Integrating Thyroid Hormone Signaling in Hypothalamic Control of Metabolism: Crosstalk Between Nuclear Receptors

{kind=link}
{kind=link}
Abstract
:1. Introduction
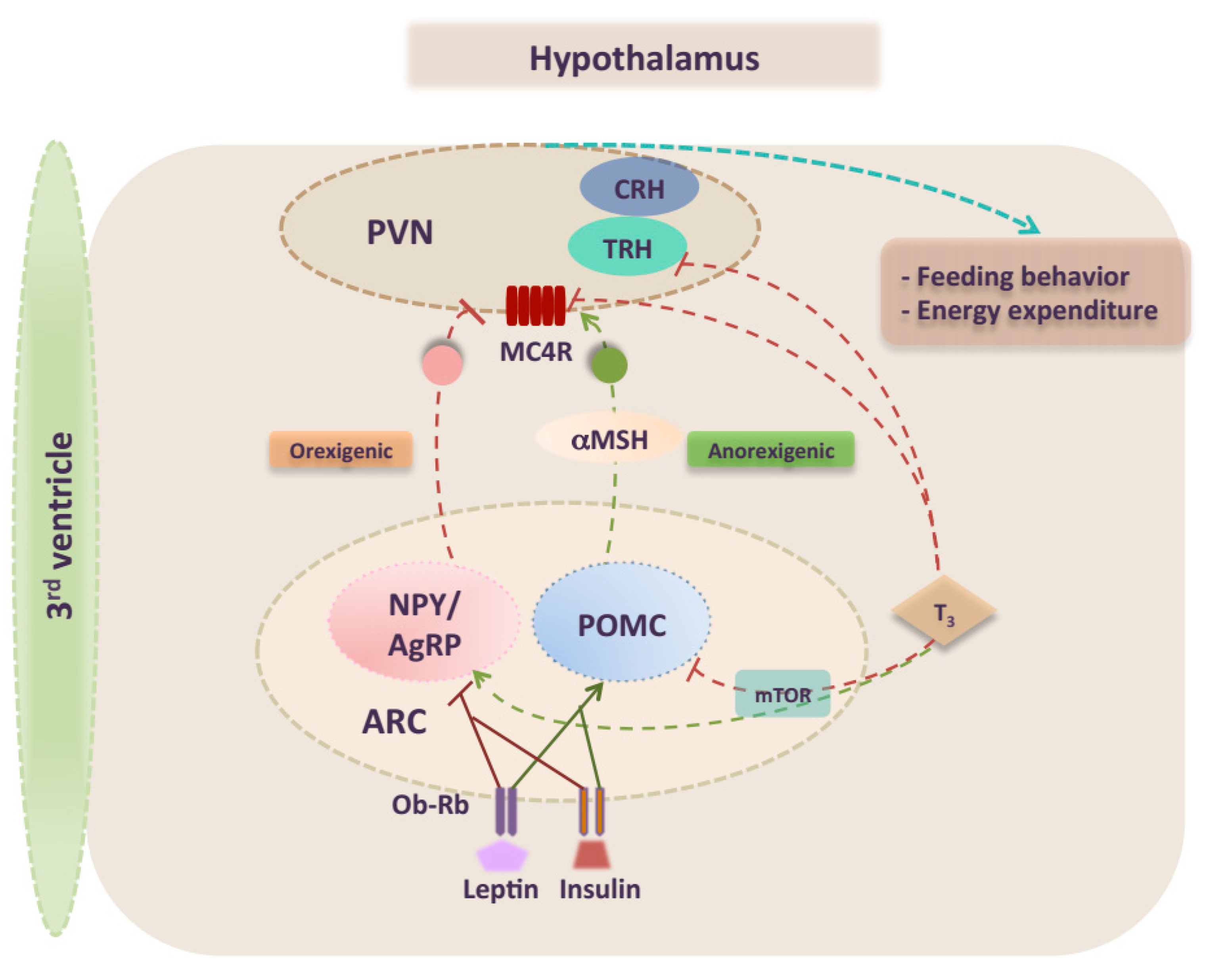
2. Overview of CNS Control of Energy Homeostasis
3. Effects of TH on Central Metabolism
4. Nuclear Receptors, Integrators of Metabolic Regulation
4.1. Thyroid Hormone Receptors
4.2. Retinoid X Receptor (RXR)
4.3. Peroxisome Proliferator-Activated Receptors (PPARs)
4.4. Liver X Receptors (LXRs)
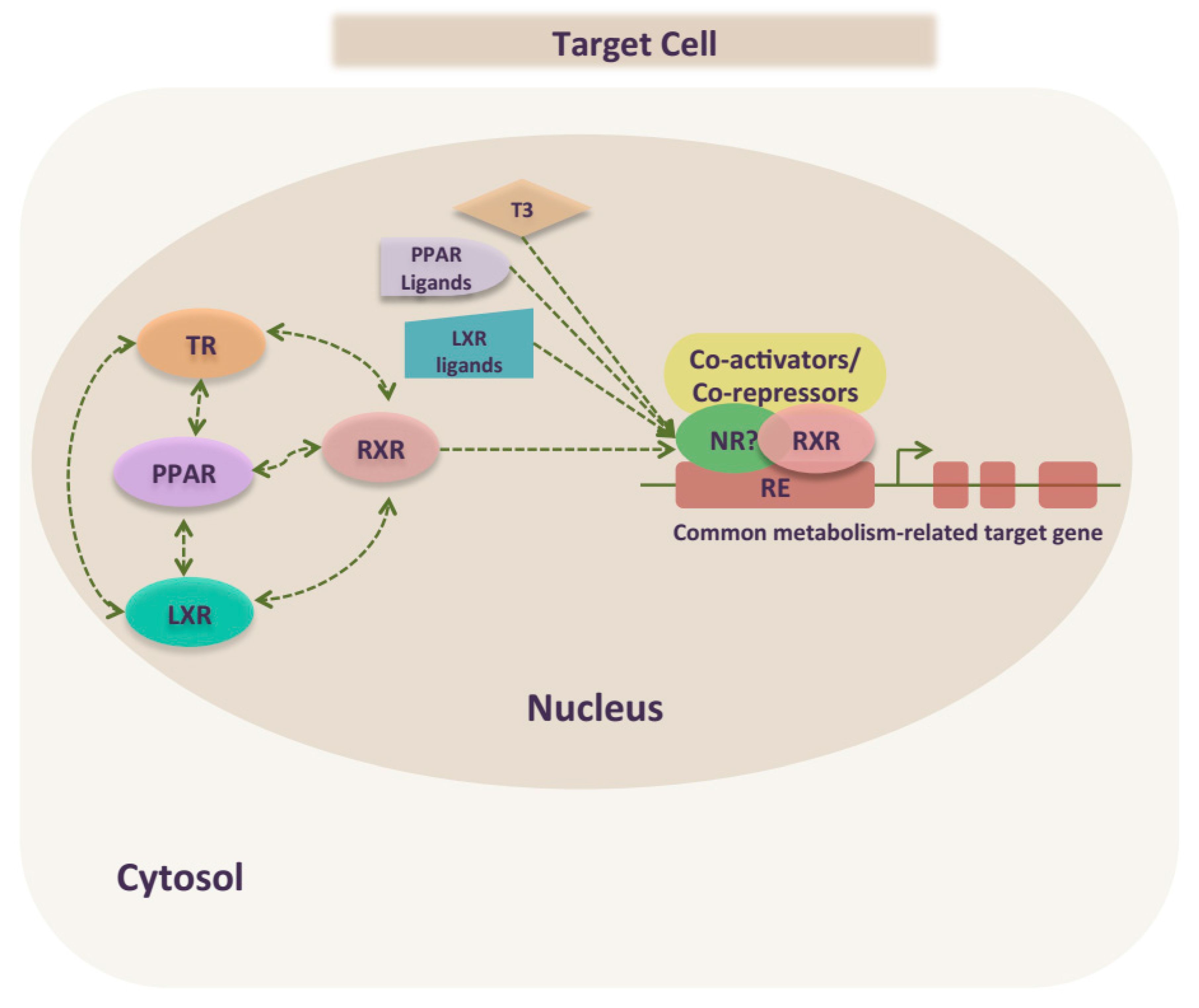
5. TR Crosstalk with PPAR and LXR to Regulate Metabolism
5.1. TR Peripheral Crosstalk in Regulating Metabolism
5.2. TR Crosstalk in the Regulation of Central Metabolism
6. Conclusions
Funding
Conflicts of Interest
References
- Lavie, C.J.; Arena, R.; Alpert, M.A.; Milani, R.V.; Ventura, H.O. Management of cardiovascular diseases in patients with obesity. Nat. Rev. Cardiol. 2018, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- González-Muniesa, P.; Mártinez-González, M.-A.; Hu, F.B.; Després, J.-P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- Jarolimova, J.; Tagoni, J.; Stern, T.A. Obesity: Its Epidemiology, Comorbidities, and Management. Primacy Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, G.; Gunter, M.J.; Polidoro, S.; Krogh, V.; Palli, D.; Panico, S.; Sacerdote, C.; Tumino, R.; Fiorito, G.; Guarrera, S.; et al. Epigenome-wide association study of adiposity and future risk of obesity-related diseases. Int. J. Obes. 2018. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.W.; Elmquist, J.K. From neuroanatomy to behavior: Central integration of peripheral signals regulating feeding behavior. Nat. Neurosci. 2012, 15, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Horvath, T.L. Neurobiology of feeding and energy expenditure. Annu. Rev. Neurosci. 2007, 30, 367–398. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.-W. Network of hypothalamic neurons that control appetite. BMB Rep. 2015, 48, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly-Amado, A.; Cansell, C.; Denis, R.G.P.; Delbes, A.-S.; Castel, J.; Martinez, S.; Luquet, S. The hypothalamic arcuate nucleus and the control of peripheral substrates. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Models Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Boelen, A.; Bisschop, P.H.; Kalsbeek, A.; Fliers, E. Hypothalamic effects of thyroid hormone. Mol. Cell. Endocrinol. 2017, 458, 143–148. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Alvarez, C.V.; Nogueiras, R.; Diéguez, C. Energy balance regulation by thyroid hormones at central level. Trends Mol. Med. 2013, 19, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Coll, A.P.; Yeo, G.S. The hypothalamus and metabolism: Integrating signals to control energy and glucose homeostasis. Curr. Opin. Pharmacol. 2013, 13, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Kim, M.-S. Molecular Mechanisms of Appetite Regulation. Diabetes Metab. J. 2012, 36, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G. Development of Hypothalamic Circuits That Control Food Intake and Energy Balance. In Appetite and Food Intake: Central Control; Harris, R.B.S., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017; ISBN 978-1-4987-2316-9. [Google Scholar]
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Perez, F.A.; Hnasko, T.S.; Palmiter, R.D. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 2005, 310, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Gropp, E.; Shanabrough, M.; Borok, E.; Xu, A.W.; Janoschek, R.; Buch, T.; Plum, L.; Balthasar, N.; Hampel, B.; Waisman, A.; et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci. 2005, 8, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 2007, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dores, R.M.; Liang, L.; Davis, P.; Thomas, A.L.; Petko, B. 60 YEARS OF POMC: Melanocortin receptors: Evolution of ligand selectivity for melanocortin peptides. J. Mol. Endocrinol. 2016, 56, T119–T133. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A. The melanocortin system and energy balance. Peptides 2006, 27, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.-J.; Yao, T.; Kong, X.; Williams, K.W.; Liu, T. Melanocortin neurons: Multiple routes to regulation of metabolism. Biochim. Biophys. Acta 2017, 1863, 2477–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.A.; Cone, R.D. Knockout studies defining different roles for melanocortin receptors in energy homeostasis. Ann. N. Y. Acad. Sci. 2003, 994, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Lensing, C.J.; Adank, D.N.; Doering, S.R.; Wilber, S.L.; Andreasen, A.; Schaub, J.W.; Xiang, Z.; Haskell-Luevano, C. The Ac-Trp-DPhe(p-I)-Arg-Trp-NH2 250-Fold Selective Melanocortin-4 Receptor (MC4R) Antagonist over the Melanocortin-3 Receptor (MC3R) Affects Energy Homeostasis in Male and Female Mice Differently. ACS Chem. Neurosci. 