Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis



Abstract
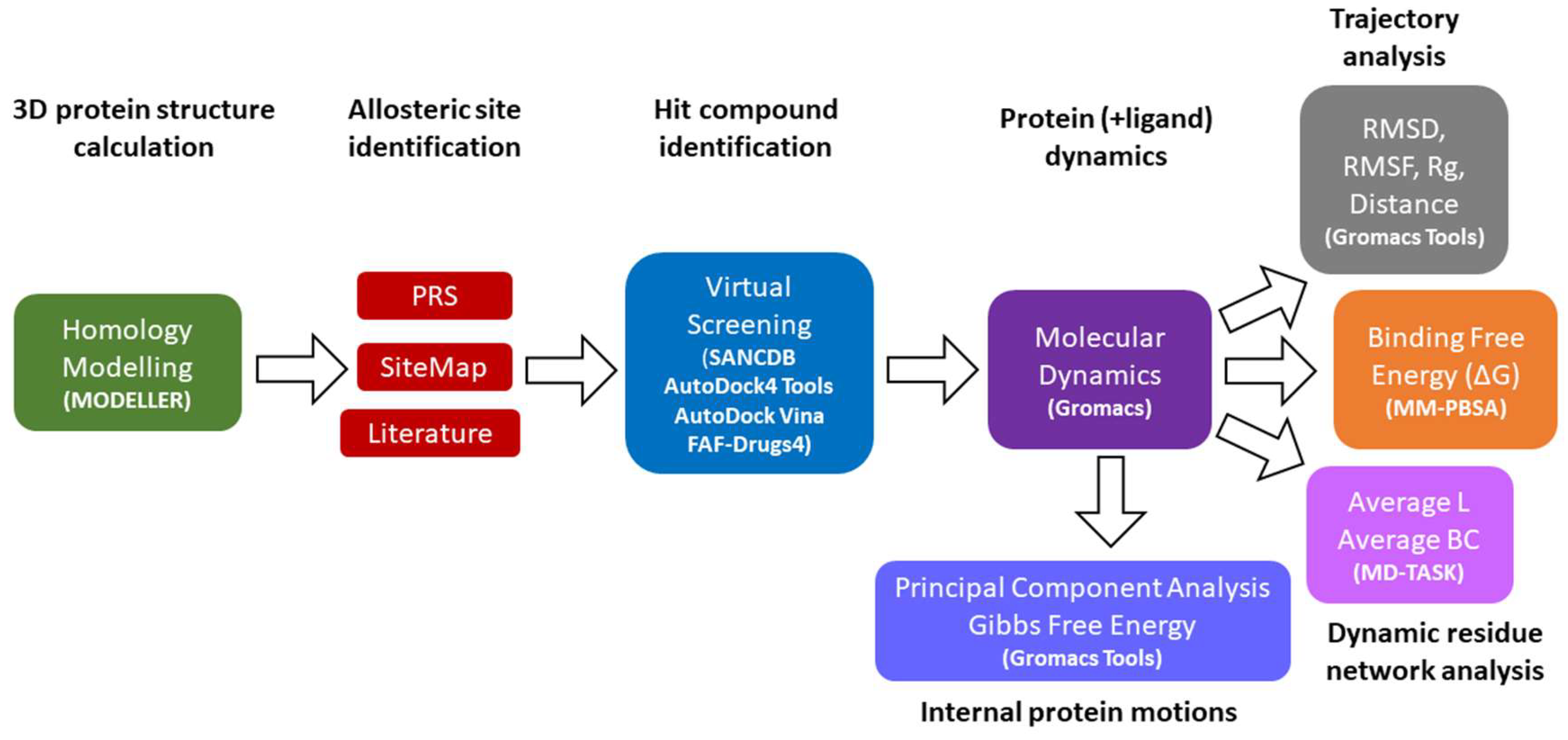
:
1. Introduction
2. Results and Discussion
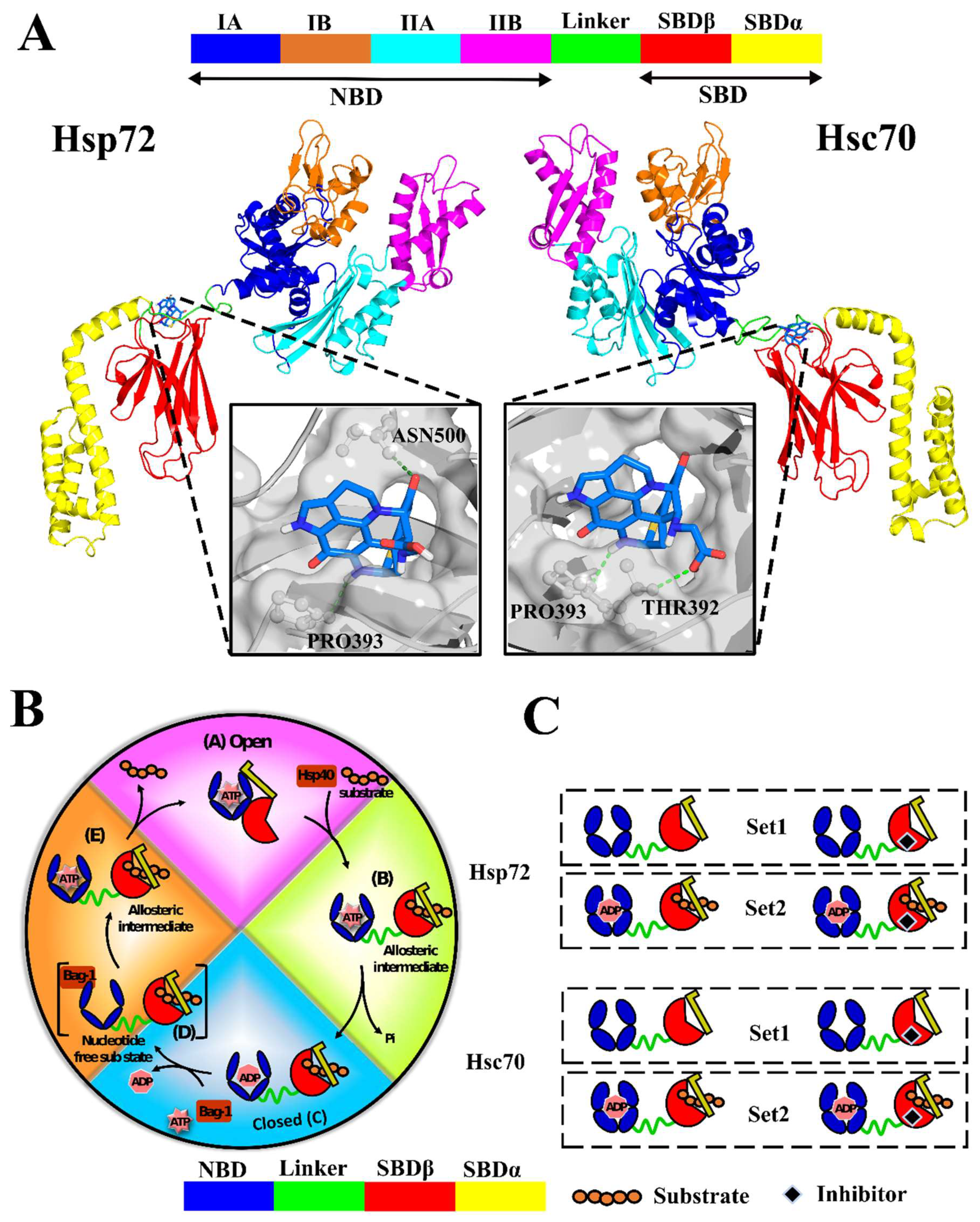
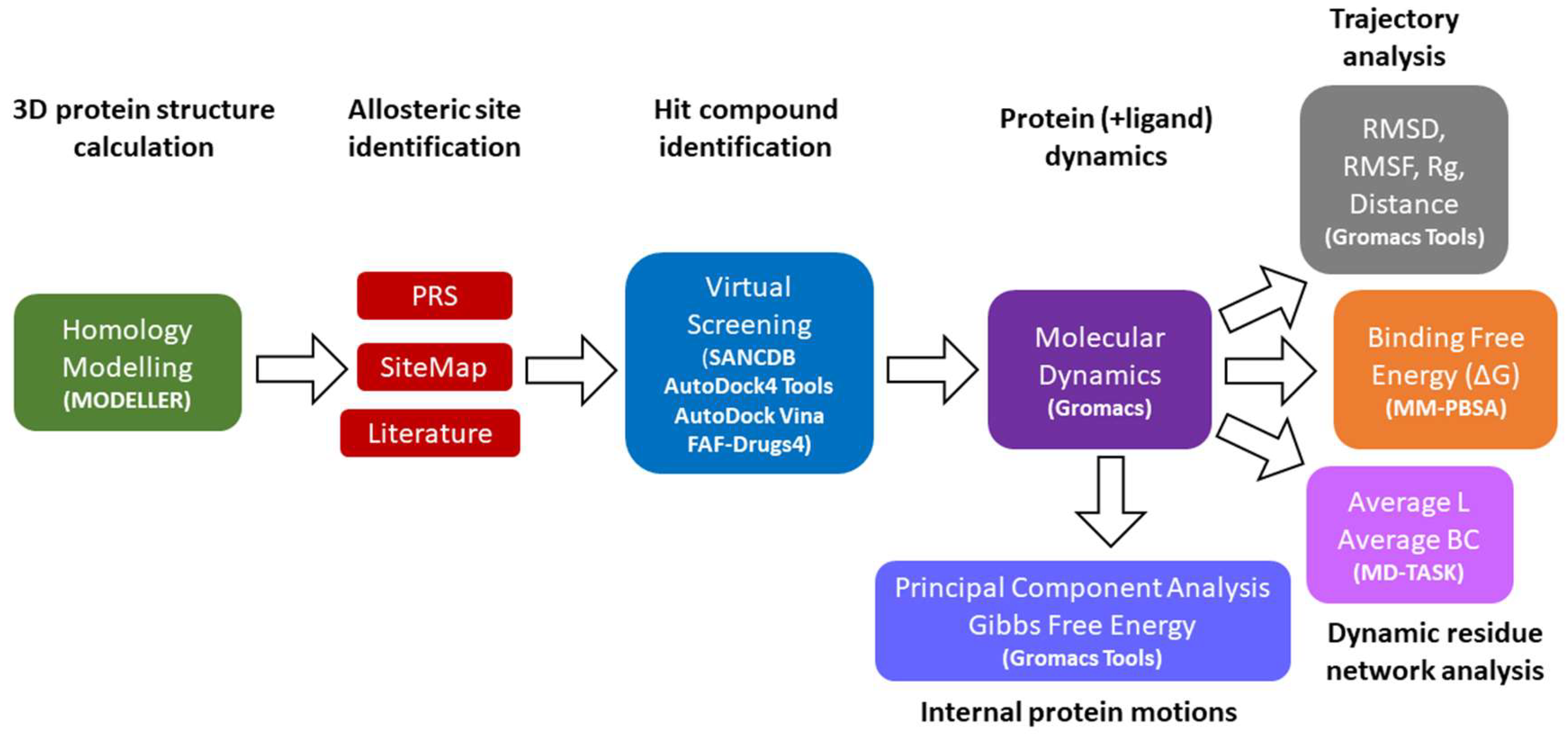
2.1. Accurate Nearly Full-Length Protein Structures Are Calculated via Homology Modeling
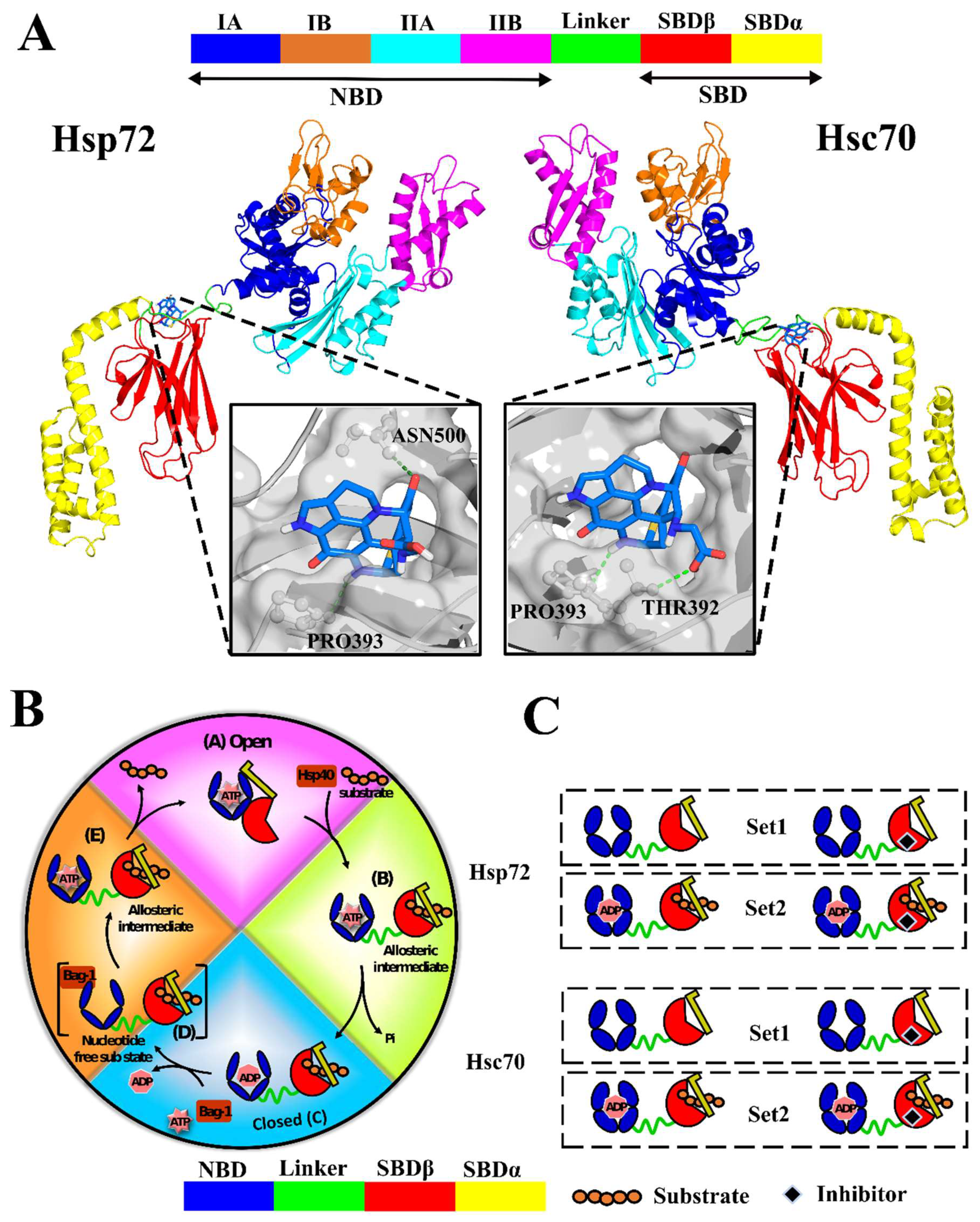
2.2. A Natural Compound Is Identified via Molecular Docking for the Selected Allosteric Site
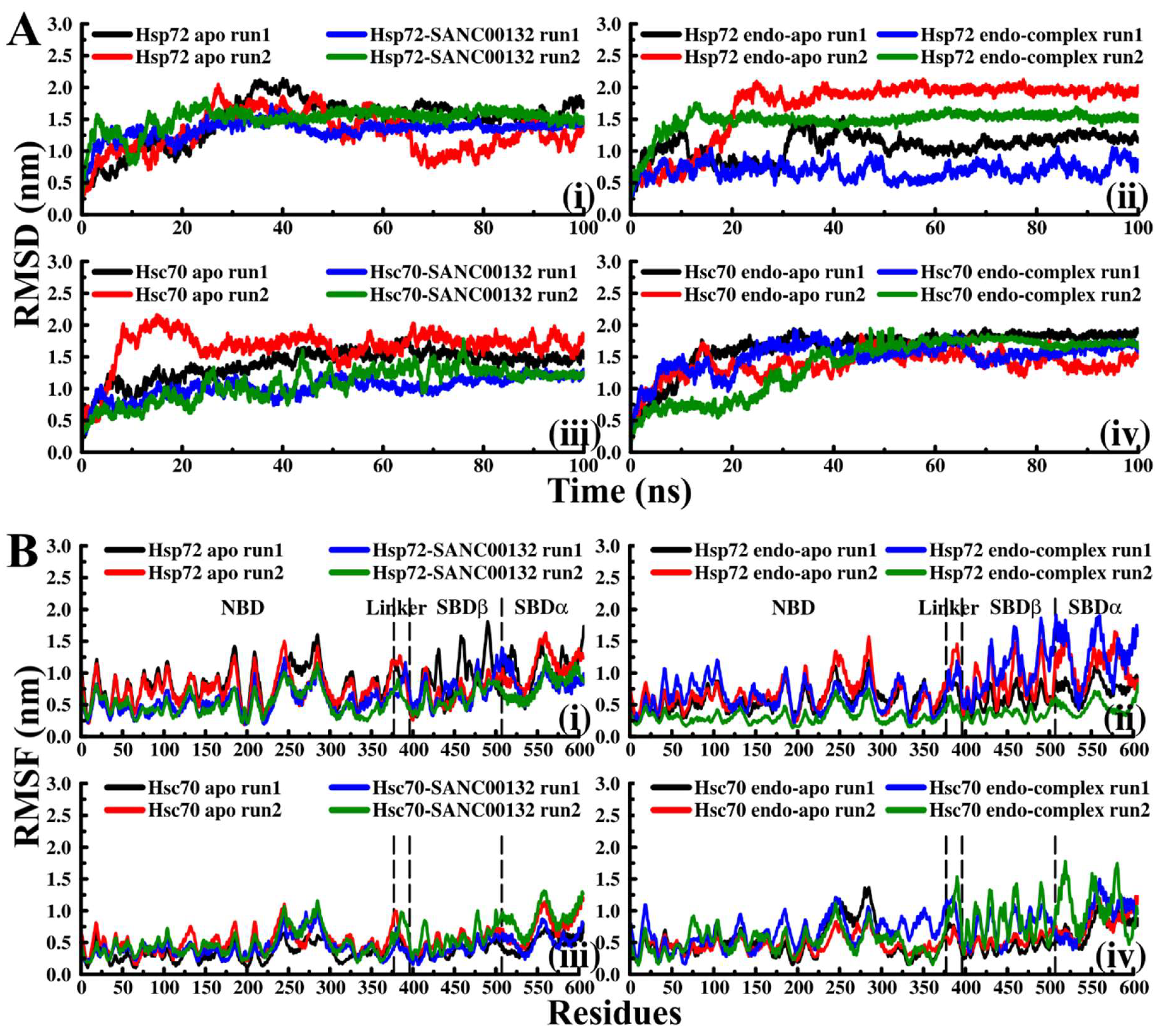
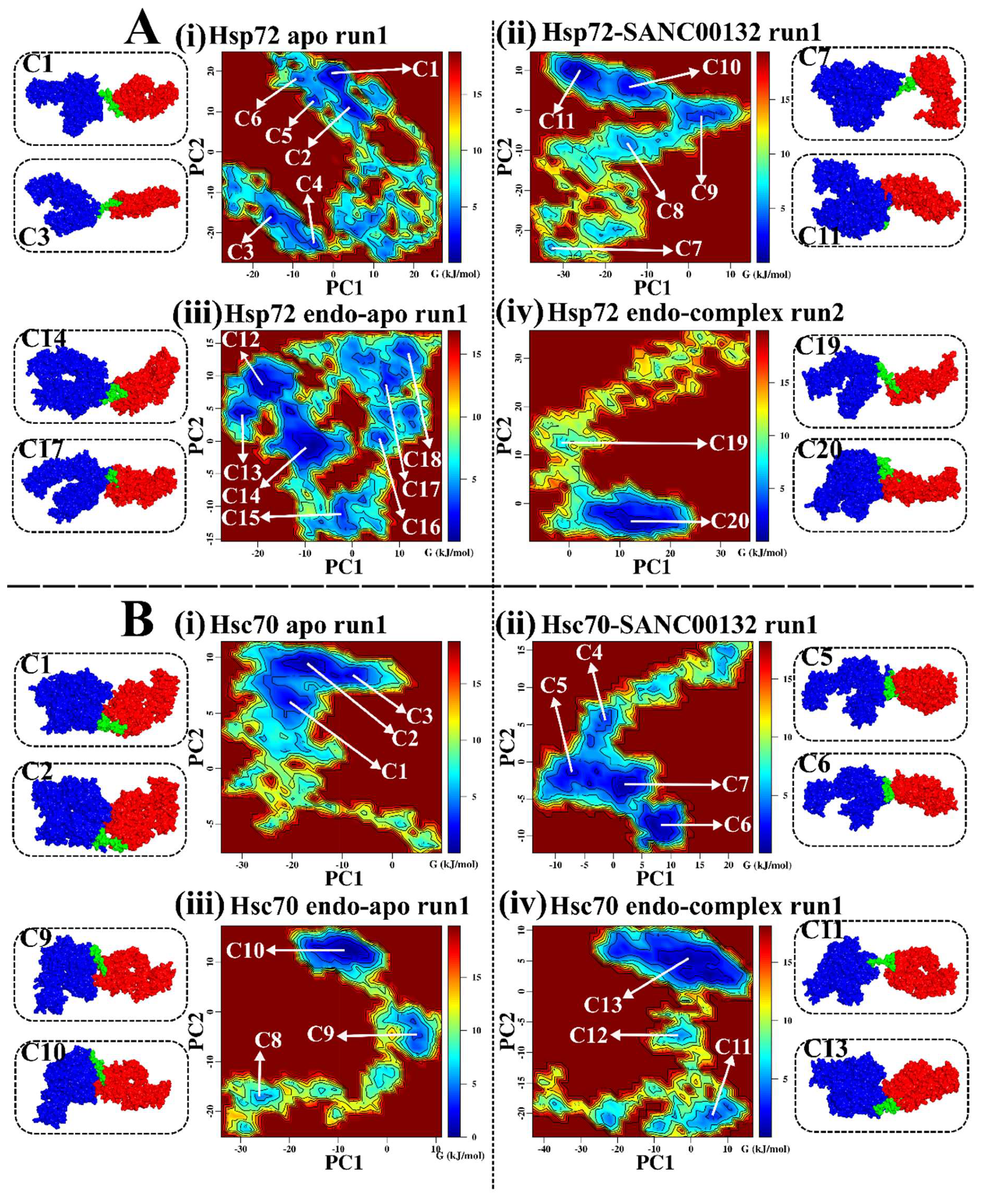
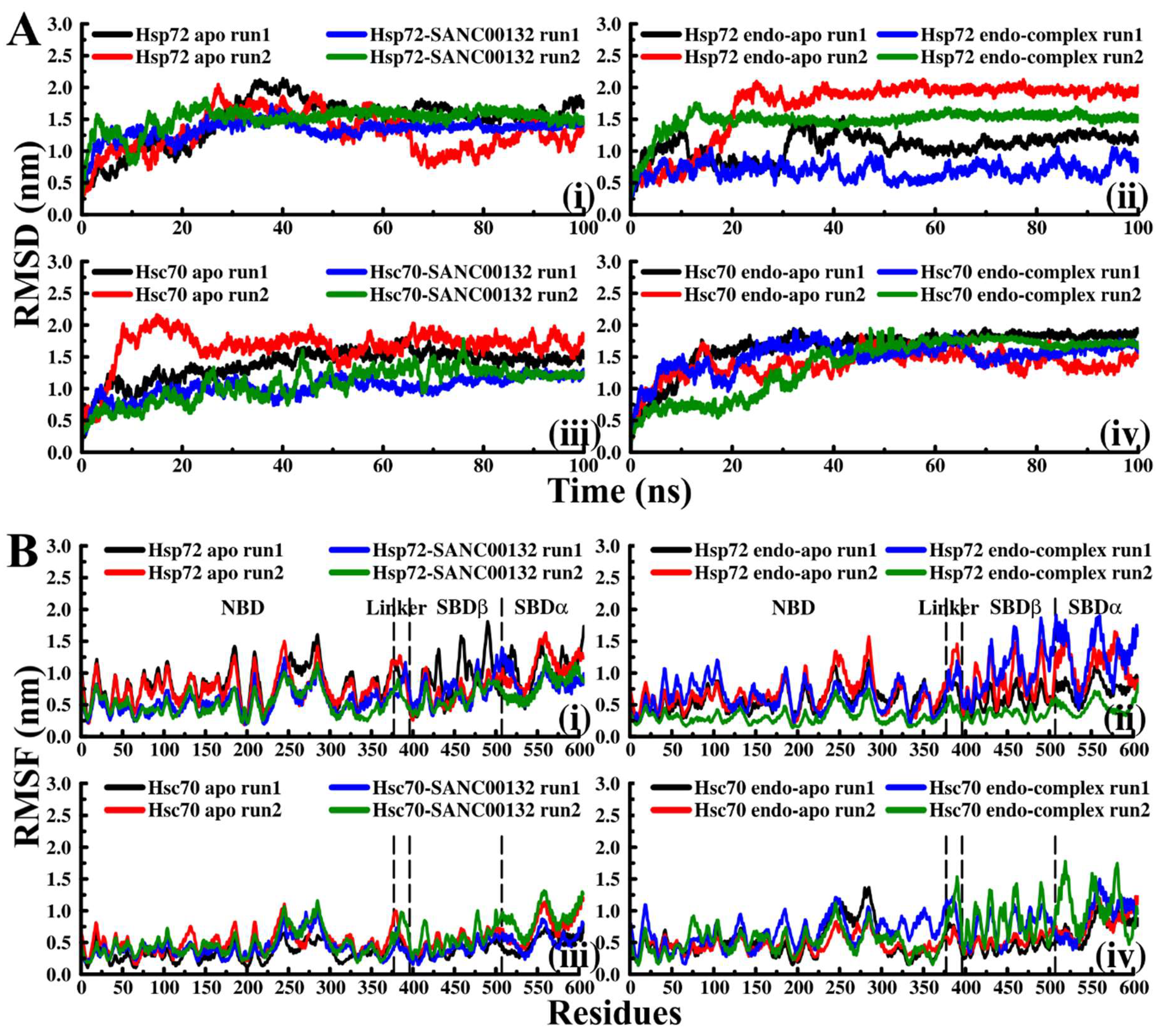
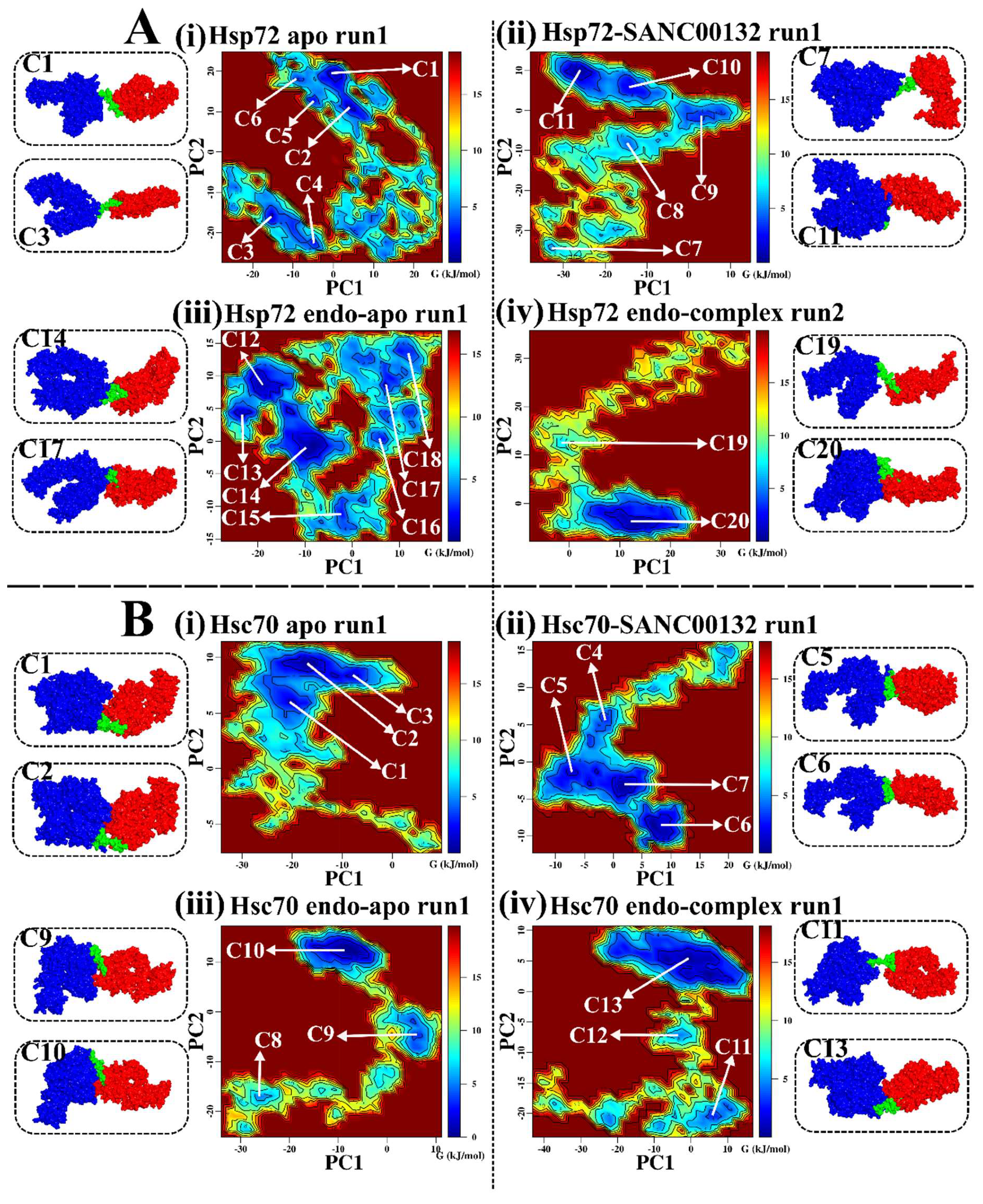
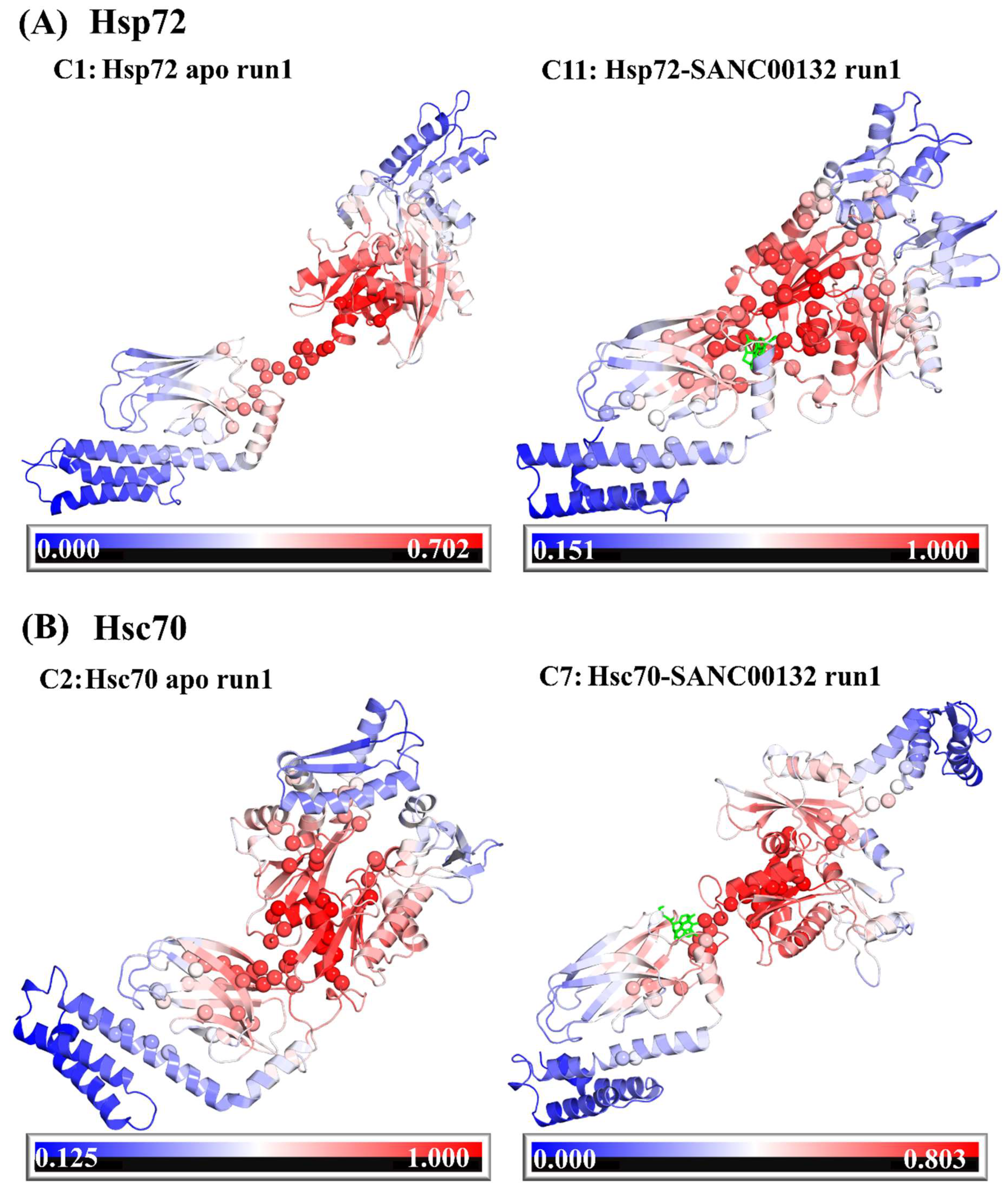
2.3. Effects of Ligand Binding on Global Protein Motions are Observed
Discorhabdin N Locks Hsp72 Domains Establishing a Rigid Closed Conformation
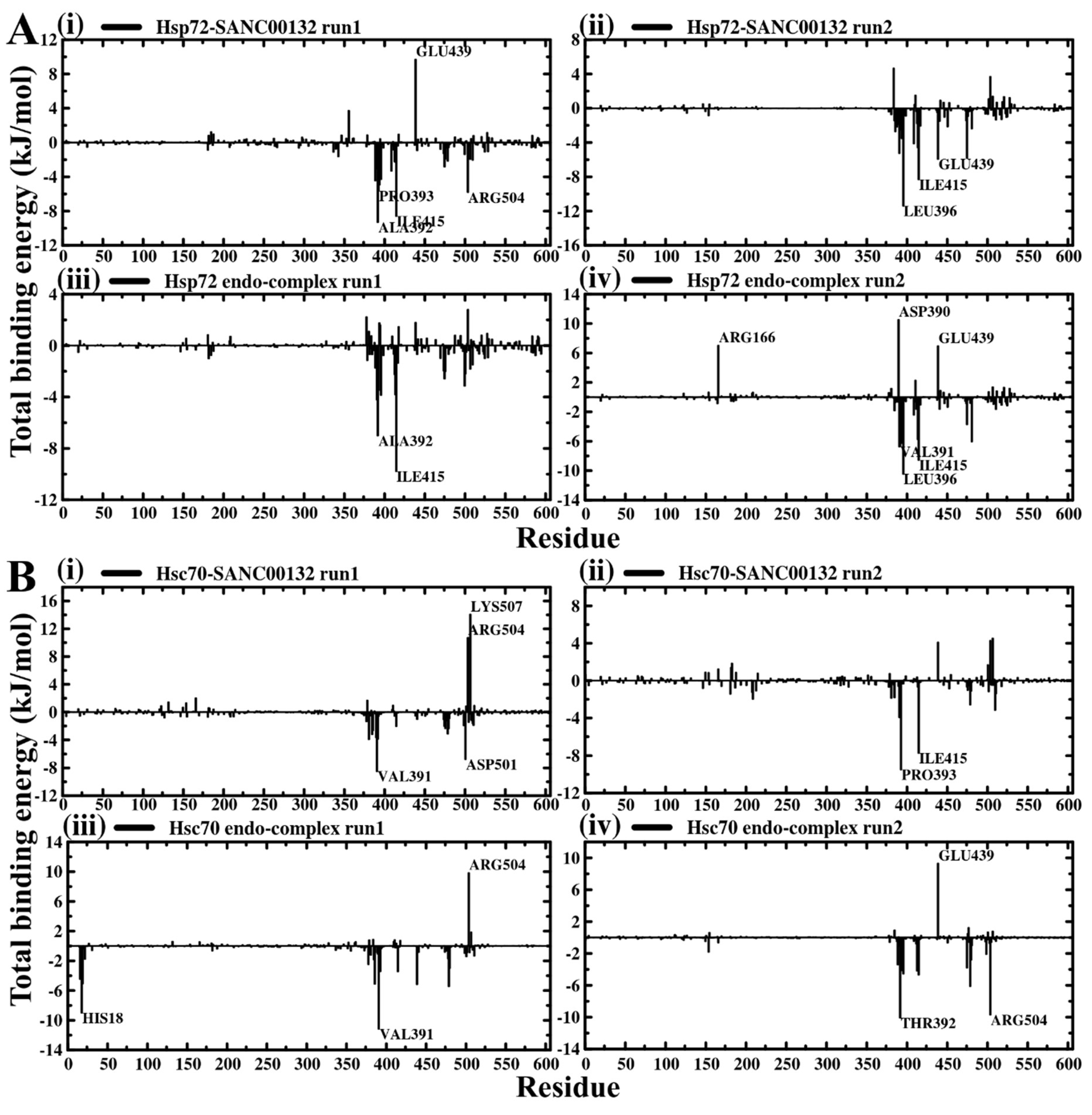
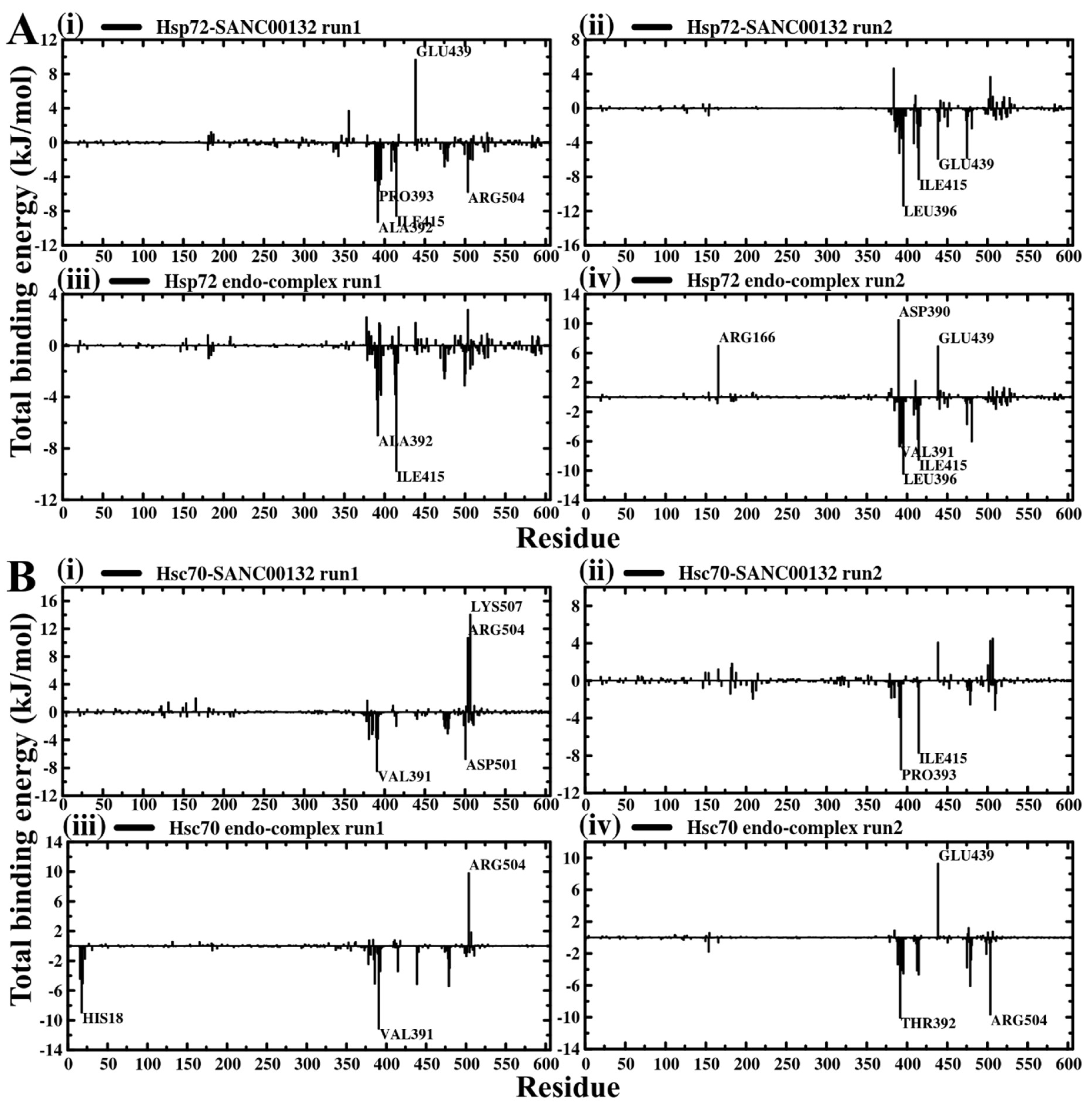
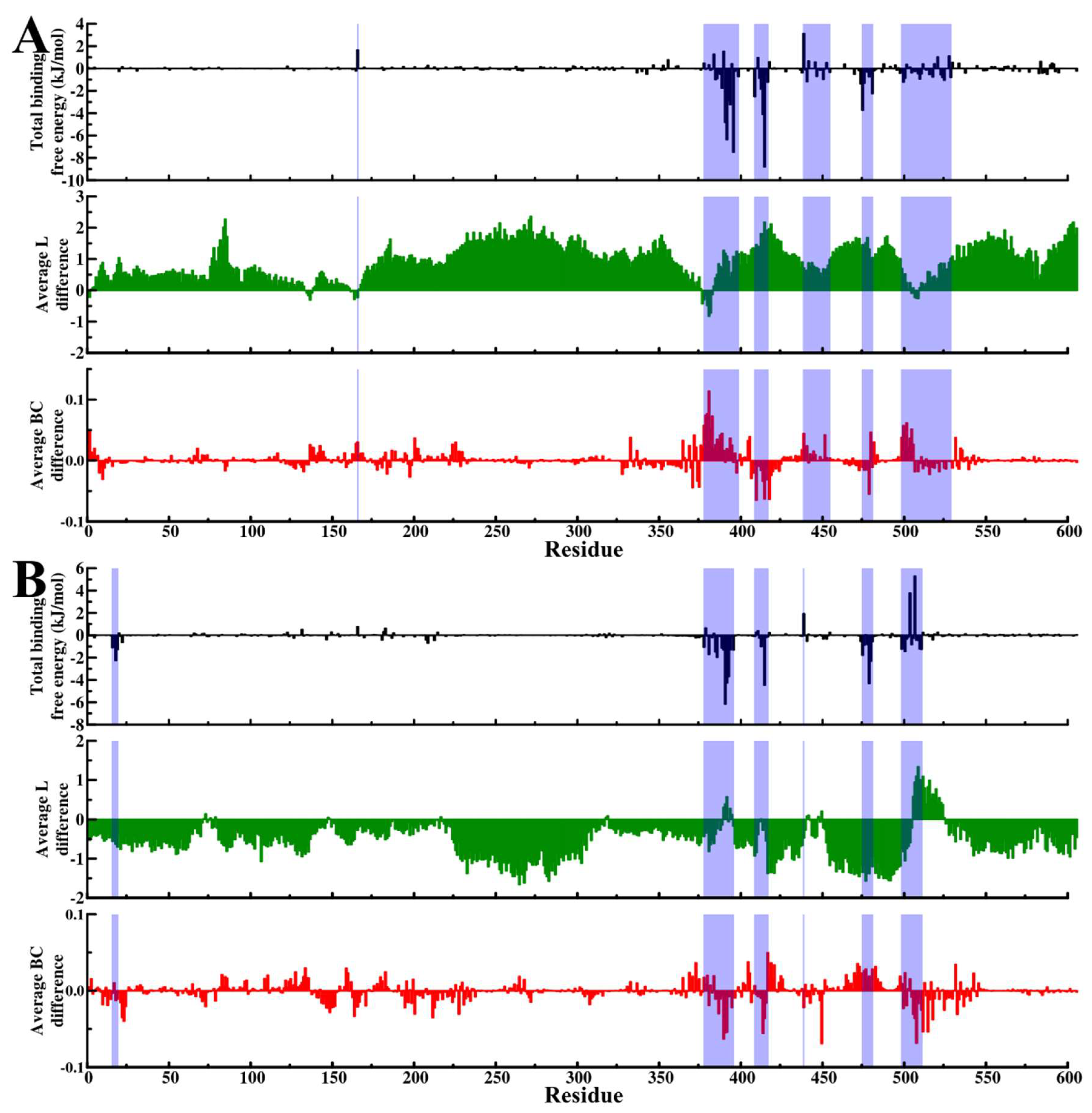
2.4. Evaluation of Binding Affinity Suggests Favorable Protein–Ligand Associations
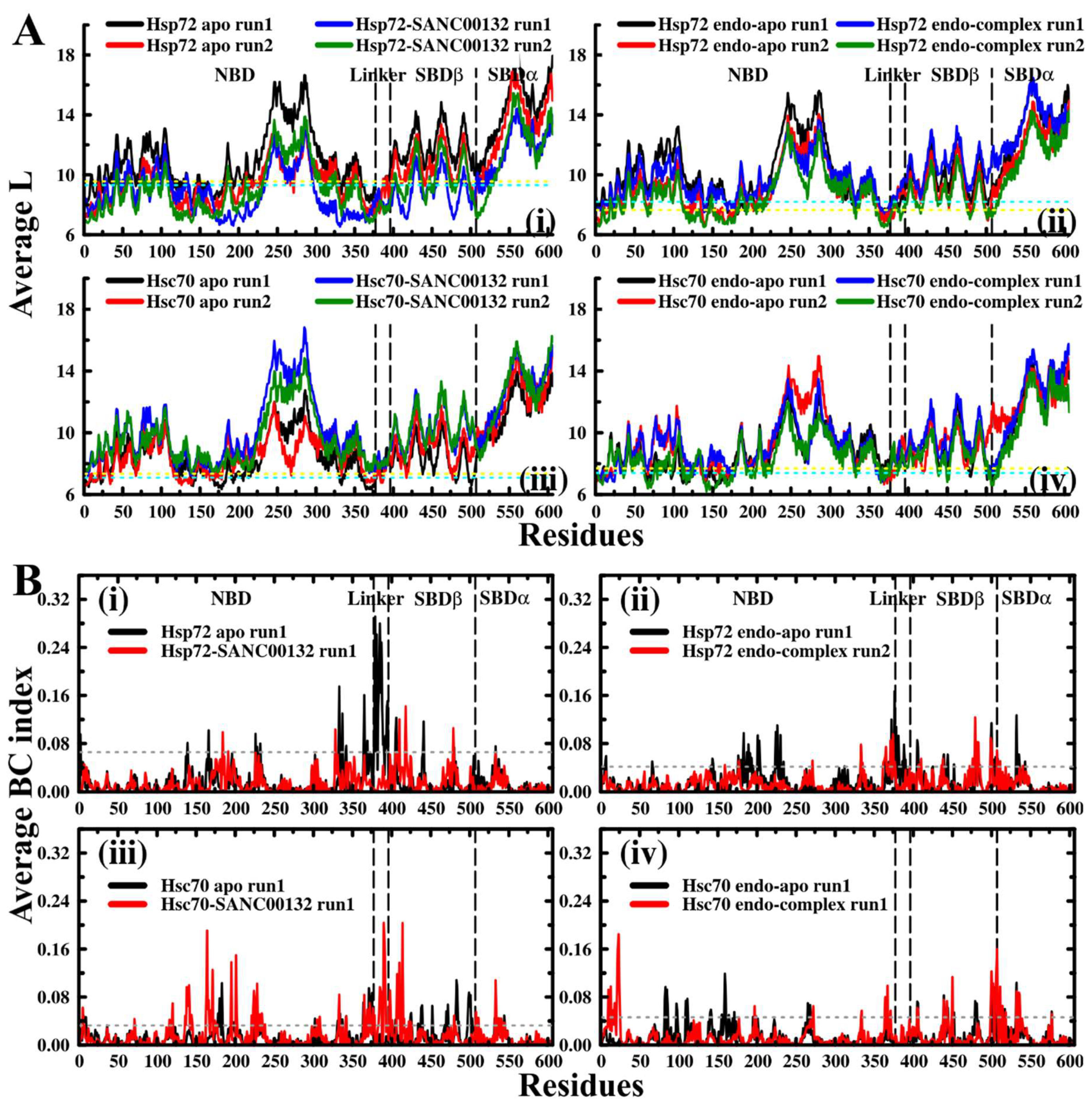
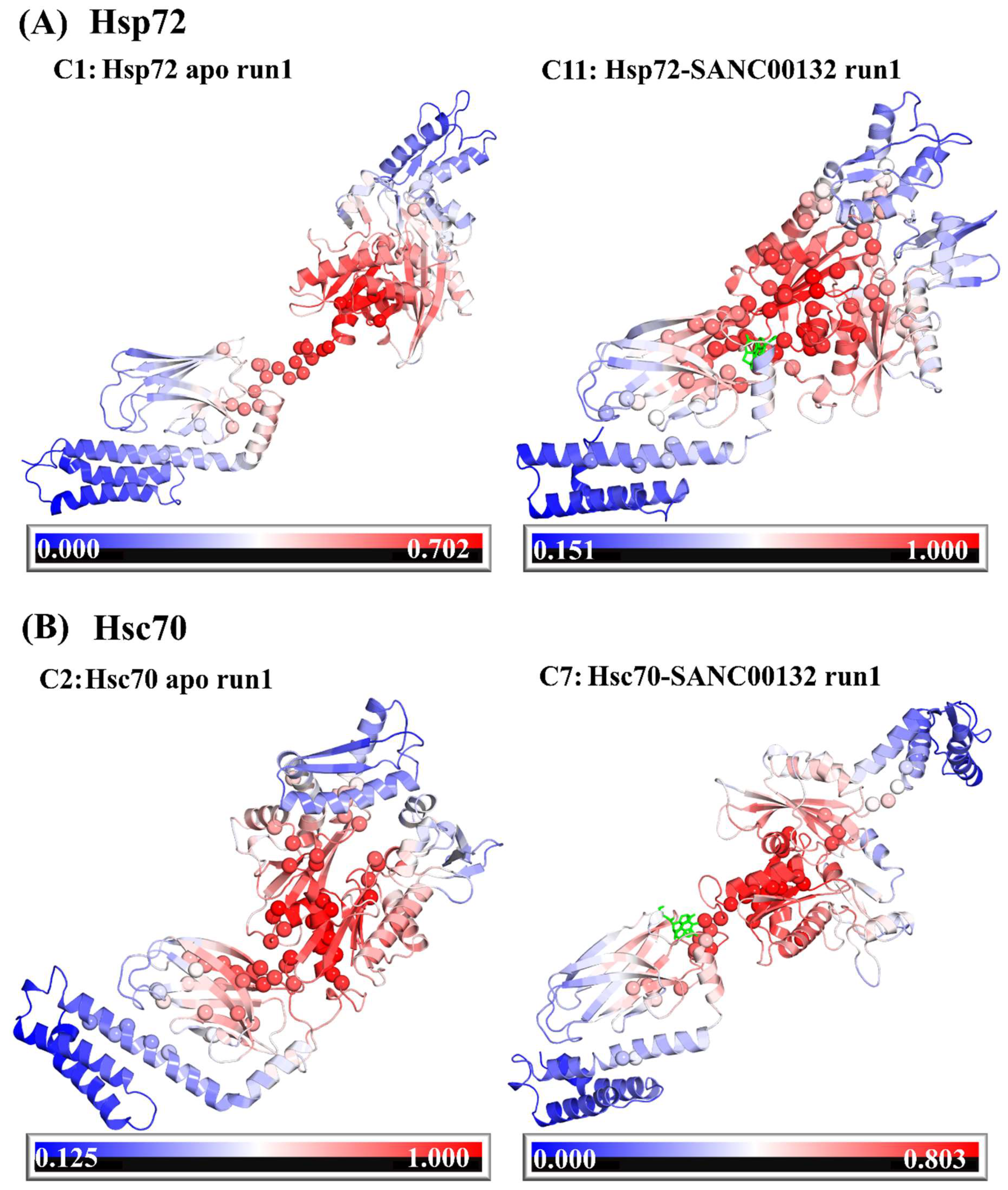
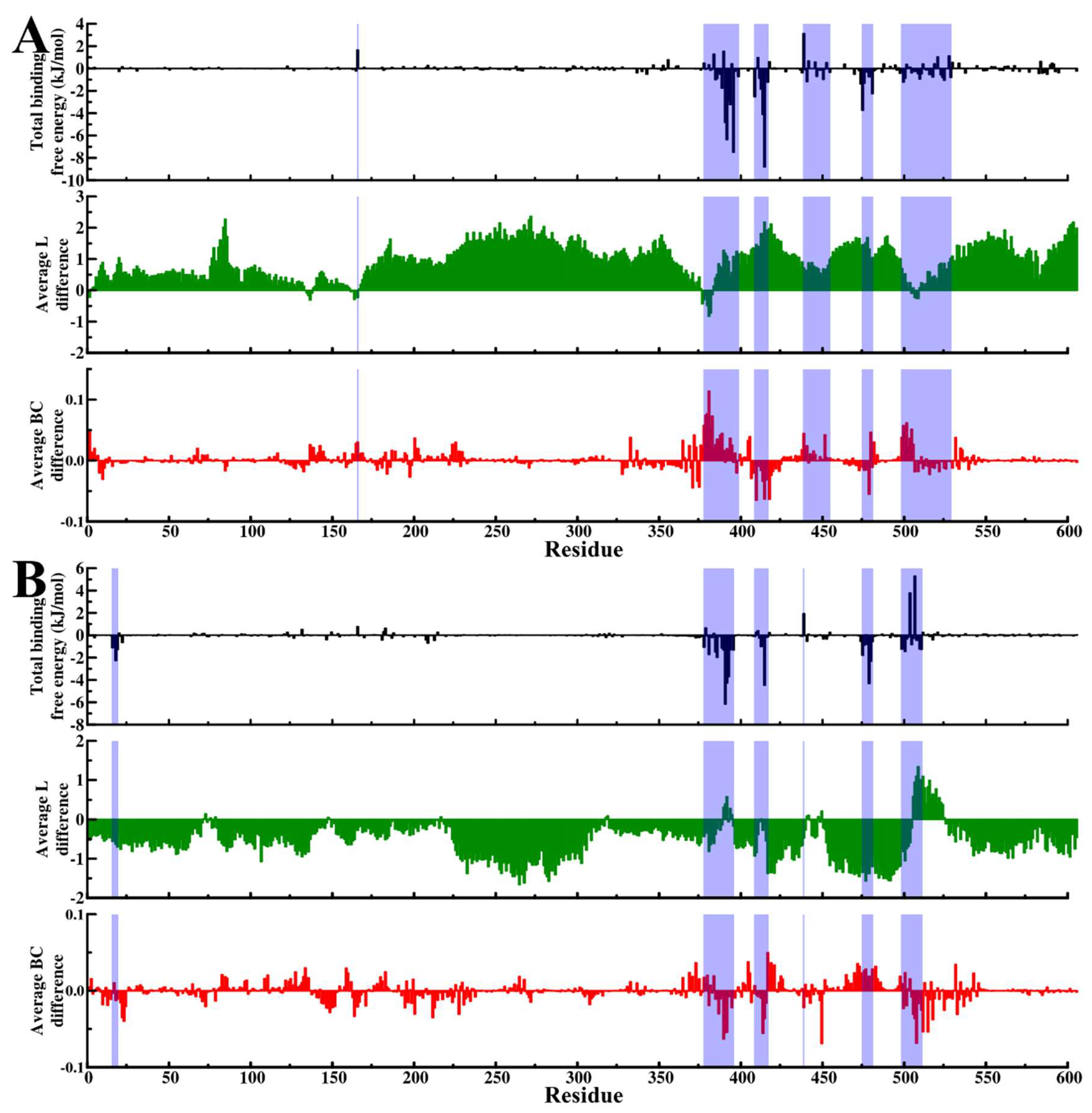
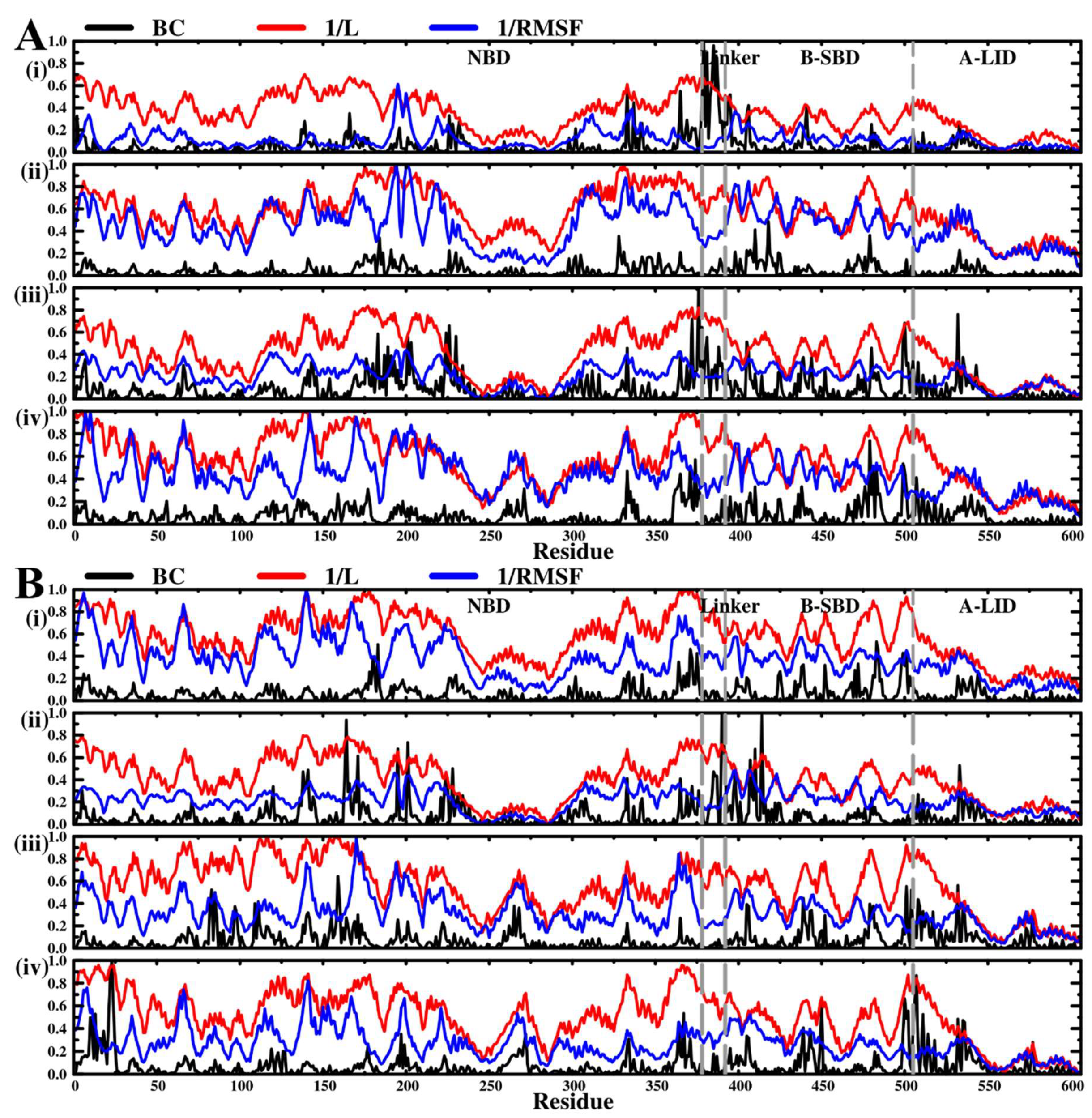
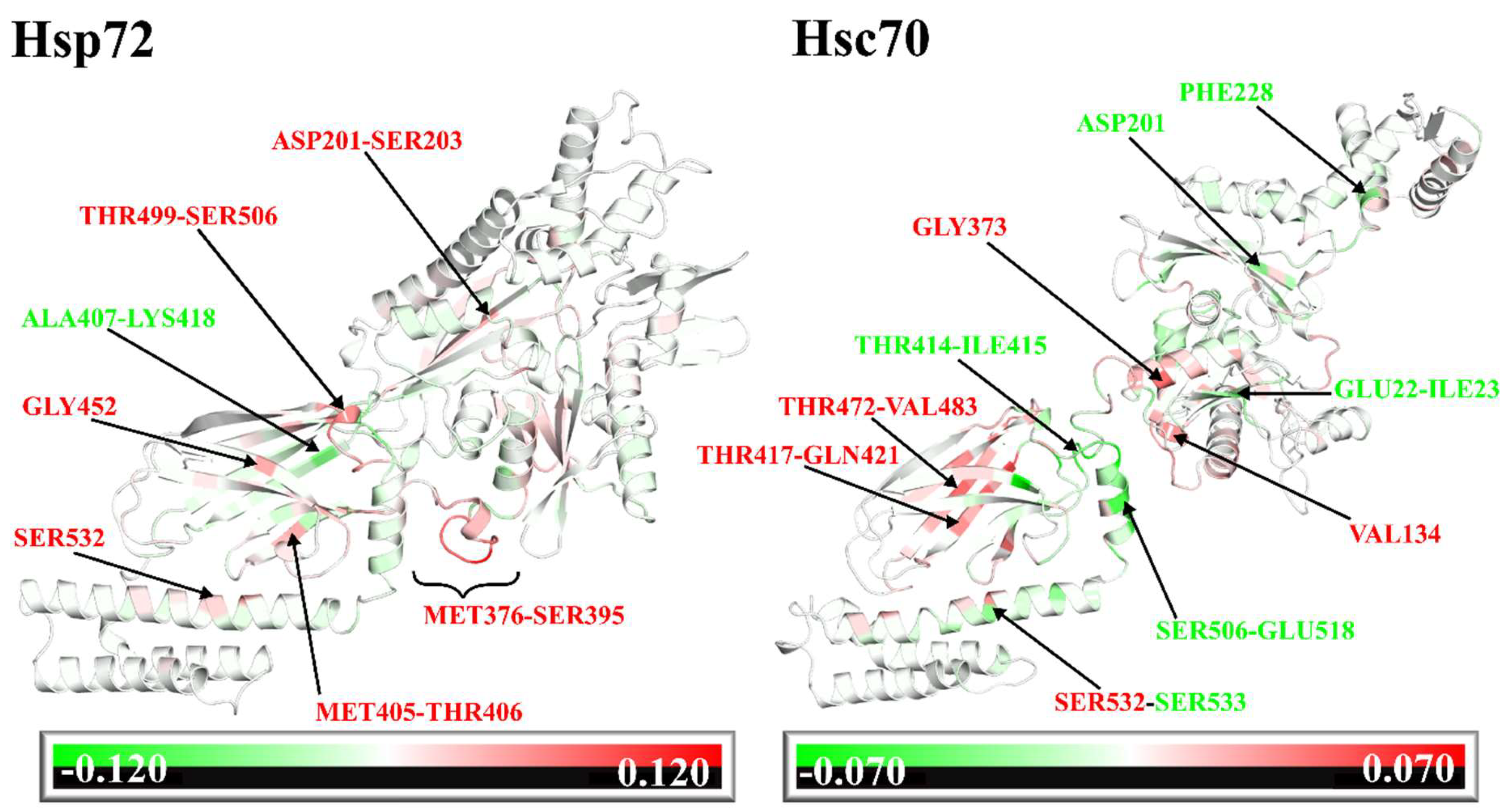
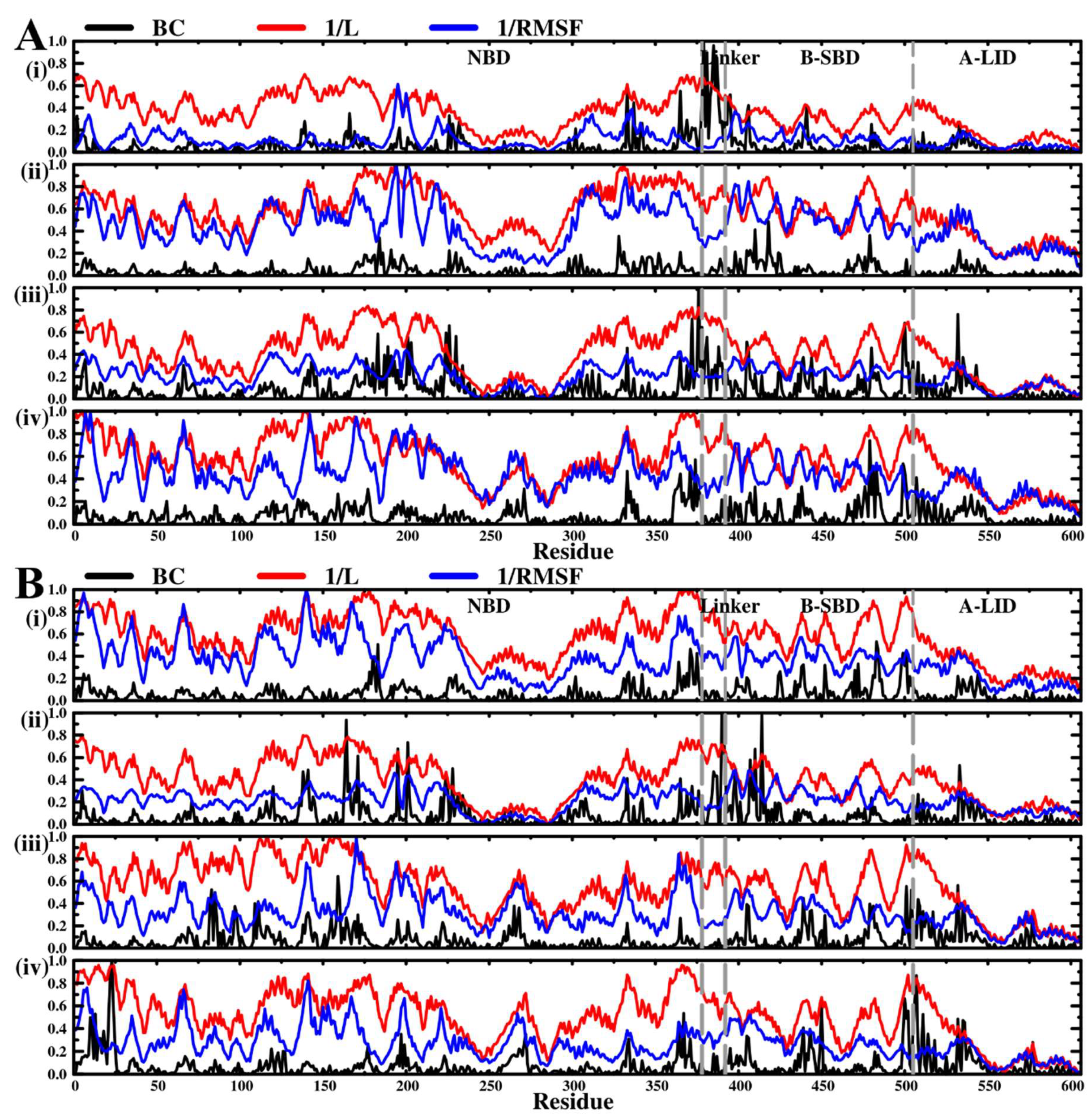
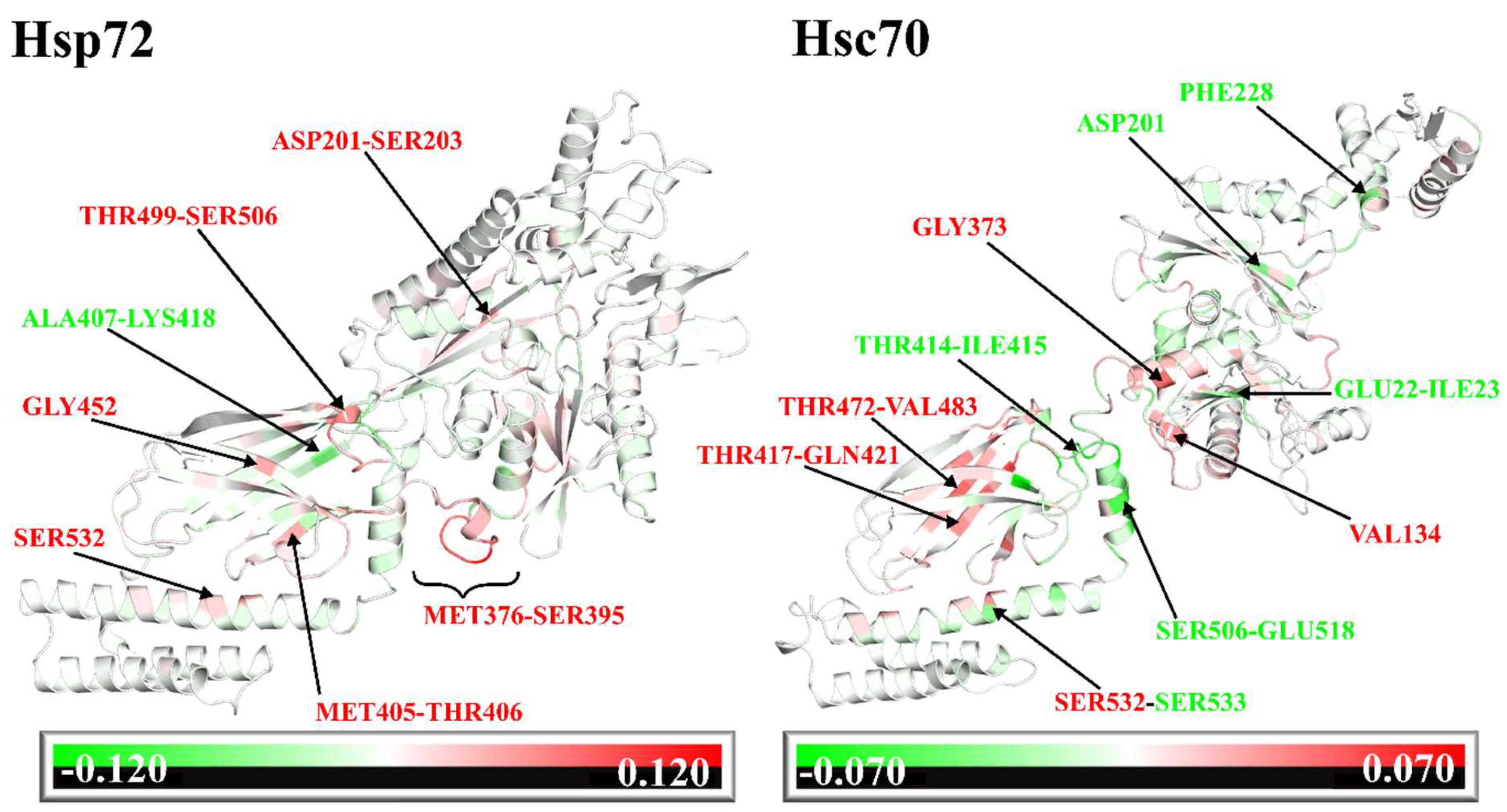
2.5. Dynamic Residue Network Analysis (DRN) Reveals Critical Communication Centers
Average L Compares to Average BC and Residue Fluctuations (RMSF)
2.6. Challenging the Capability of Discorhabdin N to Regulate Cross-Domain Communication
3. Materials and Methods
3.1. Sequence Retrieval and Homology Modelling
3.2. Virtual Screening and Druglikeness Prediction
3.3. Molecular Dynamics Studies
3.4. Trajectory Analysis
3.5. Binding Free Energy Calculations
3.6. Dynamic Residue Network Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, M.; Santarelli, R.; Gonnella, R.; Farina, A.; Trivedi, P.; Faggioni, A.; Cirone, M. Targeting of prosurvival pathways as therapeutic approaches against primary effusion lymphomas: Past, present, and Future. Biomed. Res. Int. 2015, 2015, 104912. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar]
