
Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery
by
 , and
, and
Stephani Joy Y. Macalino
† ,
,
Shaherin Basith
†,
Nina Abigail B. Clavio
,
Hyerim Chang
,
Soosung Kang
* and
Sun Choi
*
College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, Korea
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2018, 23(8), 1963; https://doi.org/10.3390/molecules23081963
Submission received: 17 July 2018
/
Revised: 3 August 2018
/
Accepted: 4 August 2018
/
Published: 6 August 2018
(This article belongs to the Special Issue Frontiers in Computational Chemistry for Drug Discovery)
Abstract
:The advent of advanced molecular modeling software, big data analytics, and high-speed processing units has led to the exponential evolution of modern drug discovery and better insights into complex biological processes and disease networks. This has progressively steered current research interests to understanding protein-protein interaction (PPI) systems that are related to a number of relevant diseases, such as cancer, neurological illnesses, metabolic disorders, etc. However, targeting PPIs are challenging due to their “undruggable” binding interfaces. In this review, we focus on the current obstacles that impede PPI drug discovery, and how recent discoveries and advances in in silico approaches can alleviate these barriers to expedite the search for potential leads, as shown in several exemplary studies. We will also discuss about currently available information on PPI compounds and systems, along with their usefulness in molecular modeling. Finally, we conclude by presenting the limits of in silico application in drug discovery and offer a perspective in the field of computer-aided PPI drug discovery.
1. Introduction
Rational drug discovery and design has progressed at a precipitous pace with the aid of in silico strategies and innovations in hardware and computational power. While still in its infancy as compared to traditional drug discovery techniques, computer-aided drug discovery (CADD) has helped deliver several success stories [1,2,3,4,5,6,7,8,9,10,11] that inspired confidence in its continuous application in the pharmaceutical industry. CADD lends cost- and labor-efficiency in the identification of potential hits for a therapeutic target before delving into extensive experimental assays. The use of computational tools requires the availability of vast amounts of information on protein and ligand structure, protein function, and an expert grasp of intermolecular forces and energies necessary for the interaction between binding partners. Its benefits encompass the different stages of drug discovery and development, including target and hit identification, structure-activity relationship studies, compound optimization, and analysis and prediction of lead pharmacokinetic properties. Nevertheless, like any other approaches, in silico methods are not infallible and are more valuable when they are employed in combination with other drug discovery tools.
In the last couple of decades, protein-protein interactions (PPIs) have become popular therapeutic targets. Proteins interact with other biologically important molecules such as peptides, proteins (homo or hetero), DNA, and RNA to carry out their functions. In general, PPIs are involved in effecting regulatory changes in response to external stimuli. Different PPI targets have been implicated in cancer, metabolic diseases, neurological disorders, and many other diseases [12,13]. The data generated through experimental techniques for these targets are often hindered in terms of manpower, cost, time, accuracy, and interactome coverage [14]. In light of this, relying only on experimental techniques may be detrimental to research efforts, as massive amounts of resources are required to fully explore the human protein interactome. Moreover, PPIs continue to be difficult targets for drug discovery efforts, due to the differences in PPI binding site properties and ligand chemical space when compared to traditional protein targets. As a solution, computational tools have been utilized to fill in the gaps, and to supplement experimental methods in the investigation of these complex targets.
In this review, we will initially discuss the significance of PPIs as therapeutic targets in the drug discovery paradigm, and how this has been dealt with so far in the research field. Next, we will expound on how various in silico methods and recent advances in this field can facilitate PPI drug discovery, as exemplified in several case studies. Lastly, the benefits and pitfalls of CADD techniques in relation to PPI drug discovery will be reviewed. Due to the breadth of PPI drug discovery, we limit the scope of this review to the computer-aided rational discovery and the development of small molecules, peptides, and peptidomimetics for PPI.
2. Relevance of PPIs in the Drug Discovery Paradigm
With an estimated 650,000 PPIs as part of the human interactome [15], it is evident that these interactions play crucial roles in various cellular processes and pathways. Dysregulation in PPIs are often found to be the primary cause of several disease pathologies, making them attractive drug targets. However, the development of PPI inhibitors and stabilizers have been hindered because of the seemingly low druggability of PPI interfaces. Extensive studies for PPI targets and modulators have been performed to understand these complex targets and identify distinct properties in its network, conformational structure, and ligand chemical space. PPI druggability continues to be poorly understood because these targets show substantial diversity as compared to protein families that have traditionally been targeted until now. Despite this, its relevance to multiple diseases and relative novelty in the drug discovery field encourage researchers to pursue these difficult targets. Moreover, the discovery of PPI inhibitors and therapeutics (Table 1) in the last few decades have ascertained that these targets are tractable and can be modulated by small molecule compounds.
2.1. Structural Features of PPIs
PPI interfaces are shallow and highly hydrophobic with large contact surfaces (>1000 Å2) [16,17] that were thought to be involved in their entirety for the formation of protein complexes. Due to this, PPI targets were portrayed as “intractable.” Later, alanine scanning experiments revealed the existence of “hot spots,” which are crucial residues in the protein-protein binding interface that act as chief contributors to the binding free energy. Hot spots correspond to structurally conserved regions primarily composed of tryptophan, isoleucine, arginine, and tyrosine, and often form clusters where compounds can potentially bind [18,19]. Though hot spots are frequently bundled closely together in a PPI interface, there are cases where regions are separated but still work together to allow tight attachment [20]. Aside from the presence of hot spots, other parameters, including contact surface area, polarity, flatness, and buriedness, have been employed to characterize PPI interfaces [21,22,23,24,25,26]. Generally, a PPI interface is split into a core and a rim region. The core region is buried, and consists of residues with higher hydrophobicity and conservation, whereas the rim region is in the adjacent solvent-accessible area with more polar and flexible residues [27,28,29,30]. Because the core regions are more buried and can form interfacial grooves, PPI inhibitors often target these areas and are therefore usually hydrophobic.
Different types of PPI complexes are found in biological systems. Obligate complexes are PPIs that require permanent interaction between protein partners or subunits to establish function, such as in the cases of the P22 Arc repressor [74] and V-ATPase [75]. In contrast, non-obligate PPIs have both stable functioning complexes and protomers in vivo [76], like the GroEL-GroES chaperonine system [77]. Obligate complex interfaces are usually more hydrophobic than non-obligate complex interfaces, which show higher polarities in order to exist as independent protomers in solution [17,78]. Protein partners can also bind to each other in a transient fashion, such as with key interactions involved in cell signaling and regulatory pathways. Weak transient PPIs are commonly formed by disordered proteins or oligomers that have several states in vivo, leading to interactions that are easily formed or broken based on the conformation of each protein partner. On the other hand, strong transient complexes require a molecular trigger, such as ligand binding or post-translational modification (PTM), to instigate a change in complex structure [79]. Weak transient complexes are usually mediated by changes in pH or temperature, and have small and planar surfaces, while those influenced by strong molecular triggers are often larger and more hydrophobic [80]. PPI interfaces with a surface area larger than 1000 Å2 are noted to be more flexible and prone to induced-fit conformational changes [17,80].
PPI complexes display a wide range of affinity and stability. However, high specificity is still observed, even with proteins that have multiple partners or “protein hubs” [81]. When dealing with binding specificity of protein hubs, key aspects include molecular recognition and binding affinity. Orientation and electrostatic complementarity between protein partners are found to be crucial for identifying the correct protein partner and forming a pre-complex. Subsequently, short range interactions between partner hot spot regions establish the binding affinity for the stabilization of the complex [82]. PPI binding interfaces exhibit various means of mediating specificity of interaction, including changes in conformation, binding site properties, and the presence of specificity-determining sites.
While hot spots are considered to be the main contributors for binding affinity and stability, not all hot spots participate in specificity, as a portion are often shared among several partners that bind to the same site of a protein hub. Separate specificity-determining hot spots that can distinguish between cognate and non-cognate partners are present in PPI binding interfaces [81,83]. The presence of anchor residues and anchoring grooves have been remarked upon as critical factors for molecular recognition between protein partners [84]. This was observed in a study done by Kimura et al. [85] wherein mutation of Lys15 of the bovine pancreatic trypsin inhibitor led to a vast decrease in association rate with trypsin. On the other hand, mutating Arg17 allowed association with trypsin but resulted in a large increase in the off rate. These indicate that although both can be classified as hot spots, each residue contributes distinctive functions in the PPI complex—Lys15 is required for recognition and initial binding, while Arg17 is critical for the stabilization of the complex [85]. For facilitated recognition and complex formation, Rajamani and colleagues [84] stated that anchor residues are usually in their bound-like conformations, while the rest of the hot regions are more flexible and buried in the unbound state. Aligned with this, molecular dynamics (MD) simulations of the receptor binding site did not exhibit any significant changes in pocket conformation or backbone rearrangements, suggesting that the grooves to which the anchor residues bind are pre-defined [84]. Interaction with several different partners can also constitute the presence of multiple binding sites, corresponding to distinct protein subunits, where various ligand proteins interact to elicit specific responses or functions [86]. Recognition of a specific protein subunit is influenced by the differences in amino acid (AA) compositions in the binding interface and the inherent diversity of PPI structural features, even among members of the same protein family [14,86].
PTM is an alternative process where proteins recognize different protein partners to carry out their specific functions. A protein can be modified using different PTMs in one or different residues at the same time, or in a sequential manner, to increase its complexity and functional scope. Data statistics from dbPTM show that over 60% of PTM sites are situated in protein functional domains involved in PPIs, implying that PTMs play a crucial role in the modulation of PPI function [87]. Phosphorylation is one of the most common type of PTMs found in cell signaling mechanisms and is frequently found on heterooligomeric and transient interfaces. It can regulate protein function by influencing specific recognition, allosteric regulation, protein modularity, and binding [88]. This is in agreement with a study conducted by Duan and Walther [89], where they noted that proteins subject to PTMs, most especially phosphorylation, are observed to have central roles and a wide interaction range in the human protein interactome [90].
Structural flexibility can also greatly affect binding affinity and specificity in PPIs. The dynamic nature of proteins allows them to alter conformations based on the required interactions for binding with a partner and inducing a corresponding function. Some proteins can subtly influence both specificity and affinity by altering the orientation of only one or a few conserved residues in the binding interface, such as MDM2 [91] and proteins with the Src homology 3 (SH3) domain [92,93]. In other cases, large global or local conformational differences are observed between different functional states. Conformational adjustment upon protein partner binding can be explained via two possible mechanisms: induced-fit [94], wherein conformational change occurs upon interaction of the partners, or population-shift [95], where it is postulated that there exists a dynamic equilibrium consisting of an ensemble of conformations from which a certain portion exhibit the conformation a particular partner preferentially binds with [95,96]. This paradigm is exhibited by intrinsically disordered proteins (IDPs) or regions (IDRs), which have high structural flexibility and diverse conformations in their native functional state. As opposed to the conventional theory, where a unique sequence defines a unique three-dimensional (3D) structure and function [97,98,99], IDPs and IDRs cover a wide range of conformational space, and have distinct structural arrangements at a given time point [97,100], enabling promiscuous and transient interactions with different protein partners. The high modularity of these structures is linked to crucial roles in cellular processes, and requires tight regulation via changes in subcellular concentration, diverse conformations, and PTMs [100,101].
For the design of efficient PPI modulators, a great level of understanding of the target is needed: the type of complex formed, elucidation of binding epitopes and hot spots, determination of binding site flexibility, etc. While more information is progressively coming to light regarding PPI binding requirements, further characterization of features that synchronize molecular recognition, affinity, and specificity within the target protein interface is vital in designing therapeutics for a specific PPI and function to improve selectivity and off-target toxicity.
2.2. Characteristics of PPI Modulators
Increased understanding of druggability and molecular recognition in PPIs has undoubtedly stimulated drug discovery efforts for these previously intractable targets, offering hope in identifying small molecule candidates for PPI-regulated pathways. However, the development of PPI modulators is still at a relatively slow pace in comparison to conventional small molecule drugs, due to the challenges conferred by the structural optimization required to improve the affinity and pharmacokinetic properties of the PPI ligands. The complexity of a PPI inhibitor primarily depends on the interface structure, and this is usually equivalent to that of its targets. Analysis of known PPI modulators showed that these compounds are larger, more hydrophobic, and contain more multiple bonds and aromatic rings [102,103,104]. Interestingly, PPI ligands have a higher topological polar surface area (TPSA), even with its high hydrophobicity, as compared to traditional drugs, resulting in their tendency to form more hydrogen bonds in the PPI surface. This can be attributed to their larger sizes that allow for the attachment of more polar atoms [103]. More success is acquired for PPIs with grooves or small pockets, compared to globular interfaces. However, those with hot spot pockets that are spread out across a large interface are naturally more challenging. Because of this, PPI inhibitors also tend to have a more 3D conformation than the average inhibitor [105,106], especially for extended surfaces and disjointed pockets, due to its propensity to mimic the binding epitope of the partner protein and anchor into small pockets present in PPI interfaces [104,106,107,108]. Based on these, it is evident that PPI inhibitors occupy a different chemical space as compared to typical small molecule inhibitors, and hence, should be carefully evaluated in a different manner [103,108]. Whereas the primarily hydrophobic interacting features of PPI inhibitors can be buried in hot spot regions, the rest of their structures are often solvent-exposed due to their binding location on the protein interface. These elements can be exploited to tailor pharmacokinetic properties without negatively influencing binding affinity [14].
Studies suggest that 15–40% of the human interactome is comprised of protein-peptide interactions [109]. Natural peptides have been used as leads for PPI inhibition [110,111,112] due to their biocompatibility, low toxicity, and high modularity, which can aid in both potency and selectivity [113]. However, these ligands tend to have low bioavailability because of their high propensity for proteolysis, making them poor drug candidates. Several other approaches are utilized for the discovery, design, and optimization of PPI inhibitors: (a) using unnatural AAs and other synthetic modifications to the peptide backbone for the design of peptide mimics (i.e., peptidomimetics) to improve bioactivity and pharmacokinetic properties of peptide leads; (b) exploiting natural cyclic and macrocyclic peptides that are large enough to bind to a PPI interface while still possessing favorable properties that can overcome proteolysis and difficulties in cell permeability; (c) designing and engineering miniproteins via phage display methods to target large PPI interfaces with high specificity [114,115]; (d) fragment-based drug design (FBDD) to identify low molecular weight (MW) fragments that targets hot spot clusters [116,117].
Traditionally, potency was the chief facet used for the early evaluation of lead candidates. However, it has been established that potency alone does not completely explain bioactivity against a target, and other physicochemical properties should also be considered [118]. Advances in organic and combinatorial chemistry, as well as changes in therapeutic target profiles, resulted in the growth of both chemical and drug-like spaces over the years [119,120]. Consequently, the rubrics that were often used to evaluate drug candidates before, have significantly evolved. Since PPI inhibitors are generally bigger and more hydrophobic than conventional small molecule drugs, and it is difficult to accurately assess its drug-likeness using typical metrics like Lipinski [121] and Veber rules [122]. Recent assessment of current drugs gave rise to beyond-rule of 5 (bRO5) characterization of orally bioavailable compounds [123,124]. Physicochemical properties that were noted by this novel classification, correlate well with typical PPI modulator features. Moreover, ligand efficiency (LE) and lipophilic ligand efficiency (LLE) are now more commonly used to determine drug-likeness. LE quantifies the contribution of a molecule’s structure to binding affinity, whereas LLE estimates a compound drug-likeness by correlating its potency and lipophilicity. Both measures are used to normalize potency and physicochemical properties to better assess a series of compounds [125]. The average LE for the PPI inhibitors was found to be 0.23 kcal/mol per heavy atom, while average LLE was 1.32 kcal/mol, both of which are lower compared to the preferred values of more than 0.3 [126] and 5 [127], respectively. In comparison, the average LE and LLE of a typical drug is approximately 0.45 and 4.43, respectively [118]. Aside from these, the correlation of potency with ADMET properties [118] and physicochemical properties with promiscuity or selectivity [118,127,128,129] are also currently being tackled to further expound our understanding of drug-likeness in relation to compound activity.
3. Emerging In Silico Approaches for PPI Drug Discovery
Experimental methods are widely accepted as the standard for any analysis as they illustrate biological scenarios in either in vitro or in vivo systems. However, the immensity of the human interactome requires considerable cost, effort, and time to study. Moreover, PPI studies are highly dependent on dynamics, PTMs, and physiological conditions, causing difficulties in distinguishing true interactions from experimental artifacts and inconsistencies in findings, especially in cases involving transient interactions and IDPs. In silico methods have emerged as alternative methods or as complements to experimental techniques, to fill in the gaps regarding vital PPI information, and to provide a foundation for further analyses (Figure 1).
3.1. Discerning the PPI Network Topology
PPI targets have become a class of their own throughout the years. In contrast to well-known protein families, like GPCRs and kinases, this class of targets encompasses exceedingly diverse structures and interactions. Efforts have been made to compile information about experimentally-determined PPIs into databases or platforms. Table 2 lists these repositories, which can provide invaluable information regarding functional characterization of proteins to ascertain and weigh their roles in diseases. Primary databases report only experimentally verified protein interactions from individually published studies that were manually curated to provide researchers with accessible information. Naturally, curating PPI information is no easy task; errors and noise are often found in these repositories. To ensure the robustness of study models, careful data selection must be observed by establishing replication with different methods, interaction type, subcellular location, and other physiological aspects [130,131]. Access to such extensive datasets are provided by meta-databases, which report experimentally validated PPIs collected from multiple primary databases and integrate them into one large data model.
Network analysis and sequence-based methods are valuable strategies that have widespread application in various stages of drug discovery, including target identification, binding site prediction, and polypharmacological studies [165]. Due to the impact of PPIs as therapeutic targets for diseases of interest, it is beneficial to understand the protein interactome and its correlation with disease onset and development via comprehensive mapping of the PPI network (PPIN) topology. Studying network topologies allows for the identification of novel biomarkers and relevant disease targets that would have taken a long time to discover and validate using conventional means. Subsequently, sequence and conservation information should be studied at length to determine their importance to protein dynamics and binding, further aiding in the rational design of drug candidates. The cornucopia of experimental data that is available due to advances in structural biology and biophysical methods can be expended for PPIN analysis.
