

Plants Metabolites as In Vitro Inhibitors of SARS-CoV-2 Targets: A Systematic Review and Computational Analysis

 , ,
, ,  , , , , , , , , ,
, , , , , , , , ,  and add
Show full author list
and add
Show full author list
Abstract

1. Introduction
2. Results and Discussion
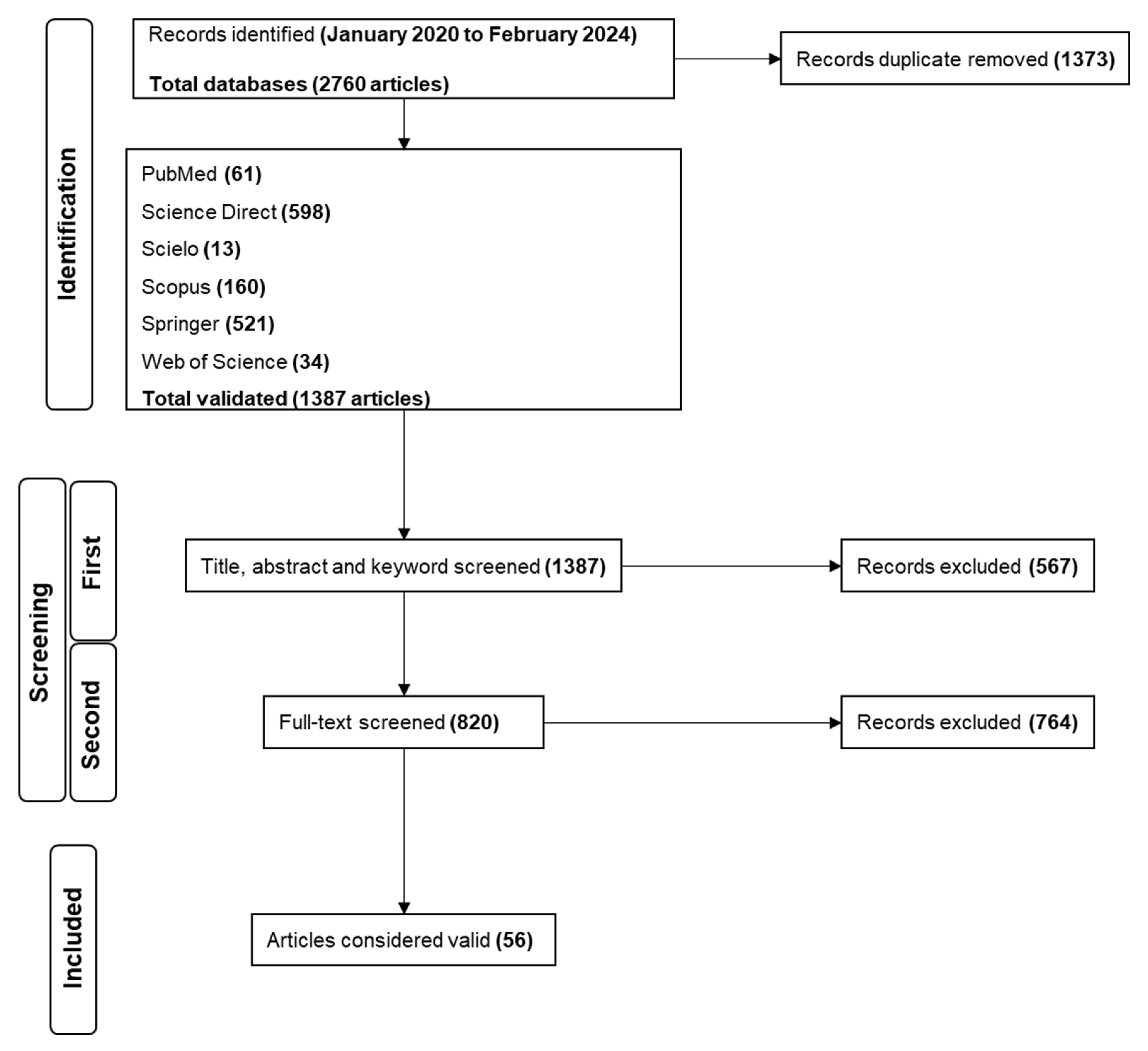
2.1. Search Results
2.2. Study Characteristics
2.2.1. Plant Compounds Evaluated as Anti-SARS-CoV-2
2.2.2. Controls and References Evaluated as Anti-SARS-CoV-2
2.2.3. SARS-CoV-2 Targets
2.2.4. Evaluation Methods for SARS-CoV-2 Targets
2.2.5. Anti-SARS-CoV-2 Effects
2.3. Plant Metabolites Against SARS-CoV-2
2.3.1. Phenolic Acids
2.3.2. Phenylethanoids
2.3.3. Chalcones
2.3.4. Flavonoids
2.3.5. Tannins
2.3.6. Coumarin
2.3.7. Stilbenes
2.3.8. Quinones
2.3.9. Lignoids
2.3.10. Alkaloids
2.3.11. Terpenes
2.3.12. Saponins
2.3.13. Other Compounds
2.4. ADMET and Drug-likeness Computational Analysis
3. Materials and Methods
3.1. Study Design
3.2. Search Strategy
3.3. Eligibility Criteria
3.3.1. Inclusion Criteria
3.3.2. Exclusion Criteria
3.4. Processing and Extraction of Data
3.5. Risk of Bias
3.6. Methodology Summary
3.7. Computational Analysis
4. Methodological Limitations of the Studies
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3CLpro | 3-Chymotrypsin-like Protease |
| ACE2 | Angiotensin-Converting Enzyme 2 |
| ADMET | Absorption, Distribution, Metabolism, Excretion, and Toxicity |
| BBB | Blood-brain barrier permeability |
| CDC | Centers for Disease Control and Prevention |
| CC50 | 50% Cytotoxic Concentration |
| COVID-19 | Coronavirus Disease 2019 |
| CoV | Coronaviruses |
| DL | Drug-likeness |
| EC50 | 50% Effective Concentration |
| E | Envelope protein |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| EV | Enterovirus |
| FRET | Fluorescence Resonance Energy Transfer |
| FU | Fraction unbound in plasma |
| H1N1 | Hemagglutinin type 1/Neuraminidase type 1 |
| HBV | Hepatitis B Virus |
| HCV | Hepatitis C virus |
| HIA | Human intestinal absorption |
| HIV | Human immunodeficiency virus type I |
| HTS | High-Throughput Screening |
| HSV | Herpes Simplex Virus |
| HTVS | High-Throughput Virtual Screening |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| IC50 | 50% Inhibitory Concentration |
