
Proteoglycan 4 (PRG4)/Lubricin and the Extracellular Matrix in Gout



Abstract
1. Introduction
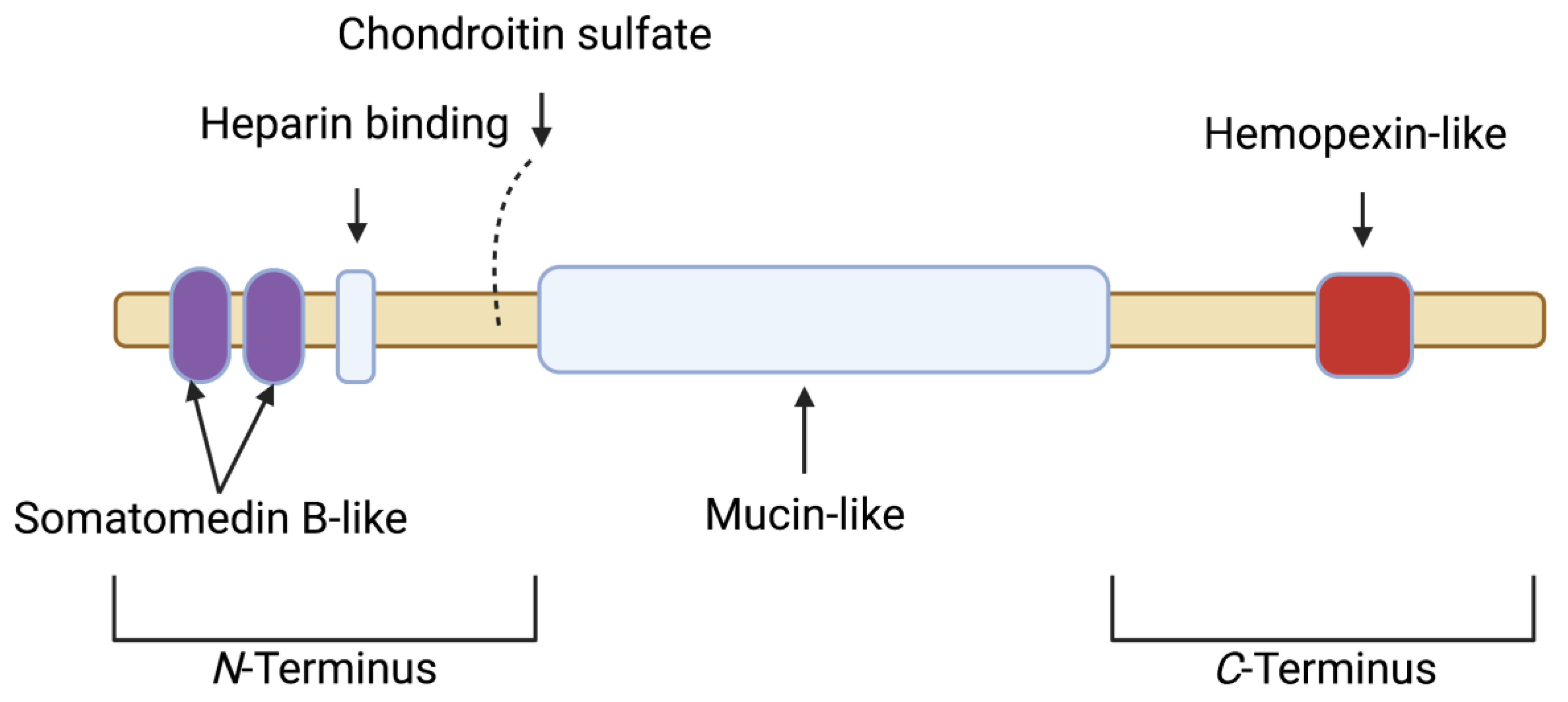
2. Proteoglycan 4 (PRG4)/Lubricin: Articular Localization, Structure, Regulation and Biological Activity
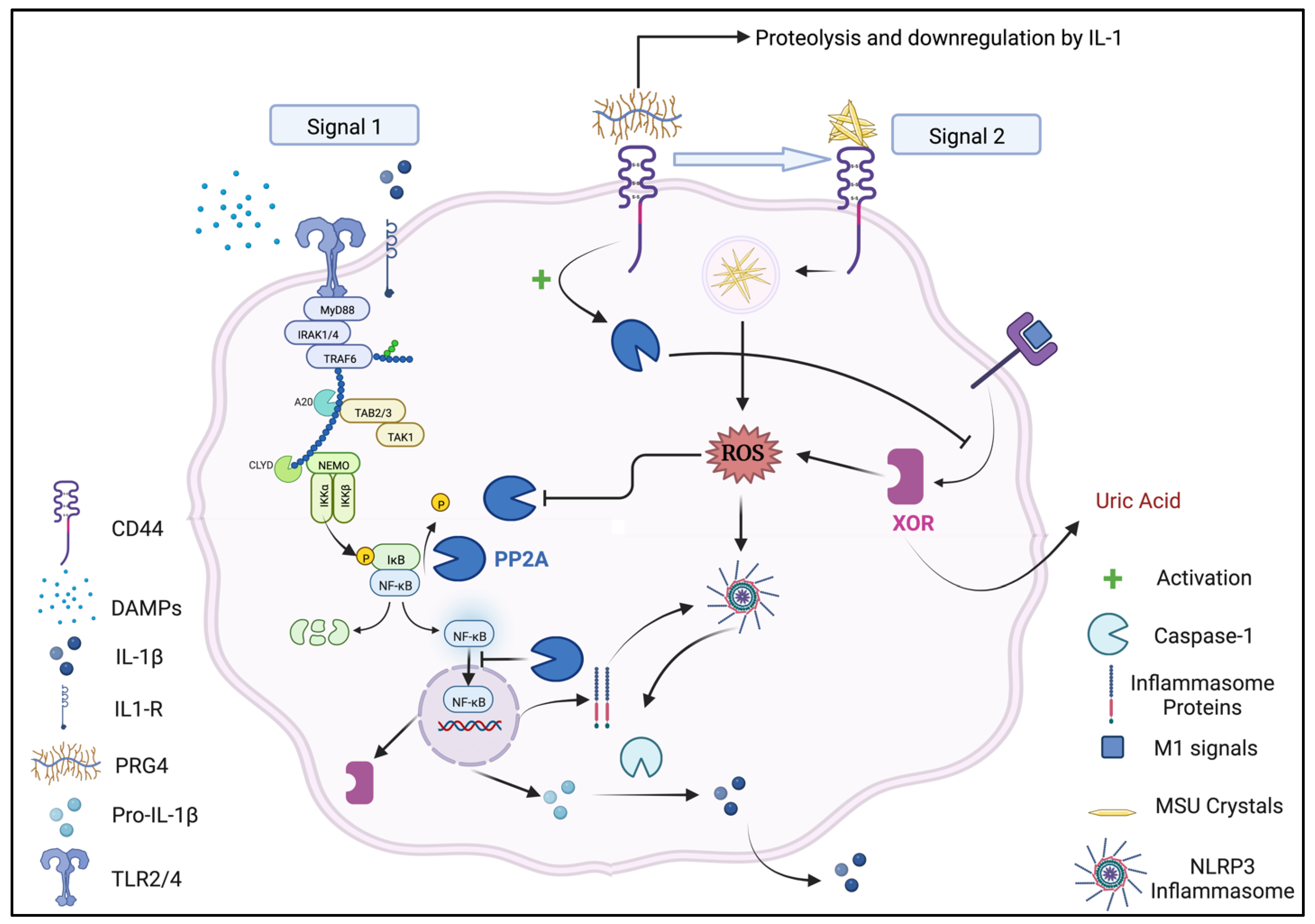
3. The PRG4/Lubricin Receptor CD44 and Protein Phosphatase 2A Signal Transduction
4. CD44 Receptors and MSU Crystals: The Role of PRG4 as a Ligand and PP2A as a Signal Transducer
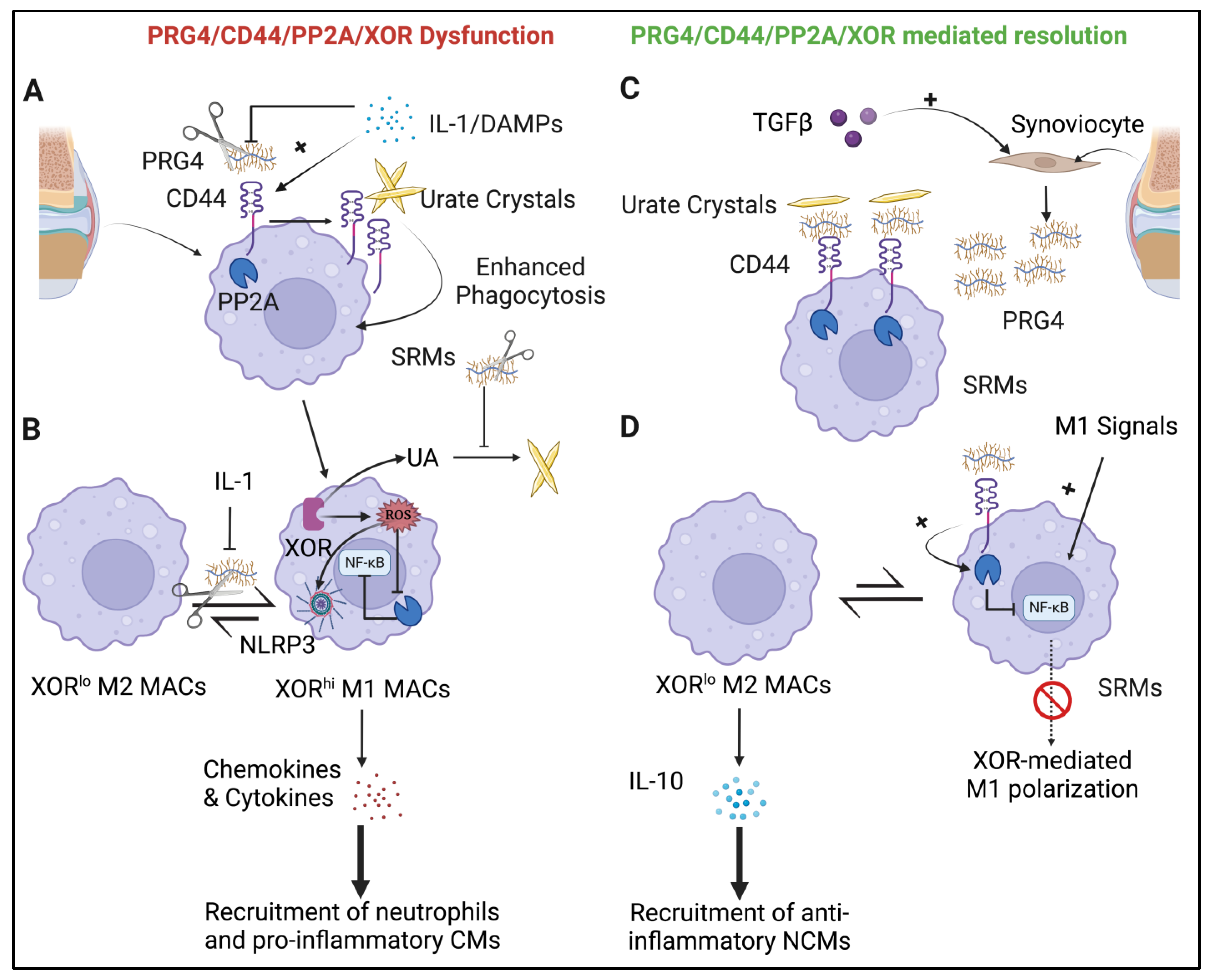
4.1. The PRG4/CD44/PP2A/XOR Circuit and Regulation of Synovitis
4.2. PRG4 Deficiency Associated with Erosive Gouty Arthritis Independent of Hyperuricemia
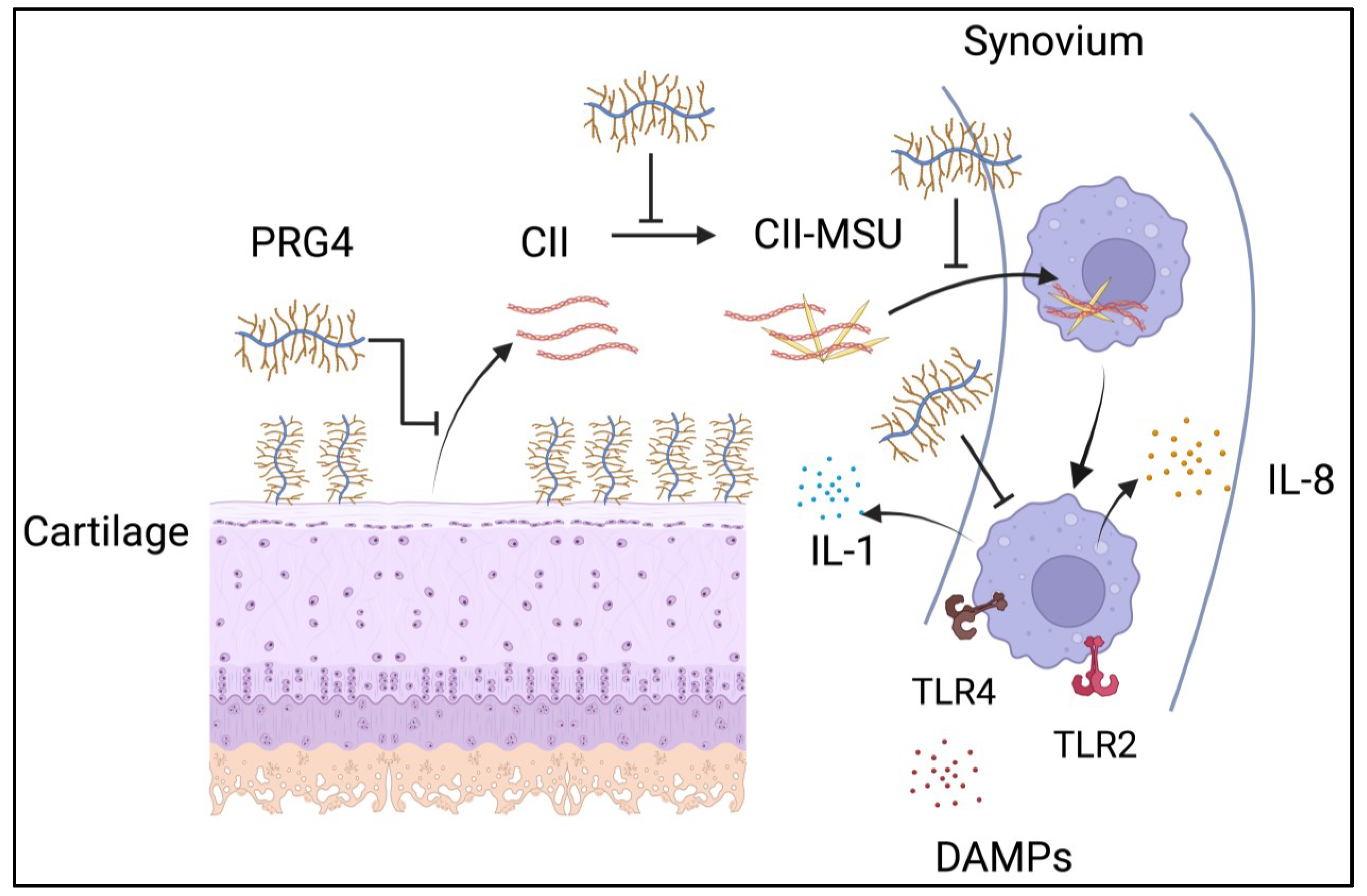
5. Cartilage Proteins and Gout Pathogenesis
6. Future Areas for Research
- What is the importance of PRG4 to synovial macrophage barrier integrity in the joint [110] in the context of acute urate crystal inflammation?
- Does disruption to the integrity of this barrier layer increase the likelihood of a gout flare and/or increase its severity?
- To what extent does XOR regulate synovial macrophage plasticity in the synovium, and in which patients will the urate produced in the joint increase the disease burden? This is quite relevant, since an erosive form of gout with urate crystal deposition was observed in an otherwise normouricemic patient [83].
- Does SZP regulate urate crystal deposition on cartilage surfaces and affect XOR expression via superficial zone articular chondrocytes?
- What is the impact of changes in the O-glycomap of PRG4 or its degradation fragments on urate crystal formation and synovial macrophage priming in gout? Potentially, there is a pro-inflammatory effect that is a consequence of these changes, since in late-stage OA, synovial PRG4 may have increased unsialylated core 1 O-glycans, which compromise its ability to bind galectin-3, a pro-inflammatory mediator [111]; meanwhile, truncated O-glycans of PRG4, commonly found in OA, promote pro-inflammatory cytokine production and may exacerbate synovitis [112]. Moreover, tryptase-mediated cleavage of PRG4 in OA SF activates TLR2 and TLR4 receptors [113].
7. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.M.; Yokose, C.; Rai, S.K.; Pillinger, M.H.; Choi, H.K. Contemporary Prevalence of Gout and Hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey, 2007–2016. Arthritis Rheumatol. 2019, 71, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef] [PubMed]
- Terkeltaub, R. What makes gouty inflammation so variable? BMC Med. 2017, 15, 158. [Google Scholar] [CrossRef]
- So, A.; Dumusc, A.; Nasi, S. The role of IL-1 in gout: From bench to bedside. Rheumatology 2018, 57 (Suppl. S1), i12–i19. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.W.; Walton, M.; Harper, J. Resident macrophages initating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 2009, 60, 281–289. [Google Scholar] [CrossRef]
- Busso, N.; So, A. Mechanisms of inflammation in gout. Arthritis Res. Ther. 2010, 12, 206. [Google Scholar] [CrossRef]
- Smith, M.D. The normal synovium. Open Rheumatol. J. 2011, 5, 100–106. [Google Scholar] [CrossRef]
- Smith, M.D.; Barg, E.; Weedon, H.; Papengelis, V.; Smeets, T.; Tak, P.P.; Kraan, M.; Coleman, M.; Ahern, M.J. Microarchitecture and protective mechanisms in synovial tissue from clinically and arthroscopically normal knee joints. Ann. Rheum. Dis. 2003, 62, 303–307. [Google Scholar] [CrossRef]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Rosenberg, J.H.; Rai, V.; Dilisio, M.F.; Agrawal, D.K. Damage-associated molecular patterns in the pathogenesis of osteoarthritis: Potentially novel therapeutic targets. Mol. Cell. Biochem. 2017, 434, 171–179. [Google Scholar] [CrossRef]