- Navrátilová, J.; Hankeová, T.; Beneš, P.; Šmarda, J. Low-Glucose Conditions of Tumor Microenvironment Enhance Cytotoxicity of Tetrathiomolybdate to Neuroblastoma Cells. Nutr. Cancer 2013, 65, 702–710. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat Shock Proteins 27 and 70: Anti-Apoptotic Proteins with Tumorigenic Properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef] [Green Version]
- Gabai, V.L.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol. Cell. Biol. 2009, 29, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 2003, 228, 111–133. [Google Scholar] [CrossRef]
- Wegele, H.; Müller, L.; Buchner, J. Hsp70 and Hsp90—A relay team for protein folding. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin, Germany, 2004; pp. 1–44. [Google Scholar]
- Garrido, C.; Ottavi, P.; Fromentin, A.; Hammann, A.; Arrigo, A.P.A.; Chauffert, B.; Mehlen, P. HSP27 as a mediator of confluence-dependent resistance to cell death induced by anticancer drugs. Cancer Res. 1997, 57, 2661–2667. [Google Scholar] [PubMed]
- Powers, M.V.; Clarke, P.A.; Workman, P. Dual Targeting of HSC70 and HSP72 Inhibits HSP90 Function and Induces Tumor-Specific Apoptosis. Cancer Cell 2008, 14, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An Interdomain Energetic Tug-of-War Creates the Allosterically Active State in Hsp70 Molecular Chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Penkler, D.; Sensoy, Ö.; Atilgan, C.; Tastan Bishop, Ö. Perturbation-Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017, 57, 1359–1374. [Google Scholar] [CrossRef]