2016, 7, 1283–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautron, L.; Elmquist, J.K. Sixteen years and counting: An update on leptin in energy balance. J. Clin. Investig. 2011, 121, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Brüning, J.C. CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 2010, 1212, 97–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, M.; Leibel, R.L. Role of leptin in energy homeostasis in humans. J. Endocrinol. 2014, 223, T83–T96. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Inoue, K.; Mori, M. The leptin-dependent and -independent melanocortin signaling system: Regulation of feeding and energy expenditure. J. Endocrinol. 2007, 193, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.A.; Hall, J.E.; do Carmo, J.M. Leptin reverses hyperglycemia and hyperphagia in insulin deficient diabetic rats by pituitary-independent central nervous system actions. PLoS ONE 2017, 12, e0184805. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.A.; Rasti, M.; Bauer, P.V.; Lam, T.K.T. Leptin enhances hypothalamic lactate dehydrogenase A (LDHA)-dependent glucose sensing to lower glucose production in high-fat-fed rats. J. Biol. Chem. 2018, 293, 4159–4166. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bugescu, R.; Mayer, T.A.; Gata-Garcia, A.; Kurt, G.; Woodworth, H.L.; Leinninger, G.M. Loss of Action via Neurotensin-Leptin Receptor Neurons Disrupts Leptin and Ghrelin-Mediated Control of Energy Balance. Endocrinology 2017, 158, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. Diet-Induced Obesity and the Mechanism of Leptin Resistance. Adv. Exp. Med. Biol. 2017, 960, 381–397. [Google Scholar] [PubMed]
- Gamber, K.M.; Huo, L.; Ha, S.; Hairston, J.E.; Greeley, S.; Bjørbæk, C. Over-expression of leptin receptors in hypothalamic POMC neurons increases susceptibility to diet-induced obesity. PLoS ONE 2012, 7, e30485. [Google Scholar] [CrossRef] [PubMed]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Yao, X.; Ying, H. Thyroid hormone action in metabolic regulation. Protein Cell 2011, 2, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjergved, L.; Jørgensen, T.; Perrild, H.; Laurberg, P.; Krejbjerg, A.; Ovesen, L.; Rasmussen, L.B.; Knudsen, N. Thyroid Function and Body Weight: A Community-Based Longitudinal Study. PLoS ONE 2014, 9, e93515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdi, H.; Kazemian, E.; Gharibzadeh, S.; Amouzegar, A.; Mehran, L.; Tohidi, M.; Rashvandi, Z.; Azizi, F. Association between Thyroid Function and Body Mass Index: A 10-Year Follow-Up. Ann. Nutr. Metab. 2017, 70, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Laurberg, P.; Knudsen, N.; Andersen, S.; Carlé, A.; Pedersen, I.B.; Karmisholt, J. Thyroid Function and Obesity. Eur. Thyroid J. 2012, 1, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbers, L.P.; Kastelein, J.J.; Sjouke, B. Thyroid Hormone Mimetics: The Past, Current Status and Future Challenges. Curr. Atheroscler. Rep. 2016, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.D.; Webb, P. Thyroid hormone mimetics: Potential applications in atherosclerosis, obesity and type 2 diabetes. Nat. Rev. Drug Discov. 2009, 8, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.M.; Smit, R.A.J.; Trompet, S.; van Heemst, D.; Noordam, R. Thyroid Signaling, Insulin Resistance, and 2 Diabetes Mellitus: A Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2017, 102, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Karbalaei, N.; Noorafshan, A.; Hoshmandi, E. Impaired glucose-stimulated insulin secretion and reduced β-cell mass in pancreatic islets of hyperthyroid rats. Exp. Physiol. 2016, 101, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, Z. Thyroid hormone potentiates insulin signaling and attenuates hyperglycemia and insulin resistance in a mouse model of type 2 diabetes. Br. J. Pharmacol. 2011, 162, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderpump, M.P.; Ahlquist, J.A.; Franklyn, J.A.; Clayton, R.N. Consensus statement for good practice and audit measures in the management of hypothyroidism and hyperthyroidism. The Research Unit of the Royal College of Physicians of London, the Endocrinology and Diabetes Committee of the Royal College of Physicians of London, and the Society for Endocrinology. BMJ 1996, 313, 539–544. [Google Scholar] [PubMed]