- Swann, D.A.; Silver, F.H.; Slayter, H.S.; Stafford, W.; Shore, E. The molecular structure and lubricating activity of lubricin isolated from bovine and human synovial fluids. Biochem. J. 1985, 225, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Habertstroh, K.; Cha, C.J. Comparison of the boundary lubricating ability of bovine synovial fluid, lubricin, and Healon. J. Biomed. Mater. Res. 1998, 49, 414–418. [Google Scholar] [CrossRef]
- Jay, G.D.; Britt, D.E.; Cha, C.J. Lubricin is a product of megakaryocyte stimulating factor gene expression by human synovial fibroblasts. J. Rheumatol. 2000, 27, 594–600. [Google Scholar] [PubMed]
- Flannery, C.R.; Hughes, C.E.; Schumacher, B.L.; Tudor, D.; Aydelotte, M.B.; Kuettner, K.E.; Caterson, B. Articular cartilage superficial zone protein (SZP) is homologous to megakaryocyte stimulating factor precursor and is a multifunctional proteoglycan with potential growth-promoting cytoprotective, and lubricating properties in cartilage metabolism. Biochem. Biophys. Res. Commun. 1999, 254, 535–541. [Google Scholar] [CrossRef]
- Jay, G.D.; Waller, K.A. The biology of lubricin: Near frictionless joint motion. Matrix Biol. 2014, 39, 17–24. [Google Scholar] [CrossRef]
- Ludwig, T.E.; Hunter, M.; Schmidt, T.A. Cartilage boundary lubrication synergism is mediated by hyaluronan concentration and PRG4 concentration and structure. BMC Musculoskelet. Disord. 2015, 16, 386. [Google Scholar] [CrossRef]
- Damen, A.; Van Donkelaar, C.; Cardinaels, R.; Brandt, J.-M.; Schmidt, T.; Ito, K. Proteoglycan 4 reduces friction more than other synovial fluid components for both cartilage-cartilage and cartilage-metal articulation. Osteoarthr. Cartil. 2021, 29, 894–904. [Google Scholar] [CrossRef]
- Waller, K.A.; Zhang, L.X.; Jay, G.D. Friction-induced mitochondrial dysregulation contributes to joint deterioration in Prg4 knockout mice. Int. J. Mol. Sci. 2017, 18, 1252. [Google Scholar] [CrossRef]
- Larson, K.; Zhang, L.; Badger, G.; Jay, G. Early genetic restoration of lubricin expression in transgenic mice mitigates chondrocyte peroxynitrite release and caspase-3 activation. Osteoarthr. Cartil. 2017, 25, 1488–1495. [Google Scholar] [CrossRef]
- Rhee, D.K.; Marcelino, J.; Baker, M.; Gong, Y.; Smits, P.; Lefebvre, V.; Jay, G.D.; Stewart, M.; Wang, H.; Warman, M.L.; et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Investig. 2005, 115, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Harris, D.A.; Cha, C.J. Boundary lubrication by lubricin is mediated by O-linked beta (1-3) Gal-GalNAc oligosaccharides. Glycoconj. J. 2001, 18, 807–815. [Google Scholar] [CrossRef]
- Zappone, B.; Greene, G.W.; Oroudjev, E.; Jay, G.D.; Israelachvili, J.N. Molecular aspects of boundary lubrication by human lubricin: Effect of disulfide bonds with enzymatic digestion. Langmuir 2008, 24, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Abubacker, S.; Ponjevic, D.; Ham, H.O.; Messersmith, P.B.; Matyas, J.R.; Schmidt, T.A. Effect of disulfide bonding and multimerization on proteoglycan 4’s cartilage boundary lubricating ability and adsorption. Connect. Tissue Res. 2016, 57, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.A.; Plaas, A.; Sandy, J. Disulfide-bonded multimers of proteoglycan 4(PRG4) are present in normal synovial fluids. Biochim. Biophys. Acta 2009, 1790, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kosinska, M.K.; Ludwig, T.E.; Liebisch, G.; Zhang, R.; Siebert, H.-C.; Wilhelm, J.; Kaesser, U.; Dettmeyer, R.B.; Klein, H.; Ishaque, B.; et al. Articular joint lubricants during osteoarthritis and rheumatoid arthritis display altered levels and molecular species. PLoS ONE 2015, 10, e0125192. [Google Scholar] [CrossRef]
- Rhee, D.K.; Marcelino, J.; Al-Mayouf, S.; Schelling, D.K.; Bartels, C.F.; Cui, Y.; Laxer, R.; Goldbach-Mansky, R.; Warman, M.L. Consequences of disease-causing mutations on lubricin protein synthesis, secretion, and post-translational processing. J. Biol. Chem. 2005, 280, 31325–31332. [Google Scholar] [CrossRef]