PPINs exhibit typical web-like structures, wherein most proteins (nodes) display interactions (edge) with only one or a few partners, while some proteins exhibit contact with multiple partners, and are hence called hubs [166]. The correlation between topology and protein function is often measured through node degree and betweenness centrality [167,168,169]. Protein hubs exhibit a high node degree, i.e., the number of edges linked to a node, through multiple associations, rendering them as prominent elements that can affect the network function [170]. Indeed, studies have suggested that protein hubs are highly essential for cellular function, and can greatly affect the whole network when dysfunctional, making them prime drug targets [171,172,173,174]. On the other hand, proteins that display high betweenness centrality are considered as bottlenecks, and they work by regulating focal points of communication within the network [170]. Targeting bottlenecks, as compared to hubs can allow careful control of the interaction network due to their apparent impact on the strength and integrity of the whole network [175,176]. Discovery of these network components have already been established as a strategic source of novel protein targets [177,178,179].
In a recent study done by Vinayagam and colleagues [179], they performed network controllability analysis using 6339 proteins involved in 64,814 PPIs to sort proteins (nodes) as indispensable, neutral, or dispensable for the directed protein interaction network. Aside from classifying proteins, they observed that indispensable proteins are often conserved among species and enriched in signaling proteins such as kinases, while neutral proteins are often found as membrane and transcription factor proteins. Indispensable proteins were also implicated in genetic and human viral diseases, along with cancer genes. More specifically, they identified 56 genes that are most often deleted or over-expressed in nine types of cancer, with 10 genes already under investigation and 46 being proposed as new potential cancer targets. Remarkably, while indispensable nodes greatly populate current Food and Drug Administration (FDA)-approved drug targets, they found that these proteins are inadequately represented in the annotated druggable genome. They postulated that network controllability analysis would be able to aid in validating the druggable genome, as well as identify novel therapeutic targets [179]. Control theory and network analysis have been applied in other similar studies to identify essential nodes for cancer [180] and viral infection [181].
PPI prediction is essential for functional annotation. Constraints in experimental approaches necessitated the application of in silico methods, such as binding site prediction (Section 3.3) and protein-protein docking (Section 3.4). However, both these methods require prior knowledge of PPI partners and the availability of 3D structural data. In cases concerning unknown PPI partners and/or structures, sequence- and coevolution-based methods can be utilized to predict the probability of interaction and residues pertinent to binding. Multiple sequence alignment (MSA) of homologs offer knowledge about residue coevolution, which can predict protein partners based on the premise that partners usually coevolve to maintain their function [170,182]. Information obtained from these types of analyses can also facilitate the 3D structure prediction of applicable PPI complexes [183]. Another approach takes advantage of orthologous associations by predicting PPIs based on interologs, wherein proteins that are observed to interact in one species, are predicted to also interact in other species if they are found to be conserved [184]. Several machine learning (ML) models are also based on sequence information, and these have been highlighted in Section 3.2.
3.2. Harnessing the Power of Machine Learning Algorithms for PPIs
To extend the concept of in silico models, intelligent and contemporary ML algorithms have been implemented in the identification of PPIs. ML is a data-driven or knowledge-based approach which requires an adequate number of training sets and features. Statistical and ML methods have been applied at varied stages: assimilation of diverse heterogenous datasets, assessment of predictions, prediction of prospective PPIs, and investigation of extrapolated PPI networks [185,186,187,188].
During the past, several ML algorithms (i.e., κ-nearest neighbor (κNN), naïve Bayesian [188,189], neural networks (NN) [190], random forest (RF), support vector machines (SVM), decision trees, logistic regression, etc.) have been applied to predict PPIs. ML methods utilize a dataset of experimentally validated PPI surfaces to train interface predictors and further employ the trained model for prediction of protein-protein interfacial residues of query proteins [191]. Prevailing ML predictors apply binary classification, where a target residue is classified as either interfacial or non-interfacial by utilizing the target features or adjacent residues to formulate predictions [191]. Moreover, the accuracy of the prediction model is dependent on the input features used for training. Hence, it is crucial to identify the various protein features that are essential for training a ML algorithm. Several protein features are utilized in the development of models for PPI predictions, either individually or in combination. However, a single feature does not support sufficient data in the prediction of PPIs. Therefore, a combination of features is essential to enhance the performance of ML methods in PPI prediction. Some of the protein features utilized in model development include AA types, co-essentiality data, evolutionary information, GO-driven frequency-based similarity and semantic similarity, MIPS functional catalogue (FunCat), physicochemical properties of AAs, position-specific scoring matrices (PSSMs), protein expression data, residue interface propensity, secondary and tertiary structural information, sequence entropy, surface shape, and solvent accessible surface area [192,193]. Once the model is properly trained with the input features, its performance is assessed with an external or test dataset. The five main steps involved in PPI predictions are depicted in Figure 2. In PPI-based ML approaches, the input of prediction models is in the form of sequence or structural features or both. However, most of the existing ML interface predictors are structure-based [191]. The merits and demerits of structure- and sequence-based methods have been well reviewed in [191]. Additionally, there are a few meta-based approaches, where raw scores from several prediction servers are integrated and re-computed to improve the prediction performance. Representative structure-, sequence-, and meta-based ML predictors for identification of PPIs are summarized in Table 3.
Jones and Thornton developed a method for the prediction of PPI interaction sites by analyzing a group of residue spots on the protein interface. Each residue patch was examined using six parameters, including the accessible surface area, hydrophobicity, planarity, protrusion, residue interface propensity, and the solvation potential. The relative combined score was calculated to predict the probability of surface patch formation in the PPIs [194]. Furthermore, the distribution of the observed patch rankings relative to all other surface patches were evaluated for each dataset [195]. Likewise, Bradford et al. developed SVM- [196] and Bayesian-based [197] classifiers using surface patches. Another group developed a PPI predictor, known as SPPIDER [198], using SVM and NN in combination with relative solvent accessibility (RSA). The authors showed that the implemented RSA-based features demonstrated superior performance to other PPI-based features. Ofran and Rost developed a NN-based method, known as ISIS [190], for the prediction of protein interacting residues from sequence. This ML approach combines AA composition of protein-protein interfaces, along with evolutionary profiles, solvent accessibility, and protein secondary structural features. A naïve Bayes classifier, PSIVER, was developed by Murakami and Mizuguchi for the prediction of PPIs using sequence-based features, such as PSSM and predicted accessibility. The conditional probabilities of each AA feature were estimated using a kernel density estimation (KDE) method [199]. Two sequence-based PPI predictors, namely LORIS [200] and SPRINGS [201], were developed by applying L1-regularized logistic regression and artificial neural network (ANN) methods based on several sequential features.
Besides supervised ML algorithms, unsupervised ML approaches were also implemented in PPI prediction. Deep learning has become a new dimension of ML field, which attempts to learn multiple layered models of inputs, known as NNs. A deep learning algorithm is capable of handling extensive raw and complex data where it operates by mimicking deep neural networks (DNNs) and learning processes of the human brain. The central idea of deep learning has been detailed in [202]. It has potent applications in decision making, natural language understanding, and image and speech recognition. This algorithm has also been applied in the field of bioinformatics and biopharma industry. Recently, Sun et al. implemented deep learning for sequence-based prediction of human PPIs [203]. They applied a stacked autoencoder (SAE) to study PPIs in humans and other species (E. coli, Drosophila, and C. elegans). The models developed using an autocovariance (AC) coding method showed the top results on 10-fold cross validation and varied external datasets ranging from 87.99% to 99.21% (prediction accuracy). Another group developed a method known as DeepPPI, which applies DNNs to obtain high-level discriminative characteristics from common protein descriptors [204]. This method showed better performance on the external data set, demonstrating an accuracy of 92.50% and a Matthews Correlation Coefficient (MCC) of 85.08%.
ML tools can also be applied in the preparation of PPI libraries or in the analysis of initial PPI hits through different filtering routes to evaluate drug-likeness or ADME/Tox properties. Information regarding PPI modulators is available in 2P2Idb [105,228], TIMBAL [229,230], and iPPI-DB [106,231], and can be used in the development of pertinent ML-based models. A decision tree strategy, known as PPI-HitProfiler [232], was built by Reynès and colleagues based on PPI inhibitors identified in literature, and it is implemented in the FAF (Free ADME-Tox Filtering tool)-Drugs webserver [233,234,235,236]. In their method, a global physicochemical depiction of PPI inhibitors was established, along with important descriptors that relate to the molecular shape (Radial Distribution Function) and the number of multiple bonds (unsaturation index), that can be used in the classification of compounds occupying the PPI chemical space. Using different PPI complexes with ligand and bioassay information, their model was able to correctly identify 70% of the validated actives and 52% of the inactives [232]. On the other hand, Hamon et al. built an SVM tool, the 2P2IHunter [237], based on PPI modulator information taken from 2P2Idb. For the model development, they distinguished molecular descriptors pertaining to an octanol-water partition coefficient, hydrophilicity, MW, unsaturation, the number of rings, H-bond donors and acceptors, TPSA, and rotatable bonds, as being essential for categorizing the potential PPI modulators from screening libraries. Their SVM model showed a high accuracy (96%) and specificity (96%), but very low sensitivity. However, it boasted a high enrichment factor of 8, making it an efficient method for filtering out non-PPI molecules from screening libraries. Thus, the application of ML algorithm in PPI predictions provides a promising approach for the deeper understanding of the vast network of protein interactions and their modulators. However, even with the success of ML-based predictors, prediction accuracy and computational efficiency of developed models can still be improved further [238].
3.3. Elucidation of Interface Characteristics and Hot Spot Contribution in PPIs
The breakthrough of hot spot residue contribution to PPI binding, in conjunction with innovations in protein structure determination in the last several years, has greatly helped in characterizing protein interfaces. Even with current efforts to fully characterize the complete human interactome using well-known genetic and biochemical techniques, numerous targets are still without structures, and the sheer number of partner interactions involved leads to difficulties in fully elucidating all of them. PPIs mainly transpire in conserved regions of protein interfaces, where hotspots are usually identified, and they can cause conformational changes in the overall protein structure [239]. However, it is unwise to generalize interface topographies due to the diversity of interface features between different PPI types. Transient interactions are often found to be pharmacologically relevant, and they have garnered a great deal of interest, especially in the development of various prediction methods. However, the dynamic capability and short duration of transient complex formation makes it more challenging to experimentally characterize than permanent complexes, resulting in fewer data available for model training and development [240,241]. Transient interfaces also tend to be less conserved and exhibit different properties compared to their permanent counterparts, suggesting the need for separate predictive strategies. Protein hubs are also difficult to assess, since their binding interfaces are shared by multiple partners [242].
Alanine scanning mutagenesis (ASM) has been an indispensable tool for the identification of hot spot residues of different relevant protein targets, but this method has only been applied in a limited number of PPIs. More to the point, experimental alanine scanning entails the mutation of several residues in the interface, along with biophysical evaluation of changes in the binding energy for each mutation, making it a costly approach [243]. Computational alanine scanning is a suitable and quick substitute for hot spot detection. The PPI complex structure of interest is required to be able to estimate the change in binding energy (ΔΔG), based on the bound and unbound state of the wild type and mutated proteins [244]. This method can be combined with MD simulations, as shown first by Massova and Kollman [245]. Modifications were added in the last several years, such as the use of distance-dependent pair potentials (e.g., DrugScorePPI [246]) and interaction entropy [247] for the computation of residue-specific free binding energies, to increase the accuracy of calculation and prediction of hot spots.
Docking methods, like Optimal Docking Area (ODA) [248] and pyDockNIP module of pyDOCK [249,250], have also been used to determine interface hot spots. ODA evaluates changes in interface energies based on the residue buriedness upon the binding of protein partners, and can be applicable in characterizing both obligate and non-obligate interactions [248]. It has been used to characterize PPI complexes, including those of metallocarboxypeptidases in complex with proteinaceous inhibitors [251], and UNC-45 chaperone protein in complex with myosin [252], providing beneficial information for drug discovery efforts. Alternatively, residue normalized-interface propensity (NIP) calculated from protein-protein docking results can be employed to identify hot spot residues on the protein surface, as is applied in pyDockNIP [249].
The current rise of the big data era has aided the return and further innovation of knowledge-based techniques, especially in the context of drug discovery. The availability of sequence and structural information has allowed the exploration and identification of distinguishing features for the analysis and prediction of hot spots. Sequence-, structural-, and physicochemical-based features have been used in the development of different data-driven models and in the characterization of relevant PPIs [253]. Protein sequences can offer preliminary description and prediction of potential binding sites for relatively unstudied proteins, especially in the absence of structural information, because of the noted high conservation of interface residues [239,254]. In combination with interaction prediction mentioned in Section 3.1, coevolution-based analysis can also predict residues that are critical for binding or, when combined with statistical physics, residues that have an allosteric or indirect effect on the complex [255]. Structural homology has also been proposed as a useful metric for interface characterization, since structures evolve at a much slower pace than sequences [256] and interface structural space is well-conserved [257]. More commonly, physicochemical and 3D structural characteristics have been applied in the characterization of PPI hot spots. Previous analyses have remarked upon the opposing characteristics of the interface, wherein both high hydrophobicity and high solvent accessibility have been observed. This conflicting observation has been resolved by the identification of core and rim regions, as mentioned in Section 2.1. These physicochemical features also correlate well with distinct geometric aspects of PPI binding sites, such as residue buriedness, side chain protrusion, and surface curvature [258]. Due to the diversity exhibited by PPIs, it is difficult to rely on any individual feature for binding site and hot spot prediction, and it has repeatedly been shown that the combination of several attributes has more success in providing adequate information for PPI surfaces [259]. Knowledge-based PPI binding site and hot spot prediction algorithms are exemplified in tools like ANCHOR [260], HomPPI [261], KFC2 (Knowledge-based FADE and Contacts) [262,263,264], HotPoint [265], FTMAP [266], MINERVA [267], and PredHS [268]. These tools and more are discussed in extensive detail in other articles [244,253].
MD simulations have also been employed in the identification of druggable PPI pockets, particularly for flexible and transient systems [269]. A typical case is shown in the BCL-XL protein, which exhibits drastic conformational differences between its unbound and peptide-bound structures. While BCL-XL is known to be experimentally druggable [270], its apo-structure appeared to have a shallow pocket and very low druggability [271]. Analysis of its MD trajectory revealed that the BCL-XL structure considerably fluctuates, resulting in the formation of a highly druggable binding region and implication that much of its druggability can be attributed to its flexible structure [271].
3.4. Exploring the PPI Interface through Macromolecular Docking and Virtual Screening
It has been estimated that the amount of interactions in the human interactome range from 130,000–650,000 [15,272,273,274]. However, there is a huge gap between the estimated PPIs and experimentally available structural data. Even though individual protein components are known, the determination of molecular complexes through experimental techniques is challenging, due to their transient nature. In the RCSB PDB, only limited structural data is available for protein-protein or -peptide complexes. Determination of 3D structures of protein-protein or -peptide complexes is necessary to understand their associated biological functions, to predict mutation effects, and to aid in the rational design of novel PPI-based drugs. However, due to its associated experimental difficulties along with incurred high costs and the time in determining complexes, fast and reliable computational approaches, such as macromolecular docking, is necessitated. Computational docking is one of the potent in silico tools in structure-based approaches, which has been broadly utilized for PPI-based drug discovery. This method aims to yield bound structures with low interaction modes (favorable ones) by sampling a very large number of possible binding modes and evaluating each conformation using scoring functions [191].
Elucidation of protein-protein or -peptide structural complexes through computational docking has evolved significantly due to the accumulating data on protein structures and interactions, improved energy functions, and powerful techniques to accelerate the sampling process [275]. Protein-protein or -peptide docking consists of two major stages: sampling and scoring/ranking. In the docking (sampling) stage, several conformations will be generated and, among those, only the potential ones are sampled. Sampling potential orientations for protein-protein binding using global or local searches is the first stage in a docking protocol. In a global search, the protein acting as the receptor is fixed and the other acting as the ligand is moved around the receptor, where all potential orientations between protein partners in the 3D space are explored. Though this search is computationally expensive, several docking algorithms such as Fast-Fourier transform (FFT) have been implemented to reduce docking complexity. Whereas, in a local search, the local protein features are matched to acquire a suitable complementarity. Additionally, if experimental data is available, then that information will be integrated in the sampling stage to improve the prediction accuracy of the docking results.
The docking phase can either be rigid or flexible. In rigid-body docking, there are no changes in the structural features. However, in flexible docking, conformational changes in the structures are considered [170]. The docking stage typically generates thousands of putative complexes. Hence, it is necessary to sort them and acquire the best docking solutions. In the ranking stage, conformations sampled from the first stage are rescored and ranked using several scoring functions. The fundamental principles of protein-protein docking have been well reviewed in [275]. The docking predictions can be evaluated by calculating the RMSD between the predicted and original complexes, and by measuring the ratio of the predicted interface residues to native ones. Due to the increasing number of docking algorithms developed to predict protein-protein or -peptide complexes, the Critical Assessment of Predicted Interactions (CAPRI) challenge was initiated to appraise the performance of different docking protocols [276]. Currently available protein-protein and -peptide docking tools are listed in Table 4 and Table 5, respectively.