- McAninch, E.A.; Bianco, A.C. Thyroid hormone signaling in energy homeostasis and energy metabolism. Ann. N. Y. Acad. Sci. 2014, 1311, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkemade, A. Central and peripheral effects of thyroid hormone signalling in the control of energy metabolism. J. Neuroendocrinol. 2010, 22, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Donangelo, I. Thyroid Hormone and Central Control of Metabolic Homeostasis. J. Endocrinol. Diabetes Obes. 2014, 2, 1047. [Google Scholar]
- Ortiga-Carvalho, T.M.; Chiamolera, M.I.; Pazos-Moura, C.C.; Wondisford, F.E. Hypothalamus-Pituitary-Thyroid Axis. Compr. Physiol. 2016, 6, 1387–1428. [Google Scholar] [PubMed]
- Fliers, E.; Boelen, A.; van Trotsenburg, A.S.P. Central regulation of the hypothalamo-pituitary-thyroid (HPT) axis: Focus on clinical aspects. Handb. Clin. Neurol. 2014, 124, 127–138. [Google Scholar] [PubMed]
- Fliers, E.; Alkemade, A.; Wiersinga, W.M.; Swaab, D.F. Hypothalamic thyroid hormone feedback in health and disease. Prog. Brain Res. 2006, 153, 189–207. [Google Scholar] [PubMed]
- Joseph-Bravo, P.; Jaimes-Hoy, L.; Uribe, R.M.; Charli, J.L. 60 YEARS OF NEUROENDOCRINOLOGY: TRH, the first hypophysiotropic releasing hormone isolated: Control of the pituitary–thyroid axis. J. Endocrinol. 2015, 226, T85–T100. [Google Scholar] [CrossRef] [PubMed]
- Fliers, E.; Kalsbeek, A.; Boelen, A. Beyond the fixed setpoint of the hypothalamus-pituitary-thyroid axis. Eur. J. Endocrinol. 2014, 171, R197–R208. [Google Scholar] [CrossRef] [PubMed]
- Nikrodhanond, A.A.; Ortiga-Carvalho, T.M.; Shibusawa, N.; Hashimoto, K.; Liao, X.H.; Refetoff, S.; Yamada, M.; Mori, M.; Wondisford, F.E. Dominant role of thyrotropin-releasing hormone in the hypothalamic-pituitary-thyroid axis. J. Biol. Chem. 2006, 281, 5000–5007. [Google Scholar] [CrossRef] [PubMed]
- Arrojo E Drigo, R.; Fonseca, T.L.; Werneck-de-Castro, J.P.S.; Bianco, A.C. Role of the type 2 iodothyronine deiodinase (D2) in the control of thyroid hormone signaling. Biochim. Biophys. Acta 2013, 1830, 3956–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werneck de Castro, J.P.; Fonseca, T.L.; Ueta, C.B.; McAninch, E.A.; Abdalla, S.; Wittmann, G.; Lechan, R.M.; Gereben, B.; Bianco, A.C. Differences in hypothalamic type 2 deiodinase ubiquitination explain localized sensitivity to thyroxine. J. Clin. Investig. 2015, 125, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Spek, A.H.; Fliers, E.; Boelen, A. The classic pathways of thyroid hormone metabolism. Mol. Cell. Endocrinol. 2017, 458, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sánchez, N.; Alvarez, C.V.; Fernø, J.; Nogueiras, R.; Diéguez, C.; López, M. Hypothalamic effects of thyroid hormones on metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Iwen, K.A.; Oelkrug, R.; Brabant, G. Effects of thyroid hormones on thermogenesis and energy partitioning. J. Mol. Endocrinol. 2018, 60, R157–R170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, A.; Ross, A.W.; Nilaweera, K.N.; Morgan, P.J.; Barrett, P. Hypothalamic Thyroid Hormone in Energy Balance Regulation. Obes. Facts 2008, 1, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Kamegai, J.; Tamura, H.; Shimizu, T.; Sugihara, H.; Oikawa, S. Triiodothyronine (T3) stimulates food intake via enhanced hypothalamic AMP-activated kinase activity. Regul. Pept. 2008, 151, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Coppola, A.; Liu, Z.-W.; Andrews, Z.B.; Paradis, E.; Roy, M.-C.; Friedman, J.M.; Ricquier, D.; Richard, D.; Horvath, T.L.; Gao, X.-B.; et al. A central thermogenic-like mechanism in feeding regulation: An interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007, 5, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Nillni, E.A. Regulation of the Hypothalamic Thyrotropin Releasing Hormone (TRH) Neuron by Neuronal and Peripheral Inputs. Front. Neuroendocrinol. 2010, 31, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Decherf, S.; Seugnet, I.; Kouidhi, S.; Lopez-Juarez, A.; Clerget-Froidevaux, M.