- Waller, K.A.; Zhang, L.X.; Elsaid, K.A.; Fleming, B.C.; Warman, M.L.; Jay, G.D. Role of lubricin and boundary lubrication in the prevention of chondrocyte apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, 5852–5857. [Google Scholar] [CrossRef]
- Hill, A.; Waller, K.A.; Cui, Y.; Allen, J.M.; Smits, P.; Zhang, L.X.; Ayturk, U.M.; Hann, S.; Lessard, S.G.; Zurakowski, D.; et al. Lubricin restoration in a mouse model of congenital deficiency. Arthritis Rheumatol. 2015, 67, 3070–3081. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Richendrfer, H.; Schmidt, T.A.; Elsaid, K.A. Proteoglycan-4 regulates fibroblast to myofibroblast transition and expression of fibrotic genes in the synovium. Arthritis Res. Ther. 2020, 22, 113. [Google Scholar] [CrossRef]
- Watkins, A.; Reesink, H.L. Lubricin in experimental and naturally occurring osteoarthritis: A systematic review. Osteoarthr. Cartil. 2020, 28, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Imbesi, R.; Giunta, S.; Szychlinska, M.A.; Loreto, C.; Castorina, S.; Mobasheri, A. Physical activity ameliorates cartilage degeneration in a rat model of aging: A study on lubricin. Scand. J. Med. Sci. Sport. 2015, 25, e222–e230. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Fleming, B.C.; Sun, X.; Teeple, E.; Wu, W.; Jay, G.D.; Elsaid, K.A.; Luo, J.; Machan, J.T.; Chen, Q. Comparison of differential biomarkers of osteoarthritis with and without posttraumatic injury in the hartley guinea pig model. J. Orthop. Res. 2010, 28, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Teeple, E.; Elsaid, K.A.; Fleming, B.C.; Jay, G.D.; Aslani, K.; Crisco, J.J.; Mechrefe, A.P. Coefficients of friction, lubricin, and cartilage damage in the anterior cruciate ligament-deficient guinea pig knee. J. Orthop. Res. 2008, 26, 231–237. [Google Scholar] [CrossRef]
- Elsaid, K.A.; Machan, J.T.; Waller, K.; Fleming, B.C.; Jay, G.D. The impact of anterior cruciate ligament injury on lubricin metabolism and the effect of inhibiting tumor necrosis factor alpha on chondroprotection in an animal model. Arthritis Rheum. 2009, 60, 2997–3006. [Google Scholar] [CrossRef]
- Elsaid, K.A.; Jay, G.D.; Chichester, C.O. Reduced expression and proteolytic susceptibility of lubricin/superficial zone protein may explain early elevation in the coefficient of friction in the joints with antigen-induced arthritis. Arthritis Rheum. 2007, 56, 108–116. [Google Scholar] [CrossRef]
- Jones, A.R.; Flannery, C.R. Bioregulation of lubricin expression by growth factors and cytokines. Eur. Cell Mater. 2007, 13, 40–45. [Google Scholar] [CrossRef]
- Schmidt, T.; Gastelum, N.; Han, E.; Nugent-Derfus, G.; Schumacher, B.; Sah, R. Differential regulation of proteoglycan 4 metabolism in cartilage by IL-1 alpha, IGF-1, and TGF-beta 1. Osteoarthr. Cartil. 2008, 16, 90–97. [Google Scholar] [CrossRef]
- McNary, S.M.; Athanasiou, K.A.; Reddi, A.H. Transforming growth factor-β induced superficial zone protein accumulation in the surface zone of articular cartilage is dependent on the cytoskeleton. Tissue Eng. Part A 2014, 20, 921–929. [Google Scholar] [CrossRef]
- Blewis, M.E.; Lao, B.J.; Schumacher, B.L.; Bugbee, W.D.; Sah, R.L.; Firestein, G.S. Interactive cytokine regulation of synoviocyte lubricant secretion. Tissue Eng. Part A 2010, 16, 1329–1337. [Google Scholar] [CrossRef]
- Cuellar, A.; Reddi, A.H. Stimulation of superficial zone protein/lubricin/PRG4 by transforming growth factor-β in superficial zone articular chondrocytes and modulation by glycosaminoglycans. Tissue Eng. Part A 2015, 21, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Thomsson, K.A.; Jin, C.; Alweddi, S.; Struglics, A.; Rolfson, O.; Björkman, L.I.; Kalamajski, S.; Schmidt, T.A.; Jay, G.D.; et al. Cathepsin g degrades both glycosylated and unglycosylated regions of lubricin, a synovial mucin. Sci. Rep. 2020, 10, 4215. [Google Scholar] [CrossRef] [PubMed]
