
Biological Retention and Accumulation of Inhaled Environmental Particles Disrupt Immune Homeostasis: Implications for Chronic Lung Disease


Abstract
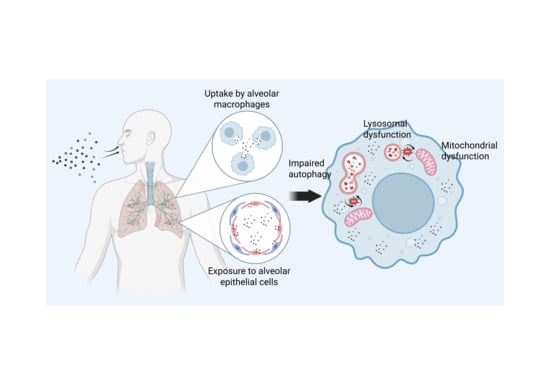
1. Introduction
2. Biological Fate and Accumulation of Environmental Particles
2.1. Deposition and Clearance of Inhaled Particles
2.2. Biological Retention and Redistribution
3. Accumulation and Functional Impairment at the Intracellular Organelle Level
3.1. Endo-Lysosomal Accumulation and Dysfunction
3.2. Mitochondrial Stress, Metabolic Reprogramming, Organelle Crosstalk, and Intracellular Trafficking
4. Disease Relevance
4.1. Chronic Lung Diseases as Accumulation-Driven Disorders
4.2. Systemic Implications
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PM2.5 | Fine particulate matter with aerodynamic diameter ≤ 2.5 µm |
| PM10 | Particulate matter with aerodynamic diameter ≤ 10 µm |
| PM | Particulate matter |
| WHO | World Health Organization |
| AHA | American Heart Association |
| COPD | Chronic obstructive pulmonary disease |
| IPF | Idiopathic pulmonary fibrosis |
| ECM | Extracellular matrix |
| SMPS | Scanning mobility particle sizer |
| APS | Aerodynamic particle sizer |
| PSLTs | Poorly soluble, low-toxicity particles |
| DC(s) | Dendritic cell(s) |
| DEP | Diesel exhaust particles |
| AM | Alveolar macrophage(s) |
| TiO2 | Titanium dioxide |
| LMP | Lysosomal membrane permeabilization |
| v-ATPase | Vacuolar-type H+-ATPase |
| ZnO | Zinc oxide |
| SiO2 | Silicon dioxide |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| BEAS-2B | Bronchial Epithelium transformed with Ad12-SV40 2B |
| ALI | Air–liquid interface |
| p62 (SQSTM1) | p62/SQSTM1 (sequestosome 1) |
| UFP | Ultrafine particles |
| OXPHOS | Oxidative phosphorylation |
| ATP | Adenosine triphosphate |
| ROS | Reactive oxygen species |
| mtROS | Mitochondrial reactive oxygen species |
| mtDNA | Mitochondrial DNA |
| PAHs | Polycyclic aromatic hydrocarbons |
| NAD | Nicotinamide adenine dinucleotide |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| AgNP | Silver nanoparticles |
| IRE1α | Inositol-requiring enzyme 1 alpha |
| XBP1 | X-box binding protein 1 |
| NOD1 | Nucleotide-binding oligomerization domain-containing protein 1 |
| NF-κB | Nuclear factor kappa B |
| MMP-12 | Matrix metalloproteinase-12 |
| Bnip3L/NIX | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like (NIX) |
References
- van Donkelaar, A.; Hammer, M.S.; Bindle, L.; Brauer, M.; Brook, J.R.; Garay, M.J.; Hsu, N.C.; Kalashnikova, O.V.; Kahn, R.A.; Lee, C.; et al. Monthly global estimates of fine particulate matter and their uncertainty. Environ. Sci. Technol. 2021, 55, 15287–15300. [Google Scholar] [CrossRef]
- Karagulian, F.; Belis, C.A.; Dora, C.F.C.; Prüss-Ustün, A.M.; Bonjour, S.; Adair-Rohani, H.; Amann, M. Contributions to cities’ ambient particulate matter (PM): A systematic review of local source contributions at global level. Atmos. Environ. 2015, 120, 475–483. [Google Scholar] [CrossRef]
- Fang, X.; Li, R.; Xu, Q.; Bottai, M.; Fang, F.; Cao, Y. A two-stage method to estimate the contribution of road traffic to PM2.5 concentrations in Beijing, China. Int. J. Environ. Res. Public Health 2016, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Deng, L.; Miao, Y.; Guo, X.; Li, Y. Particle deposition in the human lung: Health implications of particulate matter from different sources. Environ. Res. 2019, 169, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, Z.; Lau, A.K.; Lin, C.Q.; Chuang, Y.C.; Chan, J.; Jiang, W.K.; Tam, T.; Yeoh, E.-K.; Chan, T.-C.; et al. Effect of long-term exposure to fine particulate matter on lung function and chronic obstructive pulmonary disease: A longitudinal cohort study. Lancet Planet. Health 2018, 2, e114–e125. [Google Scholar] [CrossRef]