-S.; Demeneix, B.A. Thyroid hormone exerts negative feedback on hypothalamic type 4 melanocortin receptor expression. Proc. Natl. Acad. Sci. USA 2010, 107, 4471–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Martinez, M.E.; St. Germain, D.L.; Hernandez, A. Type 3 Deiodinase Role on Central Thyroid Hormone Action Affects the Leptin-Melanocortin System and Circadian Activity. Endocrinology 2016, 158, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Vella, K.R.; Ramadoss, P.; Lam, F.S.; Harris, J.C.; Ye, F.D.; Same, P.D.; O’Neill, N.F.; Maratos-Flier, E.; Hollenberg, A.N. NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab. 2011, 14, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Preston, E.; Cooney, G.J.; Wilks, D.; Baran, K.; Zhang, L.; Kraegen, E.W.; Sainsbury, A. Central neuropeptide Y infusion and melanocortin 4 receptor antagonism inhibit thyrotropic function by divergent pathways. Neuropeptides 2011, 45, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Sánchez, N.; Seoane-Collazo, P.; Contreras, C.; Varela, L.; Villarroya, J.; Rial-Pensado, E.; Buqué, X.; Aurrekoetxea, I.; Delgado, T.C.; Vázquez-Martínez, R.; et al. Hypothalamic AMPK-ER Stress-JNK1 Axis Mediates the Central Actions of Thyroid Hormones on Energy Balance. Cell Metab. 2017, 26, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.S.; Cho, H.; Cho, J.H.; Yu, S.-W.; Kim, E.-K. Hypothalamic AMPK-induced autophagy increases food intake by regulating NPY and POMC expression. Autophagy 2016, 12, 2009–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, M.; Nogueiras, R.; Tena-Sempere, M.; Diéguez, C. Hypothalamic AMPK: A canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 2016, 12, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Martínez de Morentin, P.B.; Urisarri, A.; Couce, M.L.; López, M. Molecular mechanisms of appetite and obesity: A role for brain AMPK. Clin. Sci. 2016, 130, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Varela, L.; Vázquez, M.J.; Rodríguez-Cuenca, S.; González, C.R.; Velagapudi, V.R.; Morgan, D.A.; Schoenmakers, E.; Agassandian, K.; Lage, R.; et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010, 16, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, L.; Martínez-Sánchez, N.; Gallego, R.; Vázquez, M.J.; Roa, J.; Gándara, M.; Schoenmakers, E.; Nogueiras, R.; Chatterjee, K.; Tena-Sempere, M.; et al. Hypothalamic mTOR pathway mediates thyroid hormone-induced hyperphagia in hyperthyroidism. J. Pathol. 2012, 227, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Bantubungi, K.; Prawitt, J.; Staels, B. Control of metabolism by nutrient-regulated nuclear receptors acting in the brain. J. Steroid Biochem. Mol. Biol. 2012, 130, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Downes, M.; Evans, R.M. Metabolic Regulation by Nuclear Receptors. In Innovative Medicine; Springer: Tokyo, Japan, 2015; pp. 25–37. ISBN 978-4-431-55650-3. [Google Scholar]
- Xu, Y.; O’Malley, B.W.; Elmquist, J.K. Brain nuclear receptors and body weight regulation. J. Clin. Investig. 2017, 127, 1172–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, P.M.; Ando, S.; Feng, X.; Liu, Y.; Maruvada, P.; Xia, X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol. Cell. Endocrinol. 2006, 246, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, R.T.; Tan, S.W.; Tyl, R.W. General background on the hypothalamic-pituitary-thyroid (HPT) axis. Crit. Rev. Toxicol. 2007, 37, 11–53. [Google Scholar] [CrossRef] [PubMed]
- Guissouma, H.; Ghorbel, M.T.; Seugnet, I.; Ouatas, T.; Demeneix, B.A. Physiological regulation of hypothalamic TRH transcription in vivo is T3 receptor isoform specific. FASEB J. 1998, 12, 1755–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clerget-Froidevaux, M.S.; Seugnet, I.; Demeneix, B.A. Thyroid status co-regulates thyroid hormone receptor and co-modulator genes specifically in the hypothalamus. FEBS Lett. 2004, 569, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechan, R.M.; Qi, Y.; Jackson, I.M.; Mahdavi, V. Identification of thyroid hormone receptor isoforms in thyrotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Endocrinology 1994, 135, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Guissouma, H.; Becker, N.; Seugnet, I.; Demeneix, B.A. Transcriptional repression of TRH promoter function by T3: Analysis by in vivo gene transfer. Biochem. Cell Biol. 2000, 78, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Dupré, S.M.; Guissouma, H.; Flamant, F.; Seugnet, I.; Scanlan, T.S.; Baxter, J.D.; Samarut, J.; Demeneix, B.A.; Becker, N. Both thyroid hormone receptor (TR)β1 and TR β2 isoforms contribute to the regulation of hypothalamic thyrotropin-releasing hormone. Endocrinology 2004, 145, 2337–2345. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Ahima, R.S.; Boers, M.E.; Elmquist, J.K.; Wondisford, F.E. Critical role for thyroid hormone receptor β2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J. Clin. Investig. 2001, 107, 1017–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenberg, A.N.; Monden, T.; Flynn, T.R.; Boers, M.E.; Cohen, O.; Wondisford, F.E. The human thyrotropin-releasing hormone gene is regulated by thyroid hormone through two distinct classes of negative thyroid hormone response elements. Mol. Endocrinol. 1995, 9, 540–550. [Google Scholar] [PubMed]
