
Alzheimer’s Disease Lipidome: Elevated Cortical Levels of Glycerolipids in Subjects with Mild Cognitive Impairment (MCI) but Not in Non-Demented Alzheimer’s Neuropathology (NDAN) Subjects


Abstract
1. Introduction
2. Materials and Methods
2.1. Donor Tissues
2.2. Lipidomics Analyses
2.3. Data Processing
3. Results
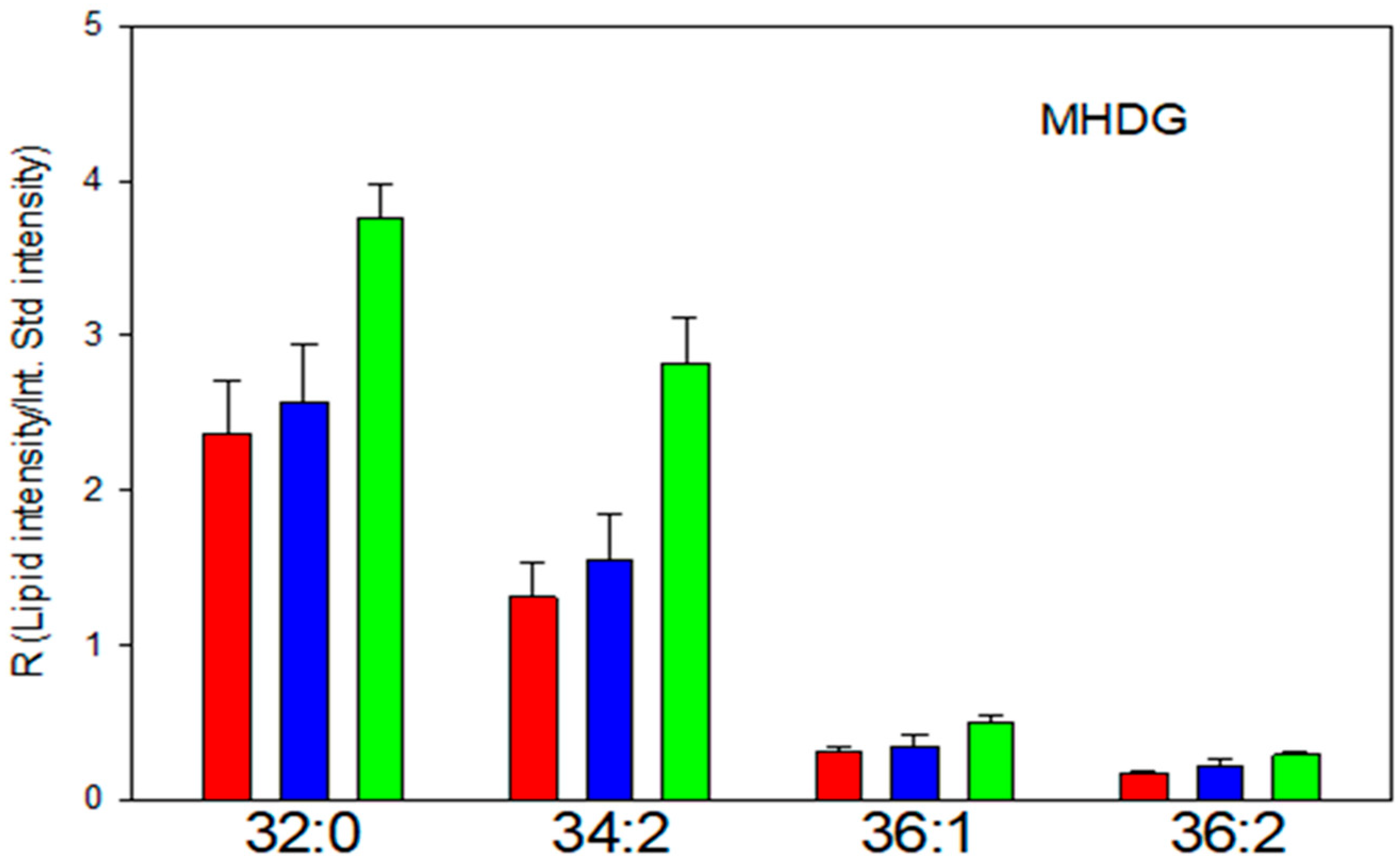
3.1. Diacyl MGDG/MHDG
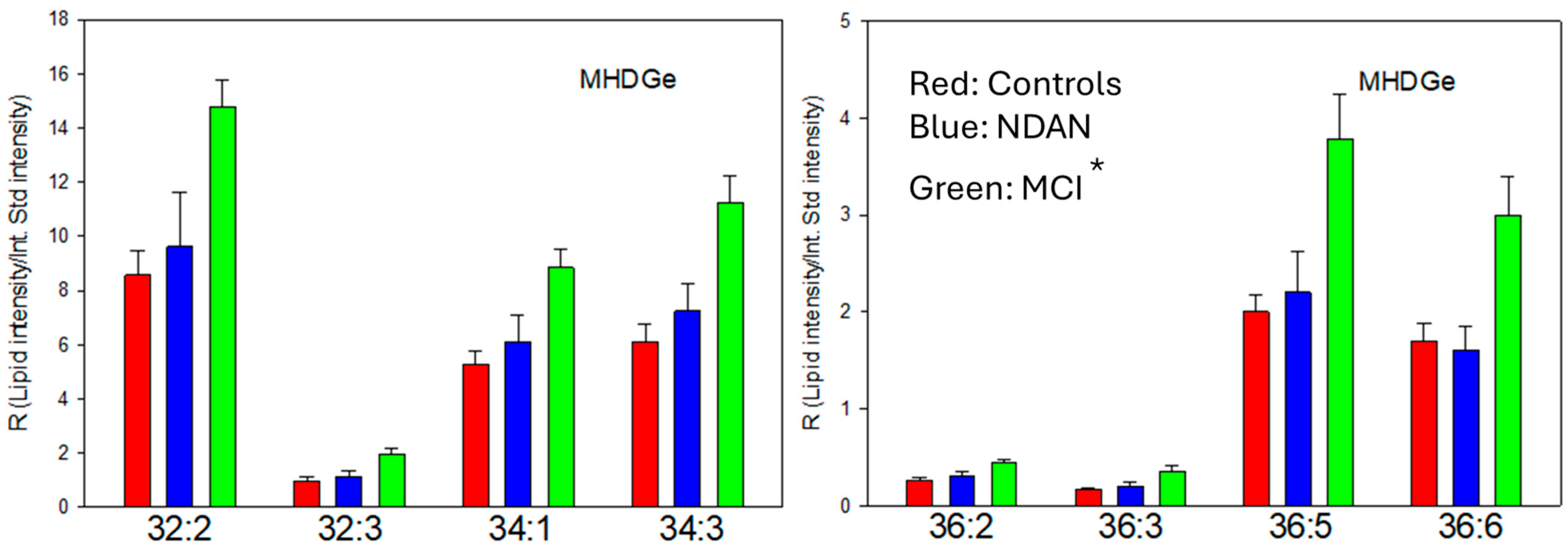
3.2. Alkyl-Acyl MGDG (MGDGe)
3.3. Alkyl-Acyl DG (DGe)
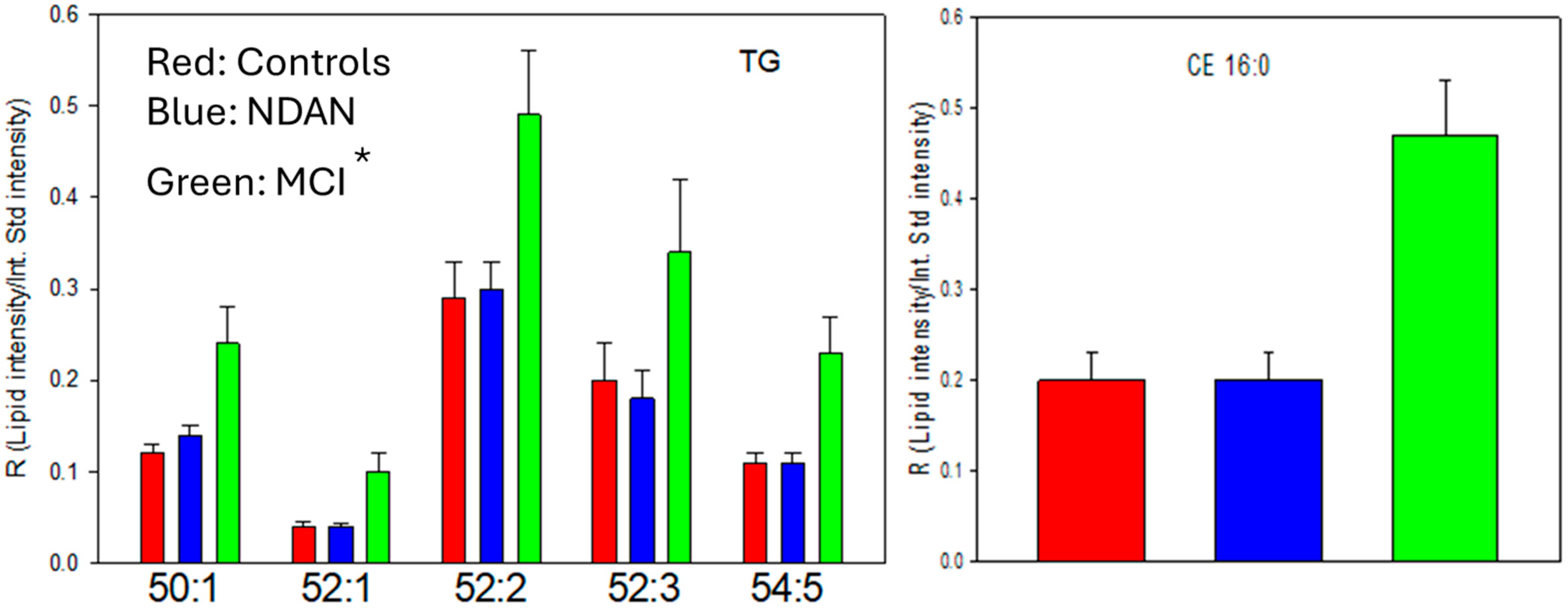
3.4. Lipid Droplets: Triacylglycerols (TGs) and Cholesterol Esters (CEs)
4. Discussion
Study Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr. 2015, 27, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Medicherla, S.; Sheikh, N.; Terry, B.; Phillipps, A.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Targeted Lipidomics of Fontal Cortex and Plasma Diacylglycerols (DAG) in Mild Cognitive Impairment and Alzheimer’s Disease: Validation of DAG Accumulation Early in the Pathophysiology of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.M.; Wang, Y.; Duan, X.; Wenk, M.R.; Kalaria, R.N.; Chen, C.P.; Lai, M.K.; Shui, G. Brain lipidomes of subcortical ischemic vascular dementia and mixed dementia. Neurobiol. Aging 2014, 35, 2369–2381. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy Body Disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Hauther, K.A.; Scarborough, J.H.; Craney, D.J.; Dudzik, B.; Cebak, J.E.; Woltjer, R.L. Human brain lipidomics: Utilities of chloride adducts in flow injection analyses (FIA). Life 2021, 11, 403. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Aytan, N.; Rajendiran, T.; Mellott, T.J.; Soni, T.; Burant, C.F.; Serrano, G.E.; Beach, T.G.; Lin, H.; Stein, T.D. Cerebral Gray and White Matter Monogalactosyl Diglyceride Levels Rise with the Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 95, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Garab, G.; Ughy, B.; Goss, R. Role of MGDG and Non-bilayer Lipid Phases in the Structure and Dynamics of Chloroplast Thylakoid Membranes. Subcell. Biochem. 2016, 86, 127–157. [Google Scholar] [PubMed]
