Analysis of DNA Methylation Patterns Associated with In Vitro Propagated Globe Artichoke Plants Using an EpiRADseq-Based Approach

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, Growth Conditions, and Samples Collection
2.2. Library Preparation
2.3. Bioinformatics Analysis
2.3.1. Demultiplexing and Cleaning of Reads
2.3.2. Differentially Methylated Loci Identification
2.3.3. DML Annotation
2.4. RNA Isolation and Gene Transcription Analysis
2.5. miRNA Target Genes Analysis
2.6. Accession Code
3. Results
3.1. Sequencing Results
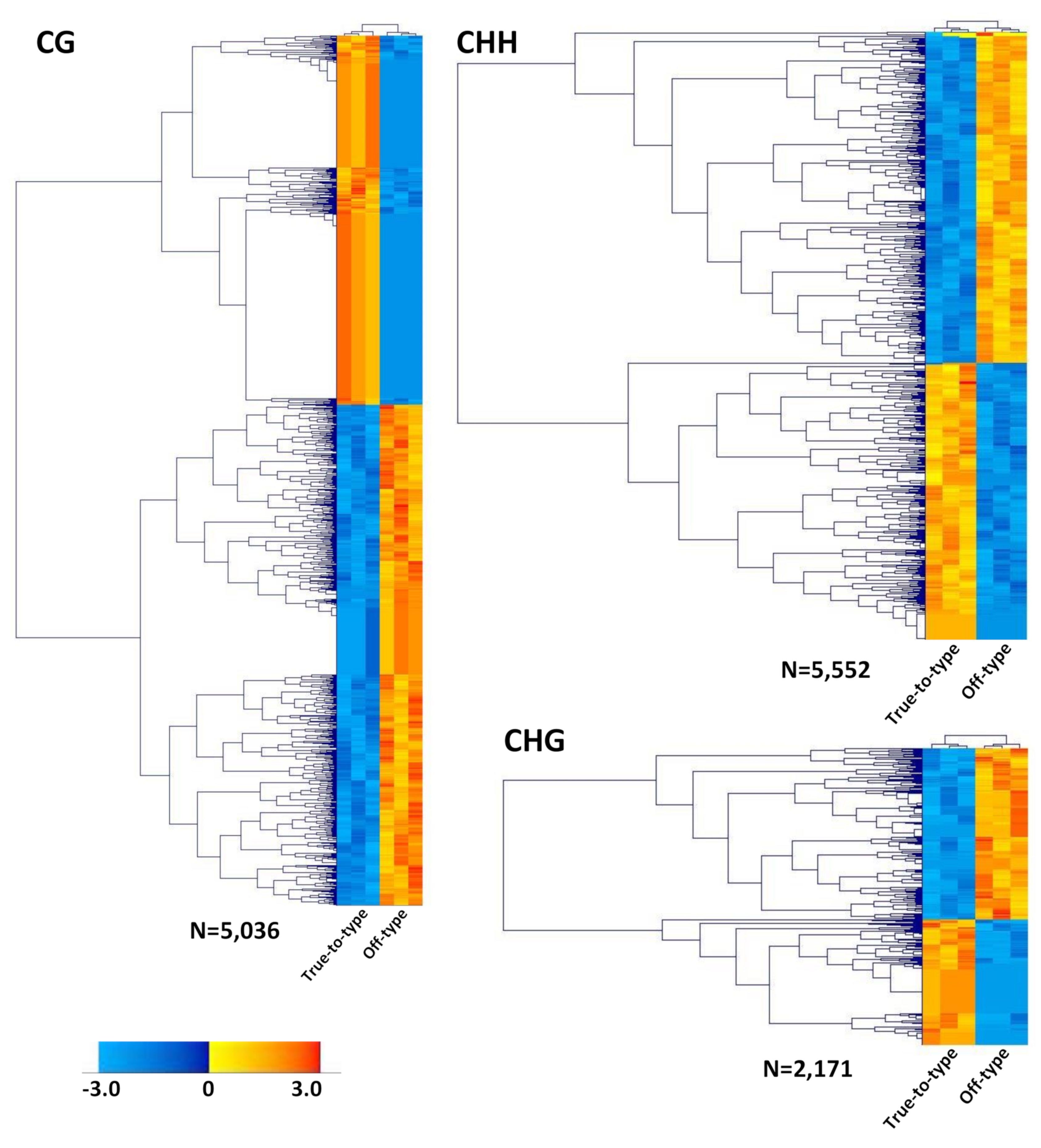
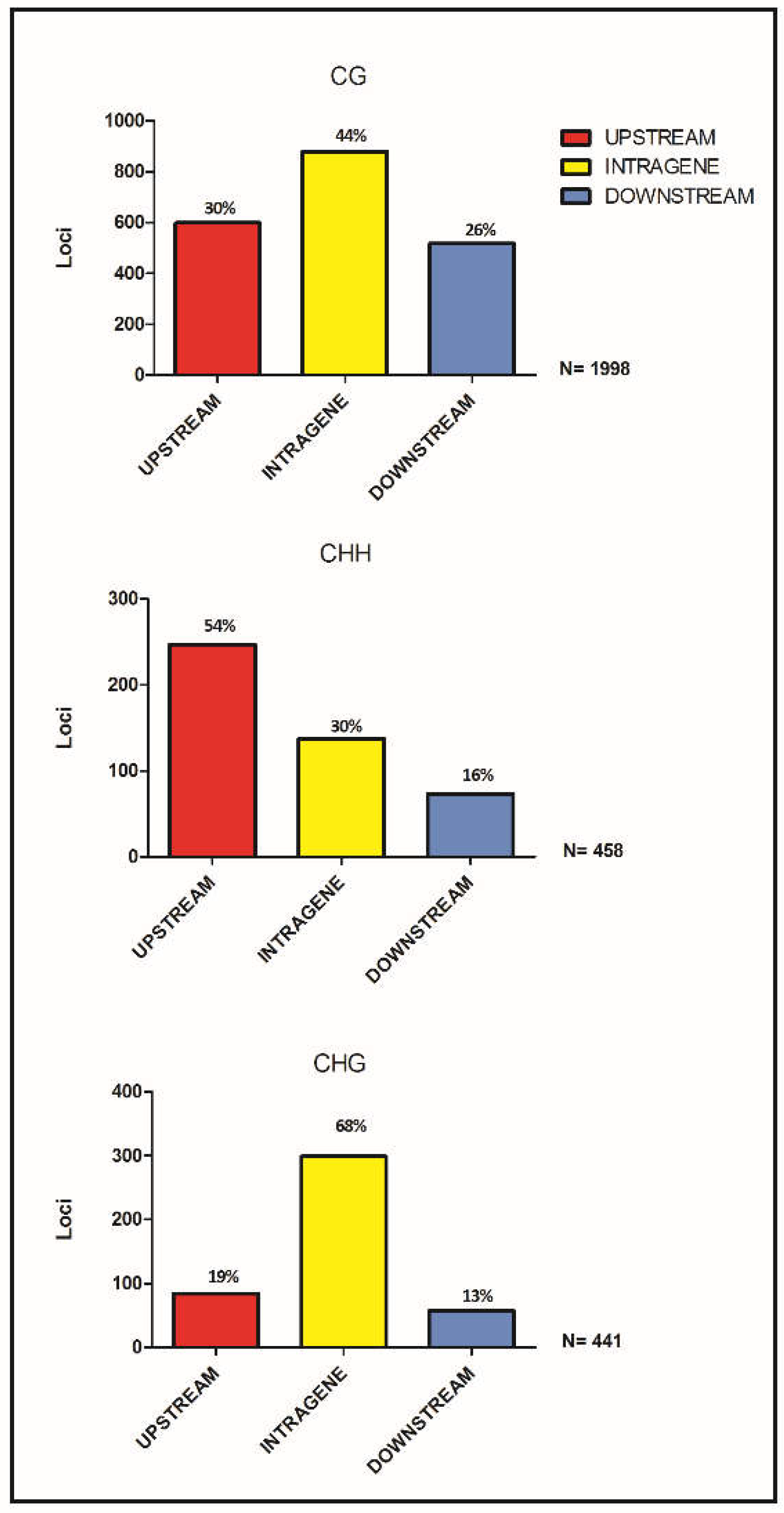
3.2. Differentially Methylated Loci
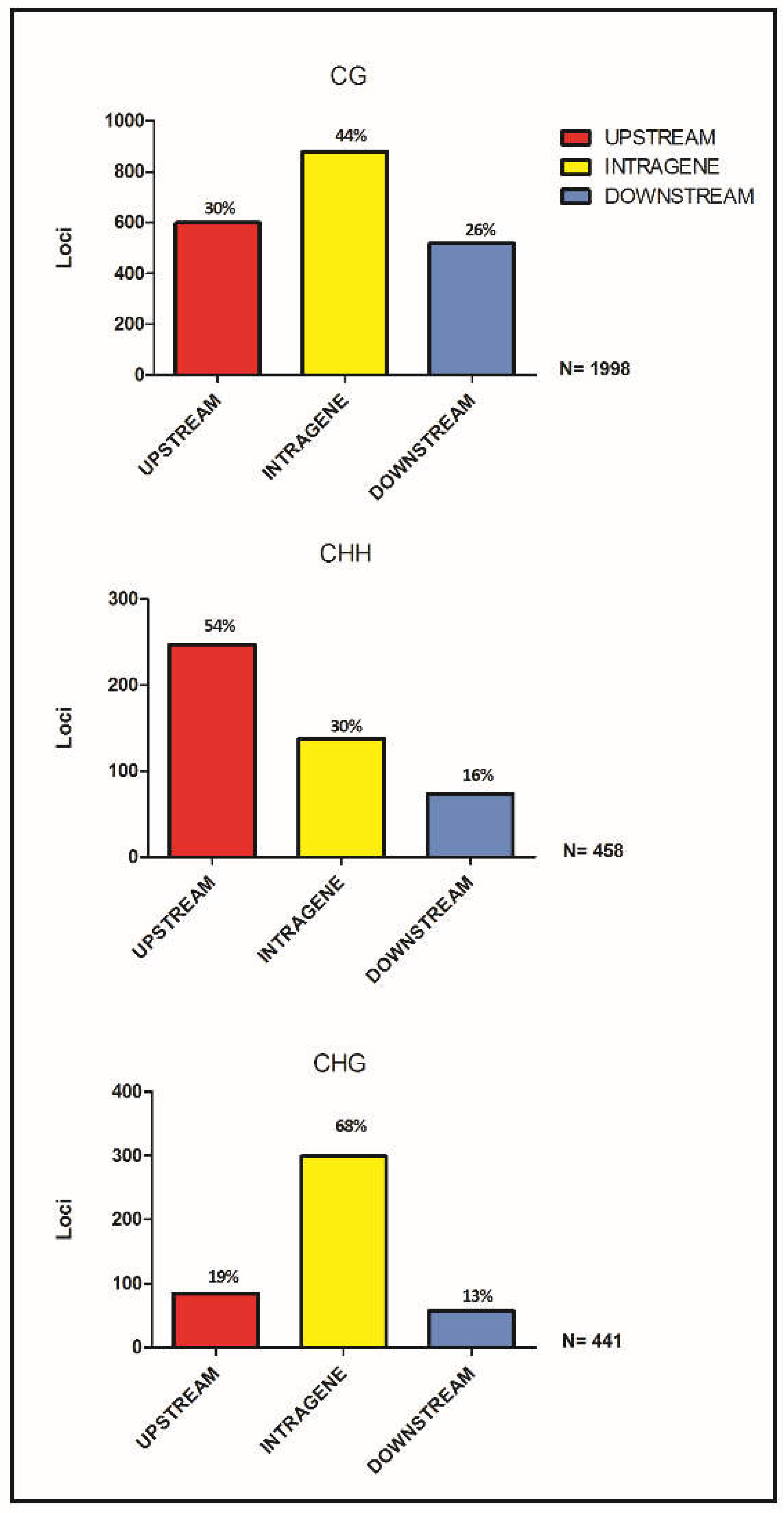
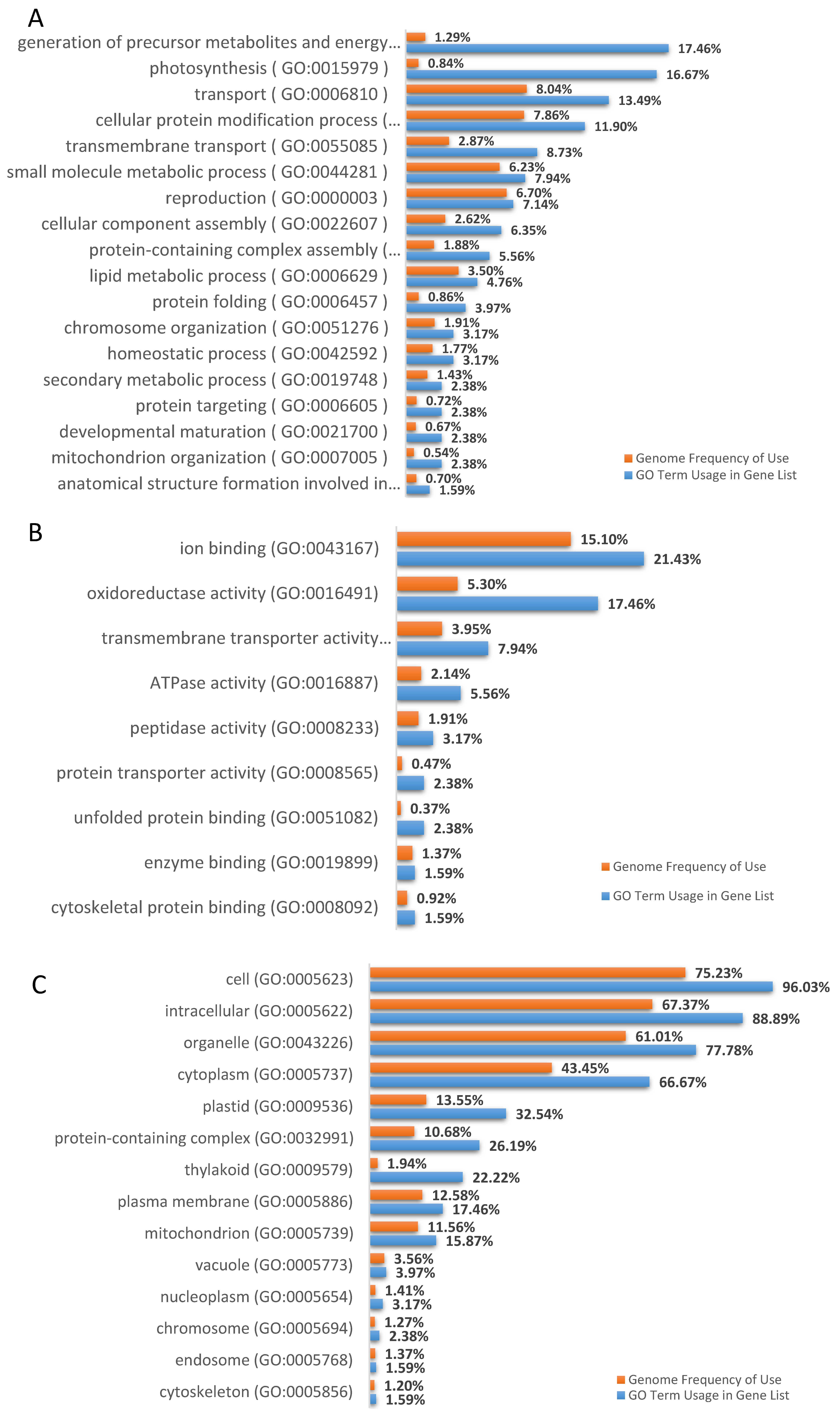
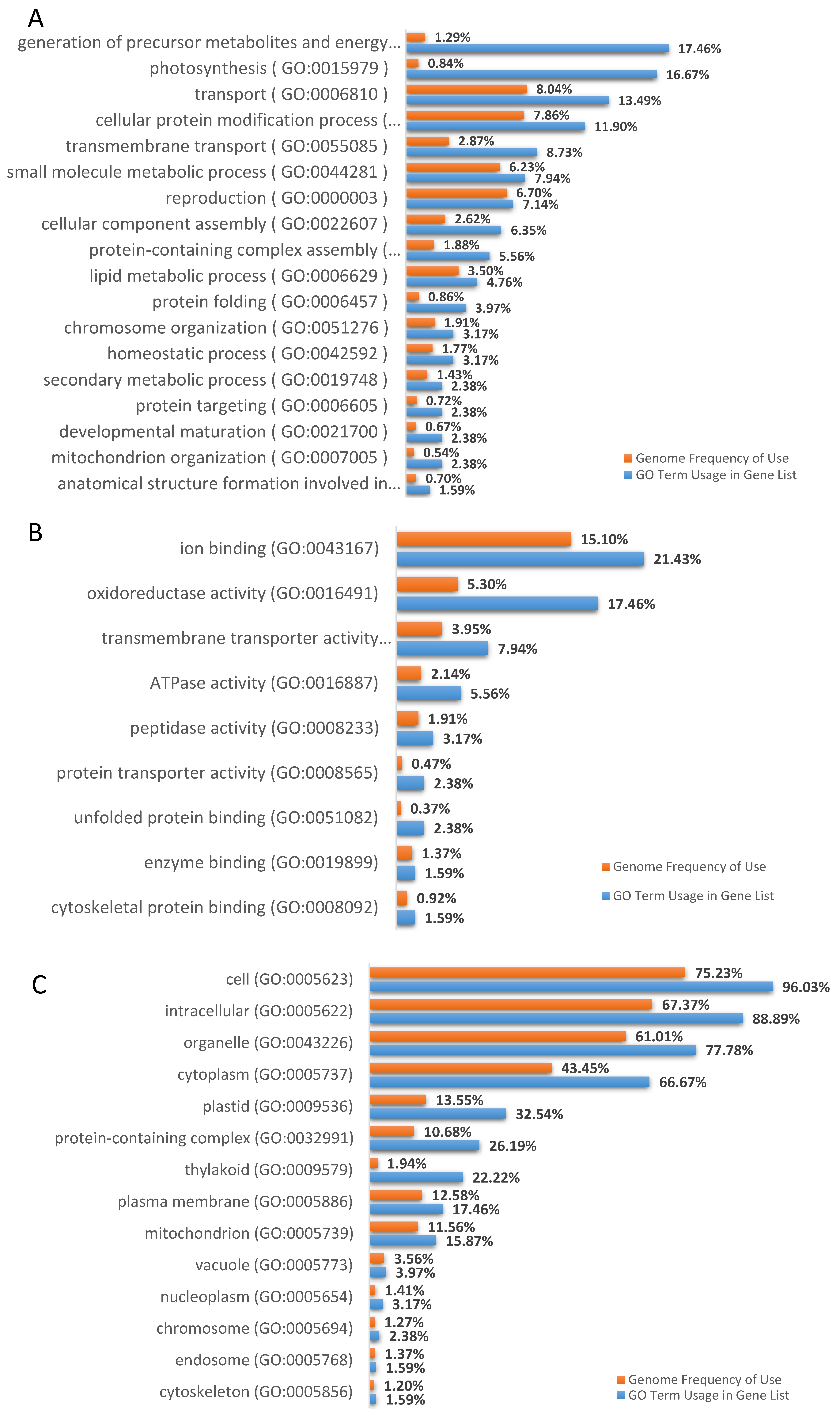
3.3. DML Annotation and GO Categorization Analysis
3.4. Gene Transcriptional Analysis
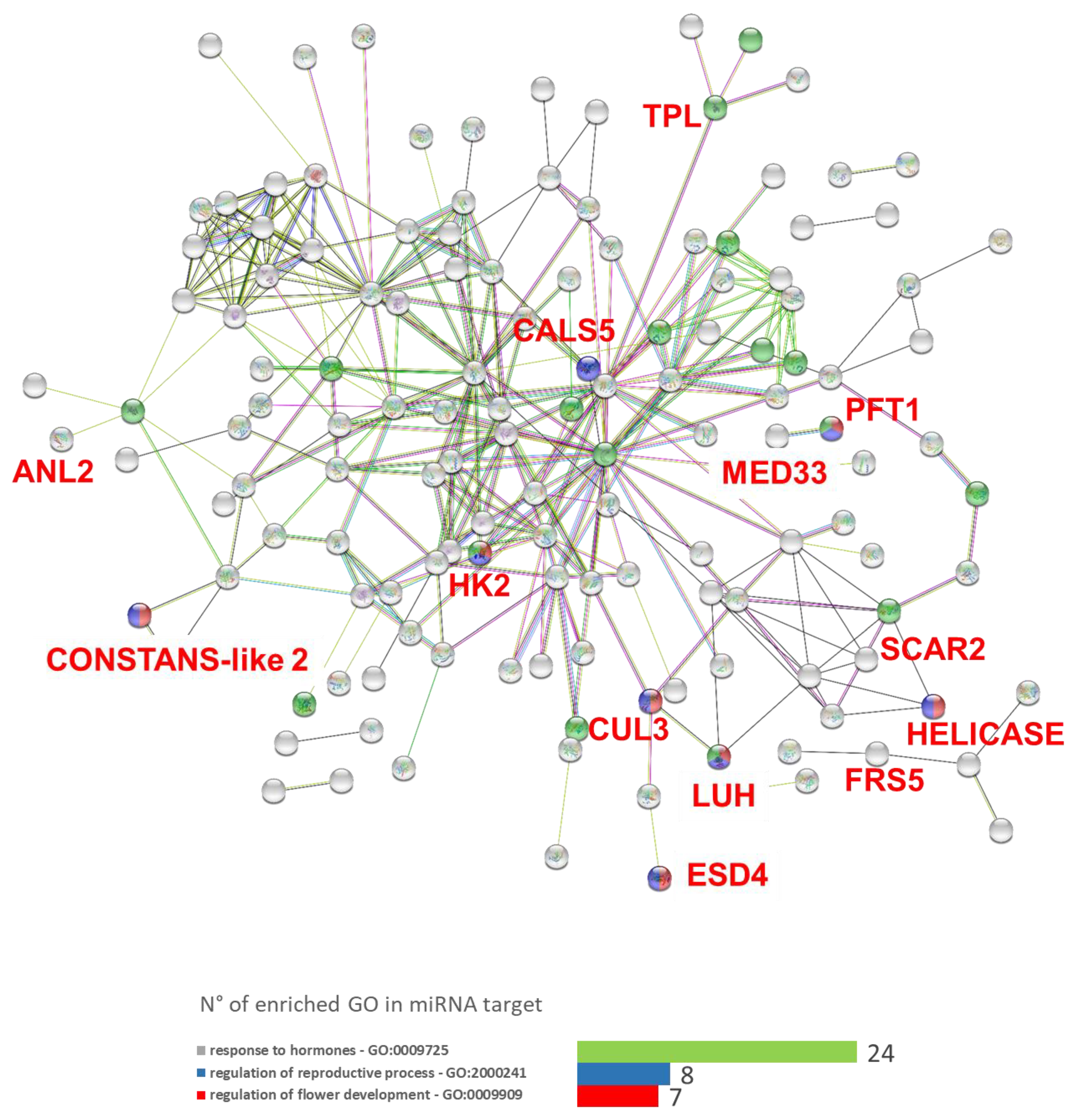
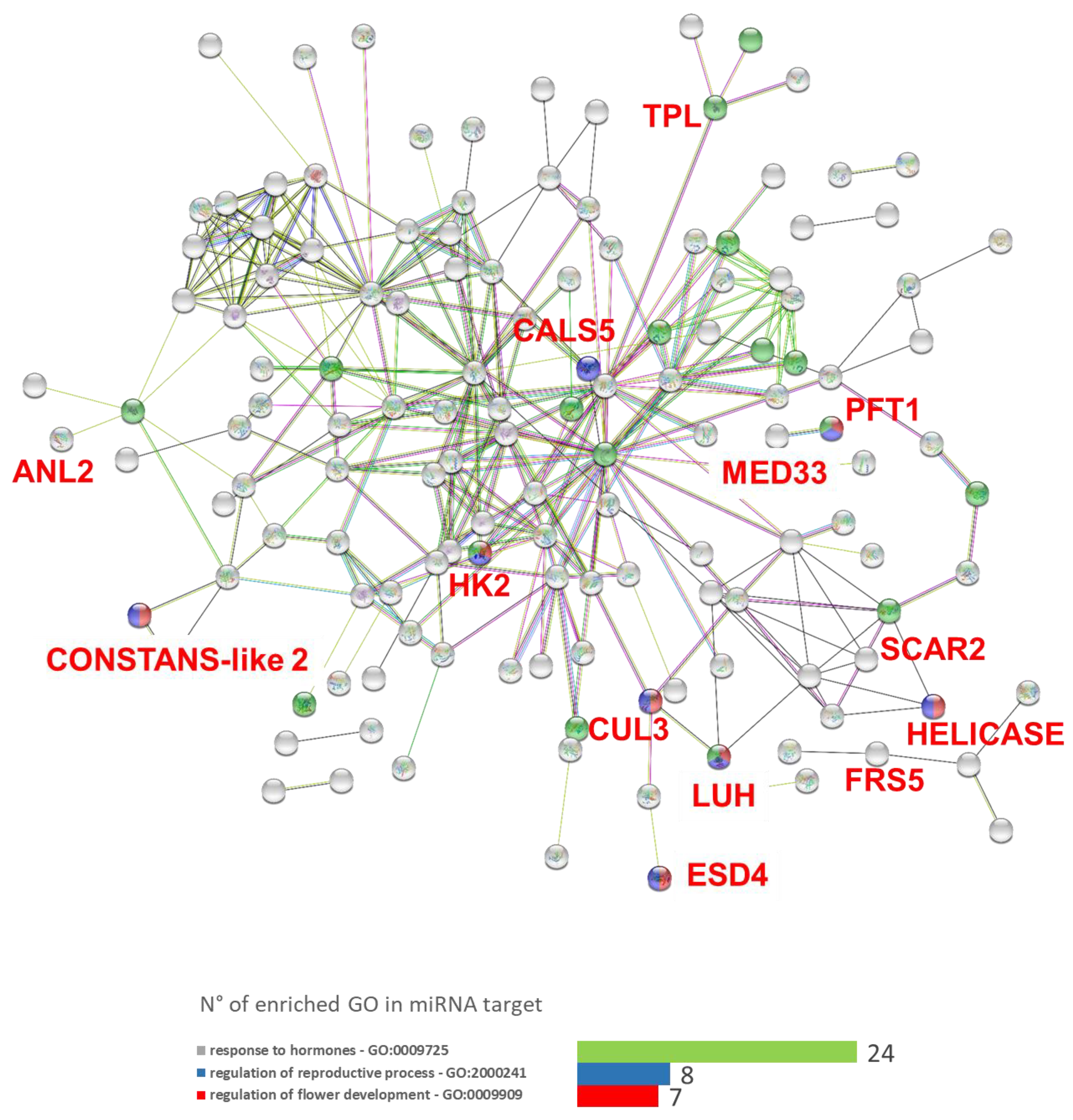
