
A Precipitation-Based Process to Generate a Solid Formulation of a Therapeutic Monoclonal Antibody: An Alternative to Lyophilization

 , , , , ,
, , , , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Monoclonal Antibody
2.2. Precipitation Process
2.3. Analytical Characterization
2.3.1. Critical Quality Attributes
2.3.2. Biological Characterization
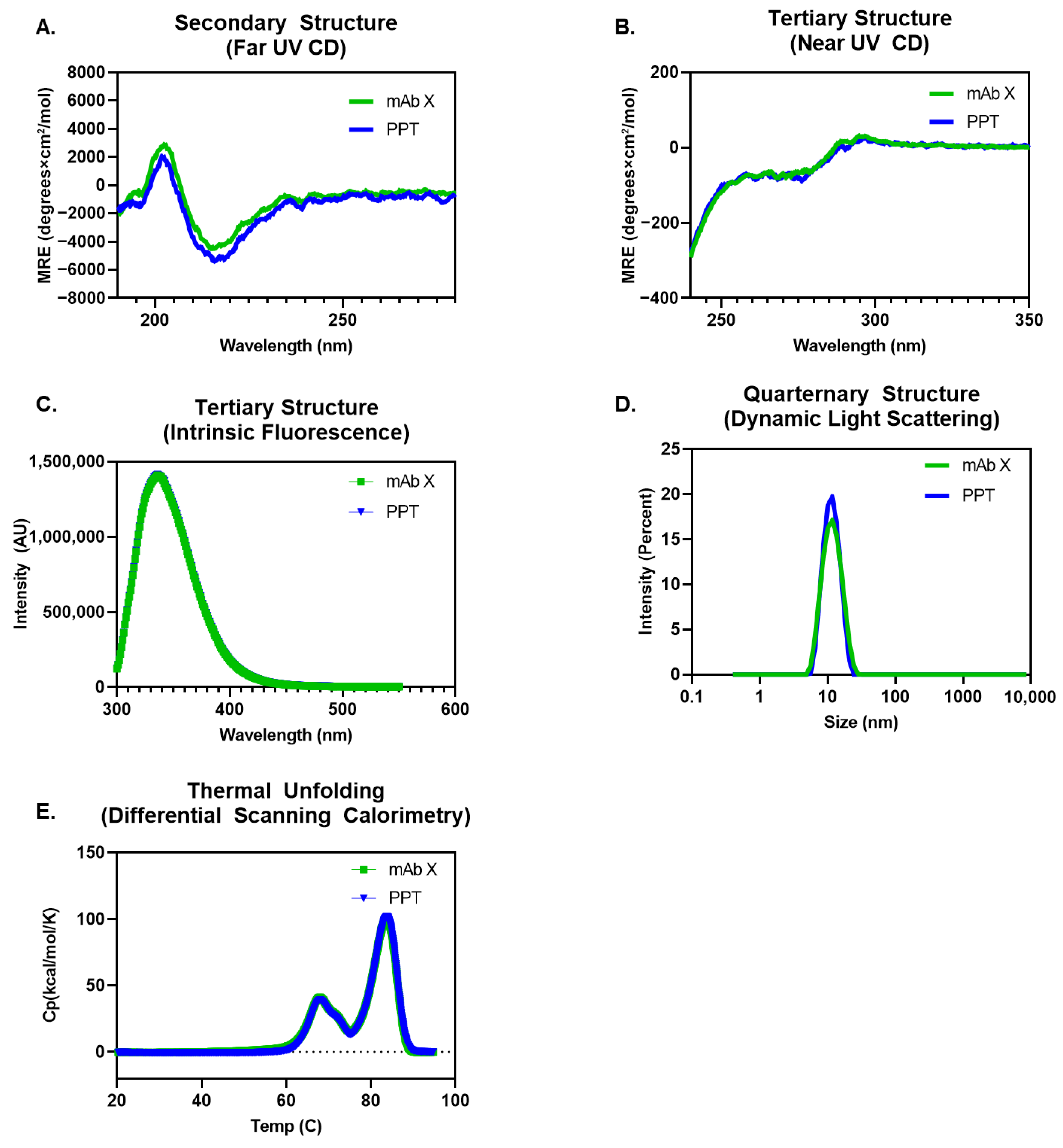
2.3.3. Biophysical Characterization
3. Results and Discussion
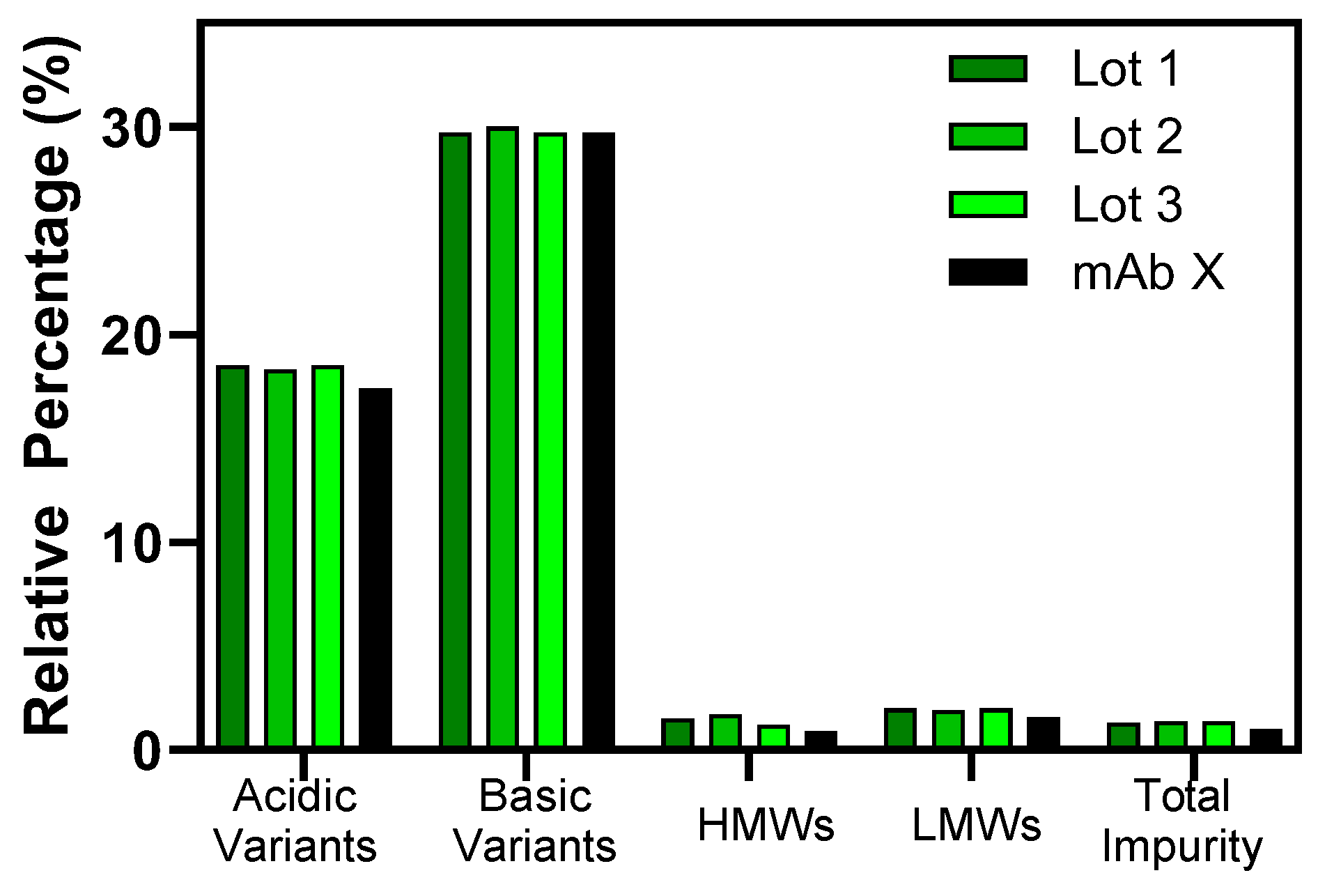
3.1. Process Reproducibility and Formulation Characterization
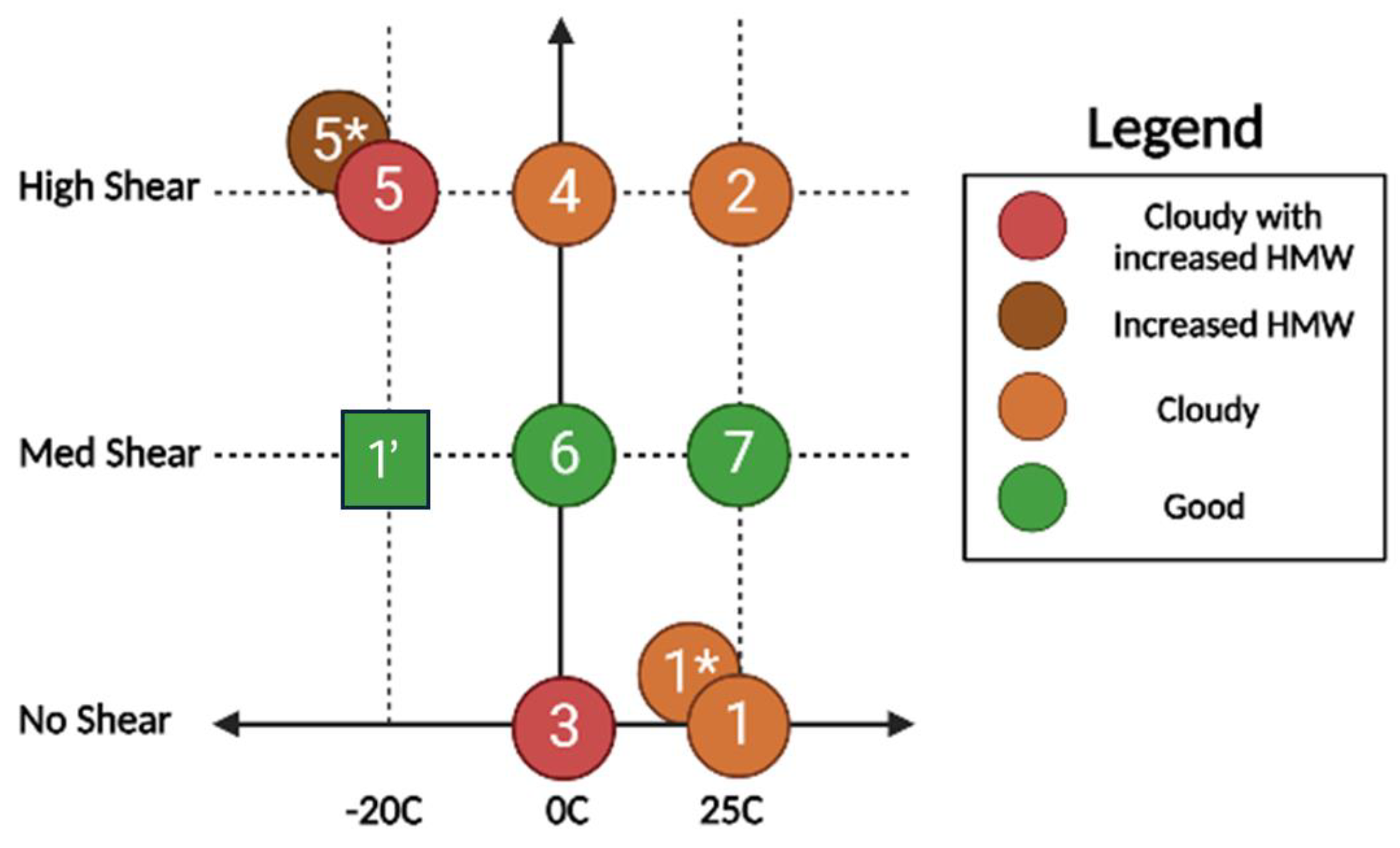
3.2. Process Characterization
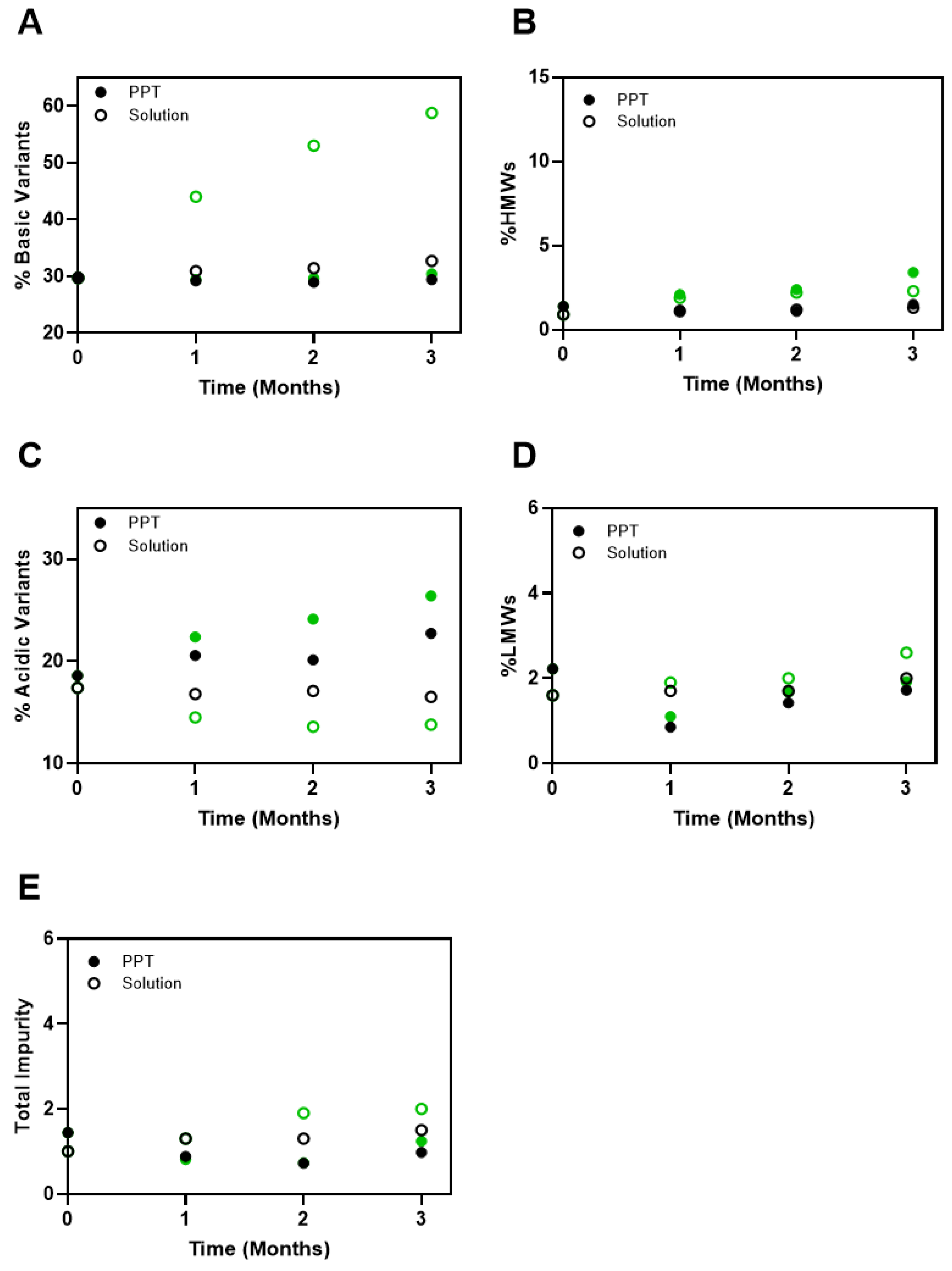
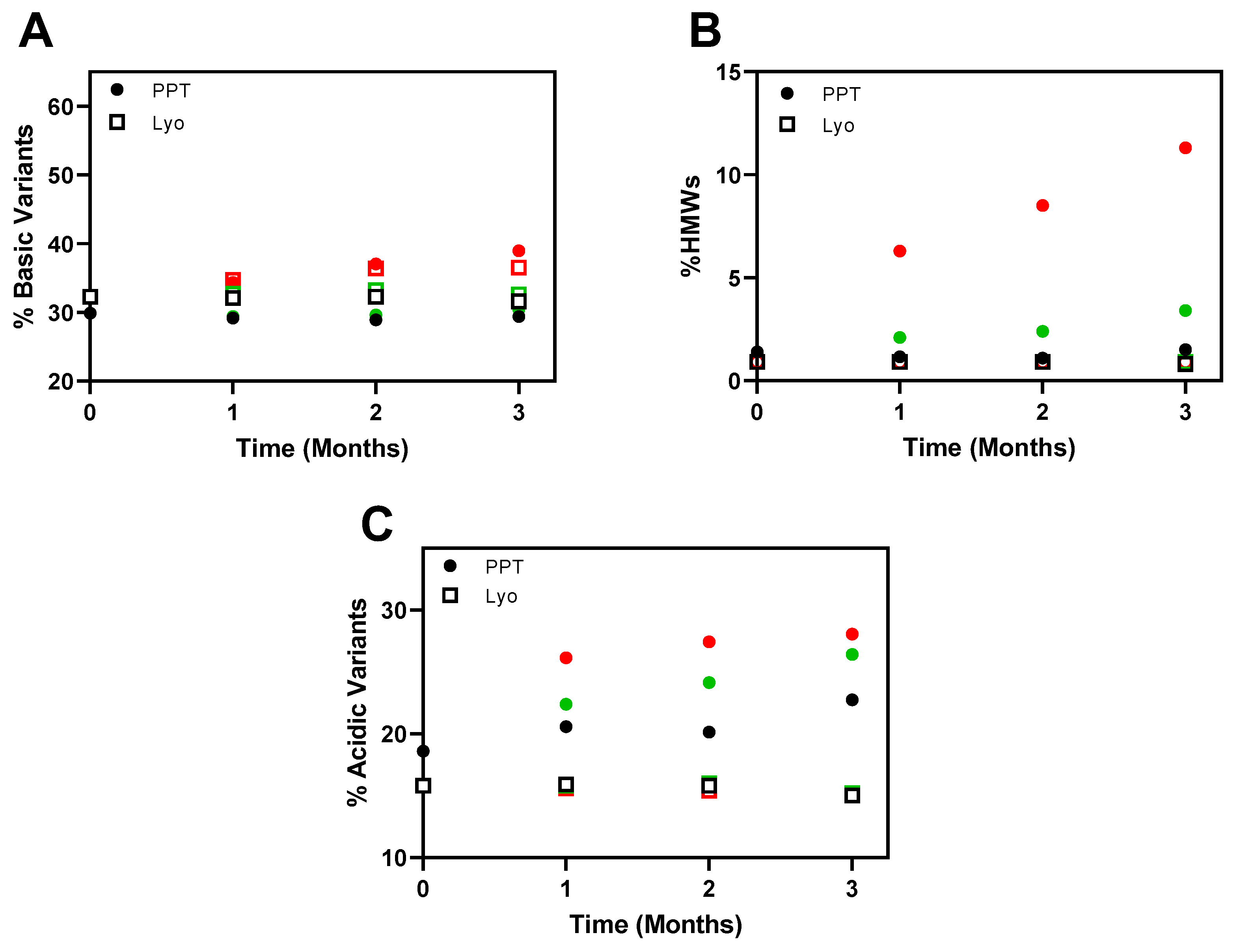
3.3. Stability of the Precipitated Formulations
4. Conclusions and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senior, M. Fresh from the biotech pipeline: Fewer approvals, but biologics gain share. Nat. Biotechnol. 2023, 41, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mutukuri, T.T.; Wilson, N.E.; Zhou, Q. Pharmaceutical protein solids: Drying technology, solid-state characterization and stability. Adv. Drug Deliv. Rev. 2021, 172, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Khamar, D.; Cullen, S.; Hayden, A.; Hughes, H. Innovative Drying Technologies for Biopharmaceuticals. Int. J. Pharm. 2021, 609, 121115. [Google Scholar] [CrossRef] [PubMed]
- Stratta, L.; Capozzi, L.C.; Franzino, S.; Pisano, R. Economic Analysis of a Freeze-Drying Cycle. Processes 2020, 8, 1399. [Google Scholar] [CrossRef]
- Heller, M.C.; Carpenter, J.F.; Randolph, T.W. Protein formulation and lyophilization cycle design: Prevention of damage due to freeze-concentration induced phase separation. Biotechnol. Bioeng. 1999, 63, 166–174. [Google Scholar] [CrossRef]
- Kasper, J.C.; Friess, W. The freezing step in lyophilization: Physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. Eur. J. Pharm. Biopharm. 2011, 78, 248–263. [Google Scholar] [CrossRef]
