
Role and Regulation of Glycogen Synthase Kinase-3 in Obesity-Associated Metabolic Perturbations

Abstract
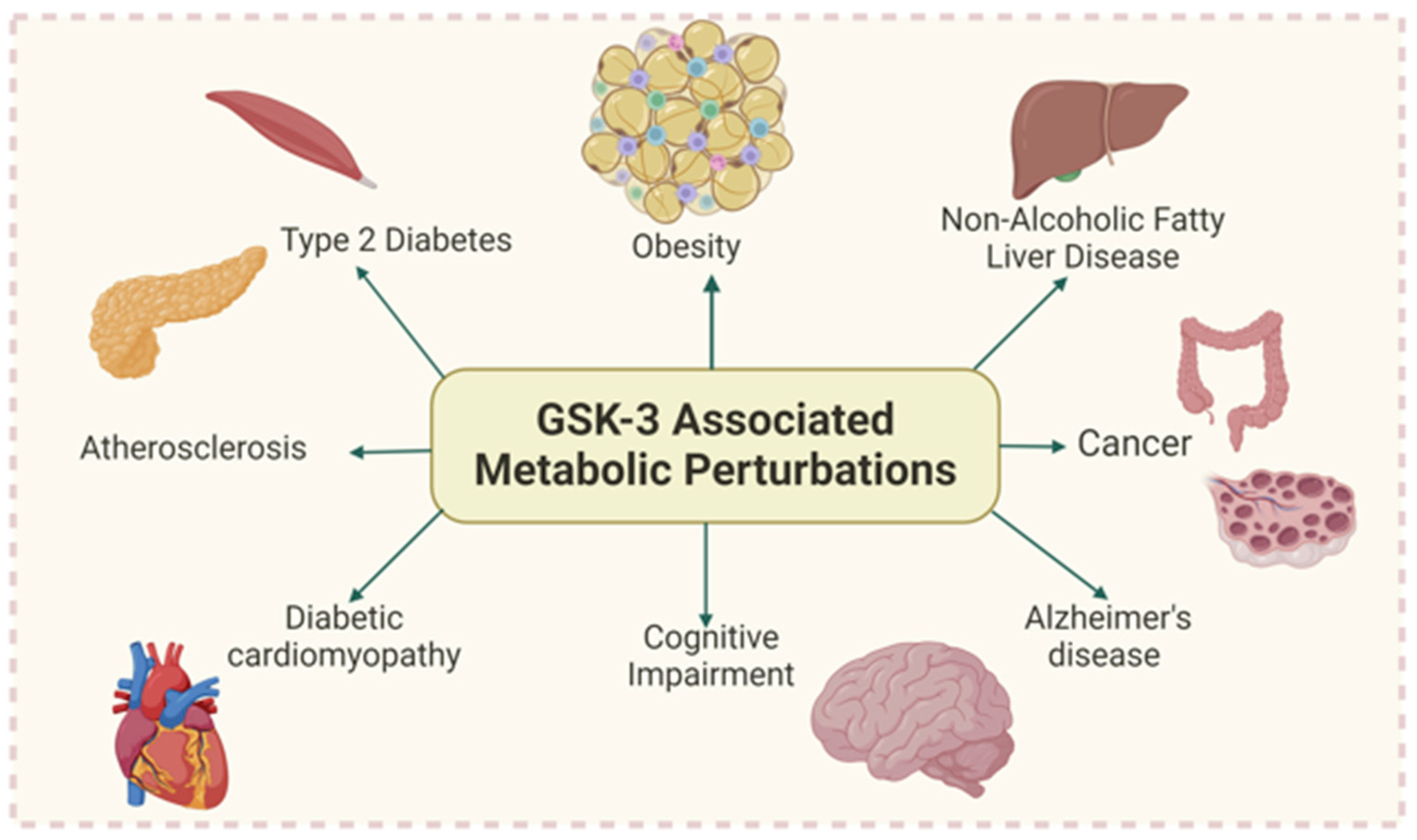
1. Introduction
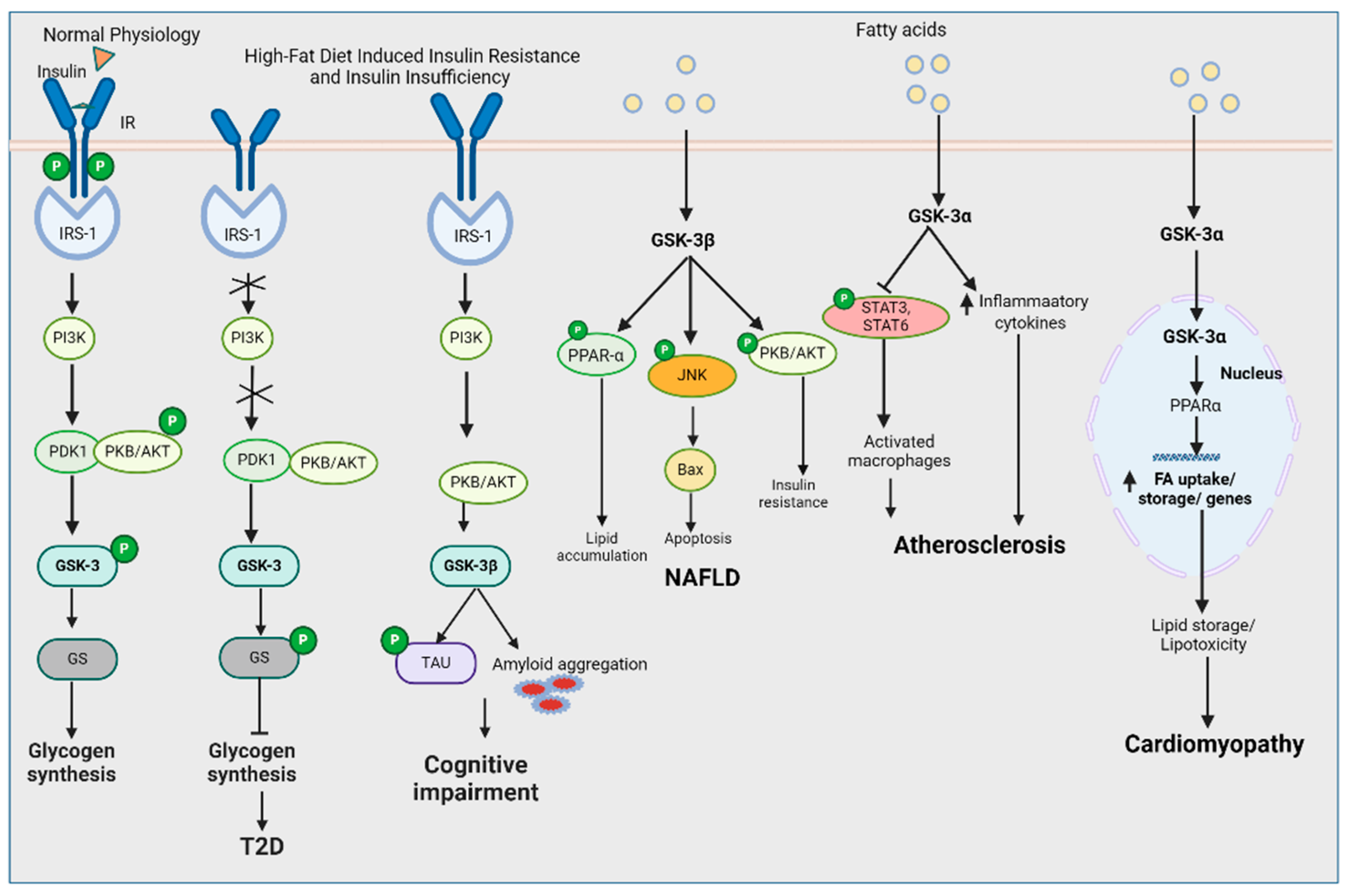
2. Regulation of GSK-3s in Heart
3. Regulation of GSK-3 in Liver
4. Regulation of GSK-3 in Skeletal Muscle
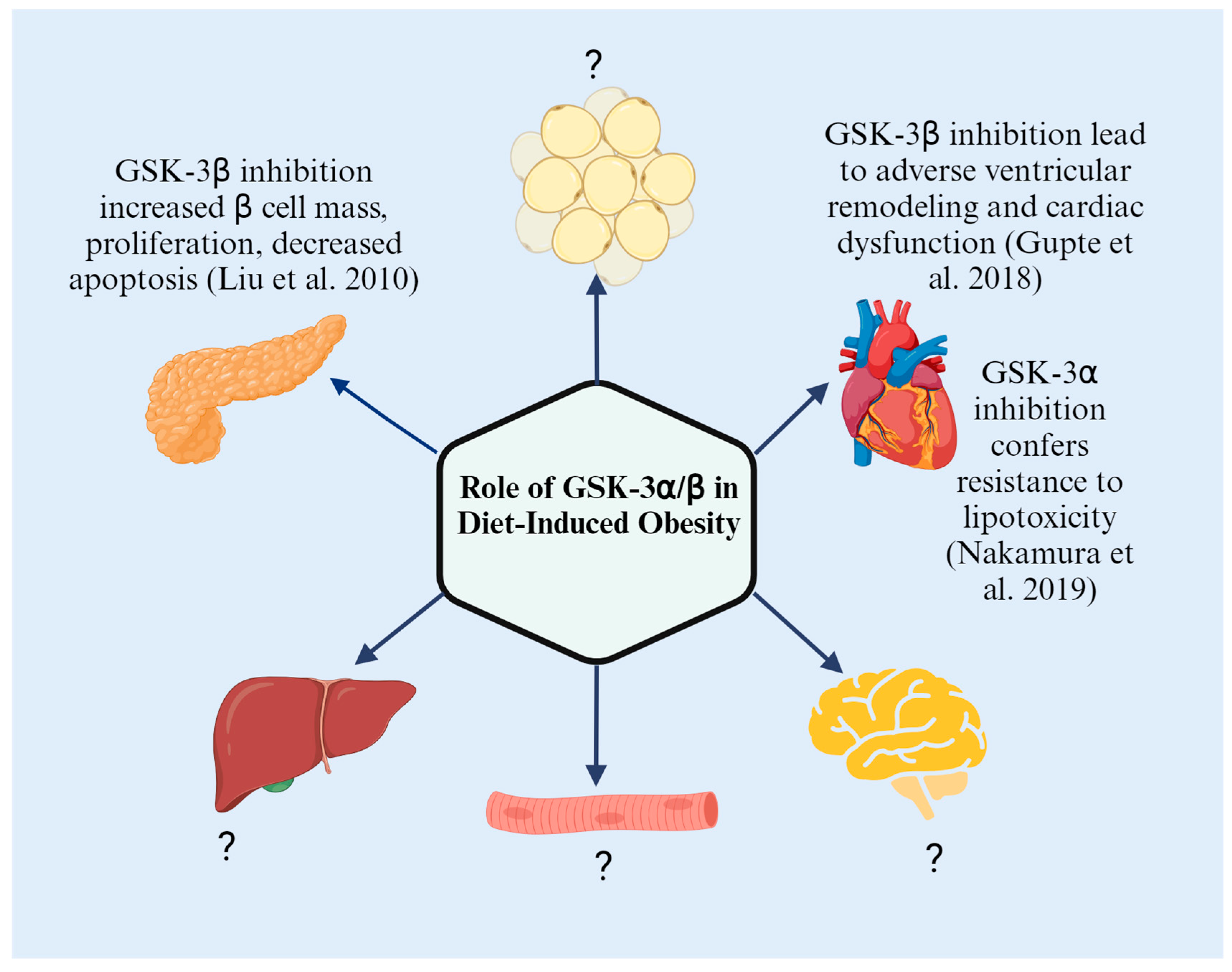
5. Regulation of GSK-3 in Pancreas
6. Regulation of GSK-3 in Brain
7. Regulation of GSK-3 in Adipose Tissue
8. Systemic Regulation of GSK-3
9. Conclusions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, S.E.; Jang, H.J.; Kang, Y.; Jung, J.G.; Han, S.J.; Kim, H.J.; Kim, D.J.; Lee, K.W. Atherosclerosis induced by a high-fat diet is alleviated by lithium chloride via reduction of VCAM expression in ApoE-deficient mice. Vasc. Pharmacol. 2010, 53, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Piche, M.E.; Tchernof, A.; Despres, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Di, L.J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef] [PubMed]
- Shakoori, A.; Mai, W.; Miyashita, K.; Yasumoto, K.; Takahashi, Y.; Ooi, A.; Kawakami, K.; Minamoto, T. Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007, 98, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Garcea, G.; Manson, M.M.; Neal, C.P.; Pattenden, C.J.; Sutton, C.D.; Dennison, A.R.; Berry, D.P. Glycogen synthase kinase-3 beta; a new target in pancreatic cancer? Curr. Cancer Drug Targets 2007, 7, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef]
- Kallamadi, P.R.; Esari, D.; Addi, U.R.; Kesavan, R.; Putcha, U.K.; Nagini, S.; Reddy, G.B. Obesity Associated with Prediabetes Increases the Risk of Breast Cancer Development and Progression—A Study on an Obese Rat Model with Impaired Glucose Tolerance. Int. J. Mol. Sci. 2023, 24, 11441. [Google Scholar] [CrossRef]
- Chandra, P.; Sachan, N.; Pal, D. Glycogen Synthase Kinase-3 (GSK-3) Inhibitors as a New Lead for Treating Breast and Ovarian Cancer. Curr. Drug Targets 2021, 22, 1548–1554. [Google Scholar] [CrossRef]
- Guil-Luna, S.; Rivas-Crespo, A.; Navarrete-Sirvent, C.; Mantrana, A.; Pera, A.; Mena-Osuna, R.; Toledano-Fonseca, M.; Garcia-Ortiz, M.V.; Villar, C.; Sanchez-Montero, M.T.; et al. Clinical significance of glycogen synthase kinase 3 (GSK-3) expression and tumor budding grade in colorectal cancer: Implications for targeted therapy. Biomed. Pharmacother. 2023, 167, 115592. [Google Scholar] [CrossRef]
- Liu, Y.; Tanabe, K.; Baronnier, D.; Patel, S.; Woodgett, J.; Cras-Meneur, C.; Permutt, M.A. Conditional ablation of Gsk-3beta in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia 2010, 53, 2600–2610. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Kinnick, T.R.; Teachey, M.K.; O’Keefe, M.P.; Ring, D.; Johnson, K.W.; Harrison, S.D. Modulation of muscle insulin resistance by selective inhibition of GSK-3 in Zucker diabetic fatty rats. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E892–E900. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich, O.; Eldar-Finkelman, H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert. Opin. Ther. Targets 2002, 6, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Schreyer, S.A.; Shinohara, M.M.; LeBoeuf, R.C.; Krebs, E.G. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 1999, 48, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Woodgett, J.R. Emerging roles of GSK-3alpha in pathophysiology: Emphasis on cardio-metabolic disorders. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118616. [Google Scholar] [CrossRef]
- Arciniegas Ruiz, S.; Rippin, I.; Eldar-Finkelman, H. Prospects in GSK-3 Signaling: From Cellular Regulation to Disease Therapy. Cells 2022, 11, 1618. [Google Scholar] [CrossRef]
- MacAulay, K.; Woodgett, J.R. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert. Opin. Ther. Targets 2008, 12, 1265–1274. [Google Scholar] [CrossRef]
- Chen, H.; Fajol, A.; Hoene, M.; Zhang, B.; Schleicher, E.D.; Lin, Y.; Calaminus, C.; Pichler, B.J.; Weigert, C.; Haring, H.U.; et al. PI3K-resistant GSK3 controls adiponectin formation and protects from metabolic syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 5754–5759. [Google Scholar] [CrossRef]
- Patel, S.; Doble, B.W.; MacAulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol. Cell Biol. 2008, 28, 6314–6328. [Google Scholar] [CrossRef]
- Frame, S.; Zheleva, D. Targeting glycogen synthase kinase-3 in insulin signalling. Expert. Opin. Ther. Targets 2006, 10, 429–444. [Google Scholar] [CrossRef]
- Umbarkar, P.; Ruiz Ramirez, S.Y.; Toro Cora, A.; Tousif, S.; Lal, H. GSK-3 at the heart of cardiometabolic diseases: Isoform-specific targeting is critical to therapeutic benefit. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166724. [Google Scholar] [CrossRef] [PubMed]
- Gupte, M.; Tousif, S.; Lemon, J.J.; Toro Cora, A.; Umbarkar, P.; Lal, H. Isoform-Specific Role of GSK-3 in High Fat Diet Induced Obesity and Glucose Intolerance. Cells 2022, 11, 559. [Google Scholar] [CrossRef] [PubMed]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M.; et al. Effects of mutations in Wnt/beta-catenin, hedgehog, Notch and PI3K pathways on GSK-3 activity-Diverse effects on cell growth, metabolism and cancer. Biochim. Biophys. Acta 2016, 1863, 2942–2976. [Google Scholar] [CrossRef] [PubMed]
- Cervello, M.; Augello, G.; Cusimano, A.; Emma, M.R.; Balasus, D.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Pivotal roles of glycogen synthase-3 in hepatocellular carcinoma. Adv. Biol. Regul. 2017, 65, 59–76. [Google Scholar] [CrossRef]
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3beta is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. [Google Scholar] [CrossRef]
- Nakamura, M.; Liu, T.; Husain, S.; Zhai, P.; Warren, J.S.; Hsu, C.P.; Matsuda, T.; Phiel, C.J.; Cox, J.E.; Tian, B.; et al. Glycogen Synthase Kinase-3alpha Promotes Fatty Acid Uptake and Lipotoxic Cardiomyopathy. Cell Metab. 2019, 29, 1119–1134.e1112. [Google Scholar] [CrossRef]
- Song, L.; De Sarno, P.; Jope, R.S. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002, 277, 44701–44708. [Google Scholar] [CrossRef]