- Dockery, D.W.; Pope, C.A.; Xu, X.; Spengler, J.D.; Ware, J.H.; Fay, M.E.; Ferris, B.G.J.; Speizer, F.E. An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 1993, 329, 1753–1759. [Google Scholar] [CrossRef]
- Pope, C.A.I.; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA 2002, 287, 1132–1141. [Google Scholar] [CrossRef]
- Pope, C.I.; Burnett, R.; Krewski, D.; Jerrett, M.; Shi, Y.; Calle, E.; Thun, M. Cardiovascular mortality and exposure to airborne fine particulate matter and cigarette smoke: Shape of the exposure-response relationship. Circulation 2009, 120, 941–948. [Google Scholar] [CrossRef]
- Giese, B.; Klaessig, F.; Park, B.; Kaegi, R.; Steinfeldt, M.; Wigger, H.; von Gleich, A.; Gottschalk, F. Risks, release and concentrations of engineered nanomaterial in the environment. Sci. Rep. 2018, 8, 1565. [Google Scholar] [CrossRef]
- Harrison, R.M.; Allan, J.; Carruthers, D.; Heal, M.R.; Lewis, A.C.; Marner, B.; Murrells, T.; Williams, A. Non-exhaust vehicle emissions of particulate matter and VOC from road traffic: A review. Atmos. Environ. 2021, 262, 118592. [Google Scholar] [CrossRef]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A.I.; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef]
- Brook, R.D.; Franklin, B.; Cascio, W.; Hong, Y.; Howard, G.; Lipsett, M.; Luepker, R.; Mittleman, M.; Samet, J.; Smith, S.C.J.; et al. Air Pollution and cardiovascular disease: A statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation 2004, 109, 2655–2671. [Google Scholar] [CrossRef]
- Pope, C.A.I.; Bhatnagar, A.; McCracken, J.P.; Abplanalp, W.; Conklin, D.J.; O’Toole, T. Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circ. Res. 2016, 119, 1204–1214. [Google Scholar] [CrossRef]
- Sigaud, S.; Goldsmith, C.-A.W.; Zhou, H.; Yang, Z.; Fedulov, A.; Imrich, A.; Kobzik, L. Air pollution particles diminish bacterial clearance in the primed lungs of mice. Toxicol. Appl. Pharmacol. 2007, 223, 1–9. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Brook, R.D. Air pollution and type 2 diabetes: Mechanistic insights. Diabetes 2012, 61, 3037–3045. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Siscovick, D.S.; Sheppard, L.; Shepherd, K.; Sullivan, J.H.; Anderson, G.L.; Kaufman, J.D. Long-term exposure to air pollution and incidence of cardiovascular events in women. N. Engl. J. Med. 2007, 356, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.; Hang, J.; Deng, Q. Particle deposition in human lung airways: Effects of airflow, particle size, and mechanisms. J. Aerosol Sci. 2020, 20, 2846–2858. [Google Scholar] [CrossRef]
- Kim, C.S.; Hu, S.-C. Total respiratory tract deposition of fine micrometer-sized particles in healthy adults: Empirical deposition equations. J. Appl. Physiol. 2006, 101, 401–412. [Google Scholar] [CrossRef]
- Rissler, J.; Nicklasson, H.; Gudmundsson, A.; Wollmer, P.; Swietlicki, S.; Löndahl, J. A set-up for respiratory tract deposition efficiency measurements (15–5000 nm) and first results for a group of children and adults. Aerosol Sci. Technol. 2017, 17, 1244–1255. [Google Scholar] [CrossRef]
- Su, W.-C.; Cheng, Y. Deposition of fiber in the human nasal airway. Aerosol Sci. Technol. 2005, 39, 888–901. [Google Scholar] [CrossRef]