- Inoue, T.; Dehmukh, D.S.; Pieringer, R.A. The association of the galactosyl diglycerides of brain with myelination. I. Changes in the concentration of monogalactosyl diglyceride in the somal and myelin fractions of brain of rats during development. J. Biol. Chem. 1971, 246, 5688–5694. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, D.S.; Inoue, T.; Pieringer, R.A. The association of the galactosyl diglycerides of brain with myelination. II. The inability of the myelin-deficient mutant, jimpy mouse, to synthesize galactosyl diglycerides effectively. J. Biol. Chem. 1971, 46, 5695–5699. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, K.; Kishimoto, Y. Changes in monogalactosyl diacylglycerols, alkylgalactolipids and cerebroside fatty acid esters in maturing rat brain measured by high-performance liquid chromatography. Biochim. Biophys. Acta 1984, 794, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Schultz, T.; Althaus, H.H. Monogalactosyl diglyceride, a marker for myelination, activates oligodendroglial protein kinase c. J. Neurochem. 1994, 62, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Wenger, D.A.; Petitpas, J.W.; Pieringer, R.A. The metabolism of glyceride glycolipids. II. Biosynthesis of monogalactosyl diglyceride from uridine diphosphate galactose and diglyceride in brain. Biochemistry 1968, 7, 3700–3707. [Google Scholar] [CrossRef] [PubMed]
- Wenger, D.A.; Subba Rao, K.; Pieringer, R.A. The metabolism of glyceride glycolipids. 3. Biosynthesis of digalactosyl diglyceride by galactosyl transferase pathways. J. Biol. Chem. 1970, 245, 2513–2519. [Google Scholar] [CrossRef] [PubMed]
- van der Bijl, P.; Strous, G.J.; Lopes-Cardozo, M.; Thomas-Oates, J.; van Meer, G. Synthesis of non-hydroxy-galactosylceramides and galactosyldiglycerides by hydroxy-ceramide galactosyltransferase. Biochem. J. 1996, 317, 589–597. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, A.T.; Moyon, S.; Wang, M.; Katsyv, I.; Song, W.M.; Zhou, X.; Dammer, E.B.; Duong, D.M.; Aaker, J.; Zhao, Y.; et al. Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Amara, S.; Barouh, N.; Lecomte, J.; Lafont, D.; Robert, S.; Villeneuve, P.; De Caro, A.; Carrière, F. Lipolysis of natural long chain and synthetic medium chain galactolipids by pancreatic lipase-related protein 2. Biochim. Biophys. Acta 2010, 1801, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Kauss, H. Turnover of galactosylglycerol and osmotic balance in ochromonas. Plant Physiol. 