renal cell carcinoma. Urol. Clin. North Am. 2004, 31, 237–247. [Google Scholar] [CrossRef]
- Qayyum, T.; Oades, G.; Horgan, P.; Aitchison, M.; Edwards, J. The epidemiology and risk factors for renal cancer. Curr. Urol. 2012, 6, 169–174. [Google Scholar] [CrossRef]
- Lucca, I.; Klatte, T.; Fajkovic, H.; de Martino, M.; Shariat, S.F. Gender differences in incidence and outcomes of urothelial and kidney cancer. Nat. Rev. Urol. 2015, 12, 585–592. [Google Scholar] [CrossRef]
- Clague, J.; Lin, J.; Cassidy, A.; Matin, S.; Tannir, N.M.; Tamboli, P.; Wood, C.G.; Wu, X. Family history and risk of renal cell carcinoma: Results from a case-control study and systematic meta-analysis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 801–807. [Google Scholar] [CrossRef]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Gruenvald, V.; Horwich, A.; Committee, E.G. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v58–v68. [Google Scholar] [CrossRef] [PubMed]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Linehan, W.M. Intratumoral heterogeneity in kidney cancer. Nat. Genet. 2014, 46, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Haas, N.B.; Nathanson, K.L. Hereditary kidney cancer syndromes. Adv. Chronic Kidney Dis. 2014, 21, 81–90. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Banks, R.E.; Tirukonda, P.; Taylor, C.; Hornigold, N.; Astuti, D.; Cohen, D.; Maher, E.R.; Stanley, A.J.; Harnden, P.; Joyce, A.; et al. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006, 66, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Shen, C.; Kaelin, W.G., Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 2013, 23, 18–25. [Google Scholar] [CrossRef]
- Schodel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef]
- Courthod, G.; Tucci, M.; Di Maio, M.; Scagliotti, G.V. Papillary renal cell carcinoma: A review of the current therapeutic landscape. Crit. Rev. Oncol. Hematol. 2015, 96, 100–112. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod. Pathol. 1997, 10, 537–544. [Google Scholar]
- Albiges, L.; Guegan, J.; Le Formal, A.; Verkarre, V.; Rioux-Leclercq, N.; Sibony, M.; Bernhard, J.C.; Camparo, P.; Merabet, Z.; Molinie, V.; et al. MET is a potential target across all papillary renal cell carcinomas: Result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin. Cancer Res. 2014, 20, 3411–3421. [Google Scholar] [CrossRef]
- Lubensky, I.A.; Schmidt, L.; Zhuang, Z.; Weirich, G.; Pack, S.; Zambrano, N.; Walther, M.M.; Choyke, P.; Linehan, W.M.; Zbar, B. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am. J. Pathol. 1999, 155, 517–526. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Haas, N.B.; Nathanson, K.L. Hereditary Renal Cancer Syndromes. Adv. Chronic Kidney Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, B.; Bottaro, D.P. Targeting the c-Met signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 3657–3660. [Google Scholar] [CrossRef]
- Farber, L.J.; Furge, K.; Teh, B.T. Renal cell carcinoma deep sequencing: Recent developments. Curr. Oncol. Rep. 2012, 14, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Sourbier, C.; Wei, M.H.; Shuch, B.; Pike, L.; Wu, M.; Rouault, T.A.; Boros, L.G.; et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE 2013, 8, e72179. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- House, S.W.; Warburg, O.; Burk, D.; Schade, A.L. On respiratory impairment in cancer cells. Science 1956, 124, 267–272. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. 2), 24S–42S. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Sudarshan, S.; Karam, J.A.; Brugarolas, J.; Thompson, R.H.; Uzzo, R.; Rini, B.; Margulis, V.; Patard, J.J.; Escudier, B.; Linehan, W.M. Metabolism of kidney cancer: From the lab to clinical practice. Eur. Urol. 2013, 63, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 2007, 39, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Soltysova, A.; Breza, J.; Takacova, M.; Feruszova, J.; Hudecova, S.; Novotna, B.; Rozborilova, E.; Pastorekova, S.; Kadasi, L.; Krizanova, O. Deregulation of energetic metabolism in the clear cell renal cell carcinoma: A multiple pathway analysis based on microarray profiling. Int. J. Oncol. 2015, 47, 287–295. [Google Scholar] [CrossRef]