3.5. microRNAs Target in Silico Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scaglione, D.; Reyes-Chin-Wo, S.; Acquadro, A.; Froenicke, L.; Portis, E.; Beitel, C.; Tirone, M.; Mauro, R.; Lo Monaco, A.; Mauromicale, G.; et al. The genome sequence of the outbreeding globe artichoke constructed de novo incorporating a phase-aware low-pass sequencing strategy of F 1 progeny. Sci. Rep. 2016, 6, 19427. [Google Scholar] [CrossRef]
- Gebhard, R. Antioxidative and protective properties of extracts from leaves of the artichoke (Cynara scolymus L.) against hydroperoxide-induced oxidative stress in cultured rat hepatocytes. Toxicol. Appl. Pharmacol. 1997, 144, 279–286. [Google Scholar] [CrossRef]
- Shimoda, H.; Ninomiya, K.; Nishida, N.; Yoshino, T.; Morikawa, T.; Matsuda, H.; Yoshikawa, M. Anti-hyperlipidemic sesquiterpenes and new sesquiterpene glycosides from the leaves of artichoke (Cynara scolymus L.): Structure requirement and mode of action. Bioorg. Med. Chem. Lett. 2003, 13, 223–228. [Google Scholar] [CrossRef]
- Schütz, K.; Kammerer, D.; Carle, R.; Schieber, A. Identification and quantification of caffeoylquinic acids and flavonoids from artichoke (Cynara scolymus L.) heads, juice, and pomace by HPLC-DAD-ESI/MS n. J. Agric. Food Chem. 2004, 52, 4090–4096. [Google Scholar] [CrossRef]
- Lattanzio, V.; Kroon, P.A.; Linsalata, V.; Cardinali, A. Globe artichoke: A functional food and source of nutraceutical ingredients. J. Funct. Foods 2009, 1, 131–144. [Google Scholar] [CrossRef]
- Pandino, G.; Lombardo, S.; Mauromicale, G. Mineral profile in globe artichoke as affected by genotype, head part and environment. J. Sci. Food Agric. 2011, 91, 302–308. [Google Scholar] [CrossRef]
- Foti, S.; Mauromicale, G.; Raccuia, S.A.; Fallico, B.; Fanella, F.; Maccarone, E. Possible alternative utilization of Cynara spp.: I. Biomass, grain yield and chemical composition of grain. Ind. Crops Prod. 1999, 10, 219–228. [Google Scholar] [CrossRef]
- González, J.F.; González-Garcı́a, C.M.; Ramiro, A.; González, J.; Sabio, E.; Gañán, J.; Rodrìguez, M.A. Combustion optimisation of biomass residue pellets for domestic heating with a mural boiler. Biomass Bioenergy 2004, 27, 145–154. [Google Scholar] [CrossRef]