- Patel, S.; Mastrogiacomo, L.; Fulmer, M.; Shi, Y.; Werstuck, G.H. Deletion of Macrophage-Specific Glycogen Synthase Kinase (GSK)-3alpha Promotes Atherosclerotic Regression in Ldlr(-/-) Mice. Int. J. Mol. Sci. 2022, 23, 9293. [Google Scholar] [CrossRef]
- Cai, X.; Zhao, Y.; Yang, Y.; Wu, X.; Zhang, L.; Ma, J.A.; Ji, J.; Bostrom, K.I.; Yao, Y. GSK3beta Inhibition Ameliorates Atherosclerotic Calcification. Int. J. Mol. Sci. 2023, 24, 11638. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Su, X.; Xu, L.; Chang, C.; Yao, Y.; Komal, S.; Cha, X.; Zang, M.; Ouyang, X.; Zhang, L.; et al. Glycogen synthase kinase-3beta inhibition alleviates activation of the NLRP3 inflammasome in myocardial infarction. J. Mol. Cell Cardiol. 2020, 149, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.M.; Qaisar, R.; Al-Tamimi, A.O.; Jayakumar, M.N.; Woodgett, J.R.; Koch, W.J.; Ahmad, F. Cardiomyocyte-GSK-3beta deficiency induces cardiac progenitor cell proliferation in the ischemic heart through paracrine mechanisms. J. Cell Physiol. 2022, 237, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Maejima, Y.; Shirakabe, A.; Yamamoto, T.; Ikeda, Y.; Sadoshima, J.; Zhai, P. Ser9 phosphorylation of GSK-3beta promotes aging in the heart through suppression of autophagy. J. Cardiovasc. Aging 2021, 1, 9. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, Q.; Yang, X.; Ren, Y.; Jiao, S.; Zhu, Q.; Guo, D.; Xia, K.; Wang, Y.; Li, C.; et al. Qishen granule attenuates cardiac fibrosis by regulating TGF-beta /Smad3 and GSK-3beta pathway. Phytomedicine 2019, 62, 152949. [Google Scholar] [CrossRef]
- Potz, B.A.; Sabe, A.A.; Elmadhun, N.Y.; Clements, R.T.; Robich, M.P.; Sodha, N.R.; Sellke, F.W. Glycogen Synthase Kinase 3beta Inhibition Improves Myocardial Angiogenesis and Perfusion in a Swine Model of Metabolic Syndrome. J. Am. Heart Assoc. 2016, 5, e003694. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, W.; Xue, W.; Tan, Y.; Hein, D.W.; Li, X.K.; Cai, L. Inactivation of GSK-3beta by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes 2009, 58, 1391–1402. [Google Scholar] [CrossRef]
- Cai, L.; Tan, Y.; Watson, S.; Wintergerst, K. Diabetic cardiomyopathy—Zinc preventive and therapeutic potentials by its anti-oxidative stress and sensitizing insulin signaling pathways. Toxicol. Appl. Pharmacol. 2023, 477, 116694. [Google Scholar] [CrossRef]
- Zhao, X.; Si, L.; Bian, J.; Pan, C.; Guo, W.; Qin, P.; Zhu, W.; Xia, Y.; Zhang, Q.; Wei, K. Adipose tissue macrophage-derived exosomes induce ferroptosis via glutathione synthesis inhibition by targeting SLC7A11 in obesity-induced cardiac injury. Free Radic. Biol. Med. 2022, 182, 232–245. [Google Scholar] [CrossRef]
- Bian, J.; Ding, Y.; Wang, S.; Jiang, Y.; Wang, M.; Wei, K.; Si, L.; Zhao, X.; Shao, Y. Celastrol confers ferroptosis resistance via AKT/GSK3beta signaling in high-fat diet-induced cardiac injury. Free Radic. Biol. Med. 2023, 200, 36–46. [Google Scholar] [CrossRef]