- Satoh, T.; Monden, T.; Ishizuka, T.; Mitsuhashi, T.; Yamada, M.; Mori, M. DNA binding and interaction with the nuclear receptor corepressor of thyroid hormone receptor are required for ligand-independent stimulation of the mouse preprothyrotropin-releasing hormone gene. Mol. Cell. Endocrinol. 1999, 154, 137–149. [Google Scholar] [CrossRef]
- Satoh, T.; Yamada, M.; Iwasaki, T.; Mori, M. Negative regulation of the gene for the preprothyrotropin-releasing hormone from the mouse by thyroid hormone requires additional factors in conjunction with thyroid hormone receptors. J. Biol. Chem. 1996, 271, 27919–27926. [Google Scholar] [CrossRef] [PubMed]
- Guissouma, H.; Dupré, S.M.; Becker, N.; Jeannin, E.; Seugnet, I.; Desvergne, B.; Demeneix, B.A. Feedback on hypothalamic TRH transcription is dependent on thyroid hormone receptor N terminus. Mol. Endocrinol. 2002, 16, 1652–1666. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.; Patterson, M.; Dhillo, W.S.; Rahman, S.A.; Ma, Y.; Holton, C.; Gogakos, A.; Yeo, G.S.H.; Lam, B.Y.H.; Polex-Wolf, J.; et al. Thyroid Hormone Receptor βin the Ventromedial Hypothalamus Is Essential for the Physiological Regulation of Food Intake and Body Weight. Cell Rep. 2017, 19, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Amorim, B.S.; Ueta, C.B.; Freitas, B.C.G.; Nassif, R.J.; de Azevedo Gouveia, C.H.; Christoffolete, M.A.; Moriscot, A.S.; Lancelloti, C.L.; Llimona, F.; Barbeiro, H.V.; et al. A TRβ-selective agonist confers resistance to diet-induced obesity. J. Endocrinol. 2009, 203, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-Y.; Schultz, J.J.; Brent, G.A. A Thyroid Hormone Receptor α Gene Mutation (P398H) Is Associated with Visceral Adiposity and Impaired Catecholamine-stimulated Lipolysis in Mice. J. Biol. Chem. 2003, 278, 38913–38920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, G.J.; Mellström, K.; Malm, J. Therapeutic potential for thyroid hormone receptor-β selective agonists for treating obesity, hyperlipidemia and diabetes. Curr. Vasc. Pharmacol. 2007, 5, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.J.; Matta-Camacho, E.; Hughes, T.S.; Srinivasan, S.; Nwachukwu, J.C.; Cavett, V.; Nowak, J.; Chalmers, M.J.; Marciano, D.P.; Kamenecka, T.M.; et al. Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat. Commun. 2015, 6, 8013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, C.G.; Silveira, R.L.; Rivalta, I.; Batista, V.S.; Skaf, M.S. Allosteric Pathways in the PPARγ-RXRα nuclear receptor complex. Sci. Rep. 2016, 6, 19940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelsdorf, D.J.; Borgmeyer, U.; Heyman, R.A.; Zhou, J.Y.; Ong, E.S.; Oro, A.E.; Kakizuka, A.; Evans, R.M. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992, 6, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Wu, D.; Li, S.; Potluri, N.; Kim, Y.; Rastinejad, F. The quaternary architecture of RARβ–RXRα heterodimer facilitates domain–domain signal transmission. Nat. Commun. 2017, 8, 868. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Umesono, K.; Chen, J.; Evans, R.M. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell 1995, 81, 541–550. [Google Scholar] [CrossRef]
- Leblanc, B.P.; Stunnenberg, H.G. 9-cis retinoic acid signaling: Changing partners causes some excitement. Genes Dev. 1995, 9, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Ye, F.; Gui, C.; Luo, H.; Cai, J.; Shen, J.; Chen, K.; Shen, X.; Jiang, H. Ligand-binding regulation of LXR/RXR and LXR/PPAR heterodimerizations: SPR technology-based kinetic analysis correlated with molecular dynamics simulation. Protein Sci. 2005, 14, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, A.I.; Mangelsdorf, D.J. Retinoid x receptor heterodimers in the metabolic syndrome. N. Engl. J. Med. 2005, 353, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, T.; Wang, F.; Tian, H.; Samuels, H.H. Functional Evidence for Retinoid X Receptor (RXR) as a Nonsilent Partner in the Thyroid Hormone Receptor/RXR Heterodimer. Mol. Cell. Biol. 2002, 22, 5782–5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, L.F.R.; Togashi, M.; Walfish, P.G.; Pessanha, R.P.; Moura, F.N.; Barra, G.B.; Nguyen, P.; Rebong, R.; Yuan, C.; Simeoni, L.A.; et al. Thyroid hormone response element organization dictates the composition of active receptor. J. Biol. Chem. 2007, 282, 12458–12466. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.I.; Sánchez-Martínez, R.; Moreno, J.L.; Martínez-Iglesias, O.A.; Palacios, D.; Aranda, A. A Permissive Retinoid X Receptor/Thyroid Hormone Receptor Heterodimer Allows Stimulation of Prolactin Gene Transcription by Thyroid Hormone and 9-cis-Retinoic Acid. Mol. Cell. Biol. 2004, 24, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Tsai, S.Y.; O’Malley, B.W.; Tsai, M.J. Ligand-dependent conformational changes in thyroid hormone and retinoic acid receptors are potentially enhanced by heterodimerization with retinoic X receptor. J. Steroid Biochem. Mol. Biol. 1993, 46, 643–661. [Google Scholar] [CrossRef]