Several studies have shown the applicability of protein-protein docking in PPI research [326,327,328]. Here, we discuss a recent study where protein-protein docking has been applied for the prediction of molecular associations between proteins. Variations in protein S-glutathionylation has been linked to several human diseases. It has been reported that glyoxalase II (GlxII), an antioxidant glutathione-dependent enzyme, plays a role in the S-glutathionylation mechanism. However, its mechanistic underpinnings remain unrevealed. In a recent study by Galeazzi et al. [329], PPIs of GlxII propensity with other cellular proteins, such as malate dehydrogenase, actin, and glyceraldehyde 3-phosphate dehydrogenase, were explored using a computational protocol involving protein-protein docking and atomistic MD simulations. The suitability of the docking programs, including ClusPro, GRAMM-X, HADDOCK, HEX, PatchDock, SymmDock, and ZDOCK was tested using a set of globular protein oligomers. A reliable protein-protein docking approach that strongly agrees with the experimental findings was developed. The docking method was applied to GlxII-protein complexes for determination of their association stabilities. Subsequently, the predicted docked complexes were subjected to MD simulations and Molecular Mechanics Poisson Boltzmann Surface Area (MM/PBSA) analysis. In silico results showed that GlxII highly interacted with actin and MDH. Therefore, this study highlighted the application of protein-protein docking approaches in the elucidation of the molecular mechanisms and thermodynamics of GlxII protein binding affinity.
Similarly, once druggability and potential binding sites in the protein interface have been established, virtual screening (VS) experiments can be performed to find potential binders that can disrupt or stabilize the target PPI. VS can be categorized as structure-based (SBVS) or ligand-based (LBVS), but the former is more useful for PPIs, since ligand information for these targets is sparse when compared to other drug targets. SB pharmacophore VS can be employed for a given target, wherein interface features are explored to generate a 3D pharmacophore model used to identify ligands with diverse scaffolds that complement the binding site and have the potential to exhibit a desired bioactivity (inhibition or stabilization) [330]. Some programs that employ the pharmacophore strategy include Catalyst [331], FLAP (Fingerprints for Ligands And Proteins) [332], HS-Pharm (Hot-Spots-guided receptor-based Pharmacophores) [333], LigandScout [334], PHASE [335], and AnchorQuery [336]. Alternatively, docking is a well-established method for SBVS, wherein ligand binding is energetically evaluated by scoring functions based on its conformation and complementarity with the binding pocket [337]. Scoring functions can be categorized as force field-, knowledge-, or empirical-based [337]. Each scoring function, in combination with docking programs, has their own strengths and weaknesses, and selecting a docking program, they should be considered carefully based on the purpose of the study, i.e., ligand binding prediction (scoring) or VS (ranking). Examples of popular protein-ligand docking programs include AutoDock [280], AutoDock Vina [338], GOLD [339], ICM [340], and Glide [341,342].
Here, we present a few case studies where VS and ligand docking protocols have been incorporated to identify small molecule PPI inhibitors or stabilizers. HIV-1 Nef is involved in infection, pathogenicity, and disease development by interacting with its own SH3 binding surface [16]. Betzi et al. [343] combined VS and high-throughput docking to identify small compounds that can bind to the HIV-1 Nef SH3 surface and prevent HIV-1 Nef/SH3 PPIs. Initially, the National Cancer Institute (NCI) diversity library was pre-filtered using 14 drug-like filters, and the identified 1420 compounds were subjected to FlexX docking protocol. The docked complexes were rescored using GFscore. The top 335 lowest energy compounds were subsequently subjected to a SH3-based pharmacophoric filter and the resulting 33 potential hits were clustered. Ten of the molecules were finally selected based on the chemical and geometrical properties for further experimental validation. Another example focuses on the Bcl-2 protein family, which are well-established targets for anticancer therapeutics due to their key roles as apoptotic regulators. Zhou et al. studied the BCL-XL structure in complex with BCL-2-associated death promoter (BAD) BH3 peptide, identifying three key hydrophobic features which were used to create a pharmacophore model. To identify scaffolds with suitable pharmacological and safety profiles, they screened an in-house database of FDA-approved drugs using the generated pharmacophore model, identifying three classes of compounds with different cores. From this, they performed molecular docking on the BCL-XL protein to predict and study the binding modes of Lipitor and Celexocib, both of which consist of bis-aryl substituted five-membered heterocyclic scaffold. Both compounds mimicked two out of three hydrophobic features required for interaction in the BCL-XL binding pocket, leading to the design of BM-501, which showed suitable affinity to both BCL-2 and BCL-XL and can function as an excellent scaffold in the design of inhibitors for the Bcl-2 protein family [344]. In a different case, Myc oncoprotein promotes cancer when it heterodimerizes with Myc-associated factor X (Max), whereas Myc homodimers suppress cancer [345]. Jian et al. utilized the available crystal structure of Max-Max homodimer to identify small compound stabilizers through VS using AutoDock. The compounds (1668) which were screened from the NCI diversity dataset were subjected to blind docking using both Max-Max homodimer and Myc-Max heterodimer structures. Subsequently, the binding sites were analyzed, and potential compounds which were specific to Max-Max homodimers were identified. These case studies are clear indications that structure-based computational approaches can be used for the discovery of small molecule modulators of PPIs.
3.5. Exploiting Hot Spot Regions for Fragment-Based Design
FBDD has been widely employed for different disease targets in the last several years to acquire new chemical entities, while efficiently scouring a much larger chemical space than traditional high-throughput screening (HTS) can cover. Compared to extensive compound libraries commonly used for VS, which comprise of millions of complex structures, fragment libraries often contain only hundreds to a few thousand low-MW structures [346]. Despite this, fragment screening reportedly displays higher hit rates and better LE than HTS efforts [346,347]. However, most of the current fragment libraries consist of mostly flat, aromatic structures, which can still limit the exploration of chemical space. Renewed interest for natural products due to their structural diversity and relatively safe pharmacological profiles steered the efforts to use them as a source of highly 3D fragment structures. The presence of sp3-character and stereogenicity in natural product-derived fragments can increase the chemical space explored for an FBDD study [348,349,350,351].
During optimization, fragment hits can undergo linking or growing strategies. For those found to bind in adjacent pockets in the protein interface, appropriate linkers can be selected to connect the two fragments together. On the other hand, fragments can also be grown synthetically to the neighboring pocket based on binding site analysis and rational design. Between the two, linking is found to be more difficult, since the best linkers must be identified to avoid detrimental effects on lead potency and pharmacokinetic properties. While fragment binders initially show weaker affinity in screens, their smaller size and low complexity make it easier to manage physicochemical properties during the optimization than a high-affinity HTS lead. Nevertheless, these weak binding fragments are discovered to form excellent interactions in the binding site, and thus, they contribute more than half of the favorable binding energy if the interactions are preserved during fragment-to-lead optimization [346].
While the expansive interface of PPIs is not very conducive for small molecule inhibitor discovery, the presence of multiple hot spot regions spread across the surface suggests the applicability of fragment-based methods. Distinct fragments can be identified for each hot spot, and can be later linked or grown into whole molecules with novel structures that are specifically designed for a particular PPI target [352]. FBDD has been successful in providing potential lead candidates for PPIs. As a continuation of the same study by Zhou et al. mentioned in the above section, they used the crystal structure of BCL-XL in complex with ABT-737 and previous assay results to identify a fragment that could be linked to BM-501. Using part of ABT-737 as a linker, they were able to rationally design low nanomolar affinity leads for BCL-2 and BCL-XL. They further employed structure-activity relationship and modeling studies to improve potency and cellular activities of their compounds [344].
It is important to note that hot spot identification and analysis of the receptor protein are important elements of fragment-based design of PPI inhibitors. Pertinent fragments must be screened and bound to essential hot spot clusters, and not to negligible areas in the PPI interface. Otherwise, while tight binding compounds can be designed from suitable fragments, it may not produce the desired outcome (i.e., disruption or stabilization of PPIs). An excellent example for this is shown by Geppert and colleagues [353], where they analyzed the nuclear magnetic resonance (NMR) structures of both the apo and holo structures of IFN-α. Potential binding cavities were identified using PocketPicker [354], and interface hot spots were predicted using iPred [355]. Remarkably, hot spot prediction was in good agreement with previous mutation studies and was able to identify the major interaction groove in the protein interface. Pharmacophore generation and VS of commercially available fragment-sized compounds was performed, resulting in the identification of six binders. From these, the highest-ranking compound displayed good affinity, despite its low molecular weight (279 Da). Probing the ligand protein hot spots can also facilitate the design of PPI inhibitors. The peptide backbone appropriately positions hot spot residues to surface pockets, which are primarily composed of hydrophobic groups, while forming a number of hydrogen bonds in other parts of the interface, such as in the cases of p53-MDM2 and XIAP-caspase 9 complexes. In this event, it is beneficial both to find fragments that can bind to the surface hot spot regions, and to find linkers that can fulfill the hydrogen bond requirements for interaction with the receptor protein [356].
3.6. Unraveling the Structural and Functional Aspects of PPIs Using MD Simulations
Despite steady progress in the growth of PPI data, comprehensive understanding of such complexes and their dynamics remains inadequate. The scarcity of 3D structural data for protein-protein or -peptide interacting complexes delays PPI research. Moreover, crystallography cannot detect the presence of druggable hot spots and transient pockets on protein-protein interfaces, due to their active motions. MD simulations surpass these limitations by facilitating in-depth analysis of structural, functional, and dynamic aspects of PPI models. They also provide valuable insights into PPI mechanisms which could be utilized in the design of PPI modulators.
MD simulation is a commonly applied in silico approach for calculating the time-dependent motion of biological molecules. The initial step in MD simulation of a PPI is the acquisition of a starting complex structure, which could be obtained through experiments (X-ray, NMR, or cryo-electron microscopy (EM)), modeling approaches (homology modeling or protein-protein docking), or PPI databases. Once the structural template is available, the next step is to prepare the system and subject it to atomistic simulations in line with software-dependent simulation procedures. Firstly, the system’s initial positions and velocities are fixed. Subsequently, the forces among all atoms are defined by using a preconfined interaction potential, where time-dependent motions of a system could be traced by solving classical equations of motions [357]. Key utilities of MD simulations in PPI exploration include structural investigations of PPIs (e.g., identification of hot spots, functional mechanisms of complexes, possible binding partners, key interacting features of complexes, and oligomerization mechanistic insights), design of PPI modulators, elucidation of macromolecular mechanisms, and refinement of low-resolution structural data (Figure 3). In the case studies below, we highlight the benefits of MD to PPI investigations.
Dixit et al. investigated the functional mechanism of heat-shock protein (Hsp) 90 using atomistic MD simulations, along with the modeling of principal correlated motions and energy landscape analysis [358]. Through this analysis, several key areas in the structure-functional depiction of controlled interactions and the center of the communication networks were identified. In another study, Ozdemir et al. unraveled the atomistic interactions of the Rho GTPases, Cdc42 and Rac1, with the scaffolding protein IQ motif-containing GTPase-activating protein 2 (IQGAP2) using all-atom MD simulations, site-directed mutagenesis, and Western blotting [359]. They were able to decipher the underlying mechanism of the different stoichiometries involved in the binding of Rac1 and Cdc42 to GRD, and IQGAP2 dimerization. It also provided insights on how Cdc42 and Rac1 mediate actin polymerization in metastasis. The outcomes of these studies assist in the design of inhibitors and additionally provide structural, stoichiometric, and working insights into the allosteric control of the complex and protein machinery dynamics.
MD simulations can also assist in PPI hot spot prediction to provide clues for the design of potential PPI inhibitors. Survivin protein is found to be over-expressed in several solid tumors; hence disruption of its interaction with substrate proteins is indispensable for the treatment of cancer. Sarvagalla et al. [360] provided an exhaustive atomistic detail of the dimer interface of survivin-chromosomal passenger complex (CPC) protein by applying a knowledge-based model for identification of hot spot residues. Subsequently, extensive MD simulations were utilized to produce an ensemble of conformations which were further utilized for the estimation of binding free energies of identified hot spots using MM/PBSA and per-residue energy decomposition. Survivin and CPC interface hot spots were identified from these analyses. Finally, a pharmacophore model based on the hot spots was generated and virtually screened against compound databases for the identification of a potential inhibitor targeting survivin-CPC interaction. Besides hot spot predictions and functional characterization of PPIs, MD simulations could be utilized for identification of plausible binding partners. It is hypothesized that Psalmopeotoxin I (PcFK1) targets subtilisin-like serine protease, PfSUB1. To investigate this hypothesis, Bastianelli et al. [361] applied bioinformatics analysis, protein-protein docking, MD simulations, and free energy calculations on these two proteins. MD simulations was utilized for refining the docked protein-protein complexes and free energy calculations. Their computational results were confirmed via experimental testing on PfSUB1 purified and active recombinant enzyme, leading to the validation that PcFK1 indeed targets PfSUB1 enzymatic activity.
It has been widely accepted that most proteins function as oligomers. However, the oligomerization process often remains unclear. In such scenarios, MD simulations can assist experimentalists in unraveling the oligomerization processes. Zhang et al. investigated the formation of small oligomers in the amyloid fibril-formation process of the peptide GNNQQNY from the yeast prion-like protein Sup35, using explicit solvent MD simulations. Their results demonstrated that primarily antiparallel dimer forms, and subsequently novel peptides may complement the assemblies in parallel arrangement, which is in line with the experimental microcrystal structure of the amyloid fibril [362]. In another recent study, interactions between Aβ1–42 oligomers with each of the four models of the full-length Amylin1–37 oligomers were explored using extensive MD simulations, resulting in the elucidation of the link between type 2 diabetes and Alzheimer’s disease [363]. Thus, MD simulation stands as an indispensable tool to complement experimental screening techniques in PPI research that dissect PPIs at an atomistic level, with its high accuracy, vigorous validation of force fields, and the extensive availability of computational resources for performing large-scale simulations of complex protein systems.
4. Pitfalls of CADD in the Discovery of PPI Inhibitors
There is no doubt that computational methods have become one of the cornerstones of drug discovery, especially with the advances in technology and the increase in experimental data in the last several years. However, as with any other drug discovery and design methods, CADD has its own strengths and weaknesses when dealing with a variety of drug targets, including PPIs.
Prediction and filtering tools are rapid and dependable strategies, particularly for targets with insufficient information at the start of a drug discovery effort. In the absence of 3D information, sequence-based methods can provide quick and useful insights about conservation and evolution information, which may also be critical for ligand design. However, information that is taken from these techniques might not translate well into 3D systems [253]. In fact, Keskin et al. remarked that conservation-based analysis is not applicable to transient interactions due to its poor conservation across species [170]. Most computational tools are dependent on the availability of structural information, such as in the case of energy- and knowledge-based techniques. Energy-based methods are built on the understanding of energy terms in complex systems. Due to the intricacy of protein structures, dynamics, and interactions, several approximations are frequently included in the development of these methods to create a balance between speed and accuracy; thus, they are more useful as estimates for prioritization rather than data that is convertible to real scenarios [364]. On the other hand, knowledge-based methods make use of currently available protein and ligand information, and models created from this technique are only as good as the data used to create it. For example, certain descriptors are more predictive for specific PPI types [253]. Furthermore, if a model is based on a particular protein family or ligand class, it cannot be reliably employed to characterize other protein families or ligand classes. This becomes a hindrance with respect to the intrinsic flexibility and diversity of PPIs.
FBDD is notably more efficient than conventional HTS, both in experimental and virtual form. However, several things are needed before pursuing this strategy. First, structural information is required for fragment screening, making it an unsuitable method for relatively unknown targets. Furthermore, information about hot spot regions is crucial, as these are the focus for any type of screening efforts. Second, binding affinity and orientation of fragments may differ from that of the complete molecule. Linker groups and attachment points must be considered conscientiously, as their addition to identified fragment hits can greatly affect both the potency and pharmacokinetic properties of the lead. Third, and the same as other computational strategies, virtual fragment hits must be validated. However, validation for these low affinity structures often requires more sensitive and sophisticated methods and expertise, as compared to conventional HTS [346].
Perhaps the most notable challenge for many computational methods is the dynamic nature of proteins, as the inclusion of flexibility is extremely computationally expensive, leading to the use of rigid protein structures by the majority of CADD modules. This drawback becomes more prominent in PPIs, whose conspicuously flat interfaces are postulated to be highly flexible. MD simulations assuages these concerns as it can approximate protein movements in a carefully monitored system. Improvements in hardware computational power have also aided increased interest and utilization of MD for the analysis of PPIs [271,365,366].
The listed shortcomings for each method should not be taken as a dissuasion of their use. In fact, this is a reinforcement that more studies are needed to further improve current computational tools. Moreover, the strengths of each method can be used to make up for the weaknesses of the others. The combination of two or more CADD tools, along with the incorporation of experimental techniques, can result in better insights and success in drug discovery endeavors.5. Conclusion and Prospects of PPI Drug Discovery.
PPIs have become prime drug targets due to their integral roles in cell signaling and regulatory pathways. While increased interest for these targets encouraged comprehensive studies about PPINs, interface conformations and interactions, and PPI modulators, much is still needed to completely characterize this vast system and to overcome hindrances associated with PPI drug discovery. Current experimental techniques have facilitated our growing understanding of PPIs, resulting in the construction of informative databases containing valuable information involving the human proteome and interactome. Unfortunately, the immensity of the human interactome renders experimental methods insufficient, necessitating the use of their computational counterparts. CADD impacts various stages of PPI drug discovery by assisting in the characterization of PPIs and its unique chemical space, thereby providing better insights into the design of PPI modulators. Innovations in computational power and algorithms, along with the growing knowledge and better parameterization of macromolecular dynamics and energetics relating to PPIs, have led to notable contributions of in silico methods in various PPI drug discovery efforts.
Overall, we can expect a remarkable future for PPI drug discovery as both in silico and experimental methods continue to evolve. While each approach has their own significance in the field of drug discovery and development, it is important to remember that one is not designed to outshine the other, but to complement it. The integration of different tools, especially in the study of diverse drug targets such as PPIs, can enhance our understanding of these targets and aid in the design of potent and effective PPI modulators.
Author Contributions
S.J.Y.M., S.B., S.K., and S.C. conceived and wrote the paper; S.J.Y.M., S.B., N.A.B.C., and S.C. conceptualized and generated the figures; S.B., N.A.B.C., and H.C. gathered literature and organized the tables; S.K. and S.C. provided critical reviews; all authors contributed to the editing of the manuscript.