- Gao, T.; Wang, J.; Xiao, M.; Wang, J.; Wang, S.; Tang, Y.; Zhang, J.; Lu, G.; Guo, H.; Guo, Y.; et al. SESN2-Mediated AKT/GSK-3beta/NRF2 Activation to Ameliorate Adriamycin Cardiotoxicity in High-Fat Diet-Induced Obese Mice. Antioxid. Redox Signal 2024, 40, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Gupte, M.; Tumuluru, S.; Sui, J.Y.; Singh, A.P.; Umbarkar, P.; Parikh, S.S.; Ahmad, F.; Zhang, Q.; Force, T.; Lal, H. Cardiomyocyte-specific deletion of GSK-3beta leads to cardiac dysfunction in a diet induced obesity model. Int. J. Cardiol. 2018, 259, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Gupte, M.; Umbarkar, P.; Singh, A.P.; Zhang, Q.; Tousif, S.; Lal, H. Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3beta) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity. Cells 2020, 9, 1120. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Wang, Y.; Shan, D.; Guo, S.; Chen, G.; Jin, L.; Zheng, W.; Feng, H.; Zeng, X.; Zhang, S.; et al. Blocking MG53(S255) Phosphorylation Protects Diabetic Heart From Ischemic Injury. Circ. Res. 2022, 131, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Macaulay, K.; Woodgett, J.R. Tissue-specific analysis of glycogen synthase kinase-3alpha (GSK-3alpha) in glucose metabolism: Effect of strain variation. PLoS ONE 2011, 6, e15845. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62 (Suppl. S1), S47–S64. [Google Scholar] [CrossRef]
- Wei, X.; Lin, L.; Yuan, Q.Q.; Wang, X.Y.; Zhang, Q.; Zhang, X.M.; Tang, K.C.; Guo, M.Y.; Dong, T.Y.; Han, W.; et al. Bavachin protects against diet-induced hepatic steatosis and obesity in mice. Acta Pharmacol. Sin. 2023, 44, 1416–1428. [Google Scholar] [CrossRef]
- Yan, F.; Li, N.; Shi, J.; Li, H.; Yue, Y.; Jiao, W.; Wang, N.; Song, Y.; Huo, G.; Li, B. Lactobacillus acidophilus alleviates type 2 diabetes by regulating hepatic glucose, lipid metabolism and gut microbiota in mice. Food Funct. 2019, 10, 5804–5815. [Google Scholar] [CrossRef]
- Wang, H.Y.; Li, Q.M.; Yu, N.J.; Chen, W.D.; Zha, X.Q.; Wu, D.L.; Pan, L.H.; Duan, J.; Luo, J.P. Dendrobium huoshanense polysaccharide regulates hepatic glucose homeostasis and pancreatic beta-cell function in type 2 diabetic mice. Carbohydr. Polym. 2019, 211, 39–48. [Google Scholar] [CrossRef]
- Fan, G.; Huang, L.; Wang, M.; Kuang, H.; Li, Y.; Yang, X. GPAT3 deficiency attenuates corticosterone-caused hepatic steatosis and oxidative stress through GSK3beta/Nrf2 signals. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167007. [Google Scholar] [CrossRef]
- Lengton, R.; Iyer, A.M.; van der Valk, E.S.; Hoogeveen, E.K.; Meijer, O.C.; van der Voorn, B.; van Rossum, E.F.C. Variation in glucocorticoid sensitivity and the relation with obesity. Obes. Rev. 2022, 23, e13401. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, K.; Xia, J.; Qian, D.; Guo, J.; Zhong, L.; Tang, D.; Chen, X.; Peng, W.; Chen, Y.; et al. Natural exosomes-like nanoparticles in mung bean sprouts possesses anti-diabetic effects via activation of PI3K/Akt/GLUT4/GSK-3beta signaling pathway. J. Nanobiotechnology 2023, 21, 349. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Wei, C.H.; Wang, T.; Eastep, J.; Li, Y.; Chen, G. Vitamin A status affects weight gain and hepatic glucose metabolism in rats fed a high-fat diet. Biochem. Cell Biol. 2019, 97, 545–553. [Google Scholar] [CrossRef]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef]