| IFN-I | Type I Interferon |
| JEP | Japanese Encephalitis Virus |
| LBBD | Ligand-Based Drug Design |
| M | Membrane protein |
| MC | Medicinal Chemistry |
| MERS-CoV | Middle East Respiratory Syndrome Coronavirus |
| N | Nucleocapsid protein |
| Nsps | Non-structural Proteins |
| ORF | Open Reading Frame |
| PAINS | Pan-Assay Interference Compounds |
| PICO | Population, Intervention, Comparator, Outcome |
| PLpro | Papain-like Protease |
| PPB | Plasma protein binding |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
| pH | Potential of Hydrogen |
| ProTox3 | Plataforma de predição de toxicidade |
| RBD | Receptor-Binding Domain |
| RBM | Receptor-Binding motif |
| RMSD | Root Mean Square Deviation |
| RNA | Ribonucleic Acid |
| RdRp | RNA-dependent RNA Polymerase |
| RTqPCR | Reverse Transcription Quantitative Polymerase Chain Reaction |
| S protein | Spike protein |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| SBDD | Structure-Based Drug Design |
| SI | Selectivity Index |
| Scielo | Scientific Electronic Library Online |
| SMILES | Simplified Molecular Input Line Entry System |
| STD NMR | Saturation Transfer Difference Nuclear Magnetic Resonance |
| SwissADME | Plataforma de predição de propriedades ADME |
| TCM | Traditional Chinese Medicine |
| TMPRSS2 | Transmembrane Protease, Serine 2 |
| TPSA | Topological Polar Surface Area |
| Toxtree | Plataforma de predição toxicológica |
| WHO | World Health Organization |
| VDss | Volume of distribution |
| VOIs | Variants of Interest |
| VOCs | Variants of Concern |
| VUMs | Variants Under Monitoring |
| ZIKV | Zika virus |
References
- Lopes, A.J.O.; Calado, G.P.; Fróes, Y.N.; Araújo, S.A.D.; França, L.M.; Paes, A.M.D.A.; Morais, S.V.D.; Rocha, C.Q.D.; Vasconcelos, C.C. Plant Metabolites as SARS-CoV-2 Inhibitors Candidates: In Silico and In Vitro Studies. Pharmaceuticals 2022, 15, 1045. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Coronavirus (COVID-19) Dashboard. Available online: http://covid19.who.int (accessed on 2 May 2024).
- World Health Organization (WHO). Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 6 March 2025).
