
Urate Biology and Biochemistry: A Year in Review 2022


{kind=link}
{kind=link}
Abstract
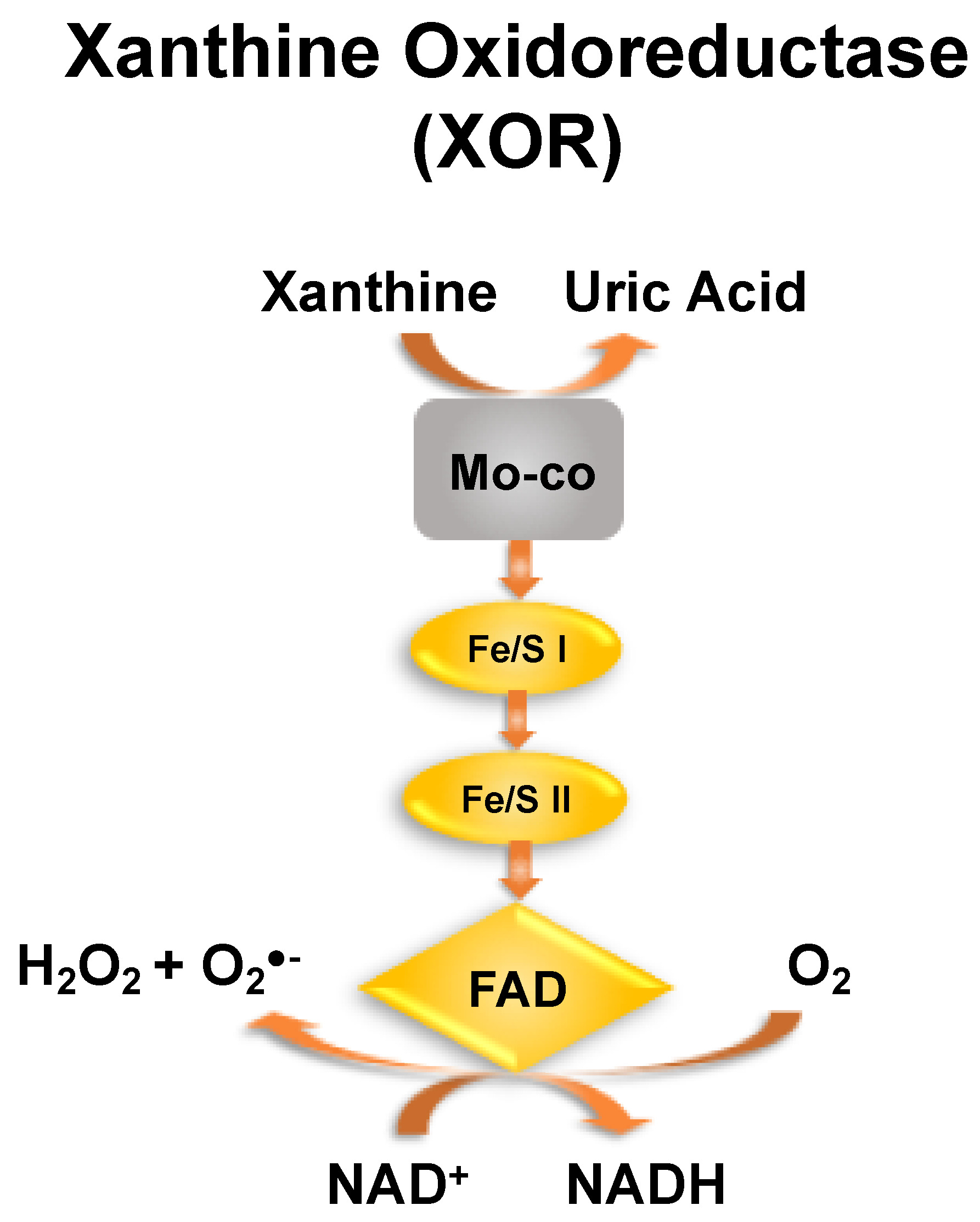
1. Introduction
2. UA and Gout
3. UA and Metabolic Dysfunction
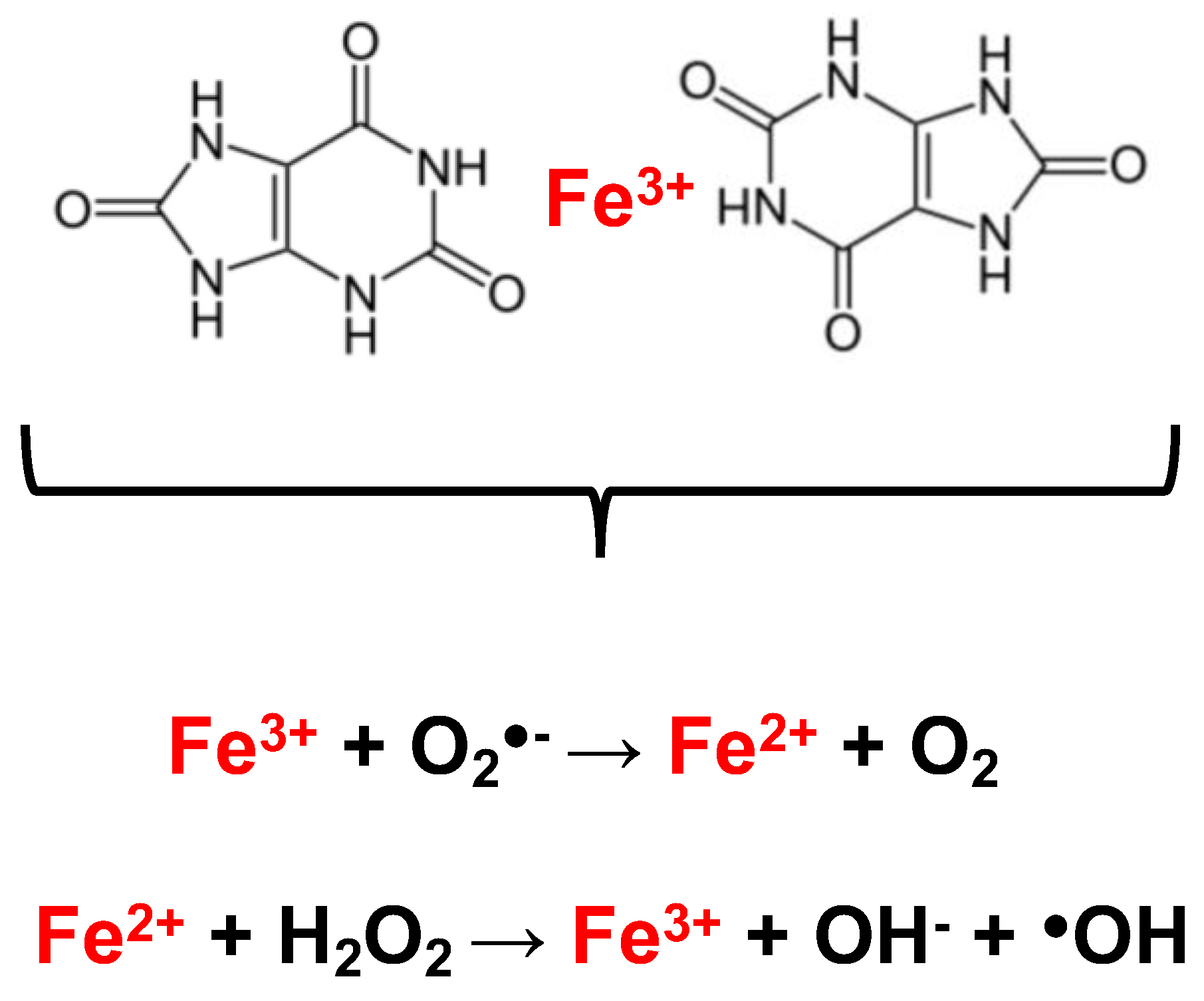
4. UA and Hemolysis
5. UA and COVID-19
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Harmon, D.B.; Mandler, W.K.; Sipula, I.J.; Dedousis, N.; Lewis, S.E.; Eckels, J.T.; Du, J.; Wang, Y.; Huckestein, B.R.; Pagano, P.J.; et al. Hepatocyte-Specific Ablation or Whole-Body Inhibition of Xanthine Oxidoreductase in Mice Corrects Obesity-Induced Systemic Hyperuricemia Without Improving Metabolic Abnormalities. Diabetes 2019, 68, 1221–1229. [Google Scholar] [CrossRef]
- Jin, M.; Yang, F.; Yang, I.; Yin, Y.; Luo, J.J.; Wang, H.; Yang, X.F. Uric acid, hyperuricemia and vascular diseases. Front. Biosci. 2012, 17, 656–669. [Google Scholar] [CrossRef]
- Fatima, T.; McKinney, C.; Major, T.J.; Stamp, L.K.; Dalbeth, N.; Iverson, C.; Merriman, T.R.; Miner, J.N. The relationship between ferritin and urate levels and risk of gout. Arthritis Res. Ther. 2018, 20, 179. [Google Scholar] [CrossRef]
- Ghio, A.J.; Kennedy, T.P.; Stonehuerner, J.; Carter, J.D.; Skinner, K.A.; Parks, D.A.; Hoidal, J.R. Iron regulates xanthine oxidase activity in the lung. Am. J. Physiol.-Lung Cell Mol. Physiol. 2002, 283, L563–L572. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Tapia, E.; Bautista-Garcia, P.; Soto, V.; Avila-Casado, C.; Vega-Campos, I.P.; Nakagawa, T.; Zhao, L.; Franco, M.; Johnson, R.J. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2008, 294, F710–F718. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Lewis, S.E.; Li, L.; Fazzari, M.; Salvatore, S.R.; Li, J.; Hileman, E.A.; Maxwell, B.A.; Schopfer, F.J.; Arteel, G.E.; Khoo, N.K.H.; et al. Obese female mice do not exhibit overt hyperuricemia despite hepatic steatosis and impaired glucose tolerance. Adv. Redox Res. 2022, 6, 100051. [Google Scholar] [CrossRef]
- Baldwin, W.; McRae, S.; Marek, G.; Wymer, D.; Pannu, V.; Baylis, C.; Johnson, R.J.; Sautin, Y.Y. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011, 60, 1258–1269. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Seno, Y.; Kushiyama, A.; Sakoda, H.; Fujishiro, M.; Katasako, A.; Mori, K.; Matsunaga, Y.; Fukushima, T.; Kanaoka, R.; et al. The xanthine oxidase inhibitor febuxostat suppresses development of nonalcoholic steatohepatitis in a rodent model. Am J. Physiol. Gastrointest Liver Physiol. 2015, 309, G42–G51. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Santos, A.M.; Junqueira, V.B.; Augusto, O. Peroxynitrite-mediated formation of free radicals in human plasma: EPR detection of ascorbyl, albumin-thiyl and uric acid-derived free radicals. Biochem. J. 1996, 314 Pt 3, 869–876. [Google Scholar] [CrossRef]
- Radi, R.; Tan, S.; Prodanov, E.; Evans, R.A.; Parks, D.A. Inhibition of xanthine oxidase by uric acid and its influence on superoxide radical production. Biochim. Biophys. Acta 1992, 1122, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Radi, R.; Gaudier, F.; Evans, R.A.; Rivera, A.; Kirk, K.A.; Parks, D.A. Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr. Res. 1993, 34, 303–307. [Google Scholar] [CrossRef]