Funding
This work was supported by the Medical Research Center (MRC) grant (No. 2018R1A5A2025286), the Mid-career Researcher Program (NRF-2017R1A2B4010084), and the Bio & Medical Technology Development Program (No. 2018M3A9A7057263) funded by the Ministry of Science and ICT (MSIT) through the National Research Foundation of Korea (NRF).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Armour, D.; de Groot, M.J.; Edwards, M.; Perros, M.; Price, D.A.; Stammen, B.L.; Wood, A. The discovery of ccr5 receptor antagonists for the treatment of hiv infection: Hit-to-lead studies. ChemMedChem 2006, 1, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Filikov, A.V.; James, T.L. Structure-based design of ligands for protein basic domains: Application to the hiv-1 tat protein. J. Comput. Aid. Mol. Des. 1998, 12, 229–240. [Google Scholar] [CrossRef]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, P.; Kokkola, T.; Suuronen, T.; Poso, A.; Jarho, E.; Lahtela-Kakkonen, M. Virtual screening approach of sirtuin inhibitors results in two new scaffolds. Eur. J. Pharm. Sci. 2015, 76, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tiwari, S.; Srivastava, K.K.; Siddiqi, M.I. Identification of novel inhibitors of mycobacterium tuberculosis pkng using pharmacophore based virtual screening, docking, molecular dynamics simulation, and their biological evaluation. J. Chem. Inf. Model. 2015, 55, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.N.; Mutyala, R.; Aparoy, P.; Reddanna, P.; Reddy, M.R. Computer aided drug design approaches to develop cyclooxygenase based novel anti-inflammatory and anti-cancer drugs. Curr. Pharm. Des. 2007, 13, 3505–3517. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Jee, J.G.; Yin, C.H.; Sandoval, C.; Jayabose, S.; Kitamura, D.; Bach, E.A.; Baeg, G.H. Nsc114792, a novel small molecule identified through structure-based computational database screening, selectively inhibits jak3. Mol. Cancer 2010, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.N.D.S.; Speck-Planche, A. Computer-aided drug design, synthesis and evaluation of new anti-cancer drugs. Curr. Top. Med. Chem. 2012, 12, 2703–2704. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.Q.; Zhou, R.; Lian, F.L.; Liu, Y.; Chen, L.M.; Shi, Z.; Zhang, N.X.; Zheng, M.Y.; Shen, B.R.; Jiang, H.L.; et al. Virtual screening and biological evaluation of novel small molecular inhibitors against protein arginine methyltransferase 1 (prmt1). Org. Biomol. Chem. 2014, 12, 9665–9673. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Li, G.H.; Zhao, D.Y.; Yu, H.Y.; Zheng, X.L.; Peng, X.D.; Zhang, X.; Fu, T.; Hu, X.Q.; Niu, M.S.; et al. Computational discovery and experimental verification of tyrosine kinase inhibitor pazopanib for the reversal of memory and cognitive deficits in rat model neurodegeneration. Chem. Sci. 2015, 6, 2812–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, V.H.; Tran, P.T.; Cui, M.; Ngo, V.T.H.; Ann, J.; Park, J.; Lee, J.; Choi, K.; Cho, H.; Kim, H.; et al. Discovery of potent human glutaminyl cyclase inhibitors as anti-alzheimer’s agents based on rational design. J. Med. Chem. 2017, 60, 2573–2590. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Zhao, Z.M. A comparative study of cancer proteins in the human protein-protein interaction network. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.J.; Aguilar, A.; Bernard, D.; Wang, S.M. Small-molecule inhibitors of the mdm2-p53 protein-protein interaction (mdm2 inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Laraia, L.; McKenzie, G.; Spring, D.R.; Venkitaraman, A.R.; Huggins, D.J. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein-protein interactions. Chem. Biol. 2015, 22, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sable, R.; Jois, S. Surfing the protein-protein interaction surface using docking methods: Application to the design of ppi inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 2002, 12, 14–20. [Google Scholar] [CrossRef]
- Moza, B.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Rahman, A.K.M.N.U.; McCormick, J.K.; Kranz, D.M.; Sundberg, E.J. Long-range cooperative binding effects in a t cell receptor variable domain. Proc. Natl. Acad. Sci. USA 2006, 103, 9867–9872. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. Principles of protein-protein recognition from structure to thermodynamics. Biochimie 1995, 77, 497–505. [Google Scholar] [CrossRef]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Argos, P. An investigation of protein subunit and domain interfaces. Protein Eng. 1988, 2, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Miller, S.; Chothia, C. Surface, subunit interfaces and interior of oligomeric proteins. J. Mol. Biol. 1988, 204, 155–164. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Protein-protein interactions: A review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Janin, J. Dissecting protein-protein recognition sites. Proteins 2002, 47, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. Dissecting subunit interfaces in homodimeric proteins. Proteins 2003, 53, 708–719. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Sternberg, M.J. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Characterization of protein-protein interfaces. Protein J. 2008, 27, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. A bh3 mimetic for killing cancer cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; de Oliveira, M.R.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase ii study of single-agent navitoclax (abt-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the bh3 mimetic bcl2 inhibitor navitoclax (abt-263) with or without rituximab, in previously untreated b-cell chronic lymphocytic leukemia. Leukemia Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef] [PubMed]
- Brachet, P.E.; Fabbro, M.; Leary, A.; Medioni, J.; Follana, P.; Lesoin, A.; Frenel, J.S.; Lacourtoisie, S.A.; Floquet, A.; Gladieff, L.; et al. A gineco phase ii study of navitoclax (abt 263) in women with platinum resistant/refractory recurrent ovarian cancer (roc). Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.N.; Rudersdorf, N.S.; Vanderwal, C.A.; Kirschbrown, W.; Holen, K.D.; Rosen, L.S. Safety, efficacy, and pharmacokinetics of navitoclax (abt-263) in combination with erlotinib in patients with advanced solid tumors. Cancer Chemoth. Pharm. 2015, 76, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of amg 232, a potent, selective, and orally bioavailable mdm2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The bet bromodomain inhibitor otx015 affects pathogenetic pathways in preclinical b-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.P.; de La Motte Rouge, T.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A. Clinical response of carcinomas harboring the brd4-nut oncoprotein to the targeted bromodomain inhibitor otx015/mk-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.Y.; Seipel, M.E.; Kapoor, G.S.; LaPorte, M.G.; Rippin, S.R.; et al. Birinapant (tl32711), a bivalent smac mimetic, targets traf2-associated ciaps, abrogates tnf-induced nf-kb activation, and is active in patient-derived xenograft models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Vetma, V.; Rozanc, J.; Charles, E.M.; Hellwig, C.T.; Alexopoulos, L.G.; Rehm, M. Examining the in vitro efficacy of the iap antagonist birinapant as a single agent or in combination with dacarbazine to induce melanoma cell death. Oncol. Res. 2017, 25, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Schilder, R.J.; Martin, L.P.; Levin, M.; Graham, M.A.; Weng, D.E.; Adjei, A.A. A phase i study of the smac-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol. Cancer Ther. 2015, 14, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Bunch, K.P.; Chen, J.Q.; Herrmann, M.A.; Lee, J.M.; Kohn, E.C.; O’Sullivan, C.C.; Jordan, E.; Houston, N.; Takebe, N.; et al. Pharmacodynamic markers and clinical results from the phase 2 study of the smac mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 2016, 122, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Paller, C.J.; Antonarakis, E.S. Cabazitaxel: A novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des. Dev. Ther. 2011, 5, 117–124. [Google Scholar]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a dihydroisoquinolinone derivative (nvp-cgm097): A highly potent and selective mdm2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti-Cancer Agent Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Stupp, R.; Van den Bent, M.J.; Erridge, S.G.; Reardon, D.A.; Hong, Y.; Wheeler, H.; Hegi, M.; Perry, J.R.; Picard, M.; Weller, M. Cilengitide in newly diagnosed glioblastoma with mgmt promoter methylation: Protocol of a multicenter, randomized, open-label, controlled phase iii trial (centric). J. Clin. Oncol. 2010, 28. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (nsclc) patients: Results of an open-label, randomized, controlled phase ii study (certo). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.P. End of the road: Confounding results of the core trial terminate the arduous journey of cilengitide for glioblastoma. Neuro-Oncology 2015, 17, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Leblanc, Y.; Cote, A.; Nasveschuk, C.G.; Taylor, A.M.; Harmange, J.C.; Audia, J.E.; Pardo, E.; et al. Discovery, design, and optimization of isoxazole azepine bet inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Cote, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a benzoisoxazoloazepine inhibitor (cpi-0610) of the bromodomain and extra-terminal (bet) family as a candidate for human clinical trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Abramson, J.; Maris, M.; Flinn, I.; Goy, A.; Mertz, J.; Sims, R.; Garner, F.; Senderowicz, A.; Younes, A. A phase i study of cpi-0610, a bromodomain and extra terminal protein (bet) inhibitor in patients with relapsed or refractory lymphoma. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Belani, C.P.; Eckardt, J. Development of docetaxel in advanced non-small-cell lung cancer. Lung Cancer 2004, 46, S3–S11. [Google Scholar] [CrossRef]
- Ford, H.E.R.; Marshall, A.; Bridgewater, J.A.; Janowitz, T.; Coxon, F.Y.; Wadsley, J.; Mansoor, W.; Fyfe, D.; Madhusudan, S.; Middleton, G.W.; et al. Docetaxel versus active symptom control for refractory oesophagogastric adenocarcinoma (cougar-02): An open-label, phase 3 randomised controlled trial. Lancet Oncol. 2014, 15, 78–86. [Google Scholar] [CrossRef]
- Feinman, H.E.; Price, D.K.; Figg, W.D. Piecing the puzzle together: Docetaxel cycles and current considerations in the treatment of metastatic castration-resistant prostate cancer. Cancer Biol. Ther. 2017, 18, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.W.; Kwon, O.K.; Chung, H.Y.; Yu, W.; Kim, J.G. Taxanes in the treatment of advanced gastric cancer. Molecules 2016, 21, 651. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Rosenthal, J.; Andreeff, M.; Zernovak, O.; Kumar, P.; Gajee, R.; Chen, S.Q.; Rosen, M.; Song, S.; Kochan, J.; et al. Phase 1 dose escalation study of mdm2 inhibitor ds-3032b in patients with hematological malignancies - preliminary results. Blood 2016, 128, 593. [Google Scholar]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Tcheng, J.E.; Talley, J.D.; O’Shea, J.C.; Gilchrist, I.C.; Kleiman, N.S.; Grines, C.L.; Davidson, C.J.; Lincoff, A.M.; Califf, R.M.; Jennings, L.K.; et al. Clinical pharmacology of higher dose eptifibatide in percutaneous coronary intervention (the pride study). Am. J. Cardiol. 2001, 88, 1097–1102. [Google Scholar] [CrossRef]
- Fung, J.J. Tacrolimus and transplantation: A decade in review. Transplantation 2004, 77, S41–S43. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Gosmini, R.; Toum, J.; Clement, C.A.; Barnathan, M.; Brusq, J.M.; Mordaunt, J.E.; Grimes, R.M.; Crowe, M.; Pineau, O.; et al. Discovery of epigenetic regulator i-bet762: Lead optimization to afford a clinical candidate inhibitor of the bet bromodomains. J. Med. Chem. 2013, 56, 7501–7515. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Kang, M.H.; Reynolds, C.P.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Keir, S.T.; Carol, H.; Lock, R.; Maris, J.M.; et al. Initial testing (stage 1) of lcl161, a smac mimetic, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2012, 58, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase i dose-escalation study of lcl161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Zuo, Y.; Yang, X.; Lu, J.; Zhan, L.L.; Xu, L.P.; Zhang, C.; Zhu, H.C.; Liu, J.; Liu, Z.M.; et al. Smac mimetic compound lcl161 sensitizes esophageal carcinoma cells to radiotherapy by inhibiting the expression of inhibitor of apoptosis protein. Tumor Biol. 2014, 35, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Martin, B.P.; Andrews, D.A.; Hogg, S.J.; Banerjee, A.; Grigoriadis, G.; Johnstone, R.W.; Shortt, J. The smac mimetic, lcl-161, reduces survival in aggressive myc-driven lymphoma while promoting susceptibility to endotoxic shock. Oncogenesis 2016, 5, e216. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.L.; Pflugfelder, S.C.; Zhang, S.; Shojaei, A.; Haque, R. Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul. Surf. 2016, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; McEachern, D.; Li, S.Q.; Ellis, M.J.; Wang, S.M. Reactivation of p53 by mdm2 inhibitor mi-77301 for the treatment of endocrine-resistant breast cancer. Mol. Cancer. Ther. 2016, 15, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Sun, W.; Zhao, Y.J.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.C.; Liu, L.; et al. Sar405838: An optimized inhibitor of mdm2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Blum, S.S.; Fung, H.B.; Bandres, J.C. Maraviroc: A ccr5-receptor antagonist for the treatment of hiv-1 infection. Clin. Ther. 2008, 30, 1228–1250. [Google Scholar] [CrossRef]
- Van Der Ryst, E. Maraviroc—A ccr5 antagonist for the treatment of hiv-1 infection. Front. Immunol. 2015, 6, 1228–1250. [Google Scholar] [CrossRef] [PubMed]
- Woodhead, A.J.; Angove, H.; Carr, M.G.; Chessari, G.; Congreve, M.; Coyle, J.E.; Cosme, J.; Graham, B.; Day, P.J.; Downham, R.; et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydrois oindol-2-yl]methanone (at13387), a novel inhibitor of the molecular chaperone hsp90 by fragment based drug design. J. Med. Chem. 2010, 53, 5956–5969. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.J.; Jiang, N.; Liu, J.J.; Zhao, C.L.; Glenn, K.; Wen, Y.; et al. Discovery of rg7112: A small-molecule mdm2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.J.; Zhang, Z.M.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of rg7388, a potent and selective p53-mdm2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- King, S.; Short, M.; Harmon, C. Glycoprotein iib/iiia inhibitors: The resurgence of tirofiban. Vasc. Pharmacol. 2016, 78, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Milla, M.E.; Sauer, R.T. P22 arc repressor: Folding kinetics of a single-domain, dimeric protein. Biochemistry 1994, 33, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, K.; Krebs, M. The v-atpase: Small cargo, large effects. Curr. Opin. Plant Biol. 2010, 13, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.; Thornton, J.M. Diversity of protein-protein interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef] [PubMed]
- Hayer-Hartl, M.; Bracher, A.; Hartl, F.U. The groel-groes chaperonin machine: A nano-cage for protein folding. Trends Biochem. Sci. 2016, 41, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient protein-protein interactions: Structural, functional, and network properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.; Thornton, J.M. Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 2003, 325, 991–1018. [Google Scholar] [CrossRef]
- Reichmann, D.; Rahat, O.; Cohen, M.; Neuvirth, H.; Schreiber, G. The molecular architecture of protein-protein binding sites. Curr. Opin. Struct. Biol. 2007, 17, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. The kinetics of protein-protein recognition. Proteins 1997, 28, 153–161. [Google Scholar] [CrossRef]
- Li, W.; Keeble, A.H.; Giffard, C.; James, R.; Moore, G.R.; Kleanthous, C. Highly discriminating protein-protein interaction specificities in the context of a conserved binding energy hotspot. J. Mol. Biol. 2004, 337, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, D.; Thiel, S.; Vajda, S.; Camacho, C.J. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.R.; Brower, R.C.; Vajda, S.; Camacho, C.J. Dynamical view of the positions of key side chains in protein-protein recognition. Biophys. J. 2001, 80, 635–642. [Google Scholar] [CrossRef]
- Kraich, M.; Klein, M.; Patino, E.; Harrer, H.; Nickel, J.; Sebald, W.; Mueller, T.D. A modular interface of il-4 allows for scalable affinity without affecting specificity for the il-4 receptor. BMC Biol. 2006, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Huang, K.Y.; Su, M.G.; Lee, T.Y.; Bretana, N.A.; Chang, W.C.; Chen, Y.J.; Chen, Y.J.; Huang, H.D. Dbptm 3.0: An informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2013, 41, D295–D305. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.Y.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS. Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Minguez, P.; Parca, L.; Diella, F.; Mende, D.R.; Kumar, R.; Helmer-Citterich, M.; Gavin, A.C.; van Noort, V.; Bork, P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Raghunathan, D.; Nicholson, J.; Hupp, T.R.; Lane, D.P.; Verma, C.S. Chemical states of the n-terminal “lid” of mdm2 regulate p53 binding: Simulations reveal complexities of modulation. Cell Cycle 2011, 10, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ballester, G.; Blanes-Mira, C.; Serrano, L. The tryptophan switch: Changing ligand-binding specificity from type i to type ii in sh3 domains. J. Mol. Biol. 2004, 335, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K.; Permi, P. Sh3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Ma, B.; Nussinov, R. Folding and binding cascades: Shifts in energy landscapes. Proc. Natl. Acad. Sci. USA 1999, 96, 9970–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, A.; Majumdar, S.; Priya, P.; De, P.; Saha, S.; Ghosh Dastidar, S. Adaptability in protein structures: Structural dynamics and implications in ligand design. J. Biomol. Struct. Dyn. 2015, 33, 298–321. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeas, R.; Basse, M.J.; Morelli, X.; Roche, P. Atomic analysis of protein-protein interfaces with known inhibitors: The 2p2i database. PLoS ONE 2010, 5, e9598. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, O.; Reynes, C.H.; Camproux, A.C.; Villoutreix, B.O. Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov. Today 2010, 15, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Kuenemann, M.A.; Bourbon, L.M.L.; Labbé, C.M.; Villoutreix, B.O.; Sperandio, O. Which three-dimensional characteristics make efficient inhibitors of protein–protein interactions? J. Chem. Inf. Model. 2014, 54, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Basse, M.J.; Betzi, S.; Bourgeas, R.; Bouzidi, S.; Chetrit, B.; Hamon, V.; Morelli, X.; Roche, P. 2p2idb: A structural database dedicated to orthosteric modulation of protein-protein interactions. Nucleic Acids Res. 2013, 41, D824–D827. [Google Scholar] [CrossRef] [PubMed]
- Labbe, C.M.; Laconde, G.; Kuenemann, M.A.; Villoutreix, B.O.; Sperandio, O. Ippi-db: A manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov. Today 2013, 18, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2p2i). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Labbe, C.M.; Lagorce, D.; Laconde, G.; Sperandio, O. A leap into the chemical space of protein-protein interaction inhibitors. Curr. Pharm. Des. 2012, 18, 4648–4667. [Google Scholar] [CrossRef] [PubMed]
- Petsalaki, E.; Russell, R.B. Peptide-mediated interactions in biological systems: New discoveries and applications. Curr. Opin. Biotechnol. 2008, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Wanner, J.; Fry, D.C.; Peng, Z.; Roberts, J. Druggability assessment of protein-protein interfaces. Future Med. Chem. 2011, 3, 2021–2038. [Google Scholar] [CrossRef] [PubMed]
- Garner, J.; Harding, M.M. Design and synthesis of alpha-helical peptides and mimetics. Org. Biomol. Chem. 2007, 5, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.A.; Wilson, A.J. Helix-mediated protein--protein interactions as targets for intervention using foldamers. Amino Acids 2011, 41, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Nevola, L.; Giralt, E. Modulating protein-protein interactions: The potential of peptides. Chem. Commun. 2015, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Zoller, F.; Markert, A.; Barthe, P.; Zhao, W.; Weichert, W.; Askoxylakis, V.; Altmann, A.; Mier, W.; Haberkorn, U. Combination of phage display and molecular grafting generates highly specific tumor-targeting miniproteins. Angew. Chem. Int. Ed. Engl. 2012, 51, 13136–13139. [Google Scholar] [CrossRef] [PubMed]
- Seoane, M.D.; Petkau-Milroy, K.; Vaz, B.; Mocklinghoff, S.; Folkertsma, S.; Milroy, L.-G.; Brunsveld, L. Structure-activity relationship studies of miniproteins targeting the androgen receptor-coactivator interaction. MedChemComm 2013, 4, 187–192. [Google Scholar] [CrossRef]