- Dokken, B.B.; Sloniger, J.A.; Henriksen, E.J. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1188–E1194. [Google Scholar] [CrossRef]
- Pearce, N.J.; Arch, J.R.; Clapham, J.C.; Coghlan, M.P.; Corcoran, S.L.; Lister, C.A.; Llano, A.; Moore, G.B.; Murphy, G.J.; Smith, S.A.; et al. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3beta on a muscle-specific promoter. Metabolism 2004, 53, 1322–1330. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; Carter, L.; Mudaliar, S.; Henry, R.R. GSK-3beta and control of glucose metabolism and insulin action in human skeletal muscle. Mol. Cell Endocrinol. 2010, 315, 153–158. [Google Scholar] [CrossRef]
- Xi, Y.L.; Li, H.X.; Chen, C.; Liu, Y.Q.; Lv, H.M.; Dong, S.Q.; Luo, E.F.; Gu, M.B.; Liu, H. Baicalin attenuates high fat diet-induced insulin resistance and ectopic fat storage in skeletal muscle, through modulating the protein kinase B/Glycogen synthase kinase 3 beta pathway. Chin. J. Nat. Med. 2016, 14, 48–55. [Google Scholar] [CrossRef]
- Pal, M.; Khan, J.; Kumar, R.; Surolia, A.; Gupta, S. Testosterone supplementation improves insulin responsiveness in HFD fed male T2DM mice and potentiates insulin signaling in the skeletal muscle and C2C12 myocyte cell line. PLoS ONE 2019, 14, e0224162. [Google Scholar] [CrossRef]
- Ma, Z.; Zhong, Z.; Zheng, Z.; Shi, X.M.; Zhang, W. Inhibition of glycogen synthase kinase-3beta attenuates glucocorticoid-induced suppression of myogenic differentiation in vitro. PLoS ONE 2014, 9, e105528. [Google Scholar] [CrossRef]
- Theeuwes, W.F.; Gosker, H.R.; Langen, R.C.J.; Pansters, N.A.M.; Schols, A.; Remels, A.H.V. Inactivation of glycogen synthase kinase 3beta (GSK-3beta) enhances mitochondrial biogenesis during myogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt B, 2913–2926. [Google Scholar] [CrossRef]
- Ma, Q.; Jiang, L.; You, Y.; Ni, H.; Ma, L.; Lin, X.; Wang, Z.; Yan, W.; Xiao, X.; Li, X.; et al. Ketogenic diet ameliorates high-fat diet-induced insulin resistance in mouse skeletal muscle by alleviating endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2024, 702, 149559. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.; Milewski, W.M.; Hara, M.; Steiner, D.F.; Dey, A. GSK-3 inactivation or depletion promotes beta-cell replication via down regulation of the CDK inhibitor, p27 (Kip1). Islets 2011, 3, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Remedi, M.S.; Pappan, K.L.; Kwon, G.; Rohatgi, N.; Marshall, C.A.; McDaniel, M.L. Glycogen synthase kinase-3 and mammalian target of rapamycin pathways contribute to DNA synthesis, cell cycle progression, and proliferation in human islets. Diabetes 2009, 58, 663–672. [Google Scholar] [CrossRef]
- Boucher, M.J.; Selander, L.; Carlsson, L.; Edlund, H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J. Biol. Chem. 2006, 281, 6395–6403. [Google Scholar] [CrossRef]