- Prinz, F.; Kánská, J.; Elcner, J.; Hájek, O.; Kummerländer, A.; Krause, M.; Jícha, M.; Lízal, F. Transport and deposition of inhaled fibers in a realistic female airway model: A combined experimental and numerical study. Comput. Biol. Med. 2025, 194, 110473. [Google Scholar] [CrossRef]
- Lizonova, D.; Nagarkar, A.; Demokritou, P.; Kelesidis, G. Effective density of inhaled environmental and engineered nanoparticles and its impact on the lung deposition and dosimetry. Part. Fibre Toxicol. 2024, 21, 7. [Google Scholar] [CrossRef]
- Sturm, R. Theoretical models for dynamic shape factors and lung deposition of small particle aggregates originating from combustion processes. Z. Med. Phys. 2010, 20, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Farkas, Á.; Füri, P.; Thén, W.; Salma, I. Effects of hygroscopic growth of ambient urban aerosol particles on their modelled regional and local deposition. Sci. Total Environ. 2022, 806, 151202. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Si, X.; Longest, W. Electrostatic charge effects on pharmaceutical aerosol deposition in human nasal–laryngeal airways. Pharmaceutics 2014, 6, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bessler, R.; Bhardwaj, S.; Malka, D.; Fishler, R.; Sznitman, J. Exploring the role of electrostatic deposition on inhaled aerosols in alveolated microchannels. Sci. Rep. 2023, 13, 23069. [Google Scholar] [CrossRef]
- Sou, T.; Bergström, C. Contemporary formulation development for inhaled pharmaceuticals. J. Pharm. Sci. 2021, 110, 66–86. [Google Scholar] [CrossRef]
- Patel, B.; Gupta, N.; Ahsan, F. Barriers that inhaled particles encounter. J. Aerosol Med. Pulm. Drug Deliv. 2024, 37, 299–306. [Google Scholar] [CrossRef]
- Lim, P.; Cervantes, M.; Pham, L.; Rothchild, A. Alveolar macrophages: Novel therapeutic targets for respiratory diseases. Expert Rev. Mol. Med. 2021, 23, e18. [Google Scholar] [CrossRef]
- Luettich, K.; Sharma, M.; Yepiskoposyan, H.; Breheny, D.; Lowe, F. An adverse outcome pathway for decreased lung function focusing on mechanisms of impaired mucociliary clearance following inhalation exposure. Front. Toxicol. 2021, 3, 750254. [Google Scholar] [CrossRef]
- Geiser, M. Update on macrophage clearance of inhaled micro- and nanoparticles. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Jeon, S.; Park, B.; Jo, S.-G.; Moon, H.-G.; Kim, S.-H.; Huh, Y.; Lee, K.; Duffin, R.; Cho, W.-S. Lung-associated lymph nodes and spleen are major secondary accumulating organs in mice following inhaled black carbon as a surrogate of ultrafine ambient particles. Environ. Int. 2025, 199, 109475. [Google Scholar] [CrossRef] [PubMed]
- Ural, B.; Caron, D.; Dogra, P.; Wells, S.; Szabo, P.; Granot, T.; Senda, T.; Poon, M.; Lam, N.; Thapa, P.; et al. Inhaled particulate accumulation with age impairs immune function and architecture in human lung lymph nodes. Nat. Med. 2022, 28, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, K.; Borm, P. Expert workshop on the hazards and risks of poorly soluble low toxicity particles. Inhal. Toxicol. 2020, 32, 53–62. [Google Scholar] [CrossRef]
- Lee, D.-K.; Kim, G.; Maruthupandy, M.; Lee, K.; Cho, W.-S. Multimodal pulmonary clearance kinetics of carbon black nanoparticles deposited in the lungs of rats: The role of alveolar macrophages. Part. Fibre Toxicol. 2024, 21, 32. [Google Scholar] [CrossRef]
- Thá, E.; Ma-Hock, L.; Rueckel, M.; Gruendling, T.; Wohlleben, W.; Reck, B.; Landsiedel, R. Lung retention, distribution and persistence of polymer particles in rats exposed via inhalation. Part. Fibre Toxicol. 2026, 23, 2. [Google Scholar] [CrossRef]