- Flannery, C.R.; Zollner, R.; Corcoran, C.; Jones, A.R.; Root, A.; Rivera-Bermúdez, M.A.; Blanchet, T.; Gleghorn, J.P.; Bonassar, L.J.; Bendele, A.M.; et al. Prevention of cartilage degeneration in a rat model of osteoarthritis by intraarticular treatment with recombinant lubricin. Arthritis Rheum. 2009, 60, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Fleming, B.C.; Watkins, B.A.; McHugh, K.A.; Anderson, S.C.; Zhang, L.X.; Teeple, E.; Waller, K.A.; Elsaid, K.A. Prevention of cartilage degeneration and restoration of chondroprotection by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheum. 2010, 62, 2382–2391. [Google Scholar] [CrossRef]
- Teeple, E.; Elsaid, K.A.; Jay, G.D.; Zhang, L.; Badger, G.J.; Akelman, M.; Bliss, T.F.; Fleming, B.C. Effects of supplemental intra-articular lubricin and hyaluronic acid on the progression of posttraumatic arthritis in the anterior cruciate ligament-deficient rat knee. Am. J. Sport. Med. 2011, 39, 164–172. [Google Scholar] [CrossRef]
- Jay, G.D.; Elsaid, K.A.; Kelly, K.A.; Anderson, S.C.; Zhang, L.; Teeple, E.; Waller, K.; Fleming, B.C. Prevention of cartilage degeneration and gait asymmetry by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheum. 2012, 64, 1162–1171. [Google Scholar] [CrossRef]
- Elsaid, K.; Zhang, L.; Waller, K.; Tofte, J.; Teeple, E.; Fleming, B.; Jay, G. The impact of forced joint exercise on lubricin biosynthesis from articular cartilage following ACL transection and intra-articular lubricin’s effect in exercised joints following ACL transection. Osteoarthr. Cartil. 2012, 20, 940–948. [Google Scholar] [CrossRef]
- Elsaid, K.A.; Zhang, L.; Shaman, Z.; Patel, C.; Schmidt, T.A.; Jay, G.D. The impact of early intra-articular administration of interleukin-1 receptor antagonist on lubricin metabolism and cartilage degeneration in an anterior cruciate ligament transection model. Osteoarthr. Cartil. 2015, 23, 114–121. [Google Scholar] [CrossRef]
- Waller, K.A.; Chin, K.E.; Jay, G.D.; Zhang, L.X.; Teeple, E.; McAllister, S.; Badger, G.J.; Schmidt, T.A.; Fleming, B.C. Intra-articular recombinant human proteoglycan-4 mitigates cartilage damage after destabilization of the medial meniscus in the Yucatan minipig. Am. J. Sport. Med. 2017, 45, 1512–1521. [Google Scholar] [CrossRef]
- Hurtig, M.; Zaghoul, I.; Sheardown, H.; Schmidt, T.A.; Liu, L.; Zhang, L.; Elsaid, K.A.; Jay, G.D. Two compartment pharmacokinetic model describes the intra-articular delivery and retention of rhPRG4 following ACL transection in the Yucatan mini pig. J. Orthop. Res. 2019, 37, 386–396. [Google Scholar] [CrossRef]
- Ruan, M.Z.C.; Erez, A.; Guse, K.; Dawson, B.; Bertin, T.; Chen, Y.; Jiang, M.-M.; Yustein, J.; Gannon, F.; Lee, B.H.L. Proteoglycan 4 expression protects against the development of osteoarthritis. Sci. Transl. Med. 2013, 5, 176ra34. [Google Scholar] [CrossRef] [PubMed]
- Ruan, M.Z.; Cerullo, V.; Cela, R.; Clarke, C.; Lundgren-Akerlund, E.; Barry, M.A.; Lee, B.H. Treatment of osteoarthritis using a helper-dependent adenoviral vector. Mol. Ther. Methods Clin. Dev. 2016, 3, 16008. [Google Scholar] [CrossRef]
- Stone, A.; Grol, M.W.; Ruan, M.Z.C.; Dawson, B.; Chen, Y.; Jiang, M.-M.; Song, I.-W.; Jayaram, P.; Cela, R.; Gannon, F.; et al. Combinatorial Prg4 and IL-1ra gene therapy protects against hyperalgesia and cartilage degeneration in post-traumatic osteoarthritis. Hum. Gene Ther. 2019, 30, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharif, A.; Jamal, M.; Zhang, L.X.; Larson, K.; Schmidt, T.A.; Jay, G.D.; Elsaid, K.A. Lubricin/proteoglycan 4 binding to CD44 receptor: A mechanism of the suppression of proinflammatory cytokine-induced synoviocyte proliferation by lubricin. Arthritis Rheumatol. 2015, 67, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Alquraini, A.; Jamal, M.; Zhang, L.; Schmidt, T.; Jay, G.D.; Elsaid, K.A. The autocrine role of proteoglycan-4 (PRG4) in modulating osteoarthritic synoviocyte proliferation and expression of matrix degrading enzymes. Arthritis Res. Ther. 2017, 19, 89. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Leonard, C.; Regmi, S.C.; De Rantere, D.; Tailor, P.; Ren, G.; Ishida, H.; Hsu, C.; Abubacker, S.; Pang, D.S.; et al. Lubricin/Proteoglycan 4 binds and regulates the activity of toll-like receptors in vitro. Sci. Rep. 2016, 6, 18910. [Google Scholar] [CrossRef]