- Winter, A.; Higueruelo, A.P.; Marsh, M.; Sigurdardottir, A.; Pitt, W.R.; Blundell, T.L. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: Applications in structure-guided drug discovery. Q. Rev. Biophys. 2012, 45, 383–426. [Google Scholar] [CrossRef] [PubMed]
- Valkov, E.; Sharpe, T.; Marsh, M.; Greive, S.; Hyvoenen, M. Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 145–179. [Google Scholar] [PubMed]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, admet and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Davis, A.M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 2004, 47, 6338–6348. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.R. The evolution of synthetic oral drug properties. Bioorg. Med. Chem. Lett. 2005, 15, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Keseru, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Fontanillo, C. Protein-protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.H.; Lopes, T.J.; Mah, N.; Shoemaker, J.E.; Matsuoka, Y.; Fontaine, J.F.; Louis-Jeune, C.; Eisfeld, A.J.; Neumann, G.; Perez-Iratxeta, C.; et al. Adding protein context to the human protein-protein interaction network to reveal meaningful interactions. PLoS Comput. Biol. 2013, 9, e1002860. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Betel, D.; Hogue, C.W.V. Bind: The biomolecular interaction network database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Chatr-aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The biogrid interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database-2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Hermjakob, H.; Montecchi-Palazzi, L.; Lewington, C.; Mudali, S.; Kerrien, S.; Orchard, S.; Vingron, M.; Roechert, B.; Roepstorff, P.; Valencia, A.; et al. Intact: An open source molecular interaction database. Nucleic Acids Res. 2004, 32, D452–D455. [Google Scholar] [CrossRef] [PubMed]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The intact molecular interaction database in 2010. Nucleic Acids Res. 2010, 38, D525–D531. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The intact molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. Mint, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.; Kovac, S.; Oesterheld, M.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Mark, P.; Stumpflen, V.; Mewes, H.W.; et al. The mips mammalian protein-protein interaction database. Bioinformatics 2005, 21, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Stransky, M.; Waegele, B.; Schmidt, T.; Doudieu, O.N.; Mpflen, V.S.; et al. Corum: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008, 36, D646–D650. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Mewes, H.W. Corum: The comprehensive resource of mammalian protein complexes-2009. Nucleic Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Pacifico, S.; Liu, G.Z.; Finley, R.L. Droid: The drosophila interactions database, a comprehensive resource for annotated gene and protein interactions. BMC Genom. 2008, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Murali, T.; Pacifico, S.; Yu, J.K.; Guest, S.; Roberts, G.G.; Finley, R.L. Droid 2011: A comprehensive, integrated resource for protein, transcription factor, rna and gene interactions for drosophila. Nucleic Acids Res. 2011, 39, D736–D743. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Lopez, D.; Gutierrez, M.A.; Lopes, K.P.; Prieto, C.; Santamaria, R.; De Las Rivas, J. Apid interactomes: Providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res. 2016, 44, W529–W535. [Google Scholar] [CrossRef] [PubMed]
- Ptak, R.G.; Fu, W.; Sanders-Beer, B.E.; Dickerson, J.E.; Pinney, J.W.; Robertson, D.L.; Rozanov, M.N.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; et al. Cataloguing the hiv type 1 human protein interaction network. Aids Res. Hum. Retrov. 2008, 24, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Sanders-Beer, B.E.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; Ptak, R.G. Human immunodeficiency virus type 1, human protein interaction database at ncbi. Nucleic Acids Res. 2009, 37, D417–D422. [Google Scholar] [CrossRef] [PubMed]
- Pinney, J.W.; Dickerson, J.E.; Fu, W.; Sanders-Beer, B.E.; Ptak, R.G.; Robertson, D.L. Hiv-host interactions: A map of viral perturbation of the host system. Aids 2009, 23, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Ako-Adjei, D.; Fu, W.; Wallin, C.; Katz, K.S.; Song, G.; Darji, D.; Brister, J.R.; Ptak, R.G.; Pruitt, K.D. Hiv-1, human interaction database: Current status and new features. Nucleic Acids Res. 2015, 43, D566–D570. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Park, B.; Kim, H.; Hong, J.; Park, J. Hpid: The human protein interaction database. Bioinformatics 2004, 20, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Nanduri, B. Hpidb—A unified resource for host-pathogen interactions. BMC Bioinform. 2010, 11, S16. [Google Scholar] [CrossRef] [PubMed]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. Hpidb 2.0: A curated database for host-pathogen interactions. Database 2016. [Google Scholar] [CrossRef] [PubMed]
- Razick, S.; Magklaras, G.; Donaldson, I.M. Irefindex: A consolidated protein interaction database with provenance. BMC Bioinform. 2008, 9, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, B.; Razick, S.; Turinsky, A.L.; Vlasblom, J.; Crowdy, E.K.; Cho, E.; Morrison, K.; Donaldson, I.M.; Wodak, S.J. Irefweb: Interactive analysis of consolidated protein interaction data and their supporting evidence. Database 2010. [Google Scholar] [CrossRef] [PubMed]
- Chautard, E.; Ballut, L.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics 2009, 25, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Chautard, E.; Fatoux-Ardore, M.; Ballut, L.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, the extracellular matrix interaction database. Nucleic Acids Res. 2011, 39, D235–D240. [Google Scholar] [CrossRef] [PubMed]
- Launay, G.; Salza, R.; Multedo, D.; Thierry-Mieg, N.; Ricard-Blum, S. Matrixdb, the extracellular matrix interaction database: Updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2015, 43, D321–D327. [Google Scholar] [CrossRef] [PubMed]
- Calderone, A.; Castagnoli, L.; Cesareni, G. Mentha: A resource for browsing integrated protein-interaction networks. Nat. Methods 2013, 10, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Beuming, T.; Skrabanek, L.; Niv, M.Y.; Mukherjee, P.; Weinstein, H. Pdzbase: A protein-protein interaction database for pdz-domains. Bioinformatics 2005, 21, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Klapa, M.I.; Tsafou, K.; Theodoridis, E.; Tsakalidis, A.; Moschonas, N.K. Reconstruction of the experimentally supported human protein interactome: What can we learn? BMC Syst. Biol. 2013, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Gioutlakis, A.; Klapa, M.I.; Moschonas, N.K. Pickle 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS ONE 2017, 12, e0186039. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.M.; Vallenius, T.; Ovaska, K.; Westermarck, J.; Makela, T.P.; Hautaniemi, S. Integrated network analysis platform for protein-protein interactions. Nat. Methods 2009, 6, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Pinese, M.; Kassahn, K.S.; Waddell, N.; Pearson, J.V.; Grimmond, S.M.; Biankin, A.V.; Hautaniemi, S.; Wu, J.M. Pina v2.0: Mining interactome modules. Nucleic Acids Res. 2012, 40, D862–D865. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tripathi, L.P.; Prathipati, P.; Mizuguchi, K. Network analysis and in silico prediction of protein-protein interactions with applications in drug discovery. Curr. Opin. Struct. Biol. 2017, 44, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Kinoshita, K.; Nakamura, H. Hub promiscuity in protein-protein interaction networks. Int. J. Mol. Sci. 2010, 11, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Raman, K. Construction and analysis of protein-protein interaction networks. Autom. Exp. 2010, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choi, S.; Hyeon, C. Mapping the intramolecular signal transduction of g-protein coupled receptors. Proteins 2014, 82, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Lee, Y.; Choi, S. Understanding g protein-coupled receptor allostery via molecular dynamics simulations: Implications for drug discovery. Methods Mol. Biol. 2018, 1762, 455–472. [Google Scholar] [PubMed]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting protein-protein interactions from the molecular to the proteome level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef] [PubMed]
- Aragues, R.; Sali, A.; Bonet, J.; Marti-Renom, M.A.; Oliva, B. Characterization of protein hubs by inferring interacting motifs from protein interactions. PLoS Comput. Biol. 2007, 3, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.M.; Lu, L.J.; Xia, Y.; Gerstein, M.B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science 2006, 314, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Wang, J.; Wang, J.; Wu, F.X.; Pan, Y. Rechecking the centrality-lethality rule in the scope of protein subcellular localization interaction networks. PLoS ONE 2015, 10, e0130743. [Google Scholar] [CrossRef] [PubMed]
- Wuchty, S. Controllability in protein interaction networks. Proc. Natl. Acad. Sci. USA 2014, 111, 7156–7160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuchty, S.; Boltz, T.; Kucuk-McGinty, H. Links between critical proteins drive the controllability of protein interaction networks. Proteomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Feldman, I.; Rzhetsky, A.; Vitkup, D. Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 4323–4328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008, 4, e32. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Gibson, T.E.; Lee, H.J.; Yilmazel, B.; Roesel, C.; Hu, Y.; Kwon, Y.; Sharma, A.; Liu, Y.Y.; Perrimon, N.; et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc. Natl. Acad. Sci. USA 2016, 113, 4976–4981. [Google Scholar] [CrossRef] [PubMed]
- Kanhaiya, K.; Czeizler, E.; Gratie, C.; Petre, I. Controlling directed protein interaction networks in cancer. Sci. Rep. 2017, 7, 10327. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, V.; Nacher, J.C.; Akutsu, T.; Ishitsuka, M.; Osadcenco, A.; Sunitha, V.; Bagler, G.; Schwartz, J.-M.; Robertson, D.L. Network controllability: Viruses are driver agents in dynamic molecular systems. bioRxiv 2018. [Google Scholar] [CrossRef]
- Pazos, F.; Valencia, A. In silico two-hybrid system for the selection of physically interacting protein pairs. Proteins 2002, 47, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Scharfe, C.P.; Rodrigues, J.P.; Green, A.G.; Kohlbacher, O.; Sander, C.; Bonvin, A.M.; Marks, D.S. Sequence co-evolution gives 3d contacts and structures of protein complexes. eLife 2014, 3, e03430. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A.J.M.; Sordella, R.; Lu, X.W.; Hartley, J.L.; Temple, G.F.; Brasch, M.A.; Thierry-Mieg, N.; Vidal, M. Protein interaction mapping in c-elegans using proteins involved in vulval development. Science 2000, 287, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Gerstein, M. Analyzing protein function on a genomic scale: The importance of gold-standard positives and negatives for network prediction. Curr. Opin. Microbiol. 2004, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Wu, B.; Jansen, R.; Gerstein, M.; Zhao, H. Information assessment on predicting protein-protein interactions. BMC Bioinform. 2004, 5, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.J.; Xia, Y.; Paccanaro, A.; Yu, H.; Gerstein, M. Assessing the limits of genomic data integration for predicting protein networks. Genome Res. 2005, 15, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.; Yu, H.; Greenbaum, D.; Kluger, Y.; Krogan, N.J.; Chung, S.; Emili, A.; Snyder, M.; Greenblatt, J.F.; Gerstein, M. A bayesian networks approach for predicting protein-protein interactions from genomic data. Science 2003, 302, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinform. 2005, 6, 100. [Google Scholar]
- Ofran, Y.; Rost, B. Isis: Interaction sites identified from sequence. Bioinformatics 2007, 23, e13–e16. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 589, 3516–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikic, M.; Tomic, S.; Vlahovicek, K. Prediction of protein-protein interaction sites in sequences and 3d structures by random forests. PLoS Comput. Biol. 2009, 5, e1000278. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, P.; Huang, D.S.; Li, J.J.; Lok, T.M.; Lyu, M.R. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. FEBS Lett. 2006, 580, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol. 1997, 272, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Analysis of protein-protein interaction sites using surface patches. J. Mol. Biol. 1997, 272, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Westhead, D.R. Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics 2005, 21, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Needham, C.J.; Bulpitt, A.J.; Westhead, D.R. Insights into protein-protein interfaces using a bayesian network prediction method. J. Mol. Biol. 2006, 362, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 2007, 66, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Mizuguchi, K. Applying the naive bayes classifier with kernel density estimation to the prediction of protein-protein interaction sites. Bioinformatics 2010, 26, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Dhole, K.; Singh, G.; Pai, P.P.; Mondal, S. Sequence-based prediction of protein-protein interaction sites with l1-logreg classifier. J. Theor. Biol. 2014, 348, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Dhole, K.; Pai, P.P.; Mondal, S. Springs: Prediction of protein-protein interaction sites using artificial neural networks. Peer. J. PrePrints. 2014, 2, e266v2. [Google Scholar]
- Du, T.; Liao, L.; Wu, C.H.; Sun, B. Prediction of residue-residue contact matrix for protein-protein interaction with fisher score features and deep learning. Methods 2016, 110, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Zhou, B.; Lai, L.H.; Pei, J.F. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinform. 2017, 18, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.Q.; Sun, S.W.; Hu, C.L.; Yao, Y.; Yan, Y.T.; Zhang, Y.P. Deepppi: Boosting prediction of protein-protein interactions with deep neural networks. J. Chem. Inf. Model. 2017, 57, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.R.; Gough, D.A. Predicting protein--protein interactions from primary structure. Bioinformatics 2001, 17, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Liu, M. Prediction of protein-protein interactions using random decision forest framework. Bioinformatics 2005, 21, 4394–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhou, H.X. Prediction of interface residues in protein-protein complexes by a consensus neural network method: Test against nmr data. Proteins 2005, 61, 21–35. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Bonvin, A.M. Cport: A consensus interface predictor and its performance in prediction-driven docking with haddock. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed]
- Dohkan, S.; Koike, A.; Takagi, T. Improving the performance of an svm-based method for predicting protein-protein interactions. In Silico Biol. 2006, 6, 515–529. [Google Scholar] [PubMed]
- Negi, S.S.; Schein, C.H.; Oezguen, N.; Power, T.D.; Braun, W. Interprosurf: A web server for predicting interacting sites on protein surfaces. Bioinformatics 2007, 23, 3397–3399. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Zhou, H.X. Meta-ppisp: A meta web server for protein-protein interaction site prediction. Bioinformatics 2007, 23, 3386–3387. [Google Scholar] [CrossRef] [PubMed]
- Minhas, F.; Geiss, B.J.; Ben-Hur, A. Pairpred: Partner-specific prediction of interacting residues from sequence and structure. Proteins 2014, 82, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Budagyan, L.; Raush, E.; Totrov, M.; Abagyan, R. Pier: Protein interface recognition for structural proteomics. Proteins 2007, 67, 400–417. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.D.; Zhang, C.; Liu, S.; Zhou, Y.Q. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006, 34, 3698–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Mizuguchi, K. Partner-aware prediction of interacting residues in protein-protein complexes from sequence data. PLoS ONE 2011, 6, e29104. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Basu, S.; Kundu, M.; Nasipuri, M.; Plewczynski, D. Ppi_svm: Prediction of protein-protein interactions using machine learning, domain-domain affinities and frequency tables. Cell Mol. Biol. Lett. 2011, 16, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Z.; Yu, L.Z.; Wen, Z.N.; Li, M.L. Using support vector machine combined with auto covariance to predict proteinprotein interactions from protein sequences. Nucleic Acids Res 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.H.; Li, K.B. Predicting protein-protein interaction sites using sequence descriptors and site propensity of neighboring amino acids. Int. J. Mol. Sci. 2016, 17, 1788. [Google Scholar] [CrossRef] [PubMed]
- Zellner, H.; Staudigel, M.; Trenner, T.; Bittkowski, M.; Wolowski, V.; Icking, C.; Merkl, R. Prescont: Predicting protein-protein interfaces utilizing four residue properties. Proteins 2012, 80, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Deng, L.; Fisher, M.; Guan, J.H.; Honig, B.; Petrey, D. Predus: A web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 2011, 39, W283–W287. [Google Scholar] [CrossRef] [PubMed]
- Baspinar, A.; Cukuroglu, E.; Nussinov, R.; Keskin, O.; Gursoy, A. Prism: A web server and repository for prediction of protein-protein interactions and modeling their 3d complexes. Nucleic Acids Res. 2014, 42, W285–W289. [Google Scholar] [CrossRef] [PubMed]
- Neuvirth, H.; Raz, R.; Schreiber, G. Promate: A structure based prediction program to identify the location of protein-protein binding sites. J. Mol. Biol. 2004, 338, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Ramasamy, S.; Raghava, G.P.S. A simple approach for predicting protein-protein interactions. Curr. Protein Pept. Sci. 2010, 11, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Jones, S. Sharp2: Protein-protein interaction predictions using patch analysis. Bioinformatics 2006, 22, 1794–1795. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.T.; Acencio, M.L.; Martins, C.; Lemke, N. The development of a universal in silico predictor of protein-protein interactions. PLoS ONE 2013, 8, e65587. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, A.D.J.; Bonvin, A.M.J.J. Whiscy: What information does surface conservation yield? Application to data-driven docking. Proteins 2006, 63, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Dobbs, D.; Honavar, V. A two-stage classifier for identification of protein-protein interface residues. Bioinformatics 2004, 20 (Suppl. 1), i371–i378. [Google Scholar] [CrossRef]
- Basse, M.J.; Betzi, S.; Morelli, X.; Roche, P. 2p2idb v2: Update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database 2016. [Google Scholar] [CrossRef] [PubMed]
- Higueruelo, A.P.; Jubb, H.; Blundell, T.L. Timbal v2: Update of a database holding small molecules modulating protein-protein interactions. Database 2013. [Google Scholar] [CrossRef] [PubMed]
- Higueruelo, A.P.; Schreyer, A.; Bickerton, G.R.; Pitt, W.R.; Groom, C.R.; Blundell, T.L. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: The timbal database. Chem. Biol. Drug Des. 2009, 74, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Labbe, C.M.; Kuenemann, M.A.; Zarzycka, B.; Vriend, G.; Nicolaes, G.A.; Lagorce, D.; Miteva, M.A.; Villoutreix, B.O.; Sperandio, O. Ippi-db: An online database of modulators of protein-protein interactions. Nucleic Acids Res. 2016, 44, D542–D547. [Google Scholar] [CrossRef] [PubMed]
- Reynes, C.; Host, H.; Camproux, A.C.; Laconde, G.; Leroux, F.; Mazars, A.; Deprez, B.; Fahraeus, R.; Villoutreix, B.O.; Sperandio, O. Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods. PLoS Comput. Biol. 2010, 6, e1000695. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.A.; Violas, S.; Montes, M.; Gomez, D.; Tuffery, P.; Villoutreix, B.O. Faf-drugs: Free adme/tox filtering of compound collections. Nucleic Acids Res. 2006, 34, W738–W744. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Galons, H.; Miteva, M.A.; Villoutreix, B.O. Faf-drugs2: Free adme/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinform. 2008, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Maupetit, J.; Baell, J.; Sperandio, O.; Tuffery, P.; Miteva, M.A.; Galons, H.; Villoutreix, B.O. The faf-drugs2 server: A multistep engine to prepare electronic chemical compound collections. Bioinformatics 2011, 27, 2018–2020. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. Faf-drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef] [PubMed]
- Hamon, V.; Bourgeas, R.; Ducrot, P.; Theret, I.; Xuereb, L.; Basse, M.J.; Brunel, J.M.; Combes, S.; Morelli, X.; Roche, P. 2p2i hunter: A tool for filtering orthosteric protein-protein interaction modulators via a dedicated support vector machine. J. R. Soc. Interface 2014, 11, 20130860. [Google Scholar] [CrossRef] [PubMed]