- Perez, L.; Ambroise, J.; Bearzatto, B.; Froidure, A.; Pilette, C.; Yakoub, Y.; Palmai-Pallag, M.; Bouzin, C.; Ryelandt, L.; Pavan, C.; et al. Unique transcriptomic responses of rat and human alveolar macrophages in an in vitro model of overload with TiO2 and carbon black. Part. Fibre Toxicol. 2025, 22, 8. [Google Scholar] [CrossRef]
- Kasai, T.; Hirai, S.; Furukawa, Y.; Misumi, K.; Takeda, T.; Goto, Y.; Takanobu, K.; Yoneyama, K.; Yamano, S.; Senoh, H.; et al. Lung carcinogenicity by whole body inhalation exposure to anatase-type nano-titanium dioxide in rats. J. Toxicol. Sci. 2024, 49, 359–383. [Google Scholar] [CrossRef]
- Borm, P. The parallels between particle induced lung overload and particle induced periprosthetic osteolysis (PPOL). Open Res. Eur. 2022, 1, 16. [Google Scholar] [CrossRef]
- Hamanaka, R.; Mutlu, G. Particulate matter air pollution: Effects on the respiratory system. J. Clin. Investig. 2025, 135, e194312. [Google Scholar] [CrossRef]
- Son, Y.; Carranza, C.; Subramhanya, S.; Gardner, C.; Black, K.; Jones, L.; Meng, Q.; Torres, M.; Vargas, A.O.; Zhang, J.; et al. Correlations between human alveolar macrophage particulate matter load, air pollution particulate matter levels, and systemic inflammation markers in Mexico City. Sci. Rep. 2025, 15, 29903. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, K. Review of lung particle overload, rat lung cancer, and the conclusions of the Edinburgh Expert Panel-It’s time to revisit cancer hazard classifications for titanium dioxide and carbon black. Front. Public Health 2022, 10, 907318. [Google Scholar] [CrossRef] [PubMed]
- Cosnier, F.; Seidel, C.; Valentino, S.; Schmid, O.; Bau, S.; Vogel, U.; Devoy, J.; Gaté, L. Retained particle surface area dose drives inflammation in rat lungs following acute, subacute, and subchronic inhalation of nanomaterials. Part. Fibre Toxicol. 2021, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Poland, C.; Duffin, R.; Weber, K.; Dekant, W.; Borm, P. Is pulmonary inflammation a valid predictor of particle induced lung pathology? The case of amorphous and crystalline silicas. Toxicol. Lett. 2024, 399, 18–30. [Google Scholar] [CrossRef]
- Qiu, J.; Ma, J.; Dong, Z.; Ren, Q.; Shan, Q.; Liu, J.; Gao, M.; Liu, G.; Zhang, S.; Qu, G.; et al. Lung megakaryocytes engulf inhaled airborne particles to promote intrapulmonary inflammation and extrapulmonary distribution. Nat. Commun. 2024, 15, 7396. [Google Scholar] [CrossRef]
- Leinardi, R.; Sanchez-Calero, C.L.; Ibouraadaten, S.; Uwambayinema, F.; Yakoub, Y.; Pavan, C.; Claus, R.; Lemaire, F.; Ronsmans, S.; Ghosh, M.; et al. Dynamic biodistribution of inhaled silica particles to extrapulmonary sites: Early and late translocation mechanisms with implication for particle biomonitoring. Environ. Int. 2025, 199, 109473. [Google Scholar] [CrossRef]
- Han, D.; Chen, R.; Kan, H.; Xu, Y. The bio-distribution, clearance pathways, and toxicity mechanisms of ambient ultrafine particles. Eco-Environ. Health 2023, 2, 95–106. [Google Scholar] [CrossRef]
- Kreyling, W.; Semmler, M.; Erbe, F.; Mayer, P.; Takenaka, S.; Schulz, H.; Oberdörster, G.; Ziesenis, A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J. Toxicol. Environ. Health A 2002, 65, 1513–1530. [Google Scholar] [CrossRef]
- Son, T.; Cho, Y.J.; Lee, H.; Cho, M.Y.; Goh, B.; Kim, H.M.; Ngoc Hoa, P.T.; Cho, S.-H.; Park, Y.-J.; Park, H.S.; et al. Monitoring in vivo behavior of size-dependent fluorescent particles as a model fine dust. J. Nanobiotechnol. 2022, 20, 227. [Google Scholar] [CrossRef]
- Yang, J.; Kim, E.K.; Park, H.J.; McDowell, A.; Kim, Y.-K. The impact of bacteria-derived ultrafine dust particles on pulmonary diseases. Exp. Mol. Med. 2020, 52, 338–347. [Google Scholar] [CrossRef]