1973, 52, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Simón-Manso, Y.; Marupaka, R.; Yan, X.; Liang, Y.; Telu, K.H.; Mirokhin, Y.; Stein, S.E. Mass Spectrometry Fingerprints of Small-Molecule Metabolites in Biofluids: Building a Spectral Library of Recurrent Spectra for Urine Analysis. Anal. Chem. 2019, 91, 12021–12029. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Alzheimer’s changes in non-demented and demented patients. Acta Neuropathol. 1995, 89, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Woltjer, R.L. Electrospray Ionization High Resolution Mass Spectrometry of the Chloride Adducts of Steroids, Mono- and Oligo-saccharides, Xyloglucans, Ceramides, Gangliosides, and Phenols. In Springer Protocols, Neuromethods: Metabolomics; Wood, P.L., Ed.; Humana Press: New York, NY, USA, 2021; Volume 159, pp. 69–76. ISBN 978-1-0716-0863-0. [Google Scholar]
- Wood, P.L.; Cebak, J.E.; Woltjer, R.L. Diacylglycerols as biomarkers of sustained immune activation in Proteinopathies associated with dementia. Clin. Chim. Acta 2018, 476, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Medici, V.; Malagoli, D.; Chiariello, A.; Cirrincione, A.; Davin, A.; Chikhladze, M.; Vasuri, F.; Legname, G.; Ferrer, I.; et al. Expression pattern of perilipins in human brain during aging and in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12756. [Google Scholar] [CrossRef] [PubMed]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [PubMed]
- Itoh, K.; Akimoto, Y.; Kondo, S.; Ichimiya, T.; Aoki, K.; Tiemeyer, M.; Nishihara, S. Glucuronylated core 1 glycans are required for precise localization of neuromuscular junctions and normal formation of basement membranes on Drosophila muscles. Dev. Biol. 2018, 436, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.R.; Reddy, B.V.; Irvine, K.D. Requirement for a core 1 galactosyltransferase in the Drosophila nervous system. Dev. Dyn. 2008, 237, 3703–3714. [Google Scholar] [CrossRef] [PubMed]
- Jhou, J.F.; Tai, H.C. The Study of Postmortem Human Synaptosomes for Understanding Alzheimer’s Disease and Other Neurological Disorders: A Review. Neurol. Ther. 2017, 6 (Suppl. S1), 57–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Roura, E.; Choi, Y.; Kim, J. Transcriptomic Analysis of the Porcine Gut in Response to Heat Stress and Dietary Soluble Fiber from Beet Pulp. Genes 2022, 13, 1456. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Li, C.; Haroutunian, V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Xie, D.; Zhong, X.P. MicroRNA-34a enhances T cell activation by targeting diacylglycerol kinase ζ. PLoS ONE 2013, 8, e77983. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Jun, S.; Rellick, S.; Quintana, D.D.; Cavendish, J.Z.; Simpkins, J.W. Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 2016, 1646, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Hozumi, Y.; Nakano, T.; Saino, S.S.; Kondo, H. Cell biology and pathophysiology of the diacylglycerol kinase family: Morphological aspects in tissues and organs. Int. Rev. Cytol. 2007, 264, 25–63. [Google Scholar] [PubMed]
- Tsuchiya, R.; Tanaka, T.; Hozumi, Y.; Nakano, T.; Okada, M.; Topham, M.K.; Iino, M.; Goto, K. Downregulation of diacylglycerol kinase ζ enhances activation of cytokine-induced NF-κB signaling pathway. Biochim. Biophys. Acta 2015, 1853, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Mérida, I.; Arranz-Nicolás, J.; Rodríguez-Rodríguez, C.; Ávila-Flores, A. Diacylglycerol kinase control of protein kinase, C. Biochem. J. 2019, 476, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Brose, N.; Betz, A.; Wegmeyer, H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr. Opin. Neurobiol. 2004, 14, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, A.; Rotem-Dai, N.; Strominger, I.; Anto, N.P.; Isakov, N.; Monsonego, A.; Livneh, E. Protein kinase C eta is activated in reactive astrocytes of an Alzheimer’s disease mouse model: Evidence for its immunoregulatory function in primary astrocytes. Glia 2021, 69, 697–714. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.Y.; Liu, X.L.; Zhang, X.Q.; Liu, Y.X.; Li, M.; Zhao, C.Z.; Zhang, X.M.; Meng, Q.H. Anti-neuroinflammation activity of acetylpuerarin mediated by a PKC-delta-dependent caspase signaling pathway: In vivo and in vitro studies. Pharmazie 2016, 71, 575–582. [Google Scholar] [PubMed]
- Morimoto, K.; Horio, J.; Satoh, H.; Sue, L.; Beach, T.; Arita, S.; Tooyama, I.; Konishi, Y. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients compared to the brains of non-demented patients with and without increasing AD pathology. J. Alzheimer’s Dis. 2011, 25, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [PubMed]
- Kok, F.K.; van Leerdam, S.L.; de Lange, E.C.M. Potential Mechanisms Underlying Resistance to Dementia in Non-Demented Individuals with Alzheimer’s Disease Neuropathology. J. Alzheimer’s Dis. 2022, 87, 51–81. [Google Scholar] [PubMed]