- Bordner, A.J.; Abagyan, R. Statistical analysis and prediction of protein-protein interfaces. Proteins 2005, 60, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Acuner Ozbabacan, S.E.; Engin, H.B.; Gursoy, A.; Keskin, O. Transient protein–protein interactions. Protein Eng. Des. Sel. 2011, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Gursoy, A.; Keskin, O. Prediction of protein-protein interactions: Unifying evolution and structure at protein interfaces. Phys. Biol. 2011, 8, 035006. [Google Scholar] [CrossRef] [PubMed]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Keskin, O.; Nussinov, R. Conformational ensembles, signal transduction and residue hot spots: Application to drug discovery. Curr. Opin. Drug Discov. Dev. 2010, 13, 527–537. [Google Scholar]
- Bradshaw, R.T.; Patel, B.H.; Tate, E.W.; Leatherbarrow, R.J.; Gould, I.R. Comparing experimental and computational alanine scanning techniques for probing a prototypical protein-protein interaction. Protein Eng. Des. Sel. 2011, 24, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Zhang, S.X. Computational prediction of protein hot spot residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Computational alanine scanning to probe protein-protein interactions: A novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Kruger, D.M.; Gohlke, H. Drugscore(ppi) webserver: Fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 2010, 38, W480–W486. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.Q.; Yan, Y.N.; Sun, Z.X.; Song, J.N.; Zhang, J.Z.H. Interaction entropy for computational alanine scanning in protein-protein binding. WIREs Comput. Mol. Sci. 2018, 8, e1342. [Google Scholar] [CrossRef]
- Fernandez-Recio, J.; Totrov, M.; Skorodumov, C.; Abagyan, R. Optimal docking area: A new method for predicting protein-protein interaction sites. Proteins 2005, 58, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, S.; Fernandez-Recio, J. Identification of hot-spot residues in protein-protein interactions by computational docking. BMC Bioinform. 2008, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.M.; Blundell, T.L.; Fernandez-Recio, J. Pydock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Vendrell, J.; Aviles, F.X.; Fernandez-Recio, J. Structural and functional characterization of binding sites in metallocarboxypeptidases based on optimal docking area analysis. Proteins 2007, 68, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F.; Jonsdottir, S.O.; Pajeva, I. Structural insight into the unc-45-myosin complex. Proteins 2013, 81, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Aumentado-Armstrong, T.T.; Istrate, B.; Murgita, R.A. Algorithmic approaches to protein-protein interaction site prediction. Algorithms Mol. Biol. 2015, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Yang, J.S.; Choi, Y.; Ryu, S.H.; Kim, S. Evolutionary conservation in multiple faces of protein interaction. Proteins 2009, 77, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Weigt, M.; White, R.A.; Szurmant, H.; Hoch, J.A.; Hwa, T. Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl. Acad. Sci. USA 2009, 106, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Ingles-Prieto, A.; Ibarra-Molero, B.; Delgado-Delgado, A.; Perez-Jimenez, R.; Fernandez, J.M.; Gaucher, E.A.; Sanchez-Ruiz, J.M.; Gavira, J.A. Conservation of protein structure over four billion years. Structure 2013, 21, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Norel, R.; Honig, B.H. Protein interface conservation across structure space. Proc. Natl. Acad. Sci. USA 2010, 107, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Feng, K.Y.; Chen, L.; Huang, T.; Cai, Y.D. Prediction of protein-protein interaction sites by random forest algorithm with mrmr and ifs. PLoS ONE 2012, 7, e43927. [Google Scholar] [CrossRef] [PubMed]
- Higa, R.H.; Tozzi, C.L. Prediction of binding hot spot residues by using structural and evolutionary parameters. Genet. Mol. Biol. 2009, 32, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meireles, L.M.C.; Domling, A.S.; Camacho, C.J. Anchor: A web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res. 2010, 38, W407–W411. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Dobbs, D.; Honavar, V. Homppi: A class of sequence homology based protein-protein interface prediction methods. BMC Bioinform. 2011, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Darnell, S.J.; LeGault, L.; Mitchell, J.C. Kfc server: Interactive forecasting of protein interaction hot spots. Nucleic Acids Res. 2008, 36, W265–W269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mitchell, J.C. Kfc2: A knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins 2011, 79, 2671–2683. [Google Scholar] [CrossRef] [PubMed]
- Darnell, S.J.; Page, D.; Mitchell, J.C. An automated decision-tree approach to predicting protein interaction hot spots. Proteins 2007, 68, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Keskin, O.; Gursoy, A. Hotpoint: Hot spot prediction server for protein interfaces. Nucleic Acids Res. 2010, 38, W402–W406. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The ftmap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Kim, D.; Lee, D. A feature-based approach to modeling protein-protein interaction hot spots. Nucleic Acids Res. 2009, 37, 2672–2687. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, Q.C.; Chen, Z.G.; Meng, Y.; Guan, J.H.; Zhou, S.G. Predhs: A web server for predicting protein-protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014, 42, W290–W295. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.K.; Karanicolas, J. Druggable protein interaction sites are more predisposed to surface pocket formation than the rest of the protein surface. PLoS Comput. Biol. 2013, 9, e1002951. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Hajduk, P.J. Effects of conformational dynamics on predicted protein druggability. ChemMedChem 2006, 1, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Rakers, C.; Bermudez, M.; Keller, B.G.; Mortier, J.; Wolber, G. Computational close up on protein-protein interactions: How to unravel the invisible using molecular dynamics simulations? WIREs Comput. Mol. Sci. 2015, 5, 345–359. [Google Scholar] [CrossRef]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. Protein-protein docking tested in blind predictions: The capri experiment. Mol. Biosyst. 2010, 6, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Lesk, V.I.; Sternberg, M.J. 3d-garden: A system for modelling protein-protein complexes based on conformational refinement of ensembles generated with the marching cubes algorithm. Bioinformatics 2008, 24, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Schindler, C.E.; Chauvot de Beauchene, I.; Zacharias, M. A web interface for easy flexible protein-protein docking with attract. Biophys. J. 2015, 108, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, D.; Huang, Y.; Liu, S.; Xiao, Y. Aspdock: Protein-protein docking algorithm using atomic solvation parameters model. BMC Bioinform. 2011, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.N.; Krippahl, L.; Wampler, J.E.; Moura, J.J. Bigger: A new (soft) docking algorithm for predicting protein interactions. Proteins 2000, 39, 372–384. [Google Scholar] [CrossRef]
- Pons, C.; Jimenez-Gonzalez, D.; Gonzalez-Alvarez, C.; Servat, H.; Cabrera-Benitez, D.; Aguilar, X.; Fernandez-Recio, J. Cell-dock: High-performance protein-protein docking. Bioinformatics 2012, 28, 2394–2396. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The cluspro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, S.; Ravikant, D.V.; Elber, R. Dock/pierr: Web server for structure prediction of protein-protein complexes. Methods Mol. Biol. 2014, 1137, 199–207. [Google Scholar] [PubMed]
- Roberts, V.A.; Thompson, E.E.; Pique, M.E.; Perez, M.S.; Ten Eyck, L.F. Dot2: Macromolecular docking with improved biophysical models. J. Comput. Chem. 2013, 34, 1743–1758. [Google Scholar] [CrossRef] [PubMed]
- Ausiello, G.; Cesareni, G.; Helmer-Citterich, M. Escher: A new docking procedure applied to the reconstruction of protein tertiary structure. Proteins 1997, 28, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, C.; Chowdhury, R.; Siddavanahalli, V. F2dock: Fast fourier protein-protein docking. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Nussinov, R.; Wolfson, H.J. Fiberdock: A web server for flexible induced-fit backbone refinement in molecular docking. Nucleic Acids Res. 2010, 38, W457–W461. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. Firedock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Aportela, E.; Lopez-Blanco, J.R.; Chacon, P. Frodock 2.0: Fast protein-protein docking server. Bioinformatics 2016, 32, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Gabb, H.A.; Jackson, R.M.; Sternberg, M.J. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol. 1997, 272, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.J.; Willett, P.; Artymiuk, P.J. Protein docking using a genetic algorithm. Proteins 2001, 44, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. Gramm-x public web server for protein-protein docking. Nucleic. Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. Haddock: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. Hdock: A web server for protein-protein and protein-DNA/rna docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Venkatraman, V. Ultra-fast fft protein docking on graphics processors. Bioinformatics 2010, 26, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Recio, J.; Totrov, M.; Abagyan, R. Icm-disco docking by global energy optimization with fully flexible side-chains. Proteins 2003, 52, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vavrusa, M.; Andreani, J.; Rey, J.; Tuffery, P.; Guerois, R. Interevdock: A docking server to predict the structure of protein-protein interactions using evolutionary information. Nucleic Acids Res. 2016, 44, W542–W549. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garcia, B.; Roel-Touris, J.; Romero-Durana, M.; Vidal, M.; Jimenez-Gonzalez, D.; Fernandez-Recio, J. Lightdock: A new multi-scale approach to protein-protein docking. Bioinformatics 2018, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kihara, D. Protein docking prediction using predicted protein-protein interface. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Ohue, M.; Shimoda, T.; Suzuki, S.; Matsuzaki, Y.; Ishida, T.; Akiyama, Y. Megadock 4.0: An ultra-high-performance protein-protein docking software for heterogeneous supercomputers. Bioinformatics 2014, 30, 3281–3283. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zeev, E.; Kowalsman, N.; Ben-Shimon, A.; Segal, D.; Atarot, T.; Noivirt, O.; Shay, T.; Eisenstein, M. Docking to single-domain and multiple-domain proteins: Old and new challenges. Proteins 2005, 60, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Patchdock and symmdock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Neveu, E.; Ritchie, D.W.; Popov, P.; Grudinin, S. Pepsi-dock: A detailed data-driven protein-protein interaction potential accelerated by polar fourier correlation. Bioinformatics 2016, 32, i693–i701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. Piper: An fft-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Pal, D. Prune and probe-two modular web services for protein-protein docking. Nucleic Acids Res. 2011, 39, W229–W234. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garcia, B.; Pons, C.; Fernandez-Recio, J. Pydockweb: A web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [PubMed]
- Lyskov, S.; Gray, J.J. The rosettadock server for local protein-protein docking. Nucleic Acids Res 2008, 36, W233–W238. [Google Scholar] [CrossRef] [PubMed]
- Terashi, G.; Takeda-Shitaka, M.; Kanou, K.; Iwadate, M.; Takaya, D.; Umeyama, H. The ske-dock server and human teams based on a combined method of shape complementarity and free energy estimation. Proteins 2007, 69, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.J.; Vajda, S. Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 10636–10641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.; Fernandez-Recio, J.; Bates, P.A. Swarmdock: A server for flexible protein-protein docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Levieux, G.; Tiger, G.; Mader, S.; Zagury, J.F.; Natkin, S.; Montes, M. Udock, the interactive docking entertainment system. Faraday Discuss. 2014, 169, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. Zdock server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shimon, A.; Niv, M.Y. Anchordock: Blind and flexible anchor-driven peptide docking. Structure 2015, 23, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. Cabs-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Moll, M.; Devaurs, D.; Jackson, K.R.; Lizee, G.; Kavraki, L.E. Dinc 2.0: A new protein-peptide docking webserver using an incremental approach. Cancer Res. 2017, 77, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta flexpepdock web server--high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. Galaxypepdock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [PubMed]
- Trellet, M.; Melquiond, A.S.; Bonvin, A.M. A unified conformational selection and induced fit approach to protein-peptide docking. PLoS ONE 2013, 8, e58769. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. Hpepdock: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.F.; Xu, X.J.; Zou, X.Q. Fully blind docking at the atomic level for protein-peptide complex structure prediction. Structure 2016, 24, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Rey, J.; Schindler, C.E.M.; Zacharias, M.; Tuffery, P. The pepattract web server for blind, large-scale peptide-protein docking. Nucleic Acids Res. 2017, 45, W361–W364. [Google Scholar] [CrossRef] [PubMed]
- Donsky, E.; Wolfson, H.J. Pepcrawler: A fast rrt-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics 2011, 27, 2836–2842. [Google Scholar] [CrossRef] [PubMed]
- Trabuco, L.G.; Lise, S.; Petsalaki, E.; Russell, R.B. Pepsite: Prediction of peptide-binding sites from protein surfaces. Nucleic Acids Res. 2012, 40, W423–W427. [Google Scholar] [CrossRef] [PubMed]
- Saladin, A.; Rey, J.; Thevenet, P.; Zacharias, M.; Moroy, G.; Tuffery, P. Pep-sitefinder: A tool for the blind identification of peptide binding sites on protein surfaces. Nucleic Acids Res. 2014, 42, W221–W226. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Govindaraj, R.G.; Choi, S. In silico approach to inhibition of signaling pathways of toll-like receptors 2 and 4 by st2l. PLoS ONE 2011, 6, e23989. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Choi, Y.M.; Lee, G.; Choi, S. Structure-function relationship of cytoplasmic and nuclear ikappab proteins: An in silico analysis. PLoS ONE 2010, 5, e15782. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Govindaraj, R.; Lee, G.; Choi, S. Molecular modeling-based evaluation of dual function of ikappabzeta ankyrin repeat domain in toll-like receptor signaling. J. Mol. Recognit. 2011, 24, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Galeazzi, R.; Laudadio, E.; Falconi, E.; Massaccesi, L.; Ercolani, L.; Mobbili, G.; Minnelli, C.; Scire, A.; Cianfruglia, L.; Armeni, T. Protein-protein interactions of human glyoxalase ii: Findings of a reliable docking protocol. Org. Biomol. Chem. 2018, 16, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Guner, O.; Clement, O.; Kurogi, Y. Pharmacophore modeling and three dimensional database searching for drug design using catalyst: Recent advances. Curr. Med. Chem. 2004, 11, 2991–3005. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and proteins (flap): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Barillari, C.; Marcou, G.; Rognan, D. Hot-spots-guided receptor-based pharmacophores (hs-pharm): A knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J. Chem. Inf. Model. 2008, 48, 1396–1410. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. Ligandscout: 3-d pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. Phase: A novel approach to pharmacophore modeling and 3d database searching. Chem. Biol. Drug. Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Domling, A.; Camacho, C.J. Anchorquery: Rapid online virtual screening for small-molecule protein-protein interaction inhibitors. Protein Sci. 2018, 27, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.H.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. Icm—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Betzi, S.; Restouin, A.; Opi, S.; Arold, S.T.; Parrot, I.; Guerlesquin, F.; Morelli, X.; Collette, Y. Protein protein interaction inhibition (2p2i) combining high throughput and virtual screening: Application to the hiv-1 nef protein. Proc. Natl. Acad. Sci. USA 2007, 104, 19256–19261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.B.; Chen, J.F.; Meagher, J.L.; Yang, C.Y.; Aguilar, A.; Liu, L.; Bai, L.C.; Gong, X.; Cai, Q.; Fang, X.L.; et al. Design of bcl-2 and bcl-xl inhibitors with subnanomolar binding affinities based upon a new scaffold. J. Med. Chem. 2012, 55, 4664–4682. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Bower, K.E.; Beuscher, A.E., 4th; Zhou, B.; Bobkov, A.A.; Olson, A.J.; Vogt, P.K. Stabilizers of the max homodimer identified in virtual ligand screening inhibit myc function. Mol. Pharmacol. 2009, 76, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Schuffenhauer, A.; Ruedisser, S.; Marzinzik, A.L.; Jahnke, W.; Blommers, M.; Selzer, P.; Jacoby, E. Library design for fragment based screening. Curr. Top. Med. Chem. 2005, 5, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.L.; Schneider, G. Scaffold architecture and pharmacophoric properties of natural products and trade drugs: Application in the design of natural product-based combinatorial libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Over, B.; Wetzel, S.; Grutter, C.; Nakai, Y.; Renner, S.; Rauh, D.; Waldmann, H. Natural-product-derived fragments for fragment-based ligand discovery. Nat. Chem. 2013, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. Pocketquery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 2012, 40, W387–392. [Google Scholar] [CrossRef] [PubMed]
- Geppert, T.; Bauer, S.; Hiss, J.A.; Conrad, E.; Reutlinger, M.; Schneider, P.; Weisel, M.; Pfeiffer, B.; Altmann, K.H.; Waibler, Z.; et al. Immunosuppressive small molecule discovered by structure-based virtual screening for inhibitors of protein-protein interactions. Angew. Chem. Int. Ed. 2012, 51, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Weisel, M.; Proschak, E.; Schneider, G. Pocketpicker: Analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Geppert, T.; Hoy, B.; Wessler, S.; Schneider, G. Context-based identification of protein-protein interfaces and “hot-spot” residues. Chem. Biol. 2011, 18, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Basith, S.; Choi, S. Recent advances in structure-based drug design targeting class a g protein-coupled receptors utilizing crystal structures and computational simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Verkhivker, G.M. Probing molecular mechanisms of the hsp90 chaperone: Biophysical modeling identifies key regulators of functional dynamics. PLoS ONE 2012, 7, e37605. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, E.S.; Jang, H.; Gursoy, A.; Keskin, O.; Li, Z.; Sacks, D.B.; Nussinov, R. Unraveling the molecular mechanism of interactions of the rho gtpases cdc42 and rac1 with the scaffolding protein iqgap2. J. Biol. Chem. 2018, 293, 3685–3699. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Cheung, C.H.A.; Tsai, J.Y.; Hsieh, H.P.; Coumar, M.S. Disruption of protein-protein interactions: Hot spot detection, structure-based virtual screening and in vitro testing for the anti-cancer drug target survivin. RSC Adv. 2016, 6, 31947–31959. [Google Scholar] [CrossRef]
- Bastianelli, G.; Bouillon, A.; Nguyen, C.; Crublet, E.; Petres, S.; Gorgette, O.; Le-Nguyen, D.; Barale, J.C.; Nilges, M. Computational reverse-engineering of a spider-venom derived peptide active against plasmodium falciparum sub1. PLoS ONE 2011, 6, e21812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Bai, H.; Lai, L. Molecular dynamics simulations on the oligomer-formation process of the gnnqqny peptide from yeast prion protein sup35. Biophys. J. 2007, 93, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Baram, M.; Atsmon-Raz, Y.; Ma, B.; Nussinov, R.; Miller, Y. Amylin-abeta oligomers at atomic resolution using molecular dynamics simulations: A link between type 2 diabetes and alzheimer’s disease. Phys. Chem. Chem. Phys. 2016, 18, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provaznik, I.; Choi, I. Computer aided drug design: Success and limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Lane, D.P.; Verma, C. Multiple peptide conformations give rise to similar binding affinities: Molecular simulations of p53-mdm2. J. Am. Chem. Soc. 2008, 130, 13514–13515. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
In silico strategies for PPI drug discovery. Diverse computational tools encompass various stages of PPI drug discovery, including the interpretation of protein network topology, the characterization of interface and hot spots, the exploration of PPI chemical spaces for lead discovery and optimization, and the elucidation of complex interactions and dynamics.