- Sharma, D.; Masison, D.C. Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept. Lett. 2009, 16, 571–581. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, X.; Burkholder, W.F.; Gragerov, A.; Ogata, C.M.; Gottesman, M.E.; Hendrickson, W.A. Structural analysis of substrate binding by the molecular chaperone DnaK. Science 1996, 272, 1606–1614. [Google Scholar] [CrossRef]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 Chaperone Machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Zhuravleva, A.; Gierasch, L.M. Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc. Natl. Acad. Sci. 2015, 112, E2865–E2873. [Google Scholar] [CrossRef] [PubMed]
- Slepenkov, S.V.; Witt, S.N. Kinetics of the Reactions of the Escherichia coli Molecular Chaperone DnaK with ATP: Evidence That a Three-Step Reaction Precedes ATP Hydrolysis. Biochemistry 1998, 37, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, R.; Brown, D.K.; Glenister, M.; Tastan Bishop, Ö. PRIMO: An Interactive Homology Modeling Pipeline. PLoS ONE 2016, 11, e0166698. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; D’Annessa, I.; Moroni, E.; Morra, G.; Paladino, A.; Rinaldi, S.; Compostella, F.; Colombo, G. Allosteric Modulators of HSP90 and HSP70: Dynamics Meets Function through Structure-Based Drug Design. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Karzai, A.W.; McMacken, R. A bipartite signaling mechanism involved in DnaJ-mediated activation of the Escherichia coli DnaK protein. J. Biol. Chem. 1996, 271, 11236–11246. [Google Scholar] [CrossRef] [PubMed]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Gierasch, L.M. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc. Natl. Acad. Sci. USA. 2011, 108, 6987–6992. [Google Scholar] [CrossRef]
- Hayer-Hartl, M.; Martin, J.; Hartl, F.; Christen, P.; Kuriyan, J. Asymmetrical interaction of GroEL and GroES in the ATPase cycle of assisted protein folding. Science 1995, 269, 836–841. [Google Scholar] [CrossRef]
- Harrison, C. GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperones 2003, 8, 218–224. [Google Scholar] [CrossRef]
- Liu, Y.; Gierasch, L.M.; Bahar, I. Role of Hsp70 ATPase Domain Intrinsic Dynamics and Sequence Evolution in Enabling its Functional Interactions with NEFs. PLoS Comput. Biol. 2010, 6, e1000931. [Google Scholar] [CrossRef]