- Nie, B.; Liu, X.; Lei, C.; Liang, X.; Zhang, D.; Zhang, J. The role of lysosomes in airborne particulate matter-induced pulmonary toxicity. Sci. Total Environ. 2024, 919, 170893. [Google Scholar] [CrossRef] [PubMed]
- Sydor, M.; Kendall, R.; Holian, A. Cholesterol content regulates silica-induced lysosomal membrane permeability. Front. Toxicol. 2023, 5, 1112822. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, Y.; Otomo, D.; Okuda, T.; Onodera, A. Sub-toxic exposure to DEPs and PM2.5 impairs dendritic cell function through intracellular particle accumulation. J. Xenobiot. 2025, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Bao, G.; Li, S.; Yang, Z.; Cheng, C.; Le, W. PM2.5 exposure triggers cell death through lysosomal membrane permeabilization and leads to ferroptosis insensitivity via the autophagy dysfunction/p62-KEAP1-NRF2 activation in neuronal cells. Ecotoxicol. Environ. Saf. 2022, 248, 114333. [Google Scholar] [CrossRef]
- Yuan, X.; Nie, Z.; Yang, J.; Shao, B.; Ma, X.; Zhang, X.; Bi, B.; Sun, L.; Liang, X.; Tie, Y.; et al. Carbon black nanoparticles induce cell necrosis through lysosomal membrane permeabilization and cause subsequent inflammatory response. Theranostics 2020, 10, 4589–4605. [Google Scholar] [CrossRef]
- Onodera, A.; Shimomura, T.; Ochi, H.; Sunada, R.; Fukutomi, E.; Hidaka, K.; Kawai, Y. The cellular accumulation of vehicle exhaust particulates changes the acidic pH environment of lysosomes in BEAS-2B airway epithelial cells. J. Xenobiot. 2023, 13, 653–661. [Google Scholar] [CrossRef]
- Zhai, X.; Wang, J.; Sun, J.; Xin, L. PM2.5 induces inflammatory responses via oxidative stress-mediated mitophagy in human bronchial epithelial cells. Toxicol. Res. 2022, 11, 195–205. [Google Scholar] [CrossRef]
- Chivé, C.; Eon-Bertho, A.; Martín-Faivre, L.; Tête, A.; Izabelle, C.; Sallenave, S.; Garcia-Verdugo, I.; Baeza-Squiban, A. Repeated exposures to PM2.5 alter the mitochondria and the influenza-mediated interferon response in an air-liquid interface bronchial epithelium. Environ. Pollut. 2025, 382, 126687. [Google Scholar] [CrossRef]
- Mussalo, L.; Lampinen, R.; Avesani, S.; Závodná, T.; Krejčík, Z.; Kalapudas, J.; Penttilä, E.; Löppönen, H.; Koivisto, A.; Malm, T.; et al. Traffic-related ultrafine particles impair mitochondrial functions in human olfactory mucosa cells—Implications for Alzheimer’s disease. Redox Biol. 2024, 75, 103272. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Z.; Yin, S.; Zhou, X.; Jiang, Y.; Shao, L. The lysosome-mitochondrion crosstalk engaged in silver nanoparticles-disturbed mitochondrial homeostasis. Sci. Total Environ. 2023, 889, 164078. [Google Scholar] [CrossRef]
- Hu, L.; Xu, C.; Tang, X.; Yu, S.; Wang, L.; Li, Q.; Zhou, X. Fine particulate matter promotes airway inflammation and mucin production by activating endoplasmic reticulum stress and the IRE1α/NOD1/NF κB pathway. Int. J. Mol. Med. 2023, 52, 96. [Google Scholar] [CrossRef]
- Uzhytchak, M.; Smolková, B.; Lunova, M.; Frtús, A.; Jirsa, M.; Dejneka, A.; Lunov, O. Lysosomal nanotoxicity: Impact of nanomedicines on lysosomal function. Adv. Drug Deliv. Rev. 2023, 197, 114828. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Fu, H.; Zhang, X.; Liu, S.; Wei, X. Lysosome toxicities induced by nanoparticle exposure and related mechanisms. Ecotoxicol. Environ. Saf. 2024, 286, 117215. [Google Scholar] [CrossRef] [PubMed]