- Ierna, A.; Mauromicale, G. Cynara cardunculus L. genotypes as a crop for energy purposes in a Mediterranean environment. Biomass Bioenergy 2010, 34, 754–760. [Google Scholar] [CrossRef]
- Nouraei, S.; Rahimmalek, M.; Saeidi, G.; Bahreininejad, B. Variation in seed oil content and fatty acid composition of globe artichoke under different irrigation regimes. J. Am. Oil Chem. Soc. 2016, 7, 953–962. [Google Scholar] [CrossRef]
- Rossi, P.; De Pauli, G. Micropropagation of artichoke (Cynara scolymus L.). In Biotechnology in Agriculture and Forestry 19, High-Tech and Micropropagation III; Bajaj, Y.P.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 118–134. [Google Scholar]
- Lanteri, S.; Di Leo, I.; Ledda, L.; Mameli, M.G.; Portis, E. RAPD variation within and among populations of globe artichoke cultivar ‘Spinoso sardo’. Plant Breed. 2001, 120, 243–246. [Google Scholar] [CrossRef]
- Spanò, R.; Bottalico, G.; Corrado, A.; Campanale, A.; Di Franco, A.; Mascia, T. A Protocol for Producing Virus-Free Artichoke Genetic Resources for Conservation, Breeding, and Production. Agriculture 2018, 8, 36. [Google Scholar] [CrossRef]
- Saccardo, F.; Micozzi, F.; Di Lernia, G.; Piccioni, C.; Barba, M.; Pagnotta, M.A. Virus free artichoke germplasm: Quali-quantitative response of globe artichoke. Acta Hort. 2007, 730, 375–380. [Google Scholar] [CrossRef]
- Acquadro, A.; Papanice, M.A.; Lanteri, S.; Bottalico, G.; Portis, E.; Campanale, A.; Finetti-Sialer, M.M.; Mascia, T.; Sumerano, P.; Gallitelli, D. Production and fingerprinting of virus-free clones in a reflowering globe artichoke. Plant Cell Tissue Organ Cult. 2010, 100, 329–337. [Google Scholar] [CrossRef]
- Tavazza, R.; Lucioli, A.; Benelli, C.; Giorgi, D.; D’Aloisio, E.; Papacchioli, V. Cryopreservation in artichoke: Towards a phytosanitary qualified germplasm collection. Ann. Appl. Biol. 2013, 163, 231–241. [Google Scholar] [CrossRef]
- Cadinu, M.; Repetto, A.M. Il carciofo e il cardo, ricerca Spinoso sardo. 2009. Available online: https://www.colturaecultura.it/capitolo/spinoso-sardo (accessed on 26 February 2019).