- Alquraini, A.; Garguilo, S.; D’souza, G.; Zhang, L.X.; Schmidt, T.A.; Jay, G.D.; Elsaid, K.A. The interaction of lubricin/proteoglycan 4 (PRG4) with toll-like receptors 2 and 4: An anti-inflammatory role of PRG4 in synovial fluid. Arthritis Res. Ther. 2015, 17, 353. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Schmidt, T.A.; Totonchy, J.; Elsaid, K.A. Proteoglycan-4 is an essential regulator of synovial macrophage polarization and inflammatory macrophage joint infiltration. Arthritis Res. Ther. 2021, 23, 241. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrlich, P. CD44: From adhesion molecules to signaling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Orian-Rousseau, V.; Sleeman, J. CD44 is a multidomain signaling platform that integrates extracellular matrix cues with growth factor and cytokine signals. Adv. Cancer Res. 2014, 123, 231054. [Google Scholar] [CrossRef]
- Senbanjo, L.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Motiani, K.; Giridhar, P.V.; Kasper, S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. 2013, 238, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rouseau, V. CD44 acts as a signaling platform controlling tumor progression and metastasis. Front. Immunol. 2015, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Miletti-González, K.E.; Murphy, K.; Kumaran, M.N.; Ravindranath, A.K.; Wernyj, R.P.; Kaur, S.; Miles, G.D.; Lim, E.; Chan, R.; Chekmareva, M.; et al. Identification of Function for CD44 Intracytoplasmic Domain (CD44-ICD). J. Biol. Chem. 2012, 287, 18995–19007. [Google Scholar] [CrossRef]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1994, 52, 189–196. [Google Scholar] [CrossRef]
- Weber, G.F.; Ashkar, S.; Glimcher, M.J.; Cantor, H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Science 1996, 271, 509–512. [Google Scholar] [CrossRef]
- Vachon, E.; Martin, R.; Plumb, J.; Kwok, V.; Vandivier, R.W.; Glogauer, M.; Kapus, A.; Wang, X.; Chow, C.-W.; Grinstein, S.; et al. CD44 is a phagocytic receptor. Blood 2006, 107, 4149–4158. [Google Scholar] [CrossRef]
- Teder, P.; Vandivier, R.W.; Jiang, D.; Liang, J.; Cohn, L.; Puré, E.; Henson, P.M.; Noble, P.W. Resolution of lung inflammation by CD44. Science 2002, 296, 155–158. [Google Scholar] [CrossRef]
- Moffat, F.L.; Han, T.; Li, Z.M.; Peck, M.D.; Falk, R.E.; Spalding, P.B.; Jy, W.; Ahn, Y.S.; Chu, A.J.; Bourguignon, L.Y.W. Involvement of CD44 and the cytoskeletal linker protein ankyrin in human neutrophil bacterial phagocytosis. J. Cell. Physiol. 1996, 168, 638–647. [Google Scholar] [CrossRef]
- Ouhtit, A.; Rizeq, B.; Saleh, H.A.; Rahman, M.; Zayed, H. Novel CD44-downstream signaling pathways mediating breast tumor invasion. Int. J. Biol. Sci. 2018, 14, 1782–1790. [Google Scholar] [CrossRef]
- Jordan, A.R.; Racine, R.R.; Hennig, M.J.P.; Lokeshwar, V.B. The role of CD44 in disease pathophysiology and targeted treatment. Front. Immunol. 2015, 6, 182. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Sontag, J.M.; Sontag, E. Protein phosphatase 2A: More than a passenger in the regulation of epithelial cell-cell junctions. Front. Cell Dev. Biol. 2019, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, C.; Haesen, D.; Sents, W.; Ivanova, E.; Janssens, V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol. Biol. 2013, 1053, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Sents, W.; Ivanova, E.; Lambercht, C.; Haesen, D.; Janssens, V. The biogenesis of active protein phosphatase 2A holozymes: A tightly regulated process creating phosphatase specificity. FEBS J. 2013, 280, 644–661. [Google Scholar] [CrossRef]