- Patel, S.M.; Nail, S.L.; Pikal, M.J.; Geidobler, R.; Winter, G.; Hawe, A.; Davagnino, J.; Rambhatla Gupta, S. Lyophilized Drug Product Cake Appearance: What Is Acceptable? J. Pharm. Sci. 2017, 106, 1706–1721. [Google Scholar] [CrossRef]
- Jameel, F.; Alexeenko, A.; Bhambhani, A.; Sacha, G.; Zhu, T.; Tchessalov, S.; Sharma, P.; Moussa, E.; Iyer, L.; Luthra, S.; et al. Recommended Best Practices for Lyophilization Validation 2021 Part II: Process Qualification and Continued Process Verification. AAPS PharmSciTech 2021, 22, 266. [Google Scholar] [CrossRef]
- Searles, J.; Ohtake, S. Strategies for Implementing New Drying & Packaging Technology for Sterile Injectable Drug Products. J. Pharm. Sci. 2021, 110, 1931–1934. [Google Scholar]
- Ohtake, S.; Izutsu, K.; Lechuga-Ballesteros, D. Drying Technologies for Biotechnology and Pharmaceutical Applications; Wiley: New York, NY, USA, 2020. [Google Scholar]
- Barcelo-Chong, C.M.; Filipe, V.; Nakach, M.; Inês Ré, M. How spray drying processing and solution composition can affect the mAbs stability in reconstituted solutions for subcutaneous injections. Part II: Exploring each protein stabilizer effect. Int. J. Pharm. 2024, 655, 124014. [Google Scholar] [CrossRef]
- Pinto, J.T.; Faulhammer, E.; Dieplinger, J.; Dekner, M.; Makert, C.; Nieder, M.; Paudel, A. Progress in spray-drying of protein pharmaceuticals: Literature analysis of trends in formulation and process attributes. Dry. Technol. 2021, 39, 1415–1446. [Google Scholar] [CrossRef]
- Tao, Y.; Chen, Y.; Howard, W.; Ibrahim, M.; Patel, S.M.; McMahon, W.P.; Kim, Y.J.; Delmar, J.A.; Davis, D. Mechanism of Insoluble Aggregate Formation in a Reconstituted Solution of Spray-Dried Protein Powder. Pharm. Res. 2023, 40, 2355–2370. [Google Scholar] [CrossRef] [PubMed]
- Anandharamakrishnan, C.; Rielly, C.D.; Stapley, A.G.F. Loss of solubility of α-lactalbumin and β-lactoglobulin during the spray drying of whey proteins. LWT—Food Sci. Technol. 2008, 41, 270–277. [Google Scholar] [CrossRef]