- Armstrong, R.A. beta-Amyloid (A beta) deposition in elderly non-demented patients and patients with Alzheimer’s disease. Neurosci. Lett. 1994, 178, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Langui, D.; Probst, A.; Ulrich, J. Alzheimer’s changes in non-demented and demented patients: A statistical approach to their relationships. Acta Neuropathol. 1995, 89, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Veerhuis, R.; Kamphorst, W.; Eikelenboom, P. Distribution of beta amyloid associated proteins in plaques in Alzheimer’s disease and in the non-demented elderly. Neurodegeneration 1995, 4, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Beta-amyloid deposition in the medial temporal lobe in elderly non-demented brains and in Alzheimer’s disease. Dementia 1995, 6, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Amyloid Biomarker Study Group; Aalten, P.; Aarsland, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L. Lipidomics of Alzheimer’s disease: Current status. Alzheimer’s Res. Ther. 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Semikasev, E.; Ahlemeyer, B.; Acker, T.; Schänzer, A.; Baumgart-Vogt, E. Rise and fall of peroxisomes during Alzheimer’s disease: A pilot study in human brains. Acta Neuropathol. Commun. 2023, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Q.; Noguti, R.; Bottino, C.M.; Vallada, H. Study of association between genetic polymorphisms of phospholipase A2 enzymes and Alzheimer’s disease. Arq. Neuropsiquiatr. 2010, 68, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Villeneuve, S.; Haase, C.M.; Madison, C.M.; Oh, H.; Landau, S.M.; Rabinovici, G.D.; Jagust, W.J. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurol. 2013, 70, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Cebak, J.E. Lipidomics Biomarker Studies: Errors, Limitations, and the Future. Biochem. Biophys. Res. Comm. 2018, 504, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Du, S.; Yao, S.Q. Bifunctional Lipid-Derived Affinity-Based Probes (AfBPs) for Analysis of Lipid-Protein Interactome. Acc. Chem. Res. 2022, 55, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Fedoryshchak, R.O.; Gorelik, A.; Shen, M.; Shchepinova, M.M.; Pérez-Dorado, I. Tate EW Discovery of lipid-mediated protein-protein interactions in living cells using metabolic labeling with photoactivatable clickable probes. Chem. Sci. 2023, 14, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Vidal, M. A global protein-lipid interactome map. Mol. Syst. Biol. 2010, 6, 443. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids 1996, 81, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Lipid polymorphism and protein-lipid interactions. Biochim. Biophys. Acta 1998, 1376, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Zick, M.; Stroupe, C.; Orr, A.; Douville, D.; Wickner, W.T. Membranes linked by trans-SNARE complexes require lipids prone to non-bilayer structure for progression to fusion. Elife 2014, 3, e01879. [Google Scholar] [CrossRef] [PubMed]
- Holz, R.W.; Zimmerberg, J. Dynamic Relationship of the SNARE Complex with a Membrane. Biophys. J. 2019, 117, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.S.; Guerini, F.R.; Arosio, B.; Galimberti, D.; Zanzottera, M.; Bianchi, A.; Nemni, R.; Clerici, M. SNARE Complex Polymorphisms Associate with Alterations of Visual Selective Attention in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Zheng, P.; Xie, Q.; Wang, C.; Pang, C. Molecular Dynamics of Apolipoprotein Genotypes APOE4 and SNARE Family Proteins and Their Impact on Alzheimer’s Disease. Life 2025, 15, 223. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ueda, Y.; Umezawa, Y. Imaging diacylglycerol dynamics at organelle membranes. Nat. Methods 2006, 3, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, T.O.; Lass, A. DAG tales: The multiple faces of diacylglycerol--stereochemistry, metabolism, and signaling. Cell Mol. Life Sci. 2015, 72, 3931–3952. [Google Scholar] [PubMed]
- Leikin, S.; Kozlov, M.M.; Fuller, N.L.; Rand, R.P. Measured effects of diacylglycerol on structural and elastic properties of phospholipid membranes. Biophys. J. 1996, 71, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.; Mérida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Age | PMI (hr) | M/F | Brak Score |
|---|---|---|---|---|---|
| Young Control (YC; <85 years) | 14 | 65.1 ± 10.7 | 26.4 ± 16.7 | 8/6 | 0 |
| NDAN (Old controls, OC; >85) | 8 | 91.5 ± 4.2 | 19.4 ± 15.5 | 4/4 | 2–4 |
| MCI | 14 | 89.3 ± 6.9 | 12.9 ± 11.6 | 4/10 | 3–6 |
| Lipid | Exact Mass | [M+NH4]+ |
|---|---|---|
| CE 16:0 | 624.5845 | 642.6183 |
| Lipid | Exact Mass | [M+NH4]+ |
| TG 50:1 (16:0/16:0/18:1) | 832.7519 | 850.7858 |
