Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis

 ,
,  ,
, 
 and
and
Abstract
1. Introduction
2. Results
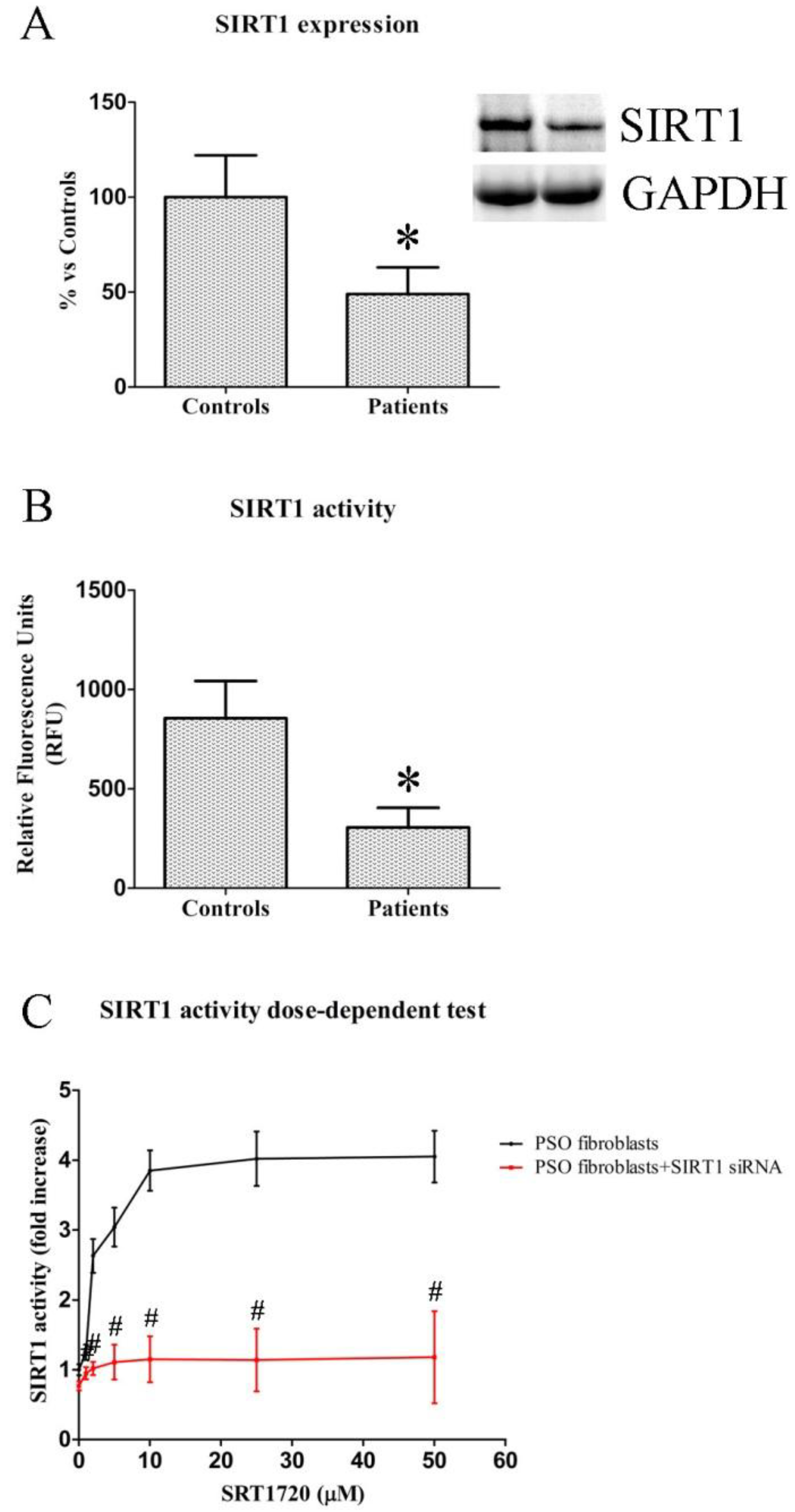
2.1. SIRT1 Expression and Activity in Fibroblasts from Healthy and Psoriatic Subjects
2.2. Dose-Dependent Effects of SIRT1720 on SIRT1 Activity in Psoriatic Fibroblasts
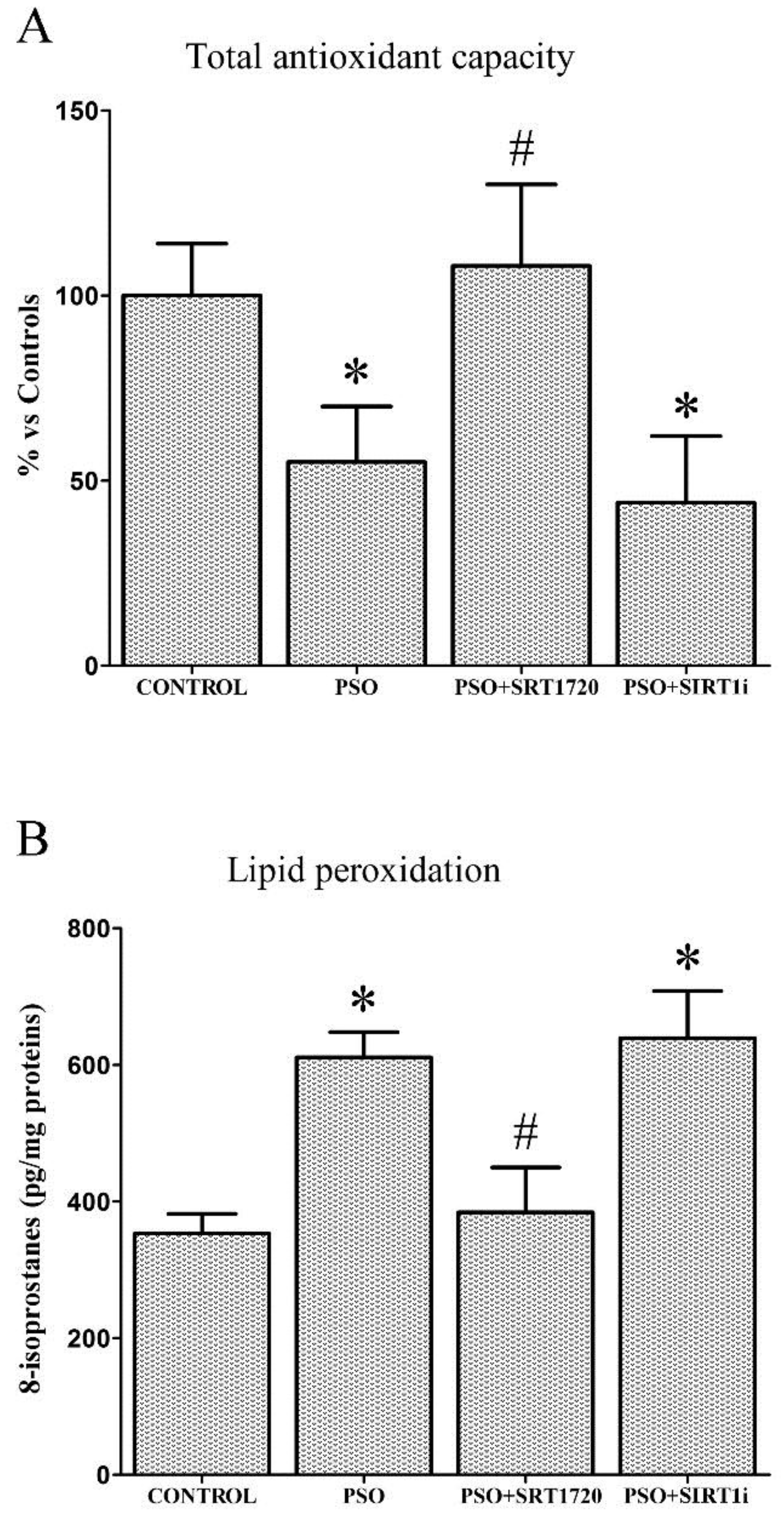
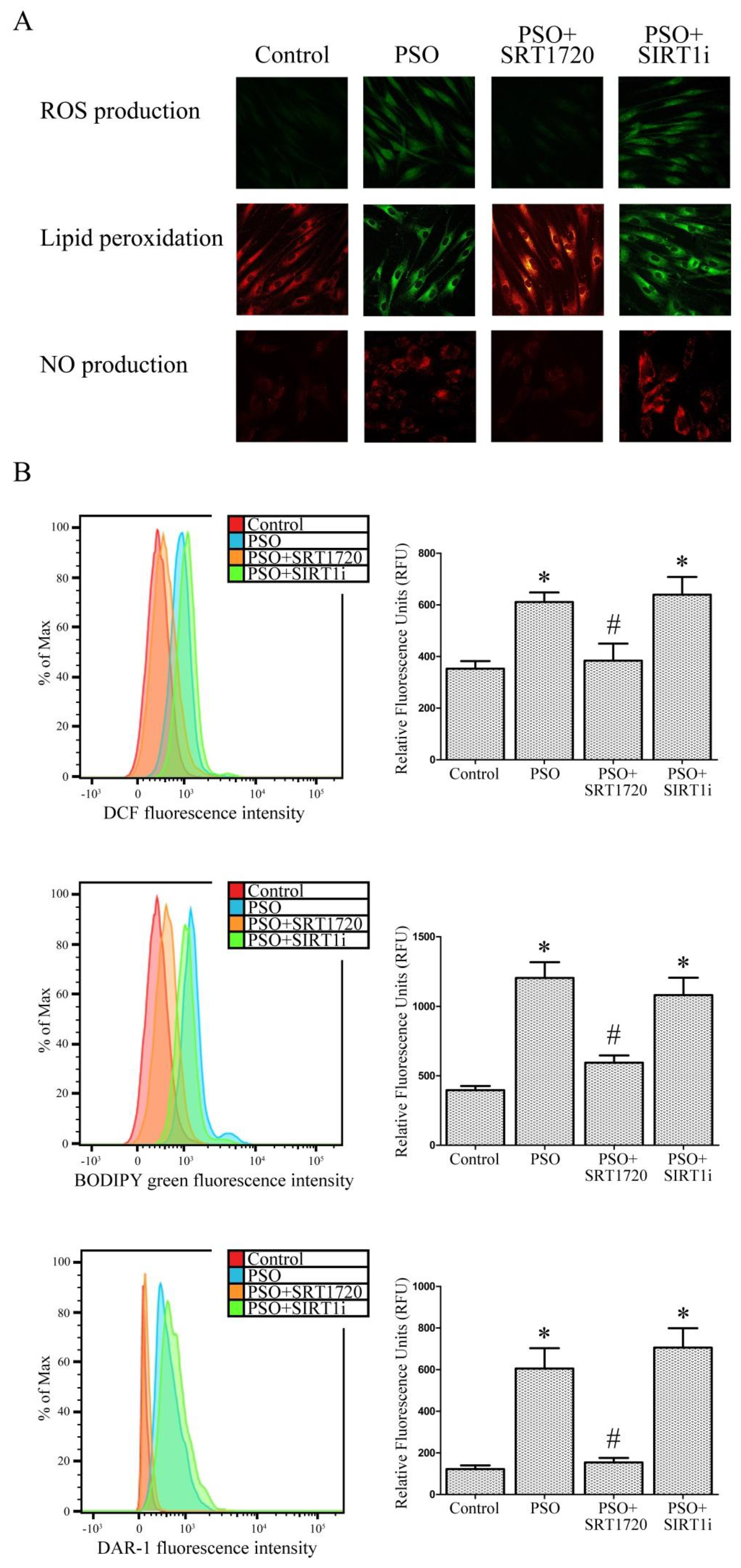
2.3. SIRT1 Activation Decreases Oxidative Stress in Fibroblasts from Psoriatic Patients
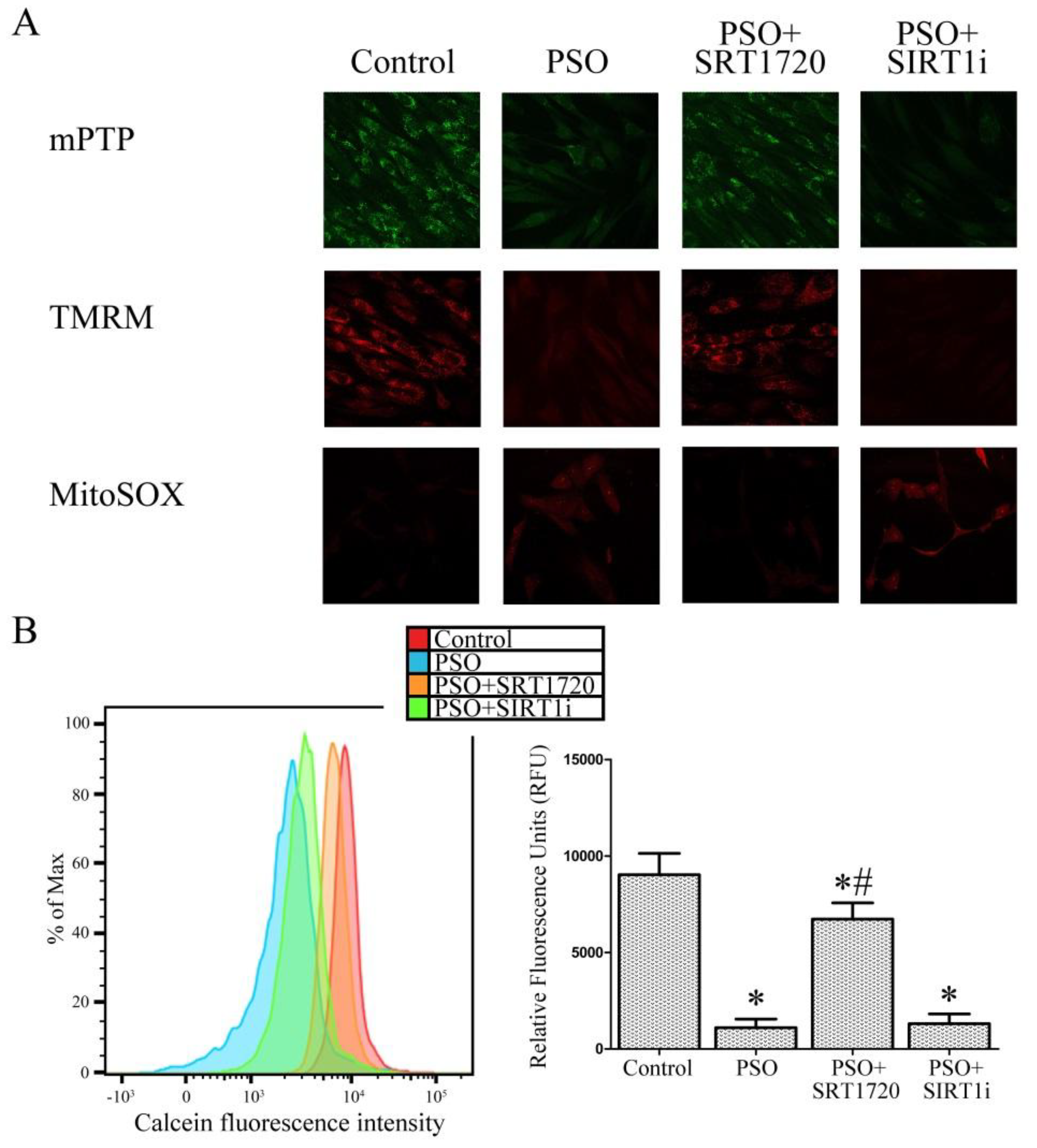
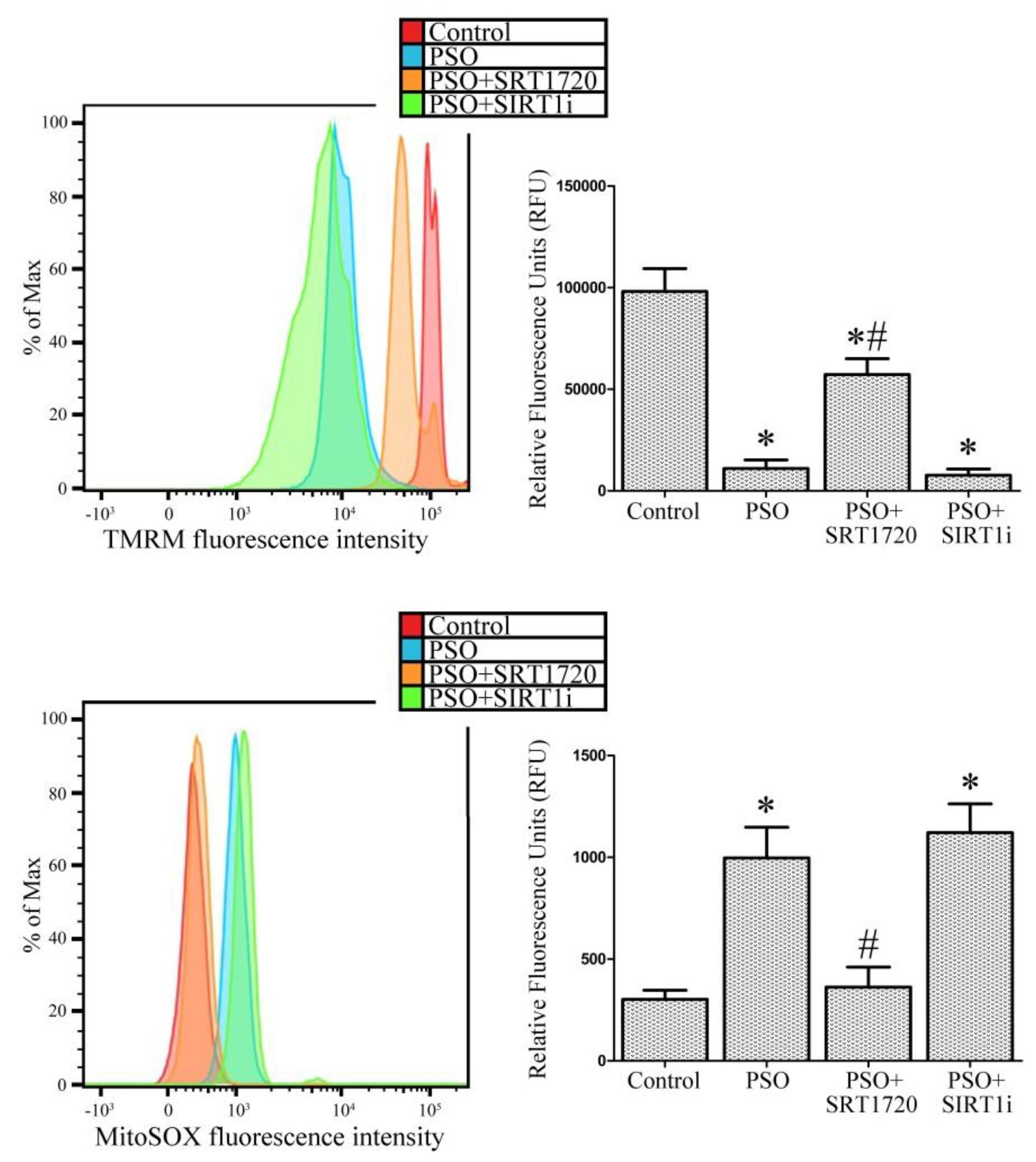
2.4. SIRT1 Activation Protects Psoriatic Fibroblasts from Mitochondrial Damage
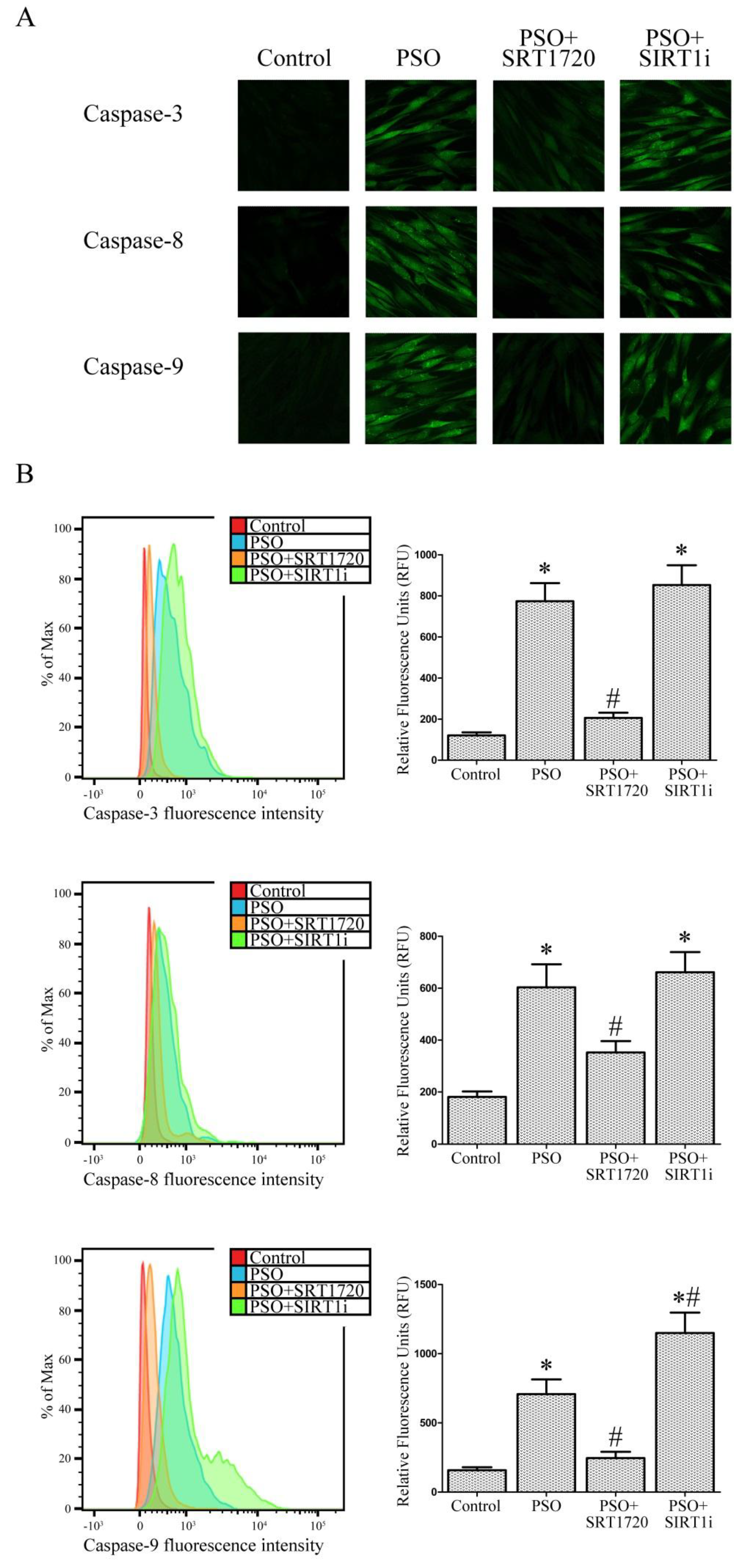
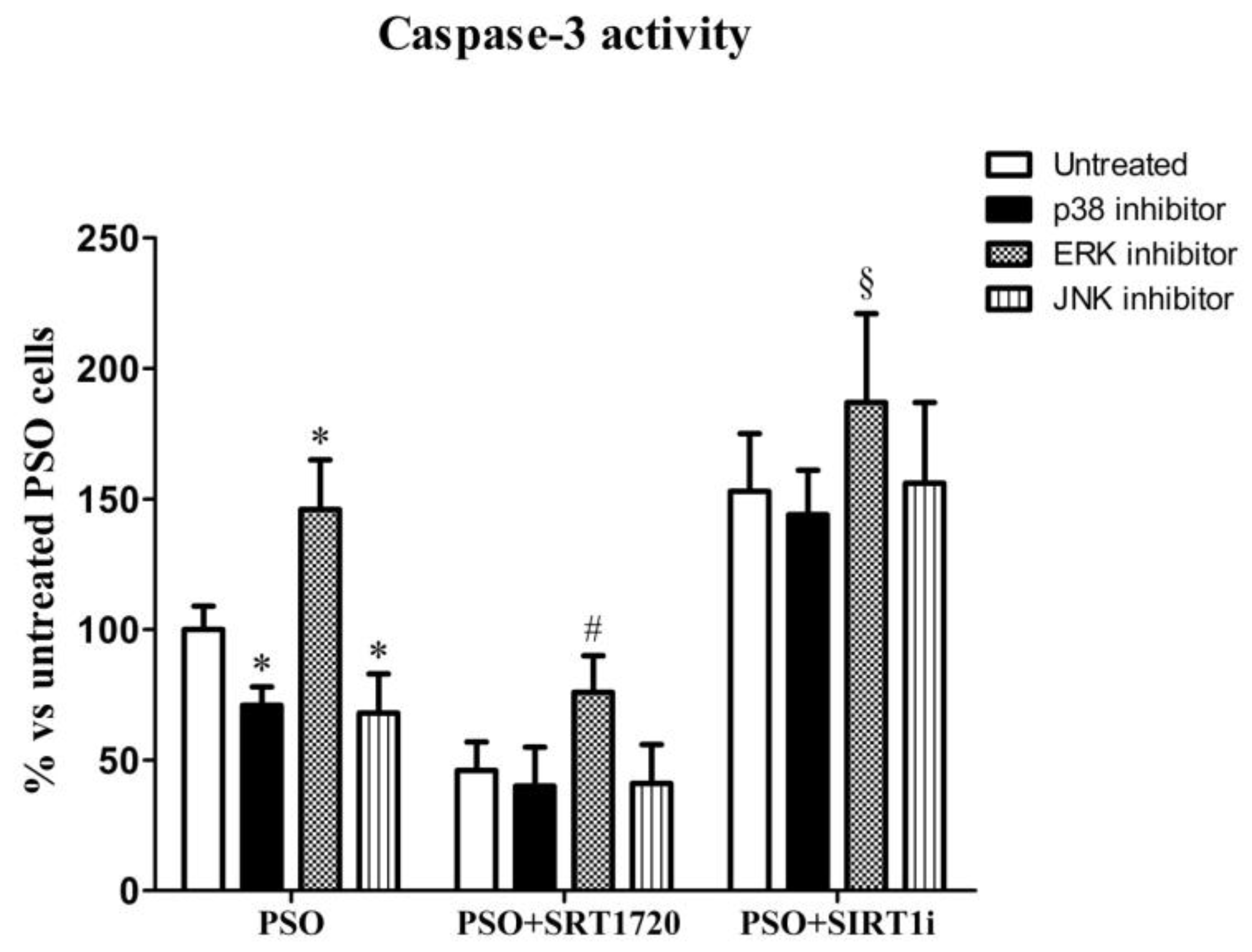
2.5. SIRT1 Activation Protects Psoriatic Fibroblasts from Apoptosis
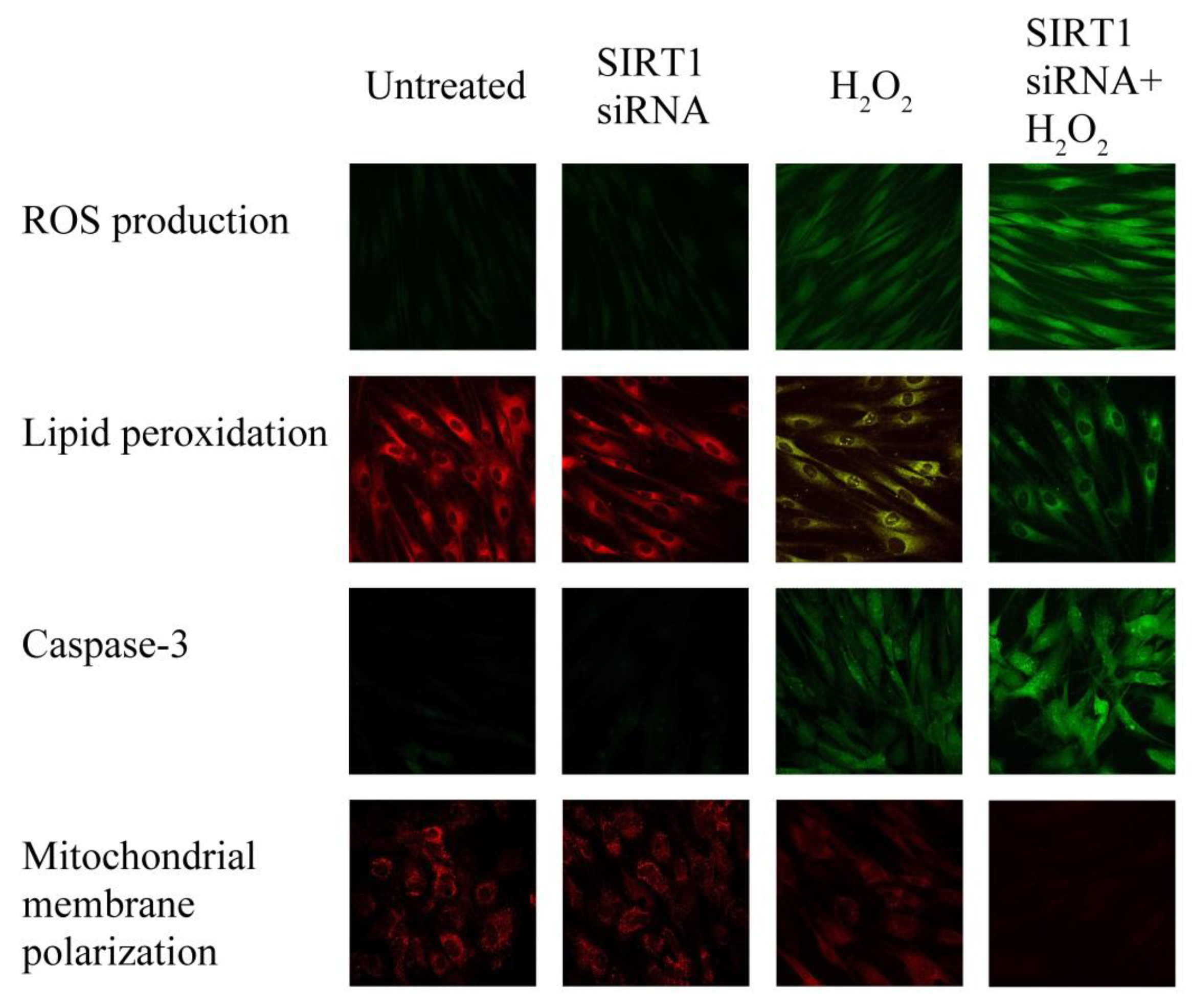
2.6. SIRT1 siRNA-Treatment of Fibroblasts from Healthy Subjects
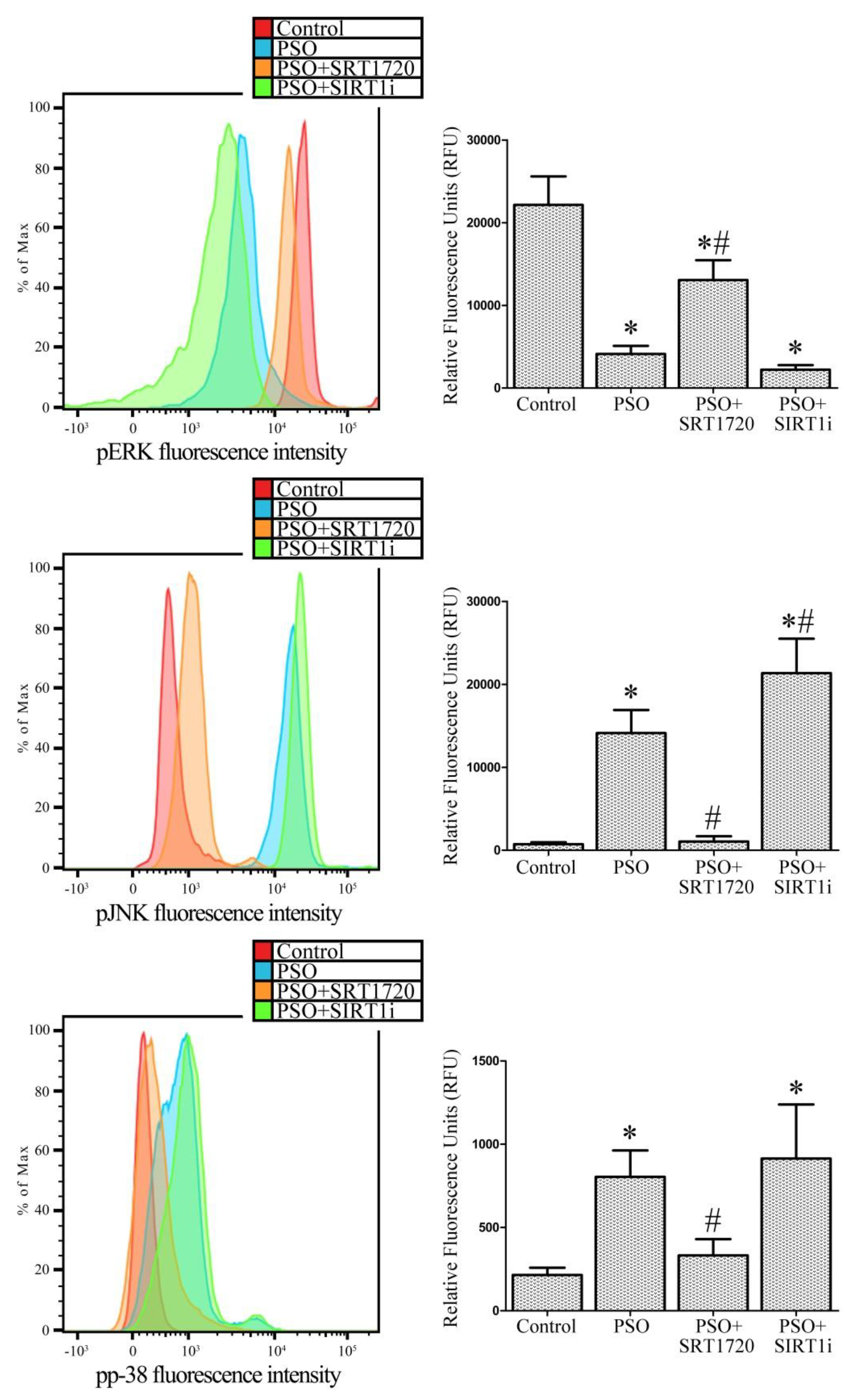
2.7. SIRT1 Activation Modulates MAPK Pathways in Fibroblasts from Psoriatic Patients
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Fibroblast Isolation and Setting Up of Cell Cultures
4.3. Preparation of Cell Homogenates
4.4. Western Blot Analysis of SIRT1
4.5. Determination of Cellular SIRT1 Activity
4.6. Cell Treatments
4.7. Determination of Total Antioxidant Capacity (TAC)
4.8. Determination of Lipid Peroxidation
4.9. Assessment of Intracellular ROS, NO Production and Mitochondrial Superoxide
4.10. Mitochondrial Number
4.11. Mitochondrial Permeability Transition Pore Opening
4.12. Mitochondrial Membrane Potential
4.13. Determination of Caspase Activity
4.14. SIRT1 RNA Interference (RNAi) Experiments
4.15. Assessment of MAPK Activity by FACS Analysis
4.16. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Boehncke, W.H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- El Ghalbzouri, A.; Lamme, E.; Ponec, M. Crucial role of fibroblasts in regulating epidermal morphogenesis. Cell Tissue Res. 2002, 310, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Wang, F.; Shao, Y.; Rittié, L.; Xia, W.; Orringer, J.S.; Voorhees, J.J.; Fisher, G.J. Enhancing structural support of the dermal microenvironment activates fibroblasts, endothelial cells, and keratinocytes in aged human skin in vivo. J. Investig. Dermatol. 