- Sanders, E.; Diehl, S. Analysis and interpretation of transcriptomic data obtained from extended Warburg effect genes in patients with clear cell renal cell carcinoma. Oncoscience 2015, 2, 151. [Google Scholar] [CrossRef]
- Lim, H.Y.; Yip, Y.M.; Chiong, E.; Tiong, H.Y.; Halliwell, B.; Esuvaranathan, K.; Wong, K.P. Metabolic signatures of renal cell carcinoma. Biochem. Biophys. Res. Commun. 2015, 460, 938–943. [Google Scholar] [CrossRef]
- Van der Mijn, J.C.; Panka, D.J.; Geissler, A.K.; Verheul, H.M.; Mier, J.W. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. 2016, 4, 14. [Google Scholar] [CrossRef]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef]
- Pinthus, J.H.; Whelan, K.F.; Gallino, D.; Lu, J.P.; Rothschild, N. Metabolic features of clear-cell renal cell carcinoma: Mechanisms and clinical implications. Can. Urol. Assoc. J. 2011, 5, 274–282. [Google Scholar] [CrossRef]
- Langbein, S.; Frederiks, W.M.; zur Hausen, A.; Popa, J.; Lehmann, J.; Weiss, C.; Alken, P.; Coy, J.F. Metastasis is promoted by a bioenergetic switch: New targets for progressive renal cell cancer. Int. J. Cancer 2008, 122, 2422–2428. [Google Scholar] [CrossRef]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J. Urol. 2008, 180, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Reis, R.M.; Baltazar, F. Lactate Transporters and pH Regulation: Potential Therapeutic Targets in Glioblastomas. Curr. Cancer Drug Targets 2016, 16, 388–399. [Google Scholar] [CrossRef]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed]
- Marchiq, I.; Pouyssegur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. (Berl.) 2016, 94, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.; Prado-Garcia, H.; Sanchez-Garcia, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; Feron, O. Lactate shuttles at a glance: From physiological paradigms to anti-cancer treatments. Dis. Model. Mech. 2011, 4, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Cantone, I.; Fisher, A.G. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol. 2013, 20, 282–289. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Brettingham-Moore, K.H.; Taberlay, P.C. Cancer Epigenetics. In Drug Discovery in Cancer Epigenetics, 1st ed.; Egger, G., Arimondo, P., Eds.; Academic Press: London, UK, 2015; pp. 41–59. [Google Scholar]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2 (Suppl. 1), S4–S11. [Google Scholar] [CrossRef]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. In Advances in Genetics; Herceg, Z., Ushijima, T., Eds.; Academic Press: London, UK, 2010; Volume 70, pp. 27–56. [Google Scholar]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Vaquero, A.; Loyola, A.; Reinberg, D. The constantly changing face of chromatin. Sci. Aging Knowl. Environ. 2003, 2003, RE4. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Cohen, I.; Poreba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Schittenhelm, J.; Honegger, J.; Schluesener, H. Prognostic relevance of global histone 3 lysine 9 acetylation in ependymal tumors. J. Neurosurg. 2013, 119, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Morris, M.R.; Latif, F. The epigenetic landscape of renal cancer. Nat. Rev. Nephrol. 2017, 13, 47–60. [Google Scholar] [CrossRef]
- Malouf, G.; Zhang, J.; Tannir, N.M.; Thompson, E.; Spano, J.-P.; Khayat, D.; Su, X. Association of CpG island methylator phenotype with clear-cell renal cell carcinoma aggressiveness. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Shenoy, N.; Vallumsetla, N.; Zou, Y.; Galeas, J.N.; Shrivastava, M.; Hu, C.; Susztak, K.; Verma, A. Role of DNA methylation in renal cell carcinoma. J. Hematol. Oncol. 2015, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.Y.; Mohtat, D.; Yu, Y.; Ko, Y.A.; Shenoy, N.; Bhattacharya, S.; Izquierdo, M.C.; Park, A.S.; Giricz, O.; Vellamkutla, N.; et al. Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin. Cancer Res. 2014, 20, 4349–4360. [Google Scholar] [CrossRef]