- Woo, H.J.; Jiang, J.; Lafer, E.M.; Sousa, R. ATP-induced conformational changes in Hsp70: Molecular dynamics and experimental validation of an in silico predicted conformation. Biochemistry 2009, 48, 11470–11477. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.F.; Schulz, E.G.; Gierasch, L.M. Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J. Biol. Chem. 2006, 281, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Gassler, C.S.; Wiederkehr, T.; Brehmer, D.; Bukau, B.; Mayer, M.P. Bag-1M accelerates nucleotide release for human Hsc70 and Hsp70 and can act concentration-dependent as positive and negative cofactor. J. Biol. Chem. 2001, 276, 32538–32544. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A Small Molecule Inhibitor of Inducible Heat Shock Protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L. Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin: Modulation of HSC70 ATPase activity without compromising DnaJ chaperone interactions. Biochem. Pharmacol. 1999, 57, 877–880. [Google Scholar] [CrossRef]
- Dal Piaz, F.; Cotugno, R.; Lepore, L.; Vassallo, A.; Malafronte, N.; Lauro, G.; Bifulco, G.; Belisario, M.A.; De Tommasi, N. Chemical proteomics reveals HSP70 1A as a target for the anticancer diterpene oridonin in Jurkat cells. J. Proteomics 2013, 82, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.Q.; Kirby, C.A.; Zhou, W.; Schuhmann, T.; Kityk, R.; Kipp, D.R.; Baird, J.; Chen, J.; Chen, Y.; Chung, F.; et al. The Novolactone Natural Product Disrupts the Allosteric Regulation of Hsp70. Chem. Biol. 2015, 22, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Pettinger, J.; Le Bihan, Y.V.; Widya, M.; van Montfort, R.L.M.; Jones, K.; Cheeseman, M.D. An Irreversible Inhibitor of HSP72 that Unexpectedly Targets Lysine-56. Angew. Chem. Int. Ed. Engl. 2017, 56, 3536–3540. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.; Westwood, I.M.; Osborne, J.D.; Matthews, T.P.; Cheeseman, M.D.; Rowlands, M.G.; Jeganathan, F.; Burke, R.; Lee, D.; Kadi, N.; et al. A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Sci. Rep. 2016, 6, 34701. [Google Scholar] [CrossRef] [Green Version]