2013, 133, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Trouba, K.J.; Hamadeh, H.K.; Amin, R.P.; Germolec, D.R. Oxidative stress and its role in skin disease. Antioxid. Redox Signal. 2002, 4, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Barygina, V.; Becatti, M.; Lotti, T.; Moretti, S.; Taddei, N.; Fiorillo, C. Treatment with low-dose cytokines reduces oxidative-mediated injury in perilesional keratinocytes from vitiligo skin. J. Dermatol. Sci. 2015, 79, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Mrowietz, U.; Rostami-Yazdi, M. Oxidative stress in the pathogenesis of psoriasis. Free Radic. Biol. Med. 2009, 47, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Gabr, S.A.; Al-Ghadir, A.H. Role of cellular oxidative stress and cytochrome c in the pathogenesis of psoriasis. Arch. Dermatol. Res. 2012, 304, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Pujari, V.M.; Ireddy, S.; Itagi, I.; Kumar, H.S. The serum levels of malondialdehyde, vitamin e and erythrocyte catalase activity in psoriasis patients. J. Clin. Diagn. Res. 2014, 8, CC14–CC16. [Google Scholar] [CrossRef] [PubMed]
- Kadam, D.P.; Suryakar, A.N.; Ankush, R.D.; Kadam, C.Y.; Deshpande, K.H. Role of oxidative stress in various stages of psoriasis. Indian J. Clin. Biochem. 2010, 25, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Basavaraj, K.H.; Vasu Devaraju, P.; Rao, K.S. Studies on serum 8-hydroxy guanosine (8-OHdG) as reliable biomarker for psoriasis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Pereira, P.; Santos-Silva, A.; Rebelo, I.; Figueiredo, A.; Quintanilha, A.; Teixeira, F. Dislipidemia and oxidative stress in mild and in severe psoriasis as a risk for cardiovascular disease. Clin. Chim. Acta 2001, 303, 33–39. [Google Scholar] [CrossRef]
- Johansen, C.; Kragballe, K.; Westergaard, M.; Henningsen, J.; Kristiansen, K.; Iversen, L. The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin. Br. J. Dermatol. 2005, 152, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.J.; Li, C.Y.; Dai, H.Y.; Cai, D.X.; Wang, K.Y.; Xu, Y.H.; Chen, L.M.; Zhou, C.L. Expression and localization of the activated mitogen-activated protein kinase in lesional psoriatic skin. Exp. Mol. Pathol. 