- Brugarolas, J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013, 19, 324–332. [Google Scholar] [CrossRef]
- Kapur, P.; Pena-Llopis, S.; Christie, A.; Zhrebker, L.; Pavia-Jimenez, A.; Rathmell, W.K.; Xie, X.J.; Brugarolas, J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159–167. [Google Scholar] [CrossRef]
- Pawlowski, R.; Muhl, S.M.; Sulser, T.; Krek, W.; Moch, H.; Schraml, P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int. J. Cancer 2013, 132, E11–E17. [Google Scholar] [CrossRef]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef]
- Ellinger, J.; Kahl, P.; Mertens, C.; Rogenhofer, S.; Hauser, S.; Hartmann, W.; Bastian, P.J.; Buttner, R.; Muller, S.C.; von Ruecker, A. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int. J. Cancer 2010, 127, 2360–2366. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef]
- Rogenhofer, S.; Kahl, P.; Holzapfel, S.; Von Ruecker, A.; Mueller, S.C.; Ellinger, J. Decreased levels of histone H3K9me1 indicate poor prognosis in patients with renal cell carcinoma. Anticancer Res. 2012, 32, 879–886. [Google Scholar]
- Rogenhofer, S.; Kahl, P.; Mertens, C.; Hauser, S.; Hartmann, W.; Buttner, R.; Muller, S.C.; von Ruecker, A.; Ellinger, J. Global histone H3 lysine 27 (H3K27) methylation levels and their prognostic relevance in renal cell carcinoma. BJU Int. 2012, 109, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, F.R.; Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Scholman, K.; Denkert, C.; Dietel, M.; et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 2008, 8, 381. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.L.; Chuang, M.J.; Wu, S.T.; Sun, G.H.; Chang, S.Y.; Yu, D.S.; Huang, S.M.; Huan, S.K.; Cheng, T.C.; Chen, T.T.; et al. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin. Cancer Res. 2009, 15, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Ku, S.; Ciamporcero, E.; Miles, K.M.; Attwood, K.; Chintala, S.; Shen, L.; Ellis, L.; Sotomayor, P.; Swetzig, W.; et al. HDAC 1 and 6 modulate cell invasion and migration in clear cell renal cell carcinoma. BMC Cancer 2016, 16, 617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cao, Y.; Zhao, W.; Guo, L.; Liu, W. HDAC6 serves as a biomarker for the prognosis of patients with renal cell carcinoma. Cancer Biomark. 2017, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Mu, X.; Liang, X.; Hu, K.; Chen, M. HDAC9 associates with distant metastasis and predicts poor prognosis in clear cell renal cell cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 4647–4653. [Google Scholar]
- Mosashvilli, D.; Kahl, P.; Mertens, C.; Holzapfel, S.; Rogenhofer, S.; Hauser, S.; Buttner, R.; Von Ruecker, A.; Muller, S.C.; Ellinger, J. Global histone acetylation levels: Prognostic relevance in patients with renal cell carcinoma. Cancer Sci. 2010, 101, 2664–2669. [Google Scholar] [CrossRef] [PubMed]
- Kanao, K.; Mikami, S.; Mizuno, R.; Shinojima, T.; Murai, M.; Oya, M. Decreased acetylation of histone H3 in renal cell carcinoma: A potential target of histone deacetylase inhibitors. J. Urol. 2008, 180, 1131–1136. [Google Scholar] [CrossRef]
- Jeh, S.U.; Park, J.J.; Lee, J.S.; Kim, D.C.; Do, J.; Lee, S.W.; Choi, S.M.; Hyun, J.S.; Seo, D.H.; Lee, C.; et al. Differential expression of the sirtuin family in renal cell carcinoma: Aspects of carcinogenesis and prognostic significance. Urol. Oncol. 2017, 35, 675.e9–675.e15. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jeronimo, C. Metabolism and epigenetic interplay in cancer: Regulation and putative therapeutic targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef]