- Rodina, A.; Patel, P.D.; Kang, Y.; Patel, Y.; Baaklini, I.; Wong, M.J.H.; Taldone, T.; Yan, P.; Yang, C.; Maharaj, R.; et al. Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem. Biol. 2013, 20, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.J.; Zhang, P.; Murphy, M.E.; Marmorstein, R.; George, D.L. Structural basis for the inhibition of HSP70 and DnaK chaperones by small-molecule targeting of a C-terminal allosteric pocket. ACS Chem. Biol. 2014, 9, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- 45 Stetz, G.; Verkhivker, G.M. Probing Allosteric Inhibition Mechanisms of the Hsp70 Chaperone Proteins Using Molecular Dynamics Simulations and Analysis of the Residue Interaction Networks. J. Chem. Inf. Model. 2016, 56, 1490–1517. [Google Scholar] [CrossRef] [PubMed]
- Penkler, D.; Tastan Bishop, Ö. Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Tastan Bishop, Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.F.; Fan, H.; Xiong, J.; Wu, S.B. Discorhabdins and Pyrroloiminoquinone-Related Alkaloids. Chem. Rev. 2011, 111, 5465–5491. [Google Scholar] [CrossRef]
- Antunes, E.M.; Beukes, D.R.; Kelly, M.; Samaai, T.; Barrows, L.R.; Marshall, K.M.; Sincich, C.; Davies-Coleman, M.T. Cytotoxic Pyrroloiminoquinones from Four New Species of South African Latrunculid Sponges. J. Nat. Prod. 2004, 67, 1268–1276. [Google Scholar] [CrossRef]
- Harris, E.; Strope, J.; Beedie, S.; Huang, P.; Goey, A.; Cook, K.; Schofield, C.; Chau, C.; Cadelis, M.; Copp, B.; et al. Preclinical Evaluation of Discorhabdins in Antiangiogenic and Antitumor Models. Mar. Drugs 2018, 16, 241. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided. Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chen, Y.F.; Shen, T.W.; Kristal, B.S.; Hsu, D.F. Consensus Scoring Criteria for Improving Enrichment in Virtual Screening. J. Chem. Inf. Model. 