2007, 83, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Uchi, H.; Hayashida, S.; Urabe, K.; Moroi, Y.; Furue, M. Differential expression of phosphorylated extracellular signal-regulated kinase 1/2, phosphorylated p38 mitogen-activated protein kinase and nuclear factor-kappaB p105/p50 in chronic inflammatory skin diseases. J. Dermatol. 2009, 36, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Haase, I.; Hobbs, R.M.; Romero, M.R.; Broad, S.; Watt, F.M. A role for mitogen-activated protein kinase activation by integrins in the pathogenesis of psoriasis. J. Clin. Investig. 2001, 108, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ibe, M.; Nakamura, S.; Ishida-Yamamoto, A.; Hashimoto, Y.; Iizuka, H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J. Dermatol. Sci. 2002, 30, 94–99. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell. Commun. Signal 2011, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Taddei, N.; Cecchi, C.; Nassi, N.; Nassi, P.A.; Fiorillo, C. SIRT1 modulates MAPK pathways in ischemic-reperfused cardiomyocytes. Cell. Mol. Life Sci. 2012, 69, 2245–2260. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Fiorillo, C.; Barygina, V.; Cecchi, C.; Lotti, T.; Prignano, F.; Silvestro, A.; Nassi, P.; Taddei, N. SIRT1 regulates MAPK pathways in vitiligo skin: Insight into the molecular pathways of cell survival. J. Cell. Mol. Med. 2014, 18, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Barygina, V.; Emmi, G.; Silvestri, E.; Taddei, N.; Lotti, T.; Fiorillo, C. SIRT1 activity is decreased in lesional psoriatic skin. Intern. Emerg. Med. 2016, 11, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 765, 725–754. [Google Scholar] [CrossRef]
- Sabat, R.; Philipp, S.; Höflich, C.; Kreutzer, S.; Wallace, E.; Asadullah, K.; Volk, H.D.; Sterry, W.; Wolk, K. Immunopathogenesis of psoriasis. Exp. Dermatol. 2007, 16, 779–798. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Conrad, C.; Tonel, G.; Gilliet, M.; Nestle, F.O. The pathogenic role of tissue-resident immune cells in psoriasis. Trends Immunol. 2007, 28, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Dimon-Gadal, S.; Gerbaud, P.; Thérond, P.; Guibourdenche, J.; Anderson, W.B.; Evain-Brion, D.; Raynaud, F. Increased oxidative damage to fibroblasts in skin with and without lesions in psoriasis. J. Investig. Dermatol. 2000, 114, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Er-raki, A.; Charveron, M.; Bonafé, J.L. Increased superoxide anion production in dermal fibroblasts of psoriatic patients. Skin Pharmacol. 