- Sontag, J.M.; Sontag, E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef]
- Baskaran, R.; Velmurugan, B.K. Protein phosphatase 2A as a therapeutic target in various disease models. Life Sci. 2018, 210, 40–46. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its abberant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef]
- Clark, A.R.; Ohlmeyer, M. Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol. Ther. 2019, 201, 181–201. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Osaki, K.; Kanamoto, M.; Nakao, Y.; Takahashi, E.; Higuchi, T.; Kamata, H. Distinct B subunits of PP2A regulate the NF-κB signaling pathway through dephosphorylation of IKKβ, IκBα and RelA. FEBS Lett. 2017, 591, 4083–4094. [Google Scholar] [CrossRef]
- Sun, L.; Pham, T.T.; Cornell, T.T.; McDonough, K.L.; McHugh, W.M.; Blatt, N.B.; Dahmer, M.K.; Shanley, T.P. Myeloid-specific gene deletion of protein phosphatase 2A magnifies MyD88- and TRIF-dependent inflammation following endotoxin challenge. J Immunol. 2017, 198, 404–416. [Google Scholar] [CrossRef]
- Bousoik, E.; Qadri, M.; Elsaid, K.A. CD44 receptor mediates crystal phagocytosis by macrophages and regulates inflammation in a murine peritoneal model of acute gout. Sci. Rep. 2020, 10, 5748. [Google Scholar] [CrossRef] [PubMed]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Wong, W.; Reginato, A.M.; Sun, C.; Schmidt, T.A.; Elsaid, K.A. Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Res. Ther. 2018, 20, 192. [Google Scholar] [CrossRef]
- Elsaid, K.; Merriman, T.R.; Rossitto, L.; Liu-Bryan, R.; Karsh, J.; Phipps-Green, A.; Jay, G.D.; Elsayed, S.; Qadri, M.; Miner, M.; et al. Amplification of inflammation by lubricin deficiency implicated in incident, erosive gout independent of hyperuricemia. Arthritis Rheumatol. 2023, 75, 794–805. [Google Scholar] [CrossRef] [PubMed]
- ElSayed, S.; Jay, G.D.; Cabezas, R.; Qadri, M.; Schmidt, T.A.; Elsaid, K.A. Recombinant human proteoglycan-4 regulates phagocytic activation of monocytes and reduces IL-1β secretion by urate crystal stimulated gout PBMCs. Front. Immunol. 2021, 12, 771677. [Google Scholar] [CrossRef] [PubMed]
- Qadri, M.; Elsayed, S.; Elsaid, K.A. Fingolimod Phosphate (FTY720-P) Activates Protein Phosphatase 2A in Human Monocytes and Inhibits Monosodium Urate Crystal-Induced IL-1β Production. J. Pharmacol. Exp. Ther. 2021, 376, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Qadri, M.; Almadani, S.; Jay, G.D.; Elsaid, K.A. Role of CD44 in regulating TLR2 activation of human macrophages and downstream expression of proinflammatory cytokines. J. Immunol. 2018, 200, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Low, I.C.C.; Loh, T.; Huang, Y.; Virshup, D.M.; Pervaiz, S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56δ stabilizes its antiapoptotic activity. Blood 2014, 124, 2223–2234. [Google Scholar] [CrossRef]
- Raman, D.; Pervaiz, S. Redox inhibition of protein phosphatase PP2A: Potential implications in oncogenesis and its progression. Redox Biol. 2019, 27, 10115. [Google Scholar] [CrossRef]
- Yee, Y.H.; Chong, S.J.F.; Kong, L.R.; Goh, B.C.; Pervaiz, S. Sustained IKKβ phosphorylation and NF-κB activation by superoxide-induced peroxynitrite-mediated nitrotyrosine modification of B56γ3 and PP2A inactivation. Redox Biol. 2021, 41, 101834. [Google Scholar] [CrossRef]
- Elsayed, S.; Elsaid, K.A. Protein phosphatase 2A regulates xanthine oxidase-derived ROS production in macrophages and influx of inflammatory monocytes in a murine gout model. Front. Pharmacol. 2022, 13, 1033520. [Google Scholar] [CrossRef]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Coras, R.; Torres, A.; Lane, N.E.; Guma, M. Synovial inflammation in osteoarthritis progression. Nat. Rev. Rheumatol. 2022, 18, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.D.; Patelli, M.; Anderson, S.R.; Prakash, N.; Rodriquez, E.J.; Bateman, H.; Sterrett, A.; Valeriano, J.; Ricca, L.R. An MRI assessment of chronic synovial-based inflammation in gout and its correlation with serum urate levels. Clin. Rheumatol. 2015, 34, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Shi, W.; Yi, S.-J.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef]