- Ma, K.C.; Castro, J.; Lambrou, A.S.; Rose, E.B.; Cook, P.W.; Batra, D.; Cubenas, C.; Hughes, L.J.; MacCannell, D.R.; Mandal, P.; et al. Genomic Surveillance for SARS-CoV-2 Variants: Circulation of Omicron XBB and JN.1 Lineages—United States, May 2023–September 2024. MMWR Morb. Mortal. Wkly. Rep. 2024, 73, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Lefrançois, T.; Malvy, D.; Atlani-Duault, L.; Benamouzig, D.; Druais, P.-L.; Yazdanpanah, Y.; Delfraissy, J.-F.; Lina, B. After 2 Years of the COVID-19 Pandemic, Translating One Health into Action Is Urgent. Lancet 2023, 401, 789–794. [Google Scholar] [CrossRef]
- Liao, H.; Lyon, C.J.; Ying, B.; Hu, T. Climate Change, Its Impact on Emerging Infectious Diseases and New Technologies to Combat the Challenge. Emerg. Microbes Infect. 2024, 13, 2356143. [Google Scholar] [CrossRef] [PubMed]
- Maccaro, A.; Audia, C.; Stokes, K.; Masud, H.; Sekalala, S.; Pecchia, L.; Piaggio, D. Pandemic Preparedness: A Scoping Review of Best and Worst Practices from COVID-19. Healthcare 2023, 11, 2572. [Google Scholar] [CrossRef]
- Kellerborg, K.; Brouwer, W.; Van Baal, P. Costs and Benefits of Interventions Aimed at Major Infectious Disease Threats: Lessons from the Literature. Eur. J. Health Econ. 2020, 21, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.S.; Ando, A.W.; Loch-Temzelides, T.; Vale, M.M.; Li, B.V.; Li, H.; Busch, J.; Chapman, C.A.; Kinnaird, M.; Nowak, K.; et al. The Costs and Benefits of Primary Prevention of Zoonotic Pandemics. Sci. Adv. 2022, 8, eabl4183. [Google Scholar] [CrossRef]
- Zhou, L.; Yan, W.; Li, S.; Yang, H.; Zhang, X.; Lu, W.; Liu, J.; Wang, Y. Cost-Effectiveness of Interventions for the Prevention and Control of COVID-19: Systematic Review of 85 Modelling Studies. J. Glob. Health 2022, 12, 05022. [Google Scholar] [CrossRef] [PubMed]
- Kaul, R.; Paul, P.; Kumar, S.; Büsselberg, D.; Dwivedi, V.D.; Chaari, A. Promising Antiviral Activities of Natural Flavonoids against SARS-CoV-2 Targets: Systematic Review. Int. J. Mol. Sci. 2021, 22, 11069. [Google Scholar] [CrossRef]
- Murali, M.; Gowtham, H.G.; Ansari, M.A.; Alomary, M.N.; Alghamdi, S.; Almehmadi, M.; Singh, S.B.; Shilpa, N.; Aiyaz, M.; Kalegowda, N.; et al. Repositioning Therapeutics for SARS-CoV-2: Virtual Screening of Plant-basedAnti-HIV Compounds as Possible Inhibitors against COVID-19 Viral RdRp. CPD 2022, 28, 969–980. [Google Scholar] [CrossRef]
- Basnet, S.; Marahatha, R.; Shrestha, A.; Bhattarai, S.; Katuwal, S.; Sharma, K.R.; Marasini, B.P.; Dahal, S.R.; Basnyat, R.C.; Patching, S.G.; et al. In Vitro and In Silico Studies for the Identification of Potent Metabolites of Some High-Altitude Medicinal Plants from Nepal Inhibiting SARS-CoV-2 Spike Protein. Molecules 2022, 27, 8957. [Google Scholar] [CrossRef] [PubMed]
- Lanrewaju, A.A.; Enitan-Folami, A.M.; Nyaga, M.M.; Sabiu, S.; Swalaha, F.M. Metabolites Profiling and Cheminformatics Bioprospection of Selected Medicinal Plants against the Main Protease and RNA-Dependent RNA Polymerase of SARS-CoV-2. J. Biomol. Struct. Dyn. 2023, 42, 6740–6760. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and Coronavirus Disease-2019 (COVID-19): The Epidemic and the Challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): A Global Pandemic and Treatment Strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Abiodun, O.A.; Ogboo, B.C.; Kola-Mustapha, A.T.; Attah, E.I.; Edemhanria, L.; Kumari, A.; Jaganathan, R.; Adelakun, N.S. Polypharmacology of Some Medicinal Plant Metabolites against SARS-CoV-2 and Host Targets: Molecular Dynamics Evaluation of NSP9 RNA Binding Protein. J. Biomol. Struct. Dyn. 2022, 40, 11467–11483. [Google Scholar] [CrossRef]