- Krishna, H.; Alizadeh, M.; Singh, D.; Singh, U.; Chauhan, N.; Eftekhari, M.; Sadh, R.K. Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Amasino, R.M. Vernalization and flowering time. Curr. Opin. Biotechnol. 2005, 16, 154–158. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E. Epigenetic modifications in plants: An evolutionary perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186. [Google Scholar] [CrossRef]
- Kinoshita, T.; Seki, M. Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 2014, 55, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Shen, L.; Gu, X.; Wang, Y.; Yu, H.; He, Y. Embryonic epigenetic reprogramming by a pioneer transcription factor in plants. Nature 2017, 551, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 1. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Pecinka, A.; Abdelsamad, A.; Vu, G.T. Hidden genetic nature of epigenetic natural variation in plants. Trends Plant Sci. 2013, 8, 625–632. [Google Scholar] [CrossRef]
- Catoni, M.; Griffiths, J.; Becker, C.; Zabet, N.R.; Bayon, C.; Dapp, M.; Lieberman-Lazarovich, M.; Weigel, D.; Paszkowski, J. DNA sequence properties that predict susceptibility to epiallelic switching. EMBO J. 2017, 36, 617–628. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.S.; Osborne, E.J.; Casale, F.P.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjamsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Huang, S.S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 124. [Google Scholar] [CrossRef] [PubMed]
- Miguel, C.; Marum, L. An epigenetic view of plant cells cultured in vitro: Somaclonal variation and beyond. J. Exp. Bot. 2011, 62, 3713–3725. [Google Scholar] [CrossRef]
- Us-Camas, R.; Rivera-Solís, G.; Duarte-Aké, F.; De-la-Pena, C. In vitro culture: An epigenetic challenge for plants. Plant Cell Tissue Organ Cult. 2014, 118, 187–201. [Google Scholar] [CrossRef]
- Lee, K.; Seo, P.J. Dynamic Epigenetic Changes during Plant Regeneration. Trends Plant Sci. 2018, 3, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Schield, D.R.; Walsh, M.R.; Card, D.C.; Andrew, A.L.; Adams, R.H.; Castoe, T.A. EpiRADseq: Scalable analysis of genome wide patterns of methylation using next-generation sequencing. Methods Ecol. Evol. 2016, 7, 60–69. [Google Scholar] [CrossRef]
- Acquadro, A.; Barchi, L.; Portis, E.; Mangino, G.; Valentino, D.; Mauromicale, G.; Lanteri, S. Genome reconstruction in Cynara cardunculus taxa gains access to chromosome-scale DNA variation. Sci. Rep. 2017, 7, 5617. [Google Scholar] [CrossRef] [PubMed]
- Gianoglio, S.; Moglia, A.; Acquadro, A.; Comino, C.; Portis, E. The genome-wide identification and transcriptional levels of DNA methyltransferases and demethylases in globe artichoke. PLoS ONE 2017, 12, e0181669. [Google Scholar] [CrossRef]
- Marconi, G.; Capomaccio, S.; Comino, C.; Acquadro, A.; Portis, E.; Porceddu, A.; Albertini, E. Methylation Content Sensitive Enzyme ddRAD (MCSeEd): A genome profiling system to address cytosine/adenine methylation changes. In preparation.
- Cadinu, M.; Repetto, A.; Frau, A.; Beneventi, S.; Meloni, S. Influence of the explant type on the phenotypic changes in micropropagated plants of artichoke. Acta Hortic. 2004. [Google Scholar] [CrossRef]
- Doyle, J.J. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.; Ruan, J.; Wu, C.I. Rainbow: An integrated tool for efficient clustering and assembling RAD-seq reads. Bioinformatics 2012, 28, 2732–2737. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Brown, M.P.; Grundy, W.N.; Lin, D.; Cristianini, N.; Sugnet, C.W.; Furey, T.S.; Ares, M., Jr.; Haussler, D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. USA 2000, 97, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gyokusen, M.; Nakazawa, Y.; Gyokusen, K. Selection of Housekeeping Genes for Transgene Expression Analysis in Eucommia ulmoides Oliver Using Real-Time RT-PCR. J. Bot. 2010, 230961. [Google Scholar] [CrossRef]
- Quian, W.; Miki, D.; Zhang, H.; Liu, Y.; Zhang, X.; Tang, K.; Kan, Y.; La, H.; Li, X.; Li, S.; et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 2012, 336, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Bartrina, I.; Jensen, H.; Novak, O.; Strnad, M.; Werner, T.; Schmülling, T. Gain-of-function mutants of the cytokinin receptors AHK2 and AHK3 regulate plant organ size, flowering time and plant longevity. Plant Physiol. 2017, 173, 1783–1797. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Liu, Y.; Xu, S.; Xiao, J.; Wang, B.; Deng, H.; Chong, K. Arabidopsis O-GlcNAc transferase SEC activates histone methyltransferase ATX1 to regulate flowering. EMBO J. 2018, 37, e98115. [Google Scholar] [CrossRef]
- Alvarez-Venegas, R.; Pien, S.; Sadder, M.; Witmer, X.; Grossniklaus, U.; Avramova, Z. ATX-1, an Arabidopsis homolog of trithorax, activates flower homeotic genes. Curr. Biol. 2003, 13, 627–637. [Google Scholar] [CrossRef]
- Bollman, K.M.; Aukerman, M.J.; Park, M.Y.; Hunter, C.; Berardini, T.Z.; Poethig, R.S. HASTY, the Arabidopsis ortholog of exportin 5/MSN5, regulates phase change and morphogenesis. Development 2003, 130, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Ledger, S.; Strayer, C.; Ashton, F.; Kay, S.A.; Putterill, J. Analysis of the function of two circadian-regulated CONSTANS-LIKE genes. Plant J. 2001, 26, 15–22. [Google Scholar] [CrossRef]
- Okushima, Y.; Overvoorde, P.J.; Arima, K.; Alonso, J.M.; Chan, A.; Chang, C.; Ecker, J.R.; Hughes, B.; Lui, A.; Nguyen, D.; et al. Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: Unique and overlapping functions of ARF7 and ARF19. Plant Cell 2005, 17, 444–463. [Google Scholar] [CrossRef]
- Wang, W.; Sijacic, P.; Xu, P.; Lian, H.; Liu, Z. Arabidopsis TSO1 and MYB3R1 form a regulatory module to coordinate cell proliferation with differentiation in shoot and root. Proc. Natl. Acad. Sci. USA 2018, 115, E3045–E3054. [Google Scholar] [CrossRef]