- Mussmann, R.; Geese, M.; Harder, F.; Kegel, S.; Andag, U.; Lomow, A.; Burk, U.; Onichtchouk, D.; Dohrmann, C.; Austen, M. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J. Biol. Chem. 2007, 282, 12030–12037. [Google Scholar] [CrossRef]
- Sacco, F.; Seelig, A.; Humphrey, S.J.; Krahmer, N.; Volta, F.; Reggio, A.; Marchetti, P.; Gerdes, J.; Mann, M. Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets. Cell Metab. 2019, 29, 1422–1432.e3. [Google Scholar] [CrossRef]
- Qian, B.; Yang, Y.; Tang, N.; Wang, J.; Sun, P.; Yang, N.; Chen, F.; Wu, T.; Sun, T.; Li, Y.; et al. M1 macrophage-derived exosomes impair beta cell insulin secretion via miR-212-5p by targeting SIRT2 and inhibiting Akt/GSK-3beta/beta-catenin pathway in mice. Diabetologia 2021, 64, 2037–2051. [Google Scholar] [CrossRef]
- Aggarwal, R.; Peng, Z.; Zeng, N.; Silva, J.; He, L.; Chen, J.; Debebe, A.; Tu, T.; Alba, M.; Chen, C.Y.; et al. Chronic Exposure to Palmitic Acid Down-Regulates AKT in Beta-Cells through Activation of mTOR. Am. J. Pathol. 2022, 192, 130–145. [Google Scholar] [CrossRef]
- Lee, J.H.; Mellado-Gil, J.M.; Bahn, Y.J.; Pathy, S.M.; Zhang, Y.E.; Rane, S.G. Protection from beta-cell apoptosis by inhibition of TGF-beta/Smad3 signaling. Cell Death Dis. 2020, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Liu, L.Z.; Xin, Q.; Zhou, J.; Zhang, X.; Zhang, R.; Wu, Z.; Yi, J.; Dong, M. Downregulation of mTORC1 and Mcl-1 by lipid-oversupply contributes to islet beta-cell apoptosis and dysfunction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2023, 1868, 159332. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Hernandez, F.; Lucas, J.J.; Avila, J. GSK-3 Mouse Models to Study Neuronal Apoptosis and Neurodegeneration. Front. Mol. Neurosci. 2011, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Muneer, A. Wnt and GSK3 Signaling Pathways in Bipolar Disorder: Clinical and Therapeutic Implications. Clin. Psychopharmacol. Neurosci. 2017, 15, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Benzler, J.; Ganjam, G.K.; Kruger, M.; Pinkenburg, O.; Kutschke, M.; Stohr, S.; Steger, J.; Koch, C.E.; Olkrug, R.; Schwartz, M.W.; et al. Hypothalamic glycogen synthase kinase 3beta has a central role in the regulation of food intake and glucose metabolism. Biochem. J. 2012, 447, 175–184. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Turenne, G.A.; Price, B.D. Glycogen synthase kinase3 beta phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biol. 2001, 2, 12. [Google Scholar] [CrossRef]
- Hernandez, F.; Borrell, J.; Guaza, C.; Avila, J.; Lucas, J.J. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 2002, 83, 1529–1533. [Google Scholar] [CrossRef]
- Engel, T.; Hernandez, F.; Avila, J.; Lucas, J.J. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 2006, 26, 5083–5090. [Google Scholar] [CrossRef]