- Yang, H.; Biermann, M.H.; Brauner, J.M.; Liu, Y.; Zhao, Y.; Herrmann, M. New insights into neutrophil extracellular traps: Mechanisms of formation and role in inflammation. Front. Immunol. 2016, 7, 302. [Google Scholar] [CrossRef]
- Lambert, C.; Zappia, J.; Sanchez, C.; Florin, A.; Dubuc, J.-E.; Henrotin, Y. The damage-associated molecular patterns (DAMPs) as potential targets to treat osteoarthritis: Perspectives from a review of the literature. Front. Med. 2020, 7, 607186. [Google Scholar] [CrossRef]
- Millerand, M.; Berenbaum, F.; Jacques, C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 2019, 37, 48–56. [Google Scholar]
- Lambert, C.; Borderie, D.; Dubuc, J.-E.; Rannou, F.; Henrotin, Y. Type II collagen peptide Coll2-1 is an actor of synovitis. Osteoarthr. Cartil. 2019, 27, 1680–1691. [Google Scholar] [CrossRef]
- Klatt, A.R.; Paul-Klausch, B.; Klinger, G.; Kühn, G.; Renno, J.H.; Banerjee, M.; Malchau, G.; Wielckens, K. A critical role for collagen II in cartilage matrix degradation: Collagen II induces pro-inflammatory cytokines and MMPs in primary human chondrocytes. J. Orthop. Res. 2009, 27, 65–70. [Google Scholar] [CrossRef]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.-J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Gondokaryono, S.P.; Ushio, H.; Niyonsaba, F.; Hara, M.; Takenaka, H.; Jayawardana, S.T.M.; Ikeda, S.; Okumura, K.; Ogawa, H. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J. Leukoc. Biol. 2007, 82, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Lees, S.; Golub, S.B.; Last, K.; Zeng, W.; Jackson, D.C.; Sutton, P.; Fosang, A.J. Bioactivity in an Aggrecan 32-mer fragment is mediated via toll-like receptor 2. Arthritis Rheumatol. 2015, 67, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Homandberg, G.A.; Hui, F. Association of proteoglycan degradation with catabolic cytokine and stromelysin release from cartilage cultured with fibronectin fragments. Arch. Biochem. Biophys. 1996, 334, 325–331. [Google Scholar] [CrossRef]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ruettger, A.; Schueler, S.; Mollenhauer, J.A.; Wiederanders, B. Cathepsins B, K, and L are regulated by a defined collagen type II peptide via activation of classical protein kinase C and p38 MAP kinase in articular chondrocytes. J. Biol. Chem. 2008, 283, 1043–1051. [Google Scholar] [CrossRef]
- Chhana, A.; Pool, B.; Wei, Y.; Choi, A.; Gao, R.; Munro, J.; Cornish, J.; Dalbeth, N. Human cartilage homogenates infleunce the crystallization of monosodium urate and inflammatory response to monosodium urate crystals: A potential link between osteoarthritis and gout. Arthritis Rheumatol. 2019, 71, 2090–2099. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, B.; Chen, Y.; Zeng, F.; Wang, W.; Chen, Z.; Cao, L.; Shi, J.; Chen, J.; Zhu, X.; et al. Type II collagen facilitates gouty arthritis by regulating MSU crystallisation and inflammatory cell recruitment. Ann. Rheum. Dis. 2023, 82, 416–427. [Google Scholar] [CrossRef]
- Yokose, C.; Chen, M.; Berhanu, A.; Pillinger, M.; Krasnokutsky, S. Gout and osteoarthritis: Associations, pathophysiology and therapeutic implications. Curr. Rheumatol. Rep. 2016, 18, 65. [Google Scholar] [CrossRef]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.Á.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef]