1993, 6, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Debets, R.; Hegmans, J.P.; Buurman, W.A.; Benner, R.; Prens, E.P. Expression of cytokines and their receptors by psoriatic fibroblasts. II. Decreased TNF receptor expression. Cytokine 1996, 8, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Sanso, S.; Higashiyama, M.; Yoshikawa, K.; Itami, S. Involvement of insulin-like growth factor-I in psoriasis as a paracrine growth factor: Dermal fibroblasts play a regulatory role in developing psoriatic lesions. Arch. Dermatol. Res. 2000, 292, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Mitsuhashi, Y.; Takagi, Y.; Hashimoto, I. Psoriatic fibroblasts enhance cornified envelope formation in normal keratinocytes in vitro. Arch. Dermatol. Res. 1997, 289, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, I.A.; Richardson, P.S.; Rees, R.C.; Rennie, I.G. Effect of TNF, IL-1, and IL-6 on proliferation of human Tenon’s capsule fibroblasts in tissue culture. Br. J. Ophthalmol. 1995, 79, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Briganti, S.; Picardo, M. Antioxidant activity, lipid peroxidation and skin diseases: What’s new. J. Eur. Acad. Dermatol. Venereol. 2003, 17, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Kökçam, I.; Naziroğlu, M. Antioxidants and lipid peroxidation status in the blood of patients with psoriasis. Clin. Chim. Acta 1999, 289, 23–31. [Google Scholar] [CrossRef]
- Barygina, V.; Becatti, M.; Lotti, T.; Taddei, N.; Fiorillo, C. Low dose cytokines reduce oxidative stress in primary lesional fibroblasts obtained from psoriatic patients. J. Dermatol. Sci. 2016, 83, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, R.R.; Kirshenbaum, L.A.; Imai, S.; Vatner, S.F.; Sadoshima, J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 2004, 95, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Bhimavarapu, A.; Mammone, T.; Maes, D.; Elliston, K.; Reich, C.; Matsui, M.S.; Guarente, L.; Loureiro, J.J. SIRT1 promotes differentiation of normal human keratinocytes. J. Investig. Dermatol. 