- Forbes, R.T.; Barry, B.W.; Elkordy, A.A. Preparation and characterisation of spray-dried and crystallised trypsin: FT-Raman study to detect protein denaturation after thermal stress. Eur. J. Pharm. Sci. 2007, 30, 315–323. [Google Scholar] [CrossRef]
- Poozesh, S.; Mezhericher, M.; Pan, Z.; Chaudhary, U.; Manikwar, P.; Stone, H.A. Rapid Room-Temperature Aerosol Dehydration Versus Spray Drying: A Novel Paradigm in Biopharmaceutical Drying Technologies. J. Pharm. Sci. 2024, 113, 974–981. [Google Scholar] [CrossRef]
- Adler, M.; Unger, M.; Lee, G. Surface Composition of Spray-Dried Particles of Bovine Serum Albumin/Trehalose/Surfactant. Pharm. Res. 2000, 17, 863–870. [Google Scholar] [CrossRef]
- Hickey, A.J.; da Rocha, S.R. Pharmaceutical Inhalation Aerosol Technology; Hickey, A.J., da Rocha, S.R., Eds.; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Abdul-Fattah, A.M.; Kalonia, D.S.; Pikal, M.J. The Challenge of Drying Method Selection for Protein Pharmaceuticals: Product Quality Implications. J. Pharm. Sci. 2007, 96, 1886–1916. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Chow, M.Y.T.; Khanal, D.; Chen, D.; Chan, H.-K. Dry powder pharmaceutical biologics for inhalation therapy. Adv. Drug Deliv. Rev. 2021, 172, 64–79. [Google Scholar] [CrossRef]
- Pan, H.W.; Seow, H.C.; Lo, J.C.K.; Guo, J.; Zhu, L.; Leung, S.W.S.; Zhang, C.; Lam, J.K.W. Spray-Dried and Spray-Freeze-Dried Powder Formulations of an Anti-Interleukin-4Rα Antibody for Pulmonary Delivery. Pharm. Res. 2022, 39, 2291–2304. [Google Scholar] [CrossRef]
- Bhambhani, A.; Stanbro, J.; Roth, D.; Sullivan, E.; Jones, M.; Evans, R.; Blue, J. Evaluation of Microwave Vacuum Drying as an Alternative to Freeze-Drying of Biologics and Vaccines: The Power of Simple Modeling to Identify a Mechanism for Faster Drying Times Achieved with Microwave. AAPS PharmSciTech 2021, 22, 52. [Google Scholar] [CrossRef]