- Liang, Z.; Gong, X.; Ye, R.; Zhao, Y.; Yu, J.; Zhao, Y.; Bao, J. Long-Term High-Fat Diet Consumption Induces Cognitive Decline Accompanied by Tau Hyper-Phosphorylation and Microglial Activation in Aging. Nutrients 2023, 15, 250. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Correction for Sen et al., Sulfhydration of AKT triggers Tau-phosphorylation by activating glycogen synthase kinase 3beta in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2114879118. [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Fenech, R.K.; Hamstra, S.I.; Finch, M.S.; Ryan, C.R.; Marko, D.M.; Roy, B.D.; Fajardo, V.A.; MacPherson, R.E.K. Low-Dose Lithium Supplementation Influences GSK3beta Activity in a Brain Region Specific Manner in C57BL6 Male Mice. J. Alzheimers Dis. 2023, 91, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Kim, K.; Cho, H.C.; Lee, J.; Kim, E.K. Silencing of hypothalamic FGF11 prevents diet-induced obesity. Mol. Brain 2022, 15, 75. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; Oh, D.K.; Christiansen, L.; Nikoulina, S.E.; Kong, A.P.; Baxi, S.; Mudaliar, S.; Henry, R.R. Tissue-specific expression and regulation of GSK-3 in human skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E891–E898. [Google Scholar] [CrossRef]
- Yan, C.; Yang, H.; Wang, Y.; Dong, Y.; Yu, F.; Wu, Y.; Wang, W.; Adaku, U.; Lutfy, K.; Friedman, T.C.; et al. Increased glycogen synthase kinase-3beta and hexose-6-phosphate dehydrogenase expression in adipose tissue may contribute to glucocorticoid-induced mouse visceral adiposity. Int. J. Obes. 2016, 40, 1233–1241. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Meng, Y.; Zhang, C.; Di, L. GSK3-activated STAT5 regulates expression of SFRPs to modulate adipogenesis. FASEB J. 2018, 32, 4714–4726. [Google Scholar] [CrossRef]
- Geromella, M.S.; Ryan, C.R.; Braun, J.L.; Finch, M.S.; Maddalena, L.A.; Bagshaw, O.; Hockey, B.L.; Moradi, F.; Fenech, R.K.; Ryoo, J.; et al. Low-dose lithium supplementation promotes adipose tissue browning and sarco(endo)plasmic reticulum Ca(2+) ATPase uncoupling in muscle. J. Biol. Chem. 2022, 298, 102568. [Google Scholar] [CrossRef]
- Zhu, P.; Zhang, J.J.; Cen, Y.; Yang, Y.; Wang, F.; Gu, K.P.; Yang, H.T.; Wang, Y.Z.; Zou, Z.Q. High Endogenously Synthesized N-3 Polyunsaturated Fatty Acids in Fat-1 Mice Attenuate High-Fat Diet-Induced Insulin Resistance by Inhibiting NLRP3 Inflammasome Activation via Akt/GSK-3beta/TXNIP Pathway. Molecules 2022, 27, 6384. [Google Scholar] [CrossRef]
- Rao, R.; Hao, C.M.; Redha, R.; Wasserman, D.H.; McGuinness, O.P.; Breyer, M.D. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia 2007, 50, 452–460. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Metabolic Perturbation | Experimental Model | Phenotype | Mechanism | Ref. |
|---|---|---|---|---|
| Lipotoxic cardiomyopathy | Cardiac-specific GSK-3α heterozygous knockouts (GSK-3α cHKO) fed HFD for 14 weeks | Improved heart function in GSK-3α cHKO mice | GSK-3α inhibition | [27] |
| Atherosclerosis | Macrophage-specific GSK-3α- in low-density lipoprotein receptor knockout (Ldlr-/-) fed HFD for 16 weeks | Atherosclerotic plaque regression, reduced plaque inflammation, increased plaque stability | GSK-3α inhibition | [29] |