2009, 129, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Uchi, H.; Morino-Koga, S.; Shi, W.; Furue, M. Resveratrol inhibition of human keratinocyte proliferation via SIRT1/ARNT/ERK dependent downregulation of aquaporin 3. J. Dermatol. Sci. 2014, 75, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, H.; El-Komy, M.; Hegazy, R.A.; Gawdat, H.I.; AlOrbani, A.M.; Shaker, O.G. Expression of sirtuins 1, 6, tumor necrosis factor, and interferon-γ in psoriatic patients. Int. J. Immunopathol. Pharmacol. 2016, 29, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Suárez-Fariñas, M.; Cueto, I.; Khacherian, A.; Matheson, R.; Parish, L.C.; Leonardi, C.; Shortino, D.; Gupta, A.; Haddad, J.; et al. A Randomized, Placebo-Controlled Study of SRT2104, a SIRT1 Activator, in Patients with Moderate to Severe Psoriasis. PLoS ONE 2015, 10, e0142081. [Google Scholar] [CrossRef] [PubMed]
- Barygina, V.V.; Becatti, M.; Soldi, G.; Prignano, F.; Lotti, T.; Nassi, P.; Wright, D.; Taddei, N.; Fiorillo, C. Altered redox status in the blood of psoriatic patients: Involvement of NADPH oxidase and role of anti-TNF-α therapy. Redox Rep. 2013, 18, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Khader, A.; Yang, W.L.; Kuncewitch, M.; Jacob, A.; Prince, J.M.; Asirvatham, J.R.; Nicastro, J.; Coppa, G.F.; Wang, P. Sirtuin 1 activation stimulates mitochondrial biogenesis and attenuates renal injury after ischemia-reperfusion. Transplantation 2014, 98, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Chen, Z.; Xu, S.; Zhang, Q.; Wang, X.; Gao, Y.; Zhao, K.S. Polydatin Protecting Kidneys against Hemorrhagic Shock-Induced Mitochondrial Dysfunction via SIRT1 Activation and p53 Deacetylation. Oxid. Med. Cell. Longev. 2016, 2016, 1737185. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Tertoolen, L.G.; de Vries-Smits, L.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1. J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Molkentin, J.D.; Paik, J.H.; DePinho, R.A.; Yutzey, K.E. FoxO transcription factors promote cardiomyce survival upon induction of oxidative stress. J. Biol. Chem. 2011, 286, 7468–7478. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Salazar, G.; Patrushev, N.; Alexander, R.W. FoxO1 mediates an autofeedback loop regulating SIRT1 expression. J. Biol. Chem. 2011, 286, 5289–5299. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sadoshima, J. Protection of the heart against ischemia/reperfusion by silent information regulator 1. Trends Cardiovasc. Med. 2011, 21, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Choi, Y.J.; Park, M.H.; Jang, E.J.; Kim, D.H.; Park, B.H.; Yu, B.P.; Chung, H.Y. Molecular Insights into SIRT1 Protection Against UVB-Induced Skin Fibroblast Senescence by Suppression of Oxidative Stress and p53 Acetylation. J. Gerontol. A Biol. Sci Med. Sci. 2015, 70, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Arasa, J.; Terencio, M.C.; Andrés, R.M.; Valcuende-Cavero, F.; Montesinos, M.C. Decreased SAPK/JNK signalling affects cytokine release and STAT3 activation in psoriatic fibroblasts. Exp. Dermatol. 2015, 24, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Soegaard-Madsen, L.; Johansen, C.; Iversen, L.; Kragballe, K. Adalimumab therapy rapidly inhibits p38 mitogen-activated protein kinase activity in lesional psoriatic skin preceding clinical improvement. Br. J. Dermatol. 2010, 162, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Elion, E.A. MAP kinase pathways. J. Cell Sci. 2005, 118, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gan, Q.; Han, L.; Li, J.; Zhang, H.; Sun, Y.; Zhang, Z.; Tong, T. SIRT1 Overexpression Antagonizes Cellular senescence with Activated ERK/S6k1 Signaling in Human Diploid Fibroblasts. PLoS ONE 2008, 3, e1710. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ohtsuki, M.; Murakami, T.; Kobayashi, T.; Sutheesophon, K.; Kitayama, H.; Kano, Y.; Kusano, E.; Nakagawa, H.; Furukawa, Y. Histone deacetylase inhibitor FK228 suppresses the Ras-MAP kinase signaling pathway by upregulating Rap1 and induces apoptosis in malignant melanoma. Oncogene 2006, 25, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Sluss, H.K.; Barrett, T.; Dérijard, B.; Davis, R.J. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol. Cell. Biol. 1994, 14, 8376–8384. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Talwar, H.S.; Lin, J.; Lin, P.; McPhillips, F.; Wang, Z.; Li, X.; Wan, Y.; Kang, S.; Voorhees, J.J. Retinoic acid inhibits induction of c-Jun protein by ultraviolet radiation that occurs subsequent to activation of mitogen-activated protein kinase pathways in human skin in vivo. J. Clin. Investig. 1998, 101, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Gazel, A.; Banno, T.; Walsh, R.; Blumenberg, M. Inhibition of JNK promotes differentiation of epidermal keratinocytes. J. Biol. Chem. 2006, 281, 20530–20541. [Google Scholar] [CrossRef] [PubMed]