- Iqhrammullah, M.; Rizki, D.R.; Purnama, A.; Duta, T.F.; Harapan, H.; Idroes, R.; Ginting, B. Antiviral Molecular Targets of Essential Oils against SARS-CoV-2: A Systematic Review. Sci. Pharm. 2023, 91, 15. [Google Scholar] [CrossRef]
- Theodoridis, S.; Drakou, E.G.; Hickler, T.; Thines, M.; Nogues-Bravo, D. Evaluating Natural Medicinal Resources and Their Exposure to Global Change. Lancet Planet. Health 2023, 7, e155–e163. [Google Scholar] [CrossRef]
- Mohammadi, M.; Yahyapour, Y.; Nasrollahian, S.; Tayefeh-Arbab, M.H.; Javanian, M.; Rajabi Fadardi, M.; Mousavi Kani, S.N.; Honarvar Bakeshloo, P.; Saghebi, R.; Rajabnia, M.; et al. A Review on Herbal Secondary Metabolites Against COVID-19 Focusing on the Genetic Variants of SARS-CoV-2. Jundishapur J. Nat. Pharm. Prod. 2022, 17, e129618. [Google Scholar] [CrossRef]
- Aribisala, J.O.; Aruwa, C.E.; Uthman, T.O.; Nurain, I.O.; Idowu, K.; Sabiu, S. Cheminformatics Bioprospection of Broad Spectrum Plant Secondary Metabolites Targeting the Spike Proteins of Omicron Variant and Wild-Type SARS-CoV-2. Metabolites 2022, 12, 982. [Google Scholar] [CrossRef]
- Low, Z.; Lani, R.; Tiong, V.; Poh, C.; AbuBakar, S.; Hassandarvish, P. COVID-19 Therapeutic Potential of Natural Products. Int. J. Mol. Sci. 2023, 24, 9589. [Google Scholar] [CrossRef]
- Gomes, B.A.; Fernandes, D.A.; Mendonça, S.C.; Campos, M.F.; Da Fonseca, T.S.; Constant, L.E.C.; De Sousa, N.F.; Priscila Barros De Menezes, R.; De Oliveira, B.A.C.; Da Silva Costa, S.; et al. Predicting the Anti-SARS-CoV-2 Potential of Isoquinoline Alkaloids from Brazilian Siparunaceae Species Using Chemometric Tools. Int. J. Mol. Sci. 2025, 26, 633. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Facchiano, A.; Carbone, V. Food Plant Secondary Metabolites Antiviral Activity and Their Possible Roles in SARS-CoV-2 Treatment: An Overview. Molecules 2023, 28, 2470. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.A.; Gomes, B.A.; Da Silva, A.F.; De Carvalho, J.A.B.; Ricardo, N.S.; Leitão, S.G.; Leitão, G.G. Brazilian Medicinal Plants and Their Metabolites as Potential Antivirals Against SARS-CoV-2: A Systematic Review of Experimental Findings. Rev. Bras. Farmacogn. 2024, 34, 883–898. [Google Scholar] [CrossRef]
- Araújo, J.; De Sousa, L.; Sousa, A.; Bastos, R.; Santos, G.; Lage, M.; Stoyanov, S.; Passos, I.; De Azevedo, R.; Rocha, J. DFT, Molecular Docking, and ADME/Tox Screening Investigations of Market-Available Drugs against SARS-CoV-2. J. Braz. Chem. Soc. 2021, 32, 1628–1641. [Google Scholar] [CrossRef]
- Srivastava, V.; Yadav, A.; Sarkar, P. Molecular Docking and ADMET Study of Bioactive Compounds of Glycyrrhiza Glabra against Main Protease of SARS-CoV2. Mater. Today Proc. 2022, 49, 2999–3007. [Google Scholar] [CrossRef]
- Khaldan, A.; Bouamrane, S.; En-Nahli, F.; El-mernissi, R.; El Khatabi, K.; Hmamouchi, R.; Maghat, H.; Ajana, M.A.; Sbai, A.; Bouachrine, M.; et al. Prediction of Potential Inhibitors of SARS-CoV-2 Using 3D-QSAR, Molecular Docking Modeling and ADMET Properties. Heliyon 2021, 7, e06603. [Google Scholar] [CrossRef]
- Goyzueta-Mamani, L.D.; Barazorda-Ccahuana, H.L.; Mena-Ulecia, K.; Chávez-Fumagalli, M.A. Antiviral Activity of Metabolites from Peruvian Plants against SARS-CoV-2: An In Silico Approach. Molecules 2021, 26, 3882. [Google Scholar] [CrossRef]
- Amparo, T.R.; Seibert, J.B.; Almeida, T.C.; Costa, F.S.F.; Silveira, B.M.; Da Silva, G.N.; Dos Santos, O.D.H.; De Souza, G.H.B. In Silico Approach of Secondary Metabolites from Brazilian Herbal Medicines to Search for Potential Drugs against SARS-CoV-2. Phytother. Res. 2021, 35, 4297–4308. [Google Scholar] [CrossRef]
- Chan, W.K.B.; Olson, K.M.; Wotring, J.W.; Sexton, J.Z.; Carlson, H.A.; Traynor, J.R. In Silico Analysis of SARS-CoV-2 Proteins as Targets for Clinically Available Drugs. Sci. Rep. 2022, 12, 5320. [Google Scholar] [CrossRef]