| TG 52:1 (16:0/18:0/18:1) | 860.7833 | 878.8176 |
| TG 52:2 (16:0/18:1/18:1) | 858.7676 | 876.8014 |
| TG 52:3 (16:0/18:1.18:2) | 856.7519 | 874.7858 |
| TG 54:5 (18:2/18:2/18:1; 16:0/18:1/20:4) | 880.7519 | 898.7858 |
| Lipid | Exact Mass | [M+NH4]+ |
| DGp 34:0 (18:0/16:0) | 580.5431 | 598.5769 |
| DGp 36:1 (18:0/18:1) | 606.5587 | 624.5925 |
| DGp 36:2 (18:1/18:1) | 604.5431 | 622.5769 |
| DGe (20:0/20:4) | 658.5900 | 676.6238 |
| Lipid | Exact Mass | [M+Cl]− |
| MHDG 32:0 (16:0/16:0) | 730.5595 | 765.5293 |
| MHDG 34:2 (16:0/18:2) | 754.5595 | 789.5293 |
| MHDG 36:1 (18:0/18:1) | 784.6065 | 819.5763 |
| MHDG 36:2 (18:0/18:2) | 792.5908 | 817.5606 |
| Lipid | Exact Mass | [M+Cl]− |
| MHDGe 32:2 | 712.5489 | 747.5188 |
| MHDGe 32:3 | 710.5333 | 745.5031 |
| MHDGe 34:1 | 742.5959 | 777.5657 |
| MHDGe 34:3 | 738.5646 | 773.5344 |
| MHDGe 36:2 | 768.6115 | 803.5814 |
| MHDGe 36:3 | 766.5959 | 801.5657 |
| MHDGe 36:5 | 763.5646 | 797.5344 |
| MHDGe 36:6 | 760.5499 | 795.5188 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wood, P.L.; Cebak, J.E.; Beger, A.W. Alzheimer’s Disease Lipidome: Elevated Cortical Levels of Glycerolipids in Subjects with Mild Cognitive Impairment (MCI) but Not in Non-Demented Alzheimer’s Neuropathology (NDAN) Subjects. J. Dement. Alzheimer's Dis. 2025, 2, 20. https://doi.org/10.3390/jdad2030020
Wood PL, Cebak JE, Beger AW. Alzheimer’s Disease Lipidome: Elevated Cortical Levels of Glycerolipids in Subjects with Mild Cognitive Impairment (MCI) but Not in Non-Demented Alzheimer’s Neuropathology (NDAN) Subjects. Journal of Dementia and Alzheimer's Disease. 2025; 2(3):20. https://doi.org/10.3390/jdad2030020
Chicago/Turabian StyleWood, Paul L., John E. Cebak, and Aaron W. Beger. 2025. "Alzheimer’s Disease Lipidome: Elevated Cortical Levels of Glycerolipids in Subjects with Mild Cognitive Impairment (MCI) but Not in Non-Demented Alzheimer’s Neuropathology (NDAN) Subjects" Journal of Dementia and Alzheimer's Disease 2, no. 3: 20. https://doi.org/10.3390/jdad2030020
APA StyleWood, P. L., Cebak, J. E., & Beger, A. W. (2025). Alzheimer’s Disease Lipidome: Elevated Cortical Levels of Glycerolipids in Subjects with Mild Cognitive Impairment (MCI) but Not in Non-Demented Alzheimer’s Neuropathology (NDAN) Subjects. Journal of Dementia and Alzheimer's Disease, 2(3), 20. https://doi.org/10.3390/jdad2030020