- Flowers, S.A.; Thomsson, K.A.; Ali, L.; Huang, S.; Mthembu, Y.; Regmi, S.C.; Holgersson, J.; Schmidt, T.A.; Rolfson, O.; Björkman, L.I.; et al. Decrease of core 2 O-glycans on synovial lubricin in osteoarthritis reduces galectin-3 mediated crosslinking. J. Biol. Chem. 2020, 295, 16023–16036. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Thomsson, K.A.; Jin, C.; Ryberg, H.; Das, N.; Struglics, A.; Rolfson, O.; Björkman, L.I.; Eisler, T.; Schmidt, T.A.; et al. Truncated lubricin glycans in osteoarthritis stimulate the synoviocyte secretion of VEGFA, IL-8 and MIP-1a: The interplay between O-linked glycosylation and inflammatory cytokines. Front. Mol. Biosci. 2022, 9, 942405. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; de Almeida, L.G.N.; Derakhshani, A.; Young, D.; Mehdinejadiani, K.; Salo, P.; Rezansoff, A.; Jay, G.D.; Sommerhoff, C.P.; Schmidt, T.A.; et al. Tryptase-b regulation of joint lubrication and inflammation via proteoglycan-4 in osteoarthritis. Nat. Commun. 2023, 14, 1910. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Model and Treatment(s) | Outcome(s) |
|---|---|---|
| Flannery et al. [43] | Rat meniscectomy; I.A. recombinant human lubricin construct with one third KEPAPTT-like sequence 3× week or 1× week for 4 weeks. | Both treatments reduce cartilage degeneration and total joint scores. |
| Jay et al. [44] | Rat ACLT; I.A. recombinant full-length lubricin, HSL or HSFL 2× week for 4 weeks. | HSL reduces cartilage degeneration scores; HSL and HSFL reduce uCTXII levels, and all lubricins enhance aggrecan synthesis. |
| Teeple et al. [45] | Rat ACLT; I.A. hyaluronan, HSFL or hyaluronan + HSFL 2× week for 4 weeks. | HSFL alone or hyaluronan + HSFL reduce radiographic and cartilage degeneration scores with no effect by hyaluronan alone. |
| Jay et al. [46] | Rat ACLT; I.A. HSL once on day 7 post-surgery and analysis at 10 weeks. | HSL enhances aggrecan synthesis, reduces uCTXII levels, and improves weight bearing in injured joints. |
| Elsaid et al. [47] | Rat ACLT + forced exercise; HSFL on day 7 post-surgery and analysis at 5 weeks | Forced exercise aggravates cartilage damage and increases uCTXII excretion; HSFL treatment protects against ACLT + forced exercise cartilage damage. |
| Elsaid et al. [48] | Rat ACLT; I.A. IL-1ra 4× week for one week; I.A. IL-1ra + rhPRG4 once on day 7 post-surgery and analysis at 5 weeks. | IL-1ra reduces synovial inflammation and increases lubricin levels in SF; rhPRG4 and IL-1ra synergistically reduce chondrocyte apoptosis. |
| Waller et al. [49] | Minipig DMM; I.A. rhPRG4, hyaluronan or rhPRG4 + hyaluronan 3× week for one week and analysis at 26 weeks post-surgery. | rhPRG4 reduces medial tibial plateau macroscopic cartilage damage, uCTXII levels, SF, and serum IL-1β. |
| Hurtig et al. [50] | Minipig ACLT; I.A. 131 I-rhPRG4 once with analysis at 10 min, 24, 72 h, 6, 13 and 20 days. | rhPRG4 joint elimination kinetics follows a two-compartment model with t1/2β of 4.81 days. |
| Study | Models and Treatment(s) | Outcome(s) |
|---|---|---|
| Qadri et al. [86] |
|
|
| Bousoik et al. [81] |
|
|
| Qadri et al. [82] |
|
|
| Elsaid et al. [83] |
|
|
| Elsayed et al. [84] |
|
|
| Qadri et al. [85] |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsaid, K.A.; Jay, G.D.; Liu-Bryan, R.; Terkeltaub, R. Proteoglycan 4 (PRG4)/Lubricin and the Extracellular Matrix in Gout. Gout Urate Cryst. Depos. Dis. 2023, 1, 122-136. https://doi.org/10.3390/gucdd1030012
Elsaid KA, Jay GD, Liu-Bryan R, Terkeltaub R. Proteoglycan 4 (PRG4)/Lubricin and the Extracellular Matrix in Gout. Gout, Urate, and Crystal Deposition Disease. 2023; 1(3):122-136. https://doi.org/10.3390/gucdd1030012
Chicago/Turabian StyleElsaid, Khaled A., Gregory D. Jay, Ru Liu-Bryan, and Robert Terkeltaub. 2023. "Proteoglycan 4 (PRG4)/Lubricin and the Extracellular Matrix in Gout" Gout, Urate, and Crystal Deposition Disease 1, no. 3: 122-136. https://doi.org/10.3390/gucdd1030012
APA StyleElsaid, K. A., Jay, G. D., Liu-Bryan, R., & Terkeltaub, R. (2023). Proteoglycan 4 (PRG4)/Lubricin and the Extracellular Matrix in Gout. Gout, Urate, and Crystal Deposition Disease, 1(3), 122-136. https://doi.org/10.3390/gucdd1030012