2005, 45, 1134–1146. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Screening-Based Translation of Public Research Encounters Painful Problems. ACS Med. Chem. Lett. 2015, 6, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the Utility of Alerts for pan-assay interference Compounds. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, J.P.; Subedi, Y.P.; Chen, L.; Chang, C.W.T. A mode of action study of cationic anthraquinone analogs: A new class of highly potent anticancer agents. Medchemcomm 2015, 6, 2012–2022. [Google Scholar] [CrossRef]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Kumar, S.; Tsai, C.J.; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng. Des. Sel. 1999, 12, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.J.; Nussinov, R. The free energy landscape in translational science: How can somatic mutations result in constitutive oncogenic activation? Phys. Chem. Chem. Phys. 2014, 16, 6332. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- BIOVA Discovery Studio. Available online: http://accelrys.com/products/collaborative-science/biovia-discovery-studio/ (accessed on 27 May 2016).
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Li, H.Y.; Wang, J.H. Folding rate prediction using complex network analysis for proteins with two- and three-state folding kinetics. J. Biomed. Sci. Eng. 2009, 02, 644–650. [Google Scholar] [CrossRef]
- del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006, 2, 2006.0019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, D.; Paci, P.; Paola, L.D.; Giuliani, A. Are Proteins Just Coiled Cords? Local and Global Analysis of Contact Maps Reveals the Backbone-Dependent Nature of Proteins. Ingenta Connect 2016, 17. [Google Scholar] [CrossRef]