- Mose, M.; Kang, Z.; Raaby, L.; Iversen, L.; Johansen, C. TNFα- and IL-17A-mediated S100A8 expression is regulated by p38 MAPK. Exp. Dermatol. 2013, 22, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.S.; Maddock, D.A.; Chen, J.P.; Heo, S.; Chiu, C.; Lai, D.; Souza, K.; Mehta, S.; Wan, Y.S. Cytokine-induced p38 activation feedback regulates the prolonged activation of AKT cell survival pathway initiated by reactive oxygen species in response to UV irradiation in human keratinocytes. Int. J. Oncol. 2001, 19, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Garmyn, M.; Mammone, T.; Pupe, A.; Gan, D.; Declercq, L.; Maes, D. Human keratinocytes respond to osmotic stress by p38 map kinase regulated induction of HSP70 and HSP27. J. Investig. Dermatol. 2001, 117, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Baliwag, J.; Barnes, D.H.; Johnston, A. Cytokines in psoriasis. Cytokine 2015, 73, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Escande, C.; Denicola, A. Potential Modulation of Sirtuins by Oxidative Stress. Oxid. Med. Cell. Longev. 2016, 2016, 9831825. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Fiorillo, C.; Becatti, M.; Pensalfini, A.; Cecchi, C.; Lanzilao, L.; Donzelli, G.; Nassi, N.; Giannini, L.; Borchi, E.; Nassi, P. Curcumin protects cardiac cells against ischemia-reperfusion injury: Effects on oxidative stress, NF-kappaB, and JNK pathways. Free Radic. Biol. Med. 2008, 45, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, C.; Becatti, M.; Attanasio, M.; Lucarini, L.; Nassi, N.; Evangelisti, L.; Porciani, M.C.; Nassi, P.; Gensini, G.F.; Abbate, R.; et al. Evidence for oxidative stress in plasma of patients with Marfan syndrome. Int. J. Cardiol. 2010, 145, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Drummen, G.P.; Gadella, B.M.; Post, J.A.; Brouwers, J.F. Mass spectrometric characterization of the oxidation of the fluorescent lipid peroxidation reporter molecule C11-BODIPY(581/591). Free Radic. Biol. Med. 2004, 36, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Pini, A.; Boccalini, G.; Baccari, M.C.; Becatti, M.; Garella, R.; Fiorillo, C.; Calosi, L.; Bani, D.; Nistri, S. Protection from cigarette smoke-induced vascular injury by recombinant human relaxin-2 (serelaxin). J. Cell. Mol. Med. 2016, 20, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Boccalini, G.; Pini, A.; Fiorillo, C.; Bencini, A.; Bani, D.; Nistri, S. Protection of coronary endothelial cells from cigarette smoke-induced oxidative stress by a new Mn(II)-containing polyamine-polycarboxilate scavenger of superoxide anion. Vascul. Pharmacol. 