- Rennick, J.; Johnston, A.; Parton, R. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, M.S.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397–5434. [Google Scholar] [CrossRef]
- Vtyurina, N.; Åberg, C.; Salvati, A. Imaging of nanoparticle uptake and kinetics of intracellular trafficking in individual cells. Nanoscale 2021, 13, 10436–10446. [Google Scholar] [CrossRef]
- Zanoni, I.; Keller, J.; Sauer, U.; Müller, P.; Ma-Hock, L.; Jensen, K.; Costa, A.; Wohlleben, W. Dissolution rate of nanomaterials determined by ions and particle size under lysosomal conditions. Chem. Res. Toxicol. 2022, 35, 963–980. [Google Scholar] [CrossRef]
- Ziglari, T.; Wang, Z.; Holian, A. Contribution of particle-induced lysosomal membrane hyperpolarization to lysosomal membrane permeabilization. Int. J. Mol. Sci. 2021, 22, 2277. [Google Scholar] [CrossRef]
- Colacurcio, D.; Nixon, R. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef]
- Syrocheva, A.O.; Gorbacheva, V.I.; Egorova, V.S.; Zamyatnin, A.A., Jr.; Parodi, A.; Kolesova, E.P. Inorganic silica nanoparticles increase lysosomal biology and protease activity. Int. J. Mol. Sci. 2025, 26, 8291. [Google Scholar] [CrossRef]
- Zhang, Z.; Yue, P.; Lu, T.; Wang, Y.; Wei, Y.; Wei, X. Role of lysosomes in physiological activities, diseases, and therapy. J. Hematol. Oncol. 2021, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Roquilly, A.; Mintern, J.; Villadangos, J. Spatiotemporal adaptations of macrophage and dendritic cell development and function. Annu. Rev. Immunol. 2022, 40, 525–557. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, S.; Stanley, J.; Eden, E. Lysosome–mitochondrial crosstalk in cellular stress and disease. Antioxidants 2025, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, M.; Ceña, V. Endocytosis: The nanoparticle and submicron nanocompounds gateway into the cell. Pharmaceutics 2020, 12, 371. [Google Scholar] [CrossRef]
- Pardo, M.; Qiu, X.; Zimmermann, R.; Rudich, Y. Particulate matter toxicity is Nrf2 and mitochondria dependent: The roles of metals and polycyclic aromatic hydrocarbons. Chem. Res. Toxicol. 2020, 33, 1110–1120. [Google Scholar] [CrossRef]
- Weinberg, S.; Chandel, N. Mitochondria reactive oxygen species signaling-dependent immune responses in macrophages and T cells. Immunity 2025, 58, 1904–1921. [Google Scholar] [CrossRef]
- Cattani-Cavalieri, I.; Trombetta-Lima, M.; Yan, H.; Manzano-Covarrubias, A.L.; Baarsma, H.A.; Oun, A.; van der Veen, M.M.; Oosterhout, E.; Dolga, A.M.; Ostrom, R.S.; et al. Schmidt Diesel exhaust particles alter mitochondrial bioenergetics and cAMP producing capacity in human bronchial epithelial cells. Front. Toxicol. 2024, 6, 1412864. [Google Scholar] [CrossRef]
- Quan, J.-H.; Gao, F.; Lee, M.; Yuk, J.-M.; Cha, G.-H.; Chu, J.-Q.; Wang, H.; Lee, Y.-H. Involvement of endoplasmic reticulum stress response and IRE1-mediated ASK1/JNK/Mcl-1 pathways in silver nanoparticle-induced apoptosis of human retinal pigment epithelial cells. Toxicology 2020, 442, 152540. [Google Scholar] [CrossRef]
- Guerra, F.; Girolimetti, G.; Beli, B.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic effect of mitochondrial and lysosomal dysfunction in Parkinson’s disease. Cells 2019, 8, 452. [Google Scholar] [CrossRef]
- Christenson, S.; Smith, B.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef]
- Caramori, G.; Casolari, P.; Barczyk, A.; Durham, A.; Di Stefano, A.; Adcock, I. COPD immunopathology. Semin. Immunopathol. 2016, 38, 497–515. [Google Scholar] [CrossRef]