- Gachomo, E.W.; Baptiste, L.J.; Kefela, T.; Saidel, W.M.; Kotchoni, S.O. The Arabidopsis CURVY1 (CVY1) gene encoding a novel receptor-like protein kinase regulates cell morphogenesis, flowering time and seed production. BMC Plant Biol. 2014, 14, 221. [Google Scholar] [CrossRef]
- Hemsley, P.A.; Hurst, C.H.; Kaliyadasa, E.; Lamb, R.; Knight, M.R.; De Cothi, E.A.; Steele, J.F.; Knight, H. The Arabidopsis mediator complex subunits MED16, MED14, and MED2 regulate mediator and RNA polymerase II recruitment to CBF-responsive cold-regulated genes. Plant Cell 2014, 26, 465–484. [Google Scholar] [CrossRef]
- Audonnet, L.; Shen, Y.; Zhou, D.X. JMJ24 antagonizes histone H3K9 demethylase IBM1/JMJ25 function and interacts with RNAi pathways for gene silencing. Gene Expr. Patterns 2017, 25, 1–7. [Google Scholar] [CrossRef]
- Jung, C.; Müller, A.E. Flowering time control and applications in plant breeding. Trends Plant Sci. 2009, 14, 563–573. [Google Scholar] [CrossRef]
- Goralogia, G.S.; Liu, T.K.; Zhao, L.; Panipinto, P.M.; Groover, E.D.; Bains, Y.S.; Imaizumi, T. Cycling Dof Factor 1 represses transcription through the Topless co-repressor to control photoperiodic flowering in Arabidopsis. Plant J. 2017, 92, 244–262. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, F.; Ruan, Y.; Li, J.; Zhang, Z.; Sun, Y.; Deng, S.; Peng, R. Similar bowtie structures and distinct largest strong components are identified in the transcriptional regulatory networks of Arabidopsis thaliana during photomorphogenesis and heat shock. Biosystems 2018, 168, 1–7. [Google Scholar] [CrossRef]
- Ma, L.; Li, G. FAR1-RELATED SEQUENCE (FRS) and FRS-RELATED FACTOR (FRF) Family Proteins in Arabidopsis Growth and Development. Front. Plant Sci. 2018, 9, 692. [Google Scholar] [CrossRef]
- Smulders, M.J.M.; De Klerk, G.J. Epigenetics in plant tissue culture. Plant Growth Regul. 2011, 63, 137–146. [Google Scholar] [CrossRef]
- Kitimu, S.R.; Taylor, J.; March, T.J.; Tairo, F.; Wilkinson, M.J.; Rodríguez López, C.M. Meristem micropropagation of cassava (Manihot esculenta) evokes genome-wide changes in DNA methylation. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef]
- Eeuwens, C.J.; Lord, S.; Donough, C.R.; Rao, V.; Vallejo, G.; Nelson, S. Effects of tissue culture conditions during embryoid multiplication on the incidence of “mantled” flowering in clonally propagated oil palm. Plant Cell Tissue Organ Cult. 2002, 70, 311–323. [Google Scholar] [CrossRef]
- Topoonyanont, N.; Debergh, P.C. Reducing bushiness in micropropagated Gerbera. Plant Cell Tissue Organ Cult. 2001, 67, 133–144. [Google Scholar] [CrossRef]
- Smith, M.K.; Drew, R.A. Current applications of tissue culture in plant propagation and improvement. Funct. Plant Biol. 1990, 17, 267–289. [Google Scholar] [CrossRef]
- Sahijram, L.; Soneji, J.; Bollamma, K. Analyzing somaclonal variation in micropropagated bananas (Musa spp.). In Vitro Cell. Dev. Biol. Plant 2003, 39, 551–556. [Google Scholar] [CrossRef]
- De Klerk, G.J.; Ter, B.J.; Bouman, H. An assay to measure the extent of variation in micropropagated plants of Begonia x hiemalis. Acta Bot. Neerl. 1990, 39, 145–151. [Google Scholar] [CrossRef]
- Villordon, A.Q.; La Bonte, D.R. Genetic variation among sweet potatoes propagated through nodal and adventitious sprouts. J. Am. Soc. Hort. Sci. 1996, 121, 170–174. [Google Scholar] [CrossRef]
- Santana, M.A.; Romay, G.; Matehus, J.; Vicente-Villardon, J.L.; Demey, J.R. Simple and low-cost strategy for micropropagation of cassava (Manihot esculenta Crantz). Afr. J. Biotechnol. 2009, 8, 3789–3897. [Google Scholar]
- Pécaut, P.; Martin, F. Non-conformity of in vitro propagated plants of early mediterranean varieties of globe artichoke (Cynara scolymus L.). Acta Hortic. 1992. [Google Scholar] [CrossRef]
- Pécaut, P.; Martin, F. Variation occurring after natural and in vitro multiplication of early Mediterranean cultivars of globe artichoke (Cynara scolymus L). Agronomie 1993, 13, 909–919. [Google Scholar] [CrossRef]
- Larkin, P.J.; Scowcroft, W.R. Somaclonal variation—A novel source of variability from cell cultures for plant improvement. Theor. Appl. Genet. 1981, 60, 197–214. [Google Scholar] [CrossRef]
- Georges, D.; Chenieux, J.C.; Ochatt, S.J. Plant regeneration from aged-callus of the woody ornamental species Lonicera japonica cv. “Hall’s Prolific”. Plant Cell Rep. 1993, 13, 91–94. [Google Scholar] [CrossRef]
- Pastor, V.; Luna, E.; Mauch-Mani, B.; Ton, J.; Flors, V. Primed plants do not forget. Environ. Exp. Bot. 2013, 94, 46–56. [Google Scholar] [CrossRef]
- Cadinu, M.; Mallica, G.M.; Lanteri, S.; Repetto, A.; Frau, A.F.; Portis, E.; Baghino, L.; Pisanu, A.B. Possibilità di rilancio per lo Spinoso Sardo. L’informatore Agrario 2007, 22, 44–46. [Google Scholar]
- Baránek, M.; Křižan, B.; Ondrušíková, E.; Pidra, M. DNA-methylation changes in grapevine somaclones following in vitro culture and thermotherapy. Plant Cell Tissue Organ Cult. 2010, 101, 11–22. [Google Scholar] [CrossRef]
- Xia, Z.; Zou, M.; Zhang, S.; Feng, B.; Wang, W. AFSM sequencing approach: A simple and rapid method for genome-wide SNP and methylation site discovery and genetic mapping. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Schulz, B.; Rolf, L.E.; Durka, W. Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 2013, 23, 3523–3537. [Google Scholar] [CrossRef]
- Alonso, C.; Perez, R.; Bazaga, P.; Medrano, M.; Herrera, C.M. MSAP markers and global cytosine methylation in plants: A literature survey and comparative analysis for a wild-growing species. Mol. Ecol. Resour. 2016, 16, 80–90. [Google Scholar] [CrossRef]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, B.; Gogol-Doring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Zilberman, D. An evolutionary case for functional gene body methylation in plants and animals. Genome Biol. 2017, 18. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Gen. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Poorter, H.; Fiorani, F.; Stitt, M.; Schurr, U.; Finck, A.; Gibon, Y.; Usadel, B.; Munns, R.; Atkin, O.K.; et al. The art of growing plants for experimental purposes: A practical guide for the plant biologist. Funct. Plant Biol. 2012, 39, 821–838. [Google Scholar] [CrossRef]