- Yen, P.M.; Sugawara, A.; Refetoff, S.; Chin, W.W. New insights on the mechanism(s) of the dominant negative effect of mutant thyroid hormone receptor in generalized resistance to thyroid hormone. J. Clin. Investig. 1992, 90, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.S.; Sharma, V.; Pugazhenthi, U.; Sladek, C.; Wood, W.M.; Haugen, B.R. A rexinoid antagonist increases the hypothalamic-pituitary-thyroid set point in mice and thyrotrope cells. Mol. Cell. Endocrinol. 2011, 339, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Hays, W.R.; Wood, W.M.; Pugazhenthi, U.; St Germain, D.L.; Bianco, A.C.; Krezel, W.; Chambon, P.; Haugen, B.R. Effects of rexinoids on thyrotrope function and the hypothalamic-pituitary-thyroid axis. Endocrinology 2006, 147, 1438–1451. [Google Scholar] [CrossRef] [PubMed]
- Decherf, S.; Seugnet, I.; Becker, N.; Demeneix, B.A.; Clerget-Froidevaux, M.-S. Retinoic X receptor subtypes exert differential effects on the regulation of Trh transcription. Mol. Cell. Endocrinol. 2013, 381, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-γ–RXR-α nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janani, C.; Kumari, B.R. PPAR γ gene—A review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor α in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARγ in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. PPARγ: A Nuclear Regulator of Metabolism, Differentiation, and Cell Growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the Promise of Insulin Sensitization in Type 2 Diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.L. Rationally designed PPARδ-specific agonists and their therapeutic potential for metabolic syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 3284–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.K.; Li, B.; Grayson, B.E.; Matter, E.K.; Woods, S.C.; Seeley, R.J. A role for central nervous system PPAR-γ in the regulation of energy balance. Nat. Med. 2011, 17, 623–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Sarruf, D.A.; Talukdar, S.; Sharma, S.; Li, P.; Bandyopadhyay, G.; Nalbandian, S.; Fan, W.; Gayen, J.R.; Mahata, S.K.; et al. Brain PPAR-γ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat. Med. 2011, 17, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Stump, M.; Guo, D.-F.; Lu, K.-T.; Mukohda, M.; Liu, X.; Rahmouni, K.; Sigmund, C.D. Effect of selective expression of dominant-negative PPARγ in pro-opiomelanocortin neurons on the control of energy balance. Physiol. Genom. 2016, 48, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Toda, C.; Jeong, J.K.; Horvath, T.L.; Diano, S. PPARγ ablation sensitizes proopiomelanocortin neurons to leptin during high-fat feeding. J. Clin. Investig. 2014, 124, 4017–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocalis, H.E.; Turney, M.K.; Printz, R.L.; Laryea, G.N.; Muglia, L.J.; Davies, S.S.; Stanwood, G.D.; McGuinness, O.P.; Niswender, K.D. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARδ) in mice leads to increased susceptibility to diet-induced obesity. PLoS ONE 2012, 7, e42981. [Google Scholar] [CrossRef] [PubMed]
- Korach-André, M.; Archer, A.; Barros, R.P.; Parini, P.; Gustafsson, J.-Å. Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity. Proc. Natl. Acad. Sci. USA 2011, 108, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Korach-André, M.; Gustafsson, J.-Å. Liver X receptors as regulators of metabolism. Biomol. Concepts 2015, 6, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; Groen, A.K.; Kuipers, F. The liver X receptor: Control of cellular lipid homeostasis and beyond Implications for drug design. Prog. Lipid Res. 2010, 49, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Breevoort, S.R.; Angdisen, J.; Fu, M.; Schmidt, D.R.; Holmstrom, S.R.; Kliewer, S.A.; Mangelsdorf, D.J.; Schulman, I.G. Liver LXRα expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Investig. 2012, 122, 1688–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gofflot, F.; Chartoire, N.; Vasseur, L.; Heikkinen, S.; Dembele, D.; Le Merrer, J.; Auwerx, J. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell 2007, 131, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Courtney, R.; Landreth, G.E. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol. Metab. 2016, 27, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzat, K.; Raoul, C.; Polge, A.; Kantar, J.; Camu, W.; Lumbroso, S. Liver X receptors: From cholesterol regulation to neuroprotection—A new barrier against neurodegeneration in amyotrophic lateral sclerosis? Cell. Mol. Life Sci. 2016, 73, 3801–3808. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Bales, K.R.; DeMattos, R.B.; Paul, S.M. Liver X receptor-mediated gene regulation and cholesterol homeostasis in brain: Relevance to Alzheimer’s disease therapeutics. Curr. Alzheimer Res. 2007, 4, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ghaddab-Zroud, R.; Seugnet, I.; Steffensen, K.R.; Demeneix, B.A.; Clerget-Froidevaux, M.-S. Liver X receptor regulation of thyrotropin-releasing hormone transcription in mouse hypothalamus is dependent on thyroid status. PLoS ONE 2014, 9, e106983. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Wu, W.; Dai, Y.; Maneix, L.; Huang, B.; Warner, M.; Gustafsson, J.-Å. Liver X receptor β controls thyroid hormone feedback in the brain and regulates browning of subcutaneous white adipose tissue. Proc. Natl. Acad. Sci. USA 2015, 112, 14006–14011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Warner, M.; Gustafsson, J.-K. Liver X receptor β: New player in the regulatory network of thyroid hormone and “browning” of white fat. Adipocyte 2016, 5, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Brent, G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010, 21, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-Y.; Heymann, R.S.; Moatamed, F.; Schultz, J.J.; Sobel, D.; Brent, G.A. A mutant thyroid hormone receptor α antagonizes peroxisome proliferator-activated receptor α signaling in vivo and impairs fatty acid oxidation. Endocrinology 2007, 148, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Cohen, R.N.; Yamada, M.; Markan, K.R.; Monden, T.; Satoh, T.; Mori, M.; Wondisford, F.E. Cross-talk between thyroid hormone receptor and liver X receptor regulatory pathways is revealed in a thyroid hormone resistance mouse model. J. Biol. Chem. 2006, 281, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.; Zavacki, A.M.; Harney, J.W.; Brent, G.A. Retinoid-X receptor (RXR) differentially augments thyroid hormone response in cell lines as a function of the response element and endogenous RXR content. Endocrinology 1995, 136, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Hyyti, O.M.; Portman, M.A. Molecular mechanisms of cross-talk between thyroid hormone and peroxisome proliferator activated receptors: Focus on the heart. Cardiovasc. Drugs Ther. 2006, 20, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Juge-Aubry, C.E.; Gorla-Bajszczak, A.; Pernin, A.; Lemberger, T.; Wahli, W.; Burger, A.G.; Meier, C.A. Peroxisome proliferator-activated receptor mediates cross-talk with thyroid hormone receptor by competition for retinoid X receptor. Possible role of a leucine zipper-like heptad repeat. J. Biol. Chem. 1995, 270, 18117–18122. [Google Scholar] [CrossRef] [PubMed]
- Araki, O.; Ying, H.; Furuya, F.; Zhu, X.; Cheng, S.-Y. Thyroid hormone receptor β bmutants: Dominant negative regulators of peroxisome proliferator-activated receptor γ action. Proc. Natl. Acad. Sci. USA 2005, 102, 16251–16256. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Suzuki, H.; Zhao, L.; Willingham, M.C.; Meltzer, P.; Cheng, S.-Y. Mutant thyroid hormone receptor βrepresses the expression and transcriptional activity of peroxisome proliferator-activated receptor γ during thyroid carcinogenesis. Cancer Res. 2003, 63, 5274–5280. [Google Scholar] [PubMed]
- Hashimoto, K.; Mori, M. Crosstalk of thyroid hormone receptor and liver X receptor in lipid metabolism and beyond. Endocr. J. 2011, 58, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Huuskonen, J.; Vishnu, M.; Pullinger, C.R.; Fielding, P.E.; Fielding, C.J. Regulation of ATP-binding cassette transporter A1 transcription by thyroid hormone receptor. Biochemistry 2004, 43, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, K.; Billon, C.; Bissler, M.; Beylot, M.; Lobaccaro, J.-M.; Vanacker, J.