- Rabhi, N.; Hannou, S.A.; Froguel, P.; Annicotte, J.-S. Cofactors as metabolic sensors driving cell adaptation in physiology and disease. Front. Endocrinol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Thangaraju, M.; Gopal, E.; Martin, P.M.; Ananth, S.; Smith, S.B.; Prasad, P.D.; Sterneck, E.; Ganapathy, V. SLC5A8 triggers tumor cell apoptosis through pyruvate-dependent inhibition of histone deacetylases. Cancer Res. 2006, 66, 11560–11564. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Ciszewski, W.M.; Kania, K.D. L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal. 2015, 13, 36. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.M.; Uribe-Lewis, S.; Madhu, B.; Honess, D.J.; Stubbs, M.; Griffiths, J.R. The action of beta-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: Evidence of a beta-hydroxybutyrate paradox. Cancer Metab. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, C.; Zhang, H. Hypoxia and cancer cell metabolism. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Kim, J.A.; Yeom, Y.I. Metabolic Signaling to Epigenetic Alterations in Cancer. Biomol. Ther. (Seoul) 2018, 26, 69–80. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef]
- Gao, X.; Lin, S.H.; Ren, F.; Li, J.T.; Chen, J.J.; Yao, C.B.; Yang, H.B.; Jiang, S.X.; Yan, G.Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Loriot, C.; Domingues, M.; Berger, A.; Menara, M.; Ruel, M.; Morin, A.; Castro-Vega, L.J.; Letouze, E.; Martinelli, C.; Bemelmans, A.P.; et al. Deciphering the molecular basis of invasiveness in Sdhb-deficient cells. Oncotarget 2015, 6, 32955–32965. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Shim, E.H.; Sudarshan, S. Another small molecule in the oncometabolite mix: L-2-Hydroxyglutarate in kidney cancer. Oncoscience 2015, 2, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Sunela, K.L.; Kataja, M.J.; Lehtinen, E.T.; Salminen, T.K.; Kujala, P.M.; Virman, J.P.; Kellokumpu-Lehtinen, P.L. Prognostic factors and long-term survival in renal cell cancer patients. Scand. J. Urol. Nephrol. 2009, 43, 454–460. [Google Scholar] [CrossRef]
- Krabbe, L.M.; Bagrodia, A.; Margulis, V.; Wood, C.G. Surgical management of renal cell carcinoma. Semin. Intervent. Radiol. 2014, 31, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Hutson, T.E.; Quinn, D.I. Cytokine therapy: A standard of care for metastatic renal cell carcinoma? Clin. Genitourin. Cancer 2005, 4, 181–186. [Google Scholar] [CrossRef]
- Erman, M.; Benekli, M.; Basaran, M.; Bavbek, S.; Buyukberber, S.; Coskun, U.; Demir, G.; Karabulut, B.; Oksuzoglu, B.; Ozkan, M.; et al. Renal cell cancer: Overview of the current therapeutic landscape. Expert Rev. Anticancer Ther. 2016, 16, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Hutson, T.E. Targeted therapy for renal cell carcinoma: A new treatment paradigm. Proc. (Bayl. Univ. Med. Cent.) 2007, 20, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Eggener, S.E.; Yossepowitch, O.; Pettus, J.A.; Snyder, M.E.; Motzer, R.J.; Russo, P. Renal cell carcinoma recurrence after nephrectomy for localized disease: Predicting survival from time of recurrence. J. Clin. Oncol. 2006, 24, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- Eggener, S.E.; Yossepowitch, O.; Kundu, S.; Motzer, R.J.; Russo, P. Risk score and metastasectomy independently impact prognosis of patients with recurrent renal cell carcinoma. J. Urol. 2008, 180, 873–878, discussion 878. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lameirinhas, A.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C. The Complex Interplay between Metabolic Reprogramming and Epigenetic Alterations in Renal Cell Carcinoma. Genes 2019, 10, 264. https://doi.org/10.3390/genes10040264
Lameirinhas A, Miranda-Gonçalves V, Henrique R, Jerónimo C. The Complex Interplay between Metabolic Reprogramming and Epigenetic Alterations in Renal Cell Carcinoma. Genes. 2019; 10(4):264. https://doi.org/10.3390/genes10040264
Chicago/Turabian StyleLameirinhas, Ana, Vera Miranda-Gonçalves, Rui Henrique, and Carmen Jerónimo. 2019. "The Complex Interplay between Metabolic Reprogramming and Epigenetic Alterations in Renal Cell Carcinoma" Genes 10, no. 4: 264. https://doi.org/10.3390/genes10040264
APA StyleLameirinhas, A., Miranda-Gonçalves, V., Henrique, R., & Jerónimo, C. (2019). The Complex Interplay between Metabolic Reprogramming and Epigenetic Alterations in Renal Cell Carcinoma. Genes, 10(4), 264. https://doi.org/10.3390/genes10040264