- Junker, A.; Muraya, M.M.; Weigelt-Fischer, K.; Arana-Ceballos, F.; Klukas, C.; Melchinger, A.E.; Meyer, R.C.; Riewe, D.; Altmann, T. Optimizing experimental procedures for quantitative evaluation of crop plant performance in high throughput phenotyping systems. Front. Plant Sci. 2015, 5, 770. [Google Scholar] [CrossRef]
- Massonnet, C.; Vile, D.; Fabre, J.; Hannah, M.A.; Caldana, C.; Lisec, J.; Beemster, G.T.; Meyer, R.C.; Messerli, G.; Gronlund, J.T.; et al. Probing the reproducibility of leaf growth and molecular phenotypes: A comparison of three Arabidopsis accessions cultivated in ten laboratories. Plant Physiol. 2010, 152, 2142–2157. [Google Scholar] [CrossRef]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, Z.; Li, J.; Minakhina, S.; Yang, M.; Padgett, R.W.; Steward, R.; Chen, X. Methylation as a crucial step in plant microRNA biogenesis. Science 2005, 307, 932–935. [Google Scholar] [CrossRef]
- Filipowicz, W.; Suvendra, N.B.; Nahum, S. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2013, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.E.; Schommer, C.; Palatnik, J.F. Control of cell proliferation by microRNAs in plants. Curr. Opin. Plant Biol. 2016, 34, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Fouracre, J.P.; Poethig, R.S. The role of small RNAs in vegetative shoot development. Curr. Opin. Plant Biol. 2016, 29, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Kidner, C.A.; Martienssen, R.A. The developmental role of microRNA in plants. Curr. Opin. Plant Biol. 2005, 8, 38–44. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Liu, D.; Song, Y.; Chen, Z.; Yu, D. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big impact on plant development. Trends Plant Sci. 2017, 12, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, L.; Wu, G. Epigenetic regulation of juvenile-to-adult transition in plants. Front. Plant Sci. 2018, 9, 1048. [Google Scholar] [CrossRef] [PubMed]
- Ori, N.; Cohen, A.R.; Etzioni, A.; Brand, A.; Yanai, O.; Shleizer, S.; Menda, N.; Amsellem, Z.; Efroni, I.; Pekker, I.; et al. Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat. Gen. 2007, 39, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Schmitz, R.J. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 2017, 18, 563–575. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus epi-RADseq | Context | True-to-typelocus | Off-typelocus | Gene name | Genomic position | Gene ID | Function | Ref. |
|---|---|---|---|---|---|---|---|---|
| E8364_L143 | ACI | - | + | IDM1 | exon | Ccrd_v2_02819_g02 | epigenetics regulation | [51] |
| E245746_L143 | FNU | - | + | AHK2 | exon | Ccrd_v2_04778_g02 | plant organ size, flowering time and plant longevity | [52] |
| E232952_L143 | ACI | + | - | ATX1 | exon | Ccrd_v2_10262_g06 | regulation of flowering | [53,54] |
| E258509_L71 | ECO | - | + | HST | exon | Ccrd_v2_10596_g06 | regulation of morphogenesis | [55] |
| E421706_L143 | ACI | - | + | COL2 | exon | Ccrd_v2_11089_g07 | circadian-clock dependent regulation of flowering time | [56] |
| E225812_L143 | FNU | - | + | ARF19 | exon | Ccrd_v2_11769_g08 | lateral organ development | [57] |
| E243304_L143 | FNU | - | + | TSO1 | exon | Ccrd_v2_12311_g08 | flower organogenesis and development | [58] |
| E339914_L127 | ECO | - | + | SCAR2 | exon | Ccrd_v2_13193_g09 | regulation of plant morphogenesis and flowering | [59] |
| E243912_L143 | FNU | - | + | MED33A | exon | Ccrd_v2_13680_g09 | transcriptional regulation of flowering time | [60] |
| E255497_L143 | ACI | - | + | JMJ25 | exon | Ccrd_v2_19643_g13 | epigenetic regulation of development | [61] |
| E172485_L143 | FNU | - | + | HAP2 | exon | Ccrd_v2_22114_g15 | plant reproduction organs | [62] |
| E256023_L143 | ECO | + | - | TPL | exon | Ccrd_v2_22902_g16 | regulation of flowering | [63] |
| E244185_L143 | ACI | + | - | ANL2 | exon | Ccrd_v2_26026_scaffold_1939 | epigenetic regulation of flowering time | [64] |
| E245348_L143 | FNU | - | + | FRS5 | exon | Ccrd_v2_10161_g06 | light-induced regulation of plant development | [65] |
| Gene Name | Transcription Level | miRNA Target | Interactome miRNA Target |
|---|---|---|---|
| ATX1 | Down-expressed in off-type | Cca-miR6107, cca-miR167, cca-miR172 | - |
| SCAR2 | Down-expressed in off-type | cca-miR319 | yes |
| TPL | Down-expressed in off-type | Cca-miR396b | yes |
| TSO1 | Down-expressed in off-type | Cca-miR395b, cca-miR395c, cca-miR6116-5p | - |
| MED33A | Down-expressed in off-type | cca-miR160a | yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerruti, E.; Comino, C.; Acquadro, A.; Marconi, G.; Repetto, A.M.; Pisanu, A.B.; Pilia, R.; Albertini, E.; Portis, E. Analysis of DNA Methylation Patterns Associated with In Vitro Propagated Globe Artichoke Plants Using an EpiRADseq-Based Approach. Genes 2019, 10, 263. https://doi.org/10.3390/genes10040263
Cerruti E, Comino C, Acquadro A, Marconi G, Repetto AM, Pisanu AB, Pilia R, Albertini E, Portis E. Analysis of DNA Methylation Patterns Associated with In Vitro Propagated Globe Artichoke Plants Using an EpiRADseq-Based Approach. Genes. 2019; 10(4):263. https://doi.org/10.3390/genes10040263
Chicago/Turabian StyleCerruti, Elisa, Cinzia Comino, Alberto Acquadro, Gianpiero Marconi, Anna Maria Repetto, Anna Barbara Pisanu, Roberto Pilia, Emidio Albertini, and Ezio Portis. 2019. "Analysis of DNA Methylation Patterns Associated with In Vitro Propagated Globe Artichoke Plants Using an EpiRADseq-Based Approach" Genes 10, no. 4: 263. https://doi.org/10.3390/genes10040263
APA StyleCerruti, E., Comino, C., Acquadro, A., Marconi, G., Repetto, A. M., Pisanu, A. B., Pilia, R., Albertini, E., & Portis, E. (2019). Analysis of DNA Methylation Patterns Associated with In Vitro Propagated Globe Artichoke Plants Using an EpiRADseq-Based Approach. Genes, 10(4), 263. https://doi.org/10.3390/genes10040263