2015, 75, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Prignano, F.; Fiorillo, C.; Pescitelli, L.; Nassi, P.; Lotti, T.; Taddei, N. The involvement of Smac/DIABLO, p53, NF-kB, and MAPK pathways in apoptosis of keratinocytes from perilesional vitiligo skin: Protective effects of curcumin and capsaicin. Antioxid. Redox Signal. 2010, 13, 1309–1321. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Body Mass Index (BMI) | Duration of Disease (Years) | PASI |
|---|---|---|---|---|
| M1 | 33 | 25 | 8 | 13 |
| M2 | 41 | 24 | 12 | 13 |
| M3 | 29 | 25 | 10 | 12 |
| M4 | 52 | 23 | 21 | 12 |
| M5 | 48 | 26 | 11 | 12 |
| M6 | 45 | 25 | 10 | 13 |
| F1 | 51 | 21 | 25 | 13 |
| F2 | 45 | 23 | 20 | 13 |
| F3 | 38 | 26 | 18 | 12 |
| F4 | 32 | 24 | 12 | 12 |
| F5 | 37 | 25 | 9 | 12 |
| F6 | 46 | 26 | 13 | 12 |
| Mean ± SD | 41.4 ± 7.6 | 24.4 ± 1.5 | 14.1 ± 5.5 | 12.4 ± 0.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becatti, M.; Barygina, V.; Mannucci, A.; Emmi, G.; Prisco, D.; Lotti, T.; Fiorillo, C.; Taddei, N. Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis. Int. J. Mol. Sci. 2018, 19, 1572. https://doi.org/10.3390/ijms19061572
Becatti M, Barygina V, Mannucci A, Emmi G, Prisco D, Lotti T, Fiorillo C, Taddei N. Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis. International Journal of Molecular Sciences. 2018; 19(6):1572. https://doi.org/10.3390/ijms19061572
Chicago/Turabian StyleBecatti, Matteo, Victoria Barygina, Amanda Mannucci, Giacomo Emmi, Domenico Prisco, Torello Lotti, Claudia Fiorillo, and Niccolò Taddei. 2018. "Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis" International Journal of Molecular Sciences 19, no. 6: 1572. https://doi.org/10.3390/ijms19061572
APA StyleBecatti, M., Barygina, V., Mannucci, A., Emmi, G., Prisco, D., Lotti, T., Fiorillo, C., & Taddei, N. (2018). Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis. International Journal of Molecular Sciences, 19(6), 1572. https://doi.org/10.3390/ijms19061572