| Atherosclerosis | Endothelial-specific GSK-3β deletion in Apoe-/- mice fed a Western diet for eight weeks | Reduced vascular calcification in atherosclerotic plaques | GSK-3β inhibition | [30] |
| Myocardial ischemia | Yorkshire swine fed high-fat/high-cholesterol diet for four weeks + GSK-3β inhibitor | Elevated myocardial perfusion ratios and capillary and arterolic density | GSK-3β inhibition | [36] |
| Cardiac dysfunction | Cardiac-specific conditional GSK-3β knockouts fed HFD for 55 weeks | Normal cardiac function | GSK-3β inhibition | [42] |
| NAFLD | C57BL/6J mice fed an HFD for 20 weeks + bavachin (natural flavonoid) | Alleviated hepatic inflammation, ameliorated HFD-induced glucose intolerance and insulin resistance | GSK-3β inhibition | [47] |
| T2D | Zucker Diabetic Fatty (ZDF) rats | Enhanced insulin action on glucose transport | GSK-3 inhibition | [12] |
| T2D | Insulin-resistant, prediabetic obese Zucker rats | Enhanced oral glucose tolerance, whole body insulin sensitivity, and improved IRS-1-dependent insulin signaling | GSK-3 inhibition | [56] |
| T2D | Mice with beta cell deficiency of GSK-3β fed HFD for 12 weeks | Improved glucose tolerance and expanded beta cell mass with increased proliferation | GSK-3β deletion | [10] |
| T2D | Global conditional deletion of GSK3α/β+HFD for 16 weeks | Improved glucose tolerance in GSK-3β KO mice | GSK-3β inhibition | [22] |
| Obesity and glucose intolerance | Lepob/ob | Hyperphagia, Glucose intolerance | Increased hypothalamic GSK-3β activity | [75] |
| Cognitive impairment | C57/BL6 HFD for 10 months (60% kcal from fat) | Cognitive impairment, Tau hyperphosphorylation | Increased GSK-3β activity | [80] |
| Alzheimer’s disease | C57/BL6 HFD for 12 weeks +lithium- supplement | Improved insulin sensitivity | Reduced GSK-3β activity in prefrontal cortex | [84] |
| Adiposity | C57/BL6J mice, Hypercortisolism | Elevated adipose GSK3β and H6pdh expression contribute to 11ß-HSD1 mediating hypercortisolism associated with visceral adiposity | GSK-3β activity | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemon, J.J.; Ogbu, C.; Gupte, M. Role and Regulation of Glycogen Synthase Kinase-3 in Obesity-Associated Metabolic Perturbations. Kinases Phosphatases 2024, 2, 279-293. https://doi.org/10.3390/kinasesphosphatases2030018
Lemon JJ, Ogbu C, Gupte M. Role and Regulation of Glycogen Synthase Kinase-3 in Obesity-Associated Metabolic Perturbations. Kinases and Phosphatases. 2024; 2(3):279-293. https://doi.org/10.3390/kinasesphosphatases2030018
Chicago/Turabian StyleLemon, Jacob J., Comfort Ogbu, and Manisha Gupte. 2024. "Role and Regulation of Glycogen Synthase Kinase-3 in Obesity-Associated Metabolic Perturbations" Kinases and Phosphatases 2, no. 3: 279-293. https://doi.org/10.3390/kinasesphosphatases2030018
APA StyleLemon, J. J., Ogbu, C., & Gupte, M. (2024). Role and Regulation of Glycogen Synthase Kinase-3 in Obesity-Associated Metabolic Perturbations. Kinases and Phosphatases, 2(3), 279-293. https://doi.org/10.3390/kinasesphosphatases2030018