- Ambros, S.; Dombrowski, J.; Boettger, D.; Kulozik, U. The Concept of Microwave Foam Drying Under Vacuum: A Gentle Preservation Method for Sensitive Biological Material. J. Food Sci. 2019, 84, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Lovalenti, P.M.; Anderl, J.; Yee, L.; Nguyen, V.; Ghavami, B.; Ohtake, S.; Saxena, A.; Voss, T.; Truong-Le, V. Stabilization of Live Attenuated Influenza Vaccines by Freeze Drying, Spray Drying, and Foam Drying. Pharm. Res. 2016, 33, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Piettre, M.; Vila, A. Sur la separation des proteines du serum. Comptes Rendus Hebd. Seances Acad. Sci 1920, 170, 1466–1468. [Google Scholar]
- Barritault, D.; Expert-Bezançon, A.; Guérin, M.-F.; Hayes, D. The Use of Acetone Precipitation in the Isolation of Ribosomal Proteins. Eur. J. Biochem. 1976, 63, 131–135. [Google Scholar] [CrossRef]
- Crowell, A.M.J.; Wall, M.J.; Doucette, A.A. Maximizing recovery of water-soluble proteins through acetone precipitation. Anal. Chim. Acta 2013, 796, 48–54. [Google Scholar] [CrossRef]
- Jiang, L.; He, L.; Fountoulakis, M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. J. Chromatogr. A 2004, 1023, 317–320. [Google Scholar] [CrossRef]
- Aniket; Gaul, D.A.; Rickard, D.L.; Needham, D. MicroglassificationTM: A Novel Technique for Protein Dehydration. J. Pharm. Sci. 2014, 103, 810–820. [Google Scholar] [CrossRef]
- Aniket; Gaul, D.A.; Bitterfield, D.L.; Su, J.T.; Li, V.M.; Singh, I.; Morton, J.; Needham, D. Enzyme Dehydration Using Microglassification™ Preserves the Protein’s Structure and Function. J. Pharm. Sci. 2015, 104, 640–651. [Google Scholar] [CrossRef]
- Chandrababu, K.B.; Kannan, A.; Savage, J.R.; Stadmiller, S.; Ryle, A.E.; Cheung, C.; Kelley, R.F.; Maa, Y.-f.; Saggu, M.; Bitterfield, D.L. Stability Comparison Between Microglassification and Lyophilization Using a Monoclonal Antibody. J. Pharm. Sci. 2023, 113, 1054–1060. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Hirano, A.; Arakawa, T.; Shiraki, K. Mechanistic insights into protein precipitation by alcohol. Int. J. Biol. Macromol. 2012, 50, 865–871. [Google Scholar] [CrossRef]