Figure 1.
In silico strategies for PPI drug discovery. Diverse computational tools encompass various stages of PPI drug discovery, including the interpretation of protein network topology, the characterization of interface and hot spots, the exploration of PPI chemical spaces for lead discovery and optimization, and the elucidation of complex interactions and dynamics.
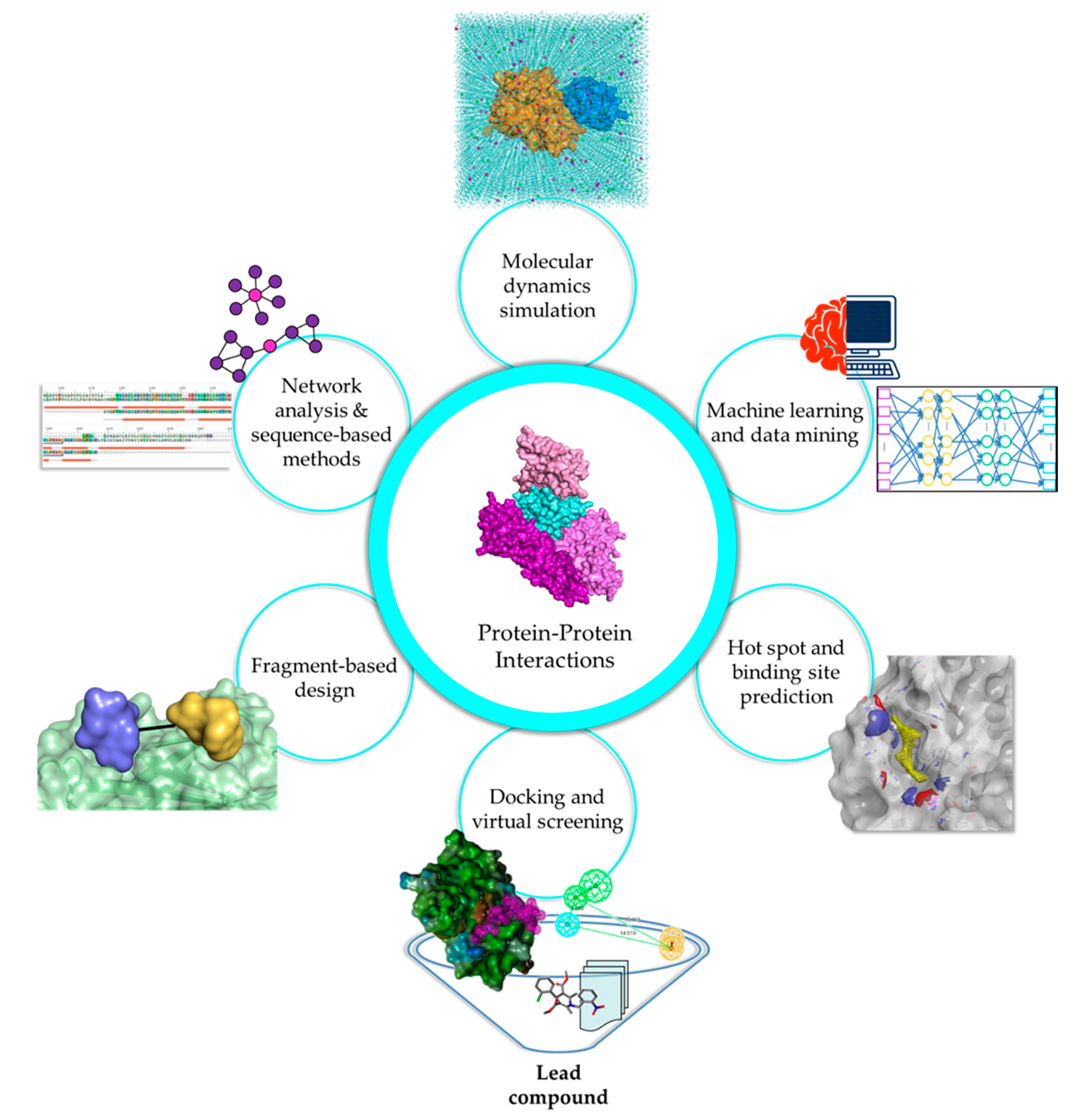
Figure 2.
General steps involved in machine-learning (ML)-based PPI predictions.

Figure 3.
Varied applications of molecular dynamics (MD) simulations in PPI research. Using MD simulations, several aspects of PPIs can be explored, such as gaining insights into their structural, functional, and mechanistic processes, design of PPI inhibitors or stabilizers, and refinement of PPI structures with lower resolutions.
Figure 3.
Varied applications of molecular dynamics (MD) simulations in PPI research. Using MD simulations, several aspects of PPIs can be explored, such as gaining insights into their structural, functional, and mechanistic processes, design of PPI inhibitors or stabilizers, and refinement of PPI structures with lower resolutions.
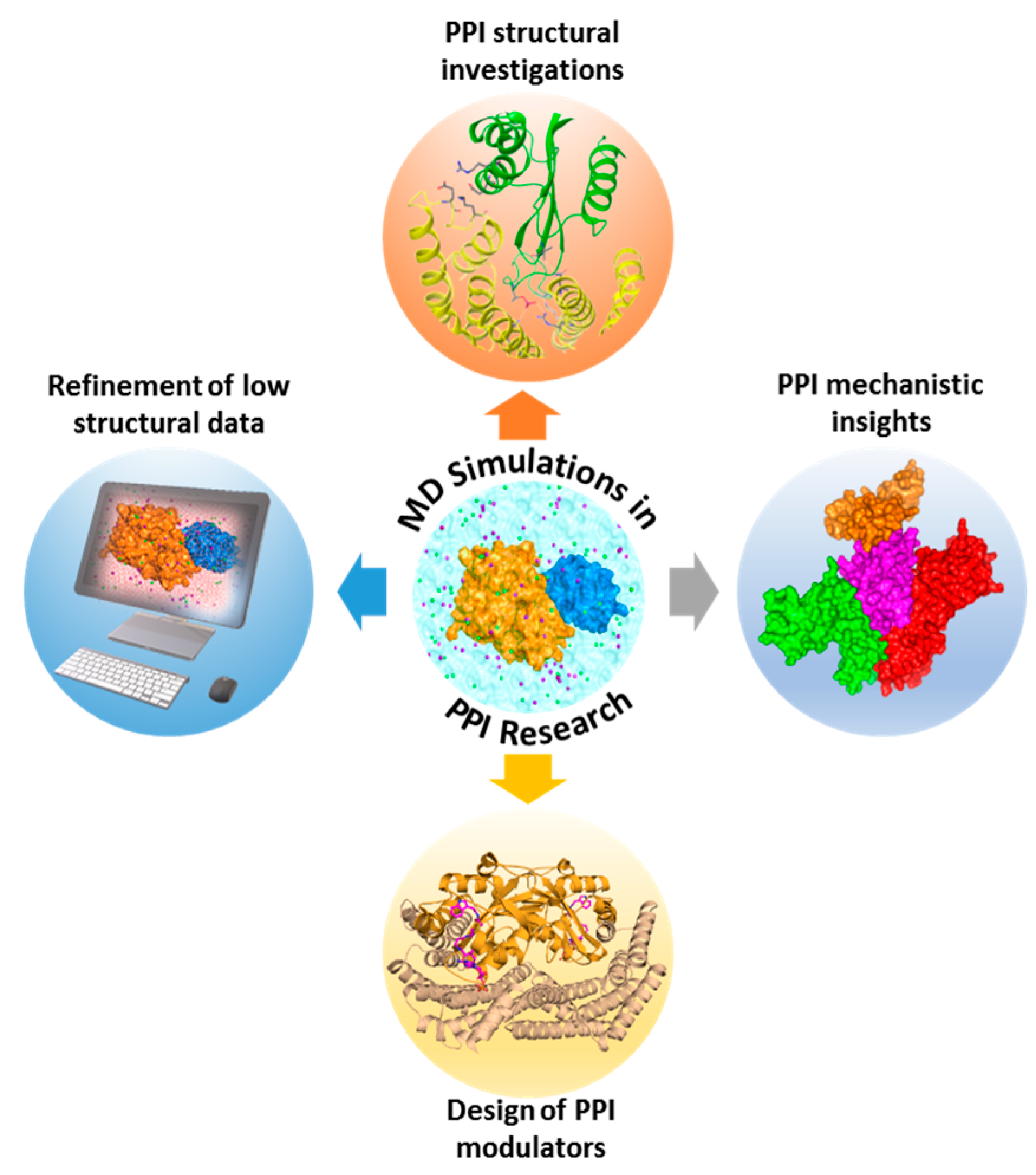

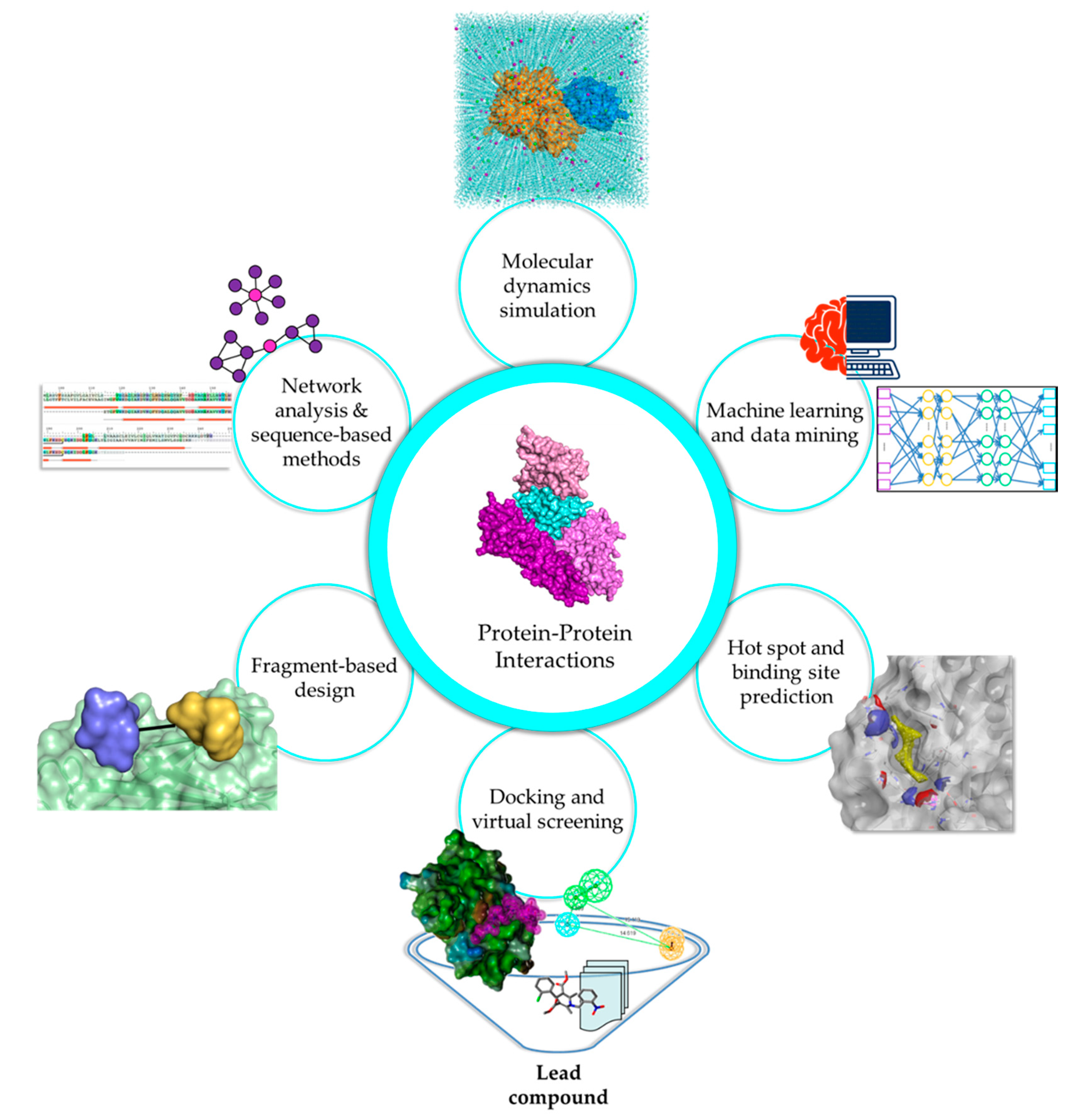{kind=link}
{kind=link}
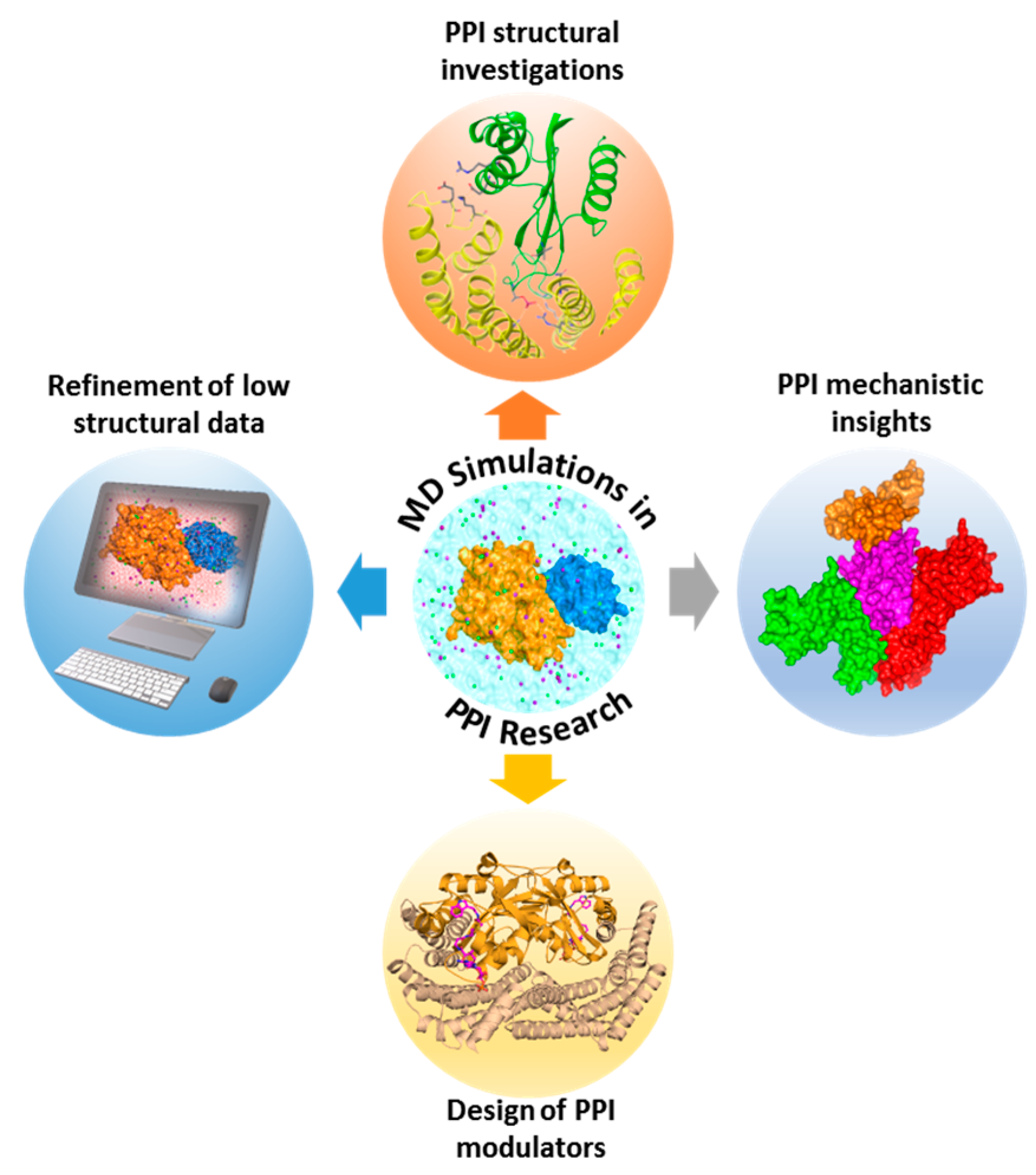{kind=link}
Table 1.