- Penkler, D.; Atilgan, C.; Tastan Bishop, Ö. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Model. 2018, 58, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Sarbeng, E.B.; Liu, Q.; Le, K.Q.; Xu, X.; Xu, H.; Yang, J.; Wong, J.L.; Vorvis, C.; Hendrickson, W.A.; et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 2013, 20, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Leu, J.I.-J.; Murphy, M.E.; George, D.L.; Marmorstein, R. Crystal Structure of the Stress-Inducible Human Heat Shock Protein 70 Substrate-Binding Domain in Complex with Peptide Substrate. PLoS ONE 2014, 9, e103518. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. IUCr PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Tastan Bishop, Ö. Structure Based Docking and Molecular Dynamic Studies of Plasmodial Cysteine Proteases against a South African Natural Compound and its Analogs. Sci. Rep. 2016, 6, 23690. [Google Scholar] [CrossRef] [PubMed]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Tastan Bishop, Ö. Analysis of non-peptidic compounds as potential malarial inhibitors against Plasmodial cysteine proteases via integrated virtual screening workflow. J. Biomol. Struct. Dyn. 2016, 34, 2084–2101. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Penkler, D.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Energy Terms | ΔGbinding (kJ·mol−1) | ||||
|---|---|---|---|---|---|---|
| ΔEvdW | ΔEelec | ΔGpolar | ΔGnonpolar | |||
| Hsp72-SANC00132 complex | Run1 | −182.161 ± 0.275 | −75.159 ± 0.289 | 167.323 ± 0.497 | −15.361 ± 0.018 | −105.338 ± 0.378 |
| Run2 | −201.464 ± 0.348 | −101.914 ± 0.243 | 194.286 ± 0.231 | −17.317 ± 0.015 | −126.402 ± 0.328 | |
| Hsp72 endo-complex | Run1 | −189.941 ± 0.311 | −67.232 ± 0.257 | 195.474 ± 0.263 | −16.650 ± 0.017 | −78.365 ± 0.321 |
| Run2 | −201.786 ± 0.365 | −131.232 ± 0.260 | 241.696 ± 0.303 | −16.901 ± 0.016 | −108.205 ± 0.330 | |
| Hsc70-SANC00132 complex | Run1 | −155.398 ± 0.331 | −64.820 ± 1.132 | 161.916 ± 1.473 | −15.631 ± 0.028 | −73.973 ± 0.547 |
| Run2 | −137.056 ± 0.277 | −77.472 ± 0.305 | 155.394 ± 0.390 | −15.298 ± 0.017 | −74.006 ± 0.542 | |
| Hsc70 endo-complex | Run1 | −196.548 ± 0.271 | −49.657 ± 0.290 | 143.562 ± 0.318 | −17.164 ± 0.017 | −119.818 ± 0.282 |