- Cass, S.; Cope, A.; Nicolau, D.; Russell, R.; Bafadhel, M. Moving the pathway goalposts: COPD as an immune-mediated inflammatory disease. Lancet Respir. Med. 2022, 10, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2022, 17, 515–546. [Google Scholar] [CrossRef]
- Baker, J.; Booth, S.; Dungwa, J.; Higham, A.; Singh, D.; Lea, S. Alveolar macrophage carbon is associated with COPD severity. ERJ Open Res. 2025, 11, 00933-2024. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P. Altered macrophage function in chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2013, 10, S180–S185. [Google Scholar] [CrossRef] [PubMed]
- Tejwani, V.; Woo, H.; Liu, C.; Tillery, A.; Gassett, A.; Kanner, R.; Hoffman, E.; Martinez, F.; Woodruff, P.; Barr, R.; et al. Black carbon content in airway macrophages is associated with increased severe exacerbations and worse COPD morbidity in SPIROMICS. Respir. Res. 2022, 23, 310. [Google Scholar] [CrossRef]
- Liu, B.; Han, Y.; Ye, Y.; Wei, X.; Li, G.; Jiang, W. Atmospheric fine particulate matter (PM2.5) induces pulmonary fibrosis by regulating different cell fates via autophagy. Sci. Total Environ. 2024, 923, 171396. [Google Scholar] [CrossRef]
- Mostovenko, E.; Canal, C.; Cho, M.; Sharma, K.; Erdely, A.; Campen, M.; Ottens, A. Indirect mediators of systemic health outcomes following nanoparticle inhalation exposure. Pharmacol. Ther. 2022, 235, 108120. [Google Scholar] [CrossRef]
- Valacchi, G.; Magnani, N.; Woodby, B.; Ferreira, S.; Evelson, P. Particulate matter induces tissue oxInflammation: From mechanism to damage. Antioxid. Redox Signal. 2020, 33, 308–326. [Google Scholar] [CrossRef]
- Aryal, A.; Harmon, A.C.; Dugas, T.R. Particulate matter air pollutants and cardiovascular disease: Strategies for intervention. Pharmacol. Ther. 2021, 223, 107890. [Google Scholar] [CrossRef]
- Sagheer, U.; Al-Kindi, S.; Abohashem, S.; Phillips, C.; Rana, J.; Bhatnagar, A.; Gulati, M.; Rajagopalan, S.; Kalra, D. Environmental pollution and cardiovascular disease: Part 1 of 2: Air pollution. JACC Adv. 2023, 3, 100805. [Google Scholar] [CrossRef]
- Krittanawong, C.; Qadeer, Y.; Hayes, R.; Wang, Z.; Thurston, G.; Virani, S.; Lavie, C. PM2.5 and cardiovascular diseases: State-of-the-art review. Int. J. Cardiol. Cardiovasc. Risk Prev. 2023, 19, 200217. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Study | Model/Methodology | Main Findings | Relevance to This Review |
|---|---|---|---|
| Particle/Exposure | |||
| Nie et al. (2024) [51] | Review | Reviewed lysosome-centered mechanisms in PM-induced pulmonary toxicity, including lysosomal accumulation, membrane destabilization, and downstream inflammatory responses | Supports the concept of endo-lysosomal dysfunction as a central early event after intracellular particle uptake |
| Airborne particulate matter | |||
| Sydor et al. (2023) [52] | Experimental cell study | Showed that silica-induced lysosomal membrane permeability is regulated by lysosomal membrane composition, especially cholesterol content | Provides mechanistic evidence linking particle exposure to lysosomal membrane permeabilization (LMP) |
| Silica particles | |||
| Nakahira et al. (2025) [53] | In vitro dendritic cell study | Demonstrated that intracellular particle accumulation impairs dendritic cell function even under sub-toxic exposure conditions | Connects intracellular particle retention with immune dysfunction rather than overt cytotoxicity alone |
| DEPs and PM2.5 under sub-toxic exposure conditions | |||
| Wei et al. (2022) [54] | In vitro neuronal cell study | Reported LMP-mediated cell death, autophagy dysfunction, and altered stress-response signaling after PM2.5 exposure | Supports the link between lysosomal injury and downstream disruption of cellular homeostasis |