Examples of protein-protein interaction (PPI) modulators in clinical trials or clinical use.
Table 1.
Examples of protein-protein interaction (PPI) modulators in clinical trials or clinical use.
| Compound | Structure | Mode of Action | Identification Method | Clinical Status | Ref. |
|---|---|---|---|---|---|
| ABT-199 (Venetoclax) |  | Bcl-2/(BAX/BAK) inhibitor | Rational design for BCL-2 | Approved for chronic lymphocytic leukemia (CLL) with 17p deletion | [31] |
| ABT-263 (Navitoclax) |  | Bcl-2/(BAX/BAK) inhibitor | High-throughput screening (HTS) and fragment-based design | Phase 1/2 for various cancer types | [32,33,34,35] |
| AMG232 |  | p53/MDM2 inhibitor | Fragment-based design | Phase 1 for cancer | [36] |
| Birabresib (OTX015) |  | Bromodomain and extra-terminal (BET)/histone peptide inhibitor | Cell assays | Phase 1 for various cancer types | [37,38] |
| Birinapant (TL32711) | 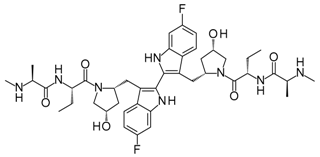 | CIAP1-BIR3/Caspase-9 and XIAP-BIR3/second mitochondrial activator of caspases (SMAC) inhibitor | Dimerized SMAC mimetics | Phase 1/2 for various cancer types | [39,40,41,42] |
| Cabazitaxel | 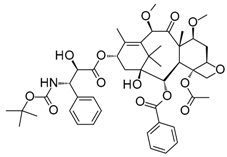 | Microtubule inhibitor | Screening of semisynthetic taxane derivatives | Approved for prostate cancer | [43] |
| CGM097 | 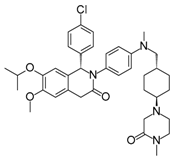 | p53/MDM2 inhibitor | Virtual screening (VS), molecular modeling, and rational design based on crystal complex structure | Phase 1 for cancer | [44] |
| Cilengitide | 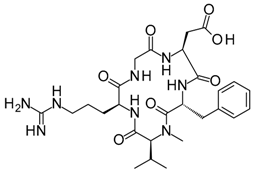 | Integrin αvβ3/αvβ5 inhibitor | Ligand-based design using Arg-Gly-Asp (RGD)-binding motif | Phase 1/2/3 for various cancer types | [45,46,47,48] |
| CPI-0610 |  | BET/histone peptide inhibitor | Structure-based drug design | Phase 1/2 for various cancer types | [49,50,51] |
| Docetaxel |  | Microtubule inhibitor | Semisynthetic taxane derivative | Approved for various cancer types | [52,53,54,55] |
| DS-3032b |  | p53/MDM2 inhibitor | Enzyme and cell assays | Phase 1 for leukemia | [56] |
| Eptifibatide |  | Glycoprotein IIb/IIIa inhibitor | Peptide-based (barbourin) design | Approved as platelet aggregation inhibitor | [57,58] |
| FK506 (Tacrolimus) |  | FK506-binding protein 12 (FKBP12)/Calcineurin inhibitor | In vitro and in vivo assays | Approved for immunosuppression/organ rejection | [59] |
| I-BET762 (Molibresib) |  | BET/histone peptide inhibitor | Cell-based HTS | Phase 2 for cancer | [60] |
| LCL161 |  | Inhibitor of apoptosis (IAP)/SMAC inhibitor | SMAC mimetics, cell assays | Phase 1/2 for various cancer types | [61,62,63,64]. |
| Lifitegrast (SAR1118) |  | Lymphocyte function-associated antigen-1 (LFA-1)/Intercellular adhesion molecule 1 ICAM-1 inhibitor | Structure-based rational design based on LFA-1 and ICAM-1 binding | Approved for dry eye | [65] |
| MI-77301 (SAR405838) |  | p53/MDM2 inhibitor | Structure-based rational design based on p53 peptide | Phase 1 for cancer | [66,67] |
| Maraviroc |  | CCR5/gp120 inhibitor | HTS | Approved for human immunodeficiency virus (HIV) | [68,69] |
| Onalespib (AT13387) |  | HSP90 inhibitor | Fragment-based design | Phase 1 for cancer | [70] |
| RG7112 |  | p53/MDM2 inhibitor | HTS and rational optimization of Nutlins | Phase 1 for cancer | [71] |
| RG7388 (Idasanutlin) | 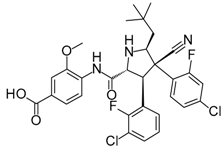 | p53/MDM2 inhibitor | Rational optimization of RG7112, biochemical and cell assays | Phase 3 for acute myeloid leukemia, phase 1/2 for other various cancer types | [72] |
| Tirofiban | 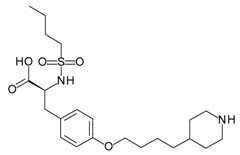 | Glycoprotein IIb/IIIa inhibitor | Ligand-based design using RGD-binding motif | Approved as platelet aggregation inhibitor | [57,73] |
Table 2.
Primary databases and meta-databases * for PPI information.
| Database | Website | Description | Number of Proteins | Number of Interactions | Ref. |
|---|---|---|---|---|---|
| BIND | http://download.baderlab.org/BINDTranslation/ | Biomolecular Interaction Network Database (Last update: 2004) | - | 198,905 | [132] |
| BioGRID | https://thebiogrid.org/ | Biological General Repository for Interaction Datasets (Last update: 2018) | - | 1,202,227 | [133,134] |
| DIP | http://dip.doe-mbi.ucla.edu/dip/ | Database of Interacting Proteins (Last update: 2017) | 28,823 | 81,762 | [135] |
| HPRD | http://www.hprd.org/ | Human Protein Reference Database (Last update: 2009) | 30,047 | 41,327 | [136] |
| IntAct | https://www.ebi.ac.uk/intact/ | IntAct Molecular Interaction Database (Last update: 2013) | 105,180 | 805,177 | [137,138,139] |
| MINT | https://mint.bio.uniroma2.it/ | Molecular INTeraction database (Last update: 2013) | 25,178 | 123,940 | [140] |
| MIPS | http://mips.helmholtz-muenchen.de/proj/ppi/ | Mammalian Protein-Protein Database (Last update: 2005) | 900 | 1800 | [141] |
| CORUM | http://mips.helmholtz-muenchen.de/corum/ | Comprehensive resource of mammalian protein complexes | - | 2837 | [142,143] |
| DroID | http://www.droidb.org/Index.jsp | Drosophila Interactions Database (Last update: 2017) | - | 262,631 | [144,145] |
| APID * | http://cicblade.dep.usal.es:8080/APID/init.action | Agile Protein Interactomes Dataserver | 90,379 | 678,441 | [146] |
| HIV Interaction DB * | https://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions/ | Interactions between HIV-1 proteins with host cell proteins, other HIV-1 proteins, or proteins from HIV-associated disease organisms | - | 2589 | [147,148,149,150] |
| HPID * | http://wilab.inha.ac.kr/hpid/ | Human Protein Interaction Database | 4642 | 719,349 | [151] |
| HPIDB * | http://hpidb.igbb.msstate.edu/index.html | Database for host-pathogen interactions (Last update: 2016) | - | 45,238 | [152,153] |
| IRefWeb * | http://wodaklab.org/iRefWeb/ | Consolidated protein interaction database with provenance | 66,701 | 1,119,604 (distinct: 263,479) | [154,155] |
| MatrixDB * | http://matrixdb.univ-lyon1.fr/ | Extracellular Matrix Interaction Database | - | 9262 | [156,157,158] |
| Mentha * | http://mentha. uniroma2.it/ | Molecular interaction database (Last update: 2018) | 89,666 | 707,003 | [159] |
| PDZBase * | http://abc.med. cornell.edu/pdzbase | Database of PPIs which involve PDZ domains | - | ~300 | [160] |
| PICKLE * | http://www.pickle.gr/ | Protein InteraCtion KnowLedgebasE | - | 120,882 | [161,162] |
| PINA * | http://omics.bjcancer.org/pina/ | Protein Interaction Network Analysis | - | 365,930 | [163,164] |
Table 3.
Alphabetical listing of machine learning predictors for the identification of PPIs.
| Tool/Server | Input Type | ML Algorithm | Features | Website URL | Ref. |
|---|---|---|---|---|---|
| Bock et al. | Structure | Support vector machine (SVM) | Primary structure and associated data | N/A | [205] |
| Chen et al. | Structure | Decision tree | Domain interaction data | Source code available upon request | [206] |
| Cons-PPISP | Structure | Neural network (NN) | Position-specific scoring matrix (PSSM), solvent accessibilities, and spatial neighbors of each residue | http://pipe.scs.fsu.edu/ppisp.html | [207] |
| CPORT * | Structure-based meta server | Scoring function | Combines six interface prediction methods: WHISCY, PIER, ProMate, cons-PPISP, SPPIDER, and PINUP into a consensus predictor | http://haddock.science.uu.nl/services/CPORT/ | [208] |
| DeepPPI | Sequence | Deep neural network (DNN) | Sequence features | http://ailab.ahu.edu.cn:8087/DeepPPI/index.html | [204] |
| Dohkan et al. | Structure | SVM | Domains and amino acid compositions | N/A | [209] |
| InterProSurf | Structure | Scoring function | Solvent accessible surface area (SASA), propensity of interface residues | http://curie.utmb.edu/prosurf.html | [210] |
| MetaPPI * | Structure | Scoring function | Raw scores from five prediction servers PPI−Pred, PPISP, PINUP, Promate, and SPPIDER | http://projects.biotec.tu-dresden.de/metappi/ | |
| Meta-PPISP * | Structure | Linear regression | Raw scores from three other servers: ProMate, PINUP, cons-PPISP | http://pipe.scs.fsu.edu/meta-ppisp.html | [211] |
| PAIRpred | Sequence or structure | Multiple pairwise kernel SVMs | Structural features: relative accessible surface area (rASA), residue depth, half sphere amino acid composition, protrusion index. Sequence features: PSSM and predicted rASA | Python code available at: http://combi.cs.colostate.edu/supplements/pairpred/ | [212] |
| PIER | Structure | Partial least square (PLS) regression | Solvent accessibility and evolutionary conservation | http://abagyan.ucsd.edu/PIER/ | [213] |
| PINUP | Structure | Empirical energy function | Side-chain energy score, residue interface propensity, and residue conservation score | http://sysbio.unl.edu/services/PINUP/ | [214] |
| PPiPP | Sequence | NN | Binary encoding of 20 amino acids and PSSM | http://mizuguchilab.org/netasa/ppipp/ | [215] |
| PPI_SVM | Structure | SVM | Physical interactions of constituent domains | N/A | [216] |
| Pred-PPI | Sequence | SVM | Conservation, electrostatic potential, hydrophobicity, propensity of interface residues, surface shape, and solvent accessible surface area | http://cic.scu.edu.cn/bioinformatics/predict_ppi/ | [217] |
| predPPIS | Sequence | SVM and Bayesian classifiers | Sequence features | http://bsaltools.ym.edu.tw/predppis/ | [218] |
| PresCont | Structure | SVM | SASA, hydrophobicity, conservation and the local environment of each amino acid on the protein surface | http://bioinf.ur.de/php/prescont.ph | [219] |
| PredUs | Structure | SVM | SASA, hydrophobicity, conservation and the local environment of each amino acid on the protein surface | http://bhapp.c2b2.columbia.edu/PredUs/ | [220] |
| PRISM | Structure | Scoring function | Geometric complementarity, conservation | http://cosbi.ku.edu.tr/prism/index.php | [221] |
| PROFisis | Sequence | NN | Sequence features | http://rostlab.org/owiki/index.php/PROFisis | [190] |
| ProMate | Structure | Composite probability | Multiple features like amino-acid propensities, pairwise amino-acid distribution, residue conservation, geometric properties, etc. | http://bioinfo41.weizmann.ac.il/promate/promate.html | [222] |
| ProPrInt | Sequence | SVM | Sequence features, PSSM | http://crdd.osdd.net/raghava/proprint/ | [223] |
| PSIVER | Sequence | Naïve Bayes classifier | PSSM, predicted solvent accessibility | http://mizuguchilab.org/PSIVER/ | [199] |
| SHARP2 | Structure | Scoring function | Solvation potential, hydrophobicity, accessible surface area, residue interface propensity, planarity and protrusion | N/A | [224] |
| SPPIDER | Sequence | SVM, NN | Fingerprints of protein interactions based on predicted relative solvent accessibility (experimental) | http://sppider.cchmc.org/ | [198] |
| Sun et al. | Sequence | DNN | Sequence features | N/A | [203] |
| UNISPPI | Sequence | Decision tree | Amino acid frequencies | N/A | [225] |
| WHISCY | Structure and multiple sequence alignment (MSA) | Scoring function | Residue conservation, interface propensity of residues | http://milou.science.uu.nl/services/WHISCY/ | [226] |
| Yan et al. | Sequence | SVM, Bayes | Interface residue neighborhoods | N/A | [227] |
* Meta-based ML predictors of PPIs.
Table 4.
Alphabetical listing of available protein-protein docking tools.
Table 5.
Alphabetical listing of available protein-peptide docking tools.
| Tool/Server | Docking Algorithm | Website URL | Type | Ref. |
|---|---|---|---|---|
| AnchorDock | Global peptide docking | N/A | Standalone | [314] |
| CABS-dock | Global peptide docking | http://biocomp.chem.uw.edu.pl/CABSdock | Online | [315] |
| DINC | Global peptide docking | http://dinc.kavrakilab.org/ | Online | [316] |
| FlexPepDock | Local peptide docking | http://flexpepdock.furmanlab.cs.huji.ac.il/ | Online | [317] |
| GalaxyPepDock | Local peptide docking | http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=PEPDOCK | Online | [318] |
| HADDOCK peptide | Local peptide docking | http://www.bonvinlab.org/software/haddock2.2/ | Standalone, online | [319] |
| HPEPDOCK | Global peptide docking | http://huanglab.phys.hust.edu.cn/hpepdock/ | Online | [320] |
| MDockPep | Global peptide docking | N/A | Standalone | [321] |
| pepATTRACT | Global peptide docking | http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::pepATTRACT | Online, standalone | [322] |
| PepCrawler | Local peptide docking | http://bioinfo3d.cs.tau.ac.il/PepCrawler/ | Online, standalone | [323] |
| PepSite | Local peptide docking | http://pepsite2.russelllab.org/ | Online | [324] |
| PEP-SiteFinder | Local peptide docking | http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-SiteFinder/ | Online | [325] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Macalino, S.J.Y.; Basith, S.; Clavio, N.A.B.; Chang, H.; Kang, S.; Choi, S. Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules 2018, 23, 1963. https://doi.org/10.3390/molecules23081963
AMA Style
Macalino SJY, Basith S, Clavio NAB, Chang H, Kang S, Choi S. Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules. 2018; 23(8):1963. https://doi.org/10.3390/molecules23081963
Chicago/Turabian StyleMacalino, Stephani Joy Y., Shaherin Basith, Nina Abigail B. Clavio, Hyerim Chang, Soosung Kang, and Sun Choi. 2018. "Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery" Molecules 23, no. 8: 1963. https://doi.org/10.3390/molecules23081963