| Run2 | −189.970 ± 0.248 | −43.352 ± 0.204 | 140.112 ± 0.252 | −16.014 ± 0.016 | −109.226 ± 0.255 | |


| Protein | L vs. RMSF (100 ns) | L vs. RMSF (last 15 ns) | L−1 vs. BC | RMSF−1 (100 ns) vs. BC | RMSF−1 (last 15 ns) vs. BC |
|---|---|---|---|---|---|
| Hsp72 apo Run1 | 0.53 | 0.45 | 0.39 | 0.12 | 0.10 |
| Hsp72 apo Run2 | 0.57 | 0.53 | 0.39 | 0.07 | 0.11 |
| Hsp72-SANC00132 Run1 | 0.43 | 0.77 | 0.51 | 0.15 | 0.37 |
| Hsp72-SANC00132 Run2 | 0.64 | 0.74 | 0.49 | 0.12 | 0.27 |
| Hsp72 endo-apo Run1 | 0.56 | 0.80 | 0.45 | 0.19 | 0.30 |
| Hsp72 endo-apoRun2 | 0.61 | 0.78 | 0.45 | 0.24 | 0.28 |
| Hsp72 endo-complex Run1 | 0.69 | 0.38 | 0.43 | 0.23 | 0.03 |
| Hsp72 endo-complex Run2 | 0.58 | 0.68 | 0.48 | 0.16 | 0.28 |
| Hsc70 apo Run1 | 0.58 | 0.87 | 0.49 | 0.10 | 0.37 |
| Hsc70 apo Run2 | 0.62 | 0.72 | 0.48 | 0.15 | 0.13 |
| Hsc70-SANC00132 Run1 | 0.74 | 0.77 | 0.40 | 0.32 | 0.30 |
| Hsc70-SANC00132 Run2 | 0.70 | 0.72 | 0.40 | 0.09 | 0.32 |
| Hsc70 endo-apo Run1 | 0.59 | 0.72 | 0.46 | 0.12 | 0.30 |
| Hsc70 endo-apo Run2 | 0.76 | 0.80 | 0.42 | 0.25 | 0.28 |
| Hsc70 endo-complex Run1 | 0.60 | 0.77 | 0.43 | 0.15 | 0.22 |
| Hsc70 endo-complex Run2 | 0.51 | 0.58 | 0.45 | 0.04 | 0.30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amusengeri, A.; Tastan Bishop, Ö. Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis. Molecules 2019, 24, 188. https://doi.org/10.3390/molecules24010188
Amusengeri A, Tastan Bishop Ö. Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis. Molecules. 2019; 24(1):188. https://doi.org/10.3390/molecules24010188
Chicago/Turabian StyleAmusengeri, Arnold, and Özlem Tastan Bishop. 2019. "Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis" Molecules 24, no. 1: 188. https://doi.org/10.3390/molecules24010188
APA StyleAmusengeri, A., & Tastan Bishop, Ö. (2019). Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis. Molecules, 24(1), 188. https://doi.org/10.3390/molecules24010188