- Cohn, E.J.; Strong, L.E.; Hughes, W.L.J.; Mulford, D.J.; Ashworth, J.N.; Melin, M.E.; Taylor, H.L. Preparation and properties of serum and plasma proteins; a system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. J. Am. Chem. Soc. 1946, 68, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, B.; Pleitt, K.; Shave, E.; Baker, K.; Lua, L.H.L. Progression of continuous downstream processing of monoclonal antibodies: Current trends and challenges. Biotechnol. Bioeng. 2018, 115, 2893–2907. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, T.-H.; Andini, E.; Coffman, J.L.; Przybycien, T.; Zydney, A.L. Enhanced filtration performance using feed-and-bleed configuration for purification of antibody precipitates. Biotechnol. Prog. 2021, 37, e3082. [Google Scholar] [CrossRef] [PubMed]
- Sommer, R.; Satzer, P.; Tscheliessnig, A.; Schulz, H.; Helk, B.; Jungbauer, A. Combined polyethylene glycol and CaCl2 precipitation for the capture and purification of recombinant antibodies. Process Biochem. 2014, 49, 2001–2009. [Google Scholar] [CrossRef]
- Li, Z.; Zydney, A.L. Effect of zinc chloride and PEG concentrations on the critical flux during tangential flow microfiltration of BSA precipitates. Biotechnol. Prog. 2017, 33, 1561–1567. [Google Scholar] [CrossRef]
- Mullerpatan, A.; Chandra, D.; Kane, E.; Karande, P.; Cramer, S. Purification of proteins using peptide-ELP based affinity precipitation. J. Biotechnol. 2020, 309, 59–67. [Google Scholar] [CrossRef]
- Swartz, A.R.; Xu, X.; Traylor, S.J.; Li, Z.J.; Chen, W. High-efficiency affinity precipitation of multiple industrial mAbs and Fc-fusion proteins from cell culture harvests using Z-ELP-E2 nanocages. Biotechnol. Bioeng. 2018, 115, 2039–2047. [Google Scholar] [CrossRef]
- Kateja, N.; Agarwal, H.; Saraswat, A.; Bhat, M.; Rathore, A.S. Continuous precipitation of process related impurities from clarified cell culture supernatant using a novel coiled flow inversion reactor (CFIR). Biotechnol. J. 2016, 11, 1320–1331. [Google Scholar] [CrossRef]
- Sommer, R.; Tscheliessnig, A.; Satzer, P.; Schulz, H.; Helk, B.; Jungbauer, A. Capture and intermediate purification of recombinant antibodies with combined precipitation methods. Biochem. Eng. J. 2015, 93, 200–211. [Google Scholar] [CrossRef]
- Brodsky, Y.; Zhang, C.; Yigzaw, Y.; Vedantham, G. Caprylic acid precipitation method for impurity reduction: An alternative to conventional chromatography for monoclonal antibody purification. Biotechnol. Bioeng. 2012, 109, 2589–2598. [Google Scholar] [CrossRef]
- Ma, J.; Hoang, H.; Myint, T.; Peram, T.; Fahrner, R.; Chou, J.H. Using precipitation by polyamines as an alternative to chromatographic separation in antibody purification processes. J. Chromatogr. B 2010, 878, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Hamzik, J.; Felo, M.; Qi, B.; Lee, J.; Ng, S.; Liebisch, G.; Shanehsaz, B.; Singh, N.; Persaud, K.; et al. Development of a novel and efficient cell culture flocculation process using a stimulus responsive polymer to streamline antibody purification processes. Biotechnol. Bioeng. 2013, 110, 2928–2937. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Arunkumar, A.; Chollangi, S.; Tan, Z.G.; Borys, M.; Li, Z.J. Clarification technologies for monoclonal antibody manufacturing processes: Current state and future perspectives. Biotechnol. Bioeng. 2016, 113, 698–716. [Google Scholar] [CrossRef] [PubMed]
- McNerney, T.; Thomas, A.; Senczuk, A.; Petty, K.; Zhao, X.; Piper, R.; Carvalho, J.; Hammond, M.; Sawant, S.; Bussiere, J. PDADMAC flocculation of Chinese hamster ovary cells: Enabling a centrifuge-less harvest process for monoclonal antibodies. mAbs 2015, 7, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, J.; Martinez-Fonts, K.; Rauscher, M.; Rivera, S.; Welsh, J.; Kandula, S. Cationic polymer precipitation for enhanced impurity removal in downstream processing. Biotechnol. Bioeng. 2023, 120, 1902–1913. [Google Scholar] [CrossRef]
- Chang, L.; Pikal, M.J. Mechanisms of protein stabilization in the solid state. J. Pharm. Sci. 2009, 98, 2886–2908. [Google Scholar] [CrossRef]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L.J. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Crowe, J.H. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry 1989, 28, 3916–3922. [Google Scholar] [CrossRef]
- Prestrelski, S.J.; Tedeschi, N.; Arakawa, T.; Carpenter, J.F. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys. J. 1993, 65, 661–671. [Google Scholar] [CrossRef]
- Arsiccio, A.; Pisano, R. Water entrapment and structure ordering as protection mechanisms for protein structural preservation. J. Chem. Phys. 2018, 148, 055102. [Google Scholar] [CrossRef]
- VanAernum, Z.L.; Sergi, J.A.; Dey, M.; Toner, T.; Kilgore, B.; Lay-Fortenbery, A.; Wang, Y.; Bian, S.; Kochert, B.A.; Bothe, J.R.; et al. Discovery and Control of Succinimide Formation and Accumulation at Aspartic Acid Residues in The Complementarity-Determining Region of a Therapeutic Monoclonal Antibody. Pharm. Res. 2023, 40, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Lamberto, D.J.; Diaz-Santana, A.; Zhou, G. Form Conversion and Solvent Entrapment during API Drying. Org. Process Res. Dev. 2017, 21, 1828–1834. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | Solvent Temperature | Mixing |
|---|---|---|
| 1′ | −20 °C | Medium |
| 1 | 25 °C | None |
| 1 * | 25 °C | None |
| 2 | 25 °C | High |
| 3 | 0 °C | None |
| 4 | 0 °C | High |
| 5 | −20 °C | High |
| 5 * | −20 °C | High |
| 6 | 0 °C | Medium |
| 7 | 25 °C | Medium |
| Assay | Sample Number | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mAb X | 1′ | 1 | 1 * | 2 | 3 | 4 | 5 | 5 * | 6 | 7 | ||
| UP-SEC | Monomer % | 99.1 | 98.6 | 98.2 | 99.0 | 99.6 | 96.8 | 98.9 | 97.9 | 94.2 | 99.3 | 99.4 |
| HMW species % | 0.9 | 1.4 | 1.9 | 1.0 | 0.4 | 3.2 | 1.1 | 2.1 | 5.8 | 0.7 | 0.6 | |
| HP-IEX | Acidic variants % | 15.8 | 18.6 | 16.5 | 17.5 | 18.1 | 17.6 | 18.1 | 18.3 | 16.9 | 18.0 | 17.9 |
| Total main % | 51.9 | 51.5 | 53.6 | 52.7 | 52.7 | 51.3 | 52.0 | 51.3 | 51.8 | 52.5 | 52.6 | |
| Basic variants % | 32.3 | 29.9 | 29.9 | 29.8 | 29.3 | 31.1 | 29.9 | 30.4 | 31.3 | 29.6 | 29.5 | |
| Reduced CE-SDS | HC + LC % | 97.9 | 98.6 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.2 | 99.3 | 99.3 |
| Total impurity % | 2.1 | 1.4 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.8 | 0.7 | 0.7 | |
| Non-reduced CE-SDS | Intact IgG % | 98.6 | 97.7 | 98.3 | 98.3 | 98.0 | 98.3 | 98.2 | 98.3 | 97.9 | 98.2 | 98.4 |
| Total LMW species % | 1.4 | 2.2 | 1.6 | 1.6 | 1.9 | 1.5 | 1.7 | 1.5 | 1.9 | 1.8 | 1.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koynov, A.A.; Lin, W.; Bothe, J.R.; Schenck, L.; Parajuli, B.; Li, Z.; Ruzanski, R.; Hoffman, N.; Frank, D.; VanAernum, Z. A Precipitation-Based Process to Generate a Solid Formulation of a Therapeutic Monoclonal Antibody: An Alternative to Lyophilization. J. Pharm. BioTech Ind. 2025, 2, 2. https://doi.org/10.3390/jpbi2010002
Koynov AA, Lin W, Bothe JR, Schenck L, Parajuli B, Li Z, Ruzanski R, Hoffman N, Frank D, VanAernum Z. A Precipitation-Based Process to Generate a Solid Formulation of a Therapeutic Monoclonal Antibody: An Alternative to Lyophilization. Journal of Pharmaceutical and BioTech Industry. 2025; 2(1):2. https://doi.org/10.3390/jpbi2010002
Chicago/Turabian StyleKoynov, Athanas A., Wei Lin, Jameson R. Bothe, Luke Schenck, Bibek Parajuli, Zhao Li, Richard Ruzanski, Natalie Hoffman, Derek Frank, and Zachary VanAernum. 2025. "A Precipitation-Based Process to Generate a Solid Formulation of a Therapeutic Monoclonal Antibody: An Alternative to Lyophilization" Journal of Pharmaceutical and BioTech Industry 2, no. 1: 2. https://doi.org/10.3390/jpbi2010002
APA StyleKoynov, A. A., Lin, W., Bothe, J. R., Schenck, L., Parajuli, B., Li, Z., Ruzanski, R., Hoffman, N., Frank, D., & VanAernum, Z. (2025). A Precipitation-Based Process to Generate a Solid Formulation of a Therapeutic Monoclonal Antibody: An Alternative to Lyophilization. Journal of Pharmaceutical and BioTech Industry, 2(1), 2. https://doi.org/10.3390/jpbi2010002