- Das, S.; Singh, A.; Samanta, S.K.; Singha Roy, A. Naturally Occurring Anthraquinones as Potential Inhibitors of SARS-CoV-2 Main Protease: An Integrated Computational Study. Biologia 2022, 77, 1121–1134. [Google Scholar] [CrossRef]
- Rieder, A.S.; Deniz, B.F.; Netto, C.A.; Wyse, A.T.S. A Review of In Silico Research, SARS-CoV-2, and Neurodegeneration: Focus on Papain-Like Protease. Neurotox. Res. 2022, 40, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Jeong, G.U.; Song, H.; Yoon, G.Y.; Kim, D.; Kwon, Y.-C. Therapeutic Strategies Against COVID-19 and Structural Characterization of SARS-CoV-2: A Review. Front. Microbiol. 2020, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef]
- Ziebuhr, J. The Coronavirus Replicase. In Coronavirus Replication and Reverse Genetics; Enjuanes, L., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 287, pp. 57–94. ISBN 978-3-540-21494-6. [Google Scholar]
- Bafna, K.; Cioffi, C.L.; Krug, R.M.; Montelione, G.T. Structural Similarities between SARS-CoV2 3CLpro and Other Viral Proteases Suggest Potential Lead Molecules for Developing Broad Spectrum Antivirals. Front. Chem. 2022, 10, 948553. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; Van Belkum, M.J.; Joyce, M.A.; et al. Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and Inhibition of the Papain-like Protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Zhai, T.; Zhang, F.; Haider, S.; Kraut, D.; Huang, Z. An Integrated Computational and Experimental Approach to Identifying Inhibitors for SARS-CoV-2 3CL Protease. Front. Mol. Biosci. 2021, 8, 661424. [Google Scholar] [CrossRef] [PubMed]
- Glaab, E.; Manoharan, G.B.; Abankwa, D. Pharmacophore Model for SARS-CoV-2 3CLpro Small-Molecule Inhibitors and in Vitro Experimental Validation of Computationally Screened Inhibitors. J. Chem. Inf. Model. 2021, 61, 4082–4096. [Google Scholar] [CrossRef]
- Kuang, Y.; Ma, X.; Shen, W.; Rao, Q.; Yang, S. Discovery of 3CLpro Inhibitor of SARS-CoV-2 Main Protease. Future Sci. OA 2023, 9, FSO853. [Google Scholar] [CrossRef]
- De Souza, A.S.; De Souza, R.F.; Guzzo, C.R. Quantitative Structure-Activity Relationships, Molecular Docking and Molecular Dynamics Simulations Reveal Drug Repurposing Candidates as Potent SARS-CoV-2 Main Protease Inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 11339–11356. [Google Scholar] [CrossRef]
- Schake, P.; Dishnica, K.; Kaiser, F.; Leberecht, C.; Haupt, V.J.; Schroeder, M. An Interaction-Based Drug Discovery Screen Explains Known SARS-CoV-2 Inhibitors and Predicts New Compound Scaffolds. Sci. Rep. 2023, 13, 9204. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef]
- Pokharkar, O.; Zyryanov, G.V.; Tsurkan, M.V. Natural Products from Marine Actinomycete Genus Salinispora Might Inhibit 3CLpro and PLpro Proteins of SARS-CoV-2: An In Silico Evidence. Microbiol. Res. 2023, 14, 1907–1941. [Google Scholar] [CrossRef]
- Silva, R.C.; Freitas, H.F.; Campos, J.M.; Kimani, N.M.; Silva, C.H.T.P.; Borges, R.S.; Pita, S.S.R.; Santos, C.B.R. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. Int. J. Mol. Sci. 2021, 22, 11739. [Google Scholar] [CrossRef] [PubMed]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 Main Protease for Treatment of COVID-19: Covalent Inhibitors Structure–Activity Relationship Insights and Evolution Perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar] [CrossRef] [PubMed]