-M.; Samarut, J. Thyroid hormone receptor β (TRβ) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J. Biol. Chem. 2010, 285, 28156–28163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poupeau, A.; Postic, C. Cross-regulation of hepatic glucose metabolism via ChREBP and nuclear receptors. Biochim. Biophys. Acta 2011, 1812, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducheix, S.; Podechard, N.; Lasserre, F.; Polizzi, A.; Pommier, A.; Murzilli, S.; Di Lisio, C.; D’Amore, S.; Bertrand-Michel, J.; Montagner, A.; et al. A systems biology approach to the hepatic role of the oxysterol receptor LXR in the regulation of lipogenesis highlights a cross-talk with PPARα. Biochimie 2013, 95, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Bu, L.; Ma, Y.; Liu, D. Concurrent Activation of Liver X Receptor and Peroxisome Proliferator-Activated Receptor α Exacerbates Hepatic Steatosis in High Fat Diet-Induced Obese Mice. PLoS ONE 2013, 8, e65641. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, E.; Benedetti, E.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Fidoamore, A.; Barone, D.; Moreno, S.; Ippoliti, R.; Cerù, M.P.; et al. Roles of PPAR transcription factors in the energetic metabolic switch occurring during adult neurogenesis. Cell Cycle 2017, 16, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, Y.; Zhang, Y.; Zhang, R.; Tang, X.; Fang, L.; Xu, Y. Recent Evidence of the Regulatory Role of PPARs in Neural Stem Cells and Their Underlying Mechanisms for Neuroprotective Effects. Curr. Stem Cell Res Ther. 2016, 11, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Rolland, B.; Bordet, R. The first theme issue on PPARs for brain disorders. Curr. Drug Targets 2013, 14, 723. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Farioli-Vecchioli, S.; Cerù, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Zhu, Y.; López, M.; Yin, L.; Wozniak, D.F.; Coleman, T.; Hu, Z.; Wolfgang, M.; Vidal-Puig, A.; Lane, M.D.; et al. Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J. Clin. Investig. 2007, 117, 2539–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouidhi, S.; Seugnet, I.; Decherf, S.; Guissouma, H.; Elgaaied, A.B.; Demeneix, B.; Clerget-Froidevaux, M.-S. Peroxisome proliferator-activated receptor-γ (PPARγ) modulates hypothalamic Trh regulation in vivo. Mol. Cell. Endocrinol. 2010, 317, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dahlman-Wright, K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2010, 204, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Stukas, S.; May, S.; Wilkinson, A.; Chan, J.; Donkin, J.; Wellington, C.L. The LXR agonist GW3965 increases apoA-I protein levels in the central nervous system independent of ABCA1. Biochim. Biophys. Acta 2012, 1821, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.S.; Suarez, L.G.; Coirini, H. Regulation of the expression of LXR in rat hypothalamic and hippocampal explants. Neurosci. Lett. 2017, 639, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ishida, E.; Hashimoto, K.; Okada, S.; Satoh, T.; Yamada, M.; Mori, M. Thyroid hormone receptor and liver X receptor competitively up-regulate human selective Alzheimer’s disease indicator-1 gene expression at the transcriptional levels. Biochem. Biophys. Res. Commun. 2013, 432, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Li, D.; Tang, X.; Bao, X.; Huang, J.; Tang, Y.; Yang, Y.; Xu, H.; Fan, X. LXR agonists: New potential therapeutic drug for neurodegenerative diseases. Mol. Neurobiol. 2013, 48, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Campolo, M.; Siracusa, R.; Cordaro, M.; Di Paola, R.; Calabrese, V.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Liver X receptors activation, through TO901317 binding, reduces neuroinflammation in Parkinson’s disease. PLoS ONE 2017, 12, e0174470. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.R.; Jakobsson, T.; Gustafsson, J.-Å. Targeting liver X receptors in inflammation. Expert Opin. Ther. Targets 2013, 17, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, S.; Henke, A.; Muse, E.D.; Cheng, B.; Welzel, G.; Chatterjee, A.K.; Wang, D.; Roland, J.; Glass, C.K.; et al. Dissociated sterol-based liver X receptor agonists as therapeutics for chronic inflammatory diseases. FASEB J. 2016, 30, 2570–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouidhi, S.; Clerget-Froidevaux, M.-S. Integrating Thyroid Hormone Signaling in Hypothalamic Control of Metabolism: Crosstalk Between Nuclear Receptors. Int. J. Mol. Sci. 2018, 19, 2017. https://doi.org/10.3390/ijms19072017
Kouidhi S, Clerget-Froidevaux M-S. Integrating Thyroid Hormone Signaling in Hypothalamic Control of Metabolism: Crosstalk Between Nuclear Receptors. International Journal of Molecular Sciences. 2018; 19(7):2017. https://doi.org/10.3390/ijms19072017
Chicago/Turabian StyleKouidhi, Soumaya, and Marie-Stéphanie Clerget-Froidevaux. 2018. "Integrating Thyroid Hormone Signaling in Hypothalamic Control of Metabolism: Crosstalk Between Nuclear Receptors" International Journal of Molecular Sciences 19, no. 7: 2017. https://doi.org/10.3390/ijms19072017