| PM2.5 | |||
| Yuan et al. (2020) [55] | Experimental cell study | Showed that carbon black nanoparticles induce LMP, necrotic cell death, and subsequent inflammatory responses | Demonstrates that lysosomal damage can initiate inflammatory and injurious downstream effects |
| Carbon black nanoparticles | |||
| Onodera et al. (2023) [56] | In vitro airway epithelial cell study | Showed that intracellular accumulation of vehicle exhaust particulates alters the acidic lysosomal environment in BEAS-2B cells | Supports the concept that retained particles disrupt lysosomal function even in epithelial cells without high-burden phagocytosis |
| Vehicle exhaust particulates | |||
| Zhai et al. (2022) [57] | In vitro bronchial epithelial cell study | Reported oxidative stress, inflammatory responses, and mitophagy-related mitochondrial dysfunction after PM2.5 exposure | Links particle exposure to mitochondria-related stress downstream of intracellular accumulation |
| PM2.5 | |||
| Chivé et al. (2025) [58] | Repeated-exposure air–liquid interface bronchial epithelium model | Showed that repeated PM2.5 exposure alters mitochondrial activity and antiviral interferon responses and is associated with p62 accumulation and impaired autophagic flux | Demonstrates how repeated particle exposure affects mitochondrial function, autophagy, and epithelial immune competence |
| PM2.5 | |||
| Mussalo et al. (2024) [59] | Human cell-based experimental study | Showed that ultrafine particles impair mitochondrial respiration, ATP production, and redox metabolism | Supports the idea that particle-induced mitochondrial dysfunction contributes to broader chronic cellular stress |
| Traffic-related ultrafine particles | |||
| Liu et al. (2023) [60] | Experimental cell study | Demonstrated that lysosome–mitochondrion crosstalk contributes to disruption of mitochondrial homeostasis after silver nanoparticle exposure | Provides mechanistic support for organelle crosstalk linking lysosomal stress to mitochondrial dysfunction |
| Silver nanoparticles | |||
| Hu et al. (2023) [61] | In vitro airway epithelial cell study | Showed that fine particulate matter promotes airway inflammation and mucin production through endoplasmic reticulum stress and the IRE1α/NOD1/NF-κB pathway | Extends the framework from lysosomal and mitochondrial dysfunction to ER stress and inflammatory signaling |
| Fine particulate matter |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Onodera, A. Biological Retention and Accumulation of Inhaled Environmental Particles Disrupt Immune Homeostasis: Implications for Chronic Lung Disease. Int. J. Environ. Med. 2026, 1, 7. https://doi.org/10.3390/ijem1020007
Onodera A. Biological Retention and Accumulation of Inhaled Environmental Particles Disrupt Immune Homeostasis: Implications for Chronic Lung Disease. International Journal of Environmental Medicine. 2026; 1(2):7. https://doi.org/10.3390/ijem1020007
Chicago/Turabian StyleOnodera, Akira. 2026. "Biological Retention and Accumulation of Inhaled Environmental Particles Disrupt Immune Homeostasis: Implications for Chronic Lung Disease" International Journal of Environmental Medicine 1, no. 2: 7. https://doi.org/10.3390/ijem1020007
APA StyleOnodera, A. (2026). Biological Retention and Accumulation of Inhaled Environmental Particles Disrupt Immune Homeostasis: Implications for Chronic Lung Disease. International Journal of Environmental Medicine, 1(2), 7. https://doi.org/10.3390/ijem1020007