- De Freitas Amorim, V.M.; De Souza, R.F.; Guzzo, C.R.; De Souza, A.S. Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins. COVID 2023, 3, 622–636. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Wasilewicz, A.; Kirchweger, B.; Bojkova, D.; Abi Saad, M.J.; Langeder, J.; Bütikofer, M.; Adelsberger, S.; Grienke, U.; Cinatl, J., Jr.; Petermann, O.; et al. Identification of Natural Products Inhibiting SARS-CoV-2 by Targeting Viral Proteases: A Combined in Silico and in Vitro Approach. J. Nat. Prod. 2023, 86, 264–275. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Li, C.; Teng, X.; Qi, Y.; Tang, B.; Shi, H.; Ma, X.; Lai, L. Conformational Flexibility of a Short Loop near the Active Site of the SARS-3CLpro Is Essential to Maintain Catalytic Activity. Sci. Rep. 2016, 6, 20918. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The Development of Coronavirus 3C-Like Protease (3CLpro) Inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef]
- Banerjee, R.; Perera, L.; Tillekeratne, L.M.V. Potential SARS-CoV-2 Main Protease Inhibitors. Drug Discov. Today 2021, 26, 804–816. [Google Scholar] [CrossRef]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the Main Protease of SARS-CoV-2 (Mpro) by Repurposing/Designing Drug-like Substances and Utilizing Nature’s Toolbox of Bioactive Compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef]
- McClain, C.B.; Vabret, N. SARS-CoV-2: The Many Pros of Targeting PLpro. Sig. Transduct. Target. Ther. 2020, 5, 223. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti–COVID-19 Drug Design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-Coronavirus Papain-like Protease: Structure, Function and Inhibition by Designed Antiviral Compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ye, G.; Si, Y.; Shen, Z.; Liu, Z.; Shi, Y.; Xiao, S.; Fu, Z.F.; Peng, G. Structure of the Multiple Functional Domains from Coronavirus Nonstructural Protein 3. Emerg. Microbes Infect. 2021, 10, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of Severe Acute Respiratory Syndrome Coronavirus Replicase Products and Characterization of Papain-Like Protease Activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of Papain-like Protease from SARS-CoV-2 and Its Complexes with Non-Covalent Inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Huynh, T.; Cornell, W.; Luan, B. In Silico Exploration of Inhibitors for SARS-CoV-2’s Papain-Like Protease. Front. Chem. 2021, 8, 624163. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef]
- Békés, M.; van der Heden van Noort, G.J.; Ekkebus, R.; Ovaa, H.; Huang, T.T.; Lima, C.D. Recognition of Lys48-Linked Di-Ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Mol. Cell 2016, 62, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.-T.K.; Baker, S.C.; et al. Regulation of IRF-3-Dependent Innate Immunity by the Papain-like Protease Domain of the Severe Acute Respiratory Syndrome Coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, Ubiquitin-like Modifiers, and Deubiquitination in Viral Infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X.; Zheng, Y.; Yang, Y.; Xing, Y.; Chen, Z. SARS Coronavirus Papain-like Protease Inhibits the Type I Interferon Signaling Pathway through Interaction with the STING-TRAF3-TBK1 Complex. Protein Cell 2014, 5, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.; Schäfer, A.; Pham, A.; Frieman, M. The SARS Coronavirus Papain like Protease Can Inhibit IRF3 at a Post Activation Step That Requires Deubiquitination Activity. Virol. J. 2014, 11, 209. [Google Scholar] [CrossRef]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the Activation and Attenuation of Innate Antiviral Immunity. JEM 2016, 213, 1–13. [Google Scholar] [CrossRef]
- Li, S.-W.; Wang, C.-Y.; Jou, Y.-J.; Yang, T.-C.; Huang, S.-H.; Wan, L.; Lin, Y.-J.; Lin, C.-W. SARS Coronavirus Papain-like Protease Induces Egr-1-Dependent up-Regulation of TGF-Β1 via ROS/P38 MAPK/STAT3 Pathway. Sci. Rep. 2016, 6, 25754. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 Proteases PLpro and 3CLpro Cleave IRF3 and Critical Modulators of Inflammatory Pathways (NLRP12 and TAB1): Implications for Disease Presentation across Species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Ma, C.; Wang, J. Validation and Invalidation of SARS-CoV-2 Papain-like Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2022, 5, 102–109. [Google Scholar] [CrossRef]
- Mahmoudvand, S.; Shokri, S. Interactions between SARS Coronavirus 2 Papain-like Protease and Immune System: A Potential Drug Target for the Treatment of COVID-19. Scand. J. Immunol. 2021, 94, e13044. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A Noncovalent Class of Papain-like Protease/Deubiquitinase Inhibitors Blocks SARS Virus Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Xia, Z.; Lambrinidis, G.; Townsend, J.A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, Y.; Lei, Y.; Wang, R.; Zhan, M.; Liu, J.; An, Y.; Zhou, Y.; Zhan, J.; Yin, F.; et al. Design and Evaluation of a Novel Peptide–Drug Conjugate Covalently Targeting SARS-CoV-2 Papain-like Protease. J. Med. Chem. 2022, 65, 876–884. [Google Scholar] [CrossRef]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879.e5. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.S.; Zhu, M.; Cloney-Clark, S.; Zhou, H.; et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Goc, A.; Sumera, W.; Rath, M.; Niedzwiecki, A. Phenolic Compounds Disrupt Spike-Mediated Receptor-Binding and Entry of SARS-CoV-2 Pseudo-Virions. PLoS ONE 2021, 16, e0253489. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, H.; Guo, L.; Xie, D.; He, H.; Zhang, H.; Liu, Y.; Peng, L.; Zheng, L.; Lu, W.; et al. Potential Inhibitors for Blocking the Interaction of the Coronavirus SARS-CoV-2 Spike Protein and Its Host Cell Receptor ACE2. J. Transl. Med. 2022, 20, 314. [Google Scholar] [CrossRef]
- Katuwal, S.; Upadhyaya, S.R.; Marahatha, R.; Shrestha, A.; Regmi, B.P.; Khadayat, K.; Basnet, S.; Basnyat, R.C.; Parajuli, N. In Silico Study of Coumarins: Wedelolactone as a Potential Inhibitor of the Spike Protein of the SARS-CoV-2 Variants. J. Trop. Med. 2023, 2023, 4771745. [Google Scholar] [CrossRef]
- Delgado-Maldonado, T.; Gonzalez-Morales, L.D.; Juarez-Saldivar, A.; Lara-Ramírez, E.E.; Rojas-Verde, G.; Moreno-Rodriguez, A.; Bandyopadhyay, D.; Rivera, G. Structure-Based Virtual Screening from Natural Products as Inhibitors of SARS-CoV-2 Spike Protein and ACE2 Receptor Binding and Their Biological Evaluation In Vitro. Med. Chem. 2024, 20, 546–553. [Google Scholar] [CrossRef]
- Elfiky, A.A. SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Targeting: An in Silico Perspective. J. Biomol. Struct. Dyn. 2020, 39, 3204–3212. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of Replicating SARS-CoV-2 Polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Lamb, Y.N. Remdesivir: First Approval. Drugs 2020, 80, 1355–1363. [Google Scholar] [CrossRef]
- Rubin, D.; Chan-Tack, K.; Farley, J.; Sherwat, A. FDA Approval of Remdesivir—A Step in the Right Direction. N. Engl. J. Med. 2020, 383, 2598–2600. [Google Scholar] [CrossRef]
- Mishra, A.; Rathore, A.S. RNA Dependent RNA Polymerase (RdRp) as a Drug Target for SARS-CoV2. J. Biomol. Struct. Dyn. 2022, 40, 6039–6051. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Suručić, R.; Travar, M.; Petković, M.; Tubić, B.; Stojiljković, M.P.; Grabež, M.; Šavikin, K.; Zdunić, G.; Škrbić, R. Pomegranate Peel Extract Polyphenols Attenuate the SARS-CoV-2 S-Glycoprotein Binding Ability to ACE2 Receptor: In Silico and in Vitro Studies. Bioorg. Chem. 2021, 114, 105145. [Google Scholar] [CrossRef]
- Alves, J.; Engel, L.; De Vasconcelos Cabral, R.; Rodrigues, E.L.; De Jesus Ribeiro, L.; Higa, L.M.; Da Costa Ferreira Júnior, O.; Castiñeiras, T.M.P.P.; De Carvalho Leitão, I.; Tanuri, A.; et al. A Bioluminescent and Homogeneous SARS-CoV-2 Spike RBD and hACE2 Interaction Assay for Antiviral Screening and Monitoring Patient Neutralizing Antibody Levels. Sci. Rep. 2021, 11, 18428. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, I.; Cassel, J.; Register, E.T.; Zhou, X.Y.; Messick, T.E.; Keeney, F.; Lu, L.D.; Beattie, K.D.; Rali, T.; Tebas, P.; et al. The Natural Stilbenoid (–)-Hopeaphenol Inhibits Cellular Entry of SARS-CoV-2 USA-WA1/2020, B.1.1.7, and B.1.351 Variants. Antimicrob. Agents Chemother. 2021, 65, e00772-21. [Google Scholar] [CrossRef]
- Invernizzi, L.; Moyo, P.; Cassel, J.; Isaacs, F.J.; Salvino, J.M.; Montaner, L.J.; Tietjen, I.; Maharaj, V. Use of Hyphenated Analytical Techniques to Identify the Bioactive Constituents of Gunnera perpensa L., a South African Medicinal Plant, Which Potently Inhibit SARS-CoV-2 Spike Glycoprotein–Host ACE2 Binding. Anal. Bioanal. Chem. 2022, 414, 3971–3985. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Huang, V.; Chao, T.-C.; Hsiao, C.-D.; Lin, A.; Chang, M.-F.; Chow, L.-P. Screening of Drugs by FRET Analysis Identifies Inhibitors of SARS-CoV 3CL Protease. Biochem. Biophys. Res. Commun. 2005, 333, 194–199. [Google Scholar] [CrossRef]
- Cihlova, B.; Huskova, A.; Böserle, J.; Nencka, R.; Boura, E.; Silhan, J. High-Throughput Fluorescent Assay for Inhibitor Screening of Proteases from RNA Viruses. Molecules 2021, 26, 3792. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence Resonance Energy Transfer (FRET) Microscopy Imaging of Live Cell Protein Localizations. J. Cell Sci. 2003, 160, 629–633. [Google Scholar] [CrossRef]
- Sabariegos, R.; Picazo, F.; Domingo, B.; Franco, S.; Martinez, M.-A.; Llopis, J. Fluorescence Resonance Energy Transfer-Based Assay for Characterization of Hepatitis C Virus NS3-4A Protease Activity in Live Cells. Antimicrob. Agents Chemother. 2009, 53, 728–734. [Google Scholar] [CrossRef]
- Ferreira, S.S.; Antunes, M.S. Re-Engineering Plant Phenylpropanoid Metabolism with the Aid of Synthetic Biosensors. Front. Plant Sci. 2021, 12, 701385. [Google Scholar] [CrossRef]
- Puttaswamy, H.; Gowtham, H.G.; Ojha, M.D.; Yadav, A.; Choudhir, G.; Raguraman, V.; Kongkham, B.; Selvaraju, K.; Shareef, S.; Gehlot, P.; et al. In Silico Studies Evidenced the Role of Structurally Diverse Plant Secondary Metabolites in Reducing SARS-CoV-2 Pathogenesis. Sci. Rep. 2020, 10, 20584. [Google Scholar] [CrossRef]
- Wang, F.; Liu, D.; Gao, D.; Yuan, J.; Zhao, J.; Yuan, S.; Cen, Y.; Lin, G.-Q.; Zhao, J.; Tian, P. Discovery of Natural Catechol Derivatives as Covalent SARS-CoV-2 3CLpro Inhibitors. Int. J. Biol. Macromol. 2024, 264, 130377. [Google Scholar] [CrossRef] [PubMed]
- Shahhamzehei, N.; Abdelfatah, S.; Efferth, T. In Silico and In Vitro Identification of Pan-Coronaviral Main Protease Inhibitors from a Large Natural Product Library. Pharmaceuticals 2022, 15, 308. [Google Scholar] [CrossRef]
- Krüger, N.; Kronenberger, T.; Xie, H.; Rocha, C.; Pöhlmann, S.; Su, H.; Xu, Y.; Laufer, S.A.; Pillaiyar, T. Discovery of Polyphenolic Natural Products as SARS-CoV-2 Mpro Inhibitors for COVID-19. Pharmaceuticals 2023, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, N.; Mehroze, A.; Sarwar, W.; Arshad, U.; Parveen, S.; Rashid, M.; Farooq, A.; Rafiq, N.; Wondmie, G.F.; Bin Jardan, Y.A.; et al. Exploration of Phenolic Acid Derivatives as Inhibitors of SARS-CoV-2 Main Protease and Receptor Binding Domain: Potential Candidates for Anti-SARS-CoV-2 Therapy. Front. Chem. 2023, 11, 1251529. [Google Scholar] [CrossRef]
- Gancia, E.; Montana, J.G.; Manallack, D.T. Theoretical Hydrogen Bonding Parameters for Drug Design. J. Mol. Graph. Model. 2001, 19, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ghiandoni, G.M.; Caldeweyher, E. Fast Calculation of Hydrogen-Bond Strengths and Free Energy of Hydration of Small Molecules. Sci. Rep. 2023, 13, 4143. [Google Scholar] [CrossRef]
- Wang, R.; Chen, X.