- Chen, J.; Mei, A.; Liu, X.; Braunstein, Z.; Wei, Y.; Wang, B.; Duan, L.; Rao, X.; Rajagopalan, S.; Dong, L.; et al. Glucagon-Like Peptide-1 Receptor Regulates Macrophage Migration in Monosodium Urate-Induced Peritoneal Inflammation. Front. Immunol. 2022, 13, 772446. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lou, Y.; Mao, Y.; Wen, X.; Fan, M.; He, Z.; Shen, Y.; Wen, C.; Shao, T. Alteration of Gut Microbiome and Correlated Amino Acid Metabolism Contribute to Hyperuricemia and Th17-Driven Inflammation in Uox-KO Mice. Front. Immunol. 2022, 13, 804306. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Stanhope, K.L.; Boucher, J.; Divanovic, S.; Lanaspa, M.A.; Johnson, R.J.; Kahn, C.R. Fructose and hepatic insulin resistance. Crit. Rev. Clin. Lab. Sci. 2020, 57, 308–322. [Google Scholar] [CrossRef]
- Zhang, X.; Mass, B.B.; Talevi, V.; Hou, R.; North, K.E.; Voruganti, V.S. Novel Insights into the Effects of Genetic Variants on Serum Urate Response to an Acute Fructose Challenge: A Pilot Study. Nutrients 2022, 14, 4030. [Google Scholar] [CrossRef]
- Hoque, K.M.; Halprin-Kuhns, V.L.; Woodward, O.M. Slc2a5 (GLUT5) upregulation in hyperuricemia drives risk for fructose induced NAFLD. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Schmidt, H.; Hahn, S.; Annarapu, G.; Carreno, M.; Wood, K.; Shiva, S.; Vitturi, D.; Kelley, E.; Straub, A. Xanthine Oxidase has a Protective Role During Heme Crisis by Binding Heme and Facilitating Degradation. Free. Radic. Biol. Med. 2022, 180, s53. [Google Scholar] [CrossRef]
- Davies, K.J.; Sevanian, A.; Muakkassah-Kelly, S.F.; Hochstein, P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem. J. 1986, 235, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.J.; Lin, W.C.; Chou, Y.C.; Yang, C.M.; Wu, H.L.; Cheng, Y.H.; Liu, P.C.; Chang, J.Y.; Chen, H.Y.; Sun, J.R. Dynamic Changes of the Blood Chemistry in Syrian Hamsters Post-Acute COVID-19. Microbiol. Spectr. 2022, 10, e0236221. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Chubachi, S.; Namkoong, H.; Otake, S.; Nakagawara, K.; Tanaka, H.; Lee, H.; Morita, A.; Watase, M.; Kusumoto, T.; et al. U-shaped association between abnormal serum uric acid levels and COVID-19 severity: Reports from the Japan COVID-19 Task Force. Int. J. Infect. Dis. 2022, 122, 747–754. [Google Scholar] [CrossRef]
- Majumder, N.; Deepak, V.; Hadique, S.; Aesoph, D.; Velayutham, M.; Ye, Q.; Mazumder, M.H.H.; Lewis, S.E.; Kodali, V.; Roohollahi, A.; et al. Redox imbalance in COVID-19 pathophysiology. Redox Biol. 2022, 56, 102465. [Google Scholar] [CrossRef]
- Parmaksiz, E.; Parmaksiz, E.T. Uric acid as a prognostic predictor in COVID-19. Pak. J. Med. Sci. 2022, 38, 2246–2252. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, R.D.; Kelley, E.E. Urate Biology and Biochemistry: A Year in Review 2022. Gout Urate Cryst. Depos. Dis. 2023, 1, 115-121. https://doi.org/10.3390/gucdd1030011
King RD, Kelley EE. Urate Biology and Biochemistry: A Year in Review 2022. Gout, Urate, and Crystal Deposition Disease. 2023; 1(3):115-121. https://doi.org/10.3390/gucdd1030011
Chicago/Turabian StyleKing, Rachel D., and Eric E. Kelley. 2023. "Urate Biology and Biochemistry: A Year in Review 2022" Gout, Urate, and Crystal Deposition Disease 1, no. 3: 115-121. https://doi.org/10.3390/gucdd1030011
APA StyleKing, R. D., & Kelley, E. E. (2023). Urate Biology and Biochemistry: A Year in Review 2022. Gout, Urate, and Crystal Deposition Disease, 1(3), 115-121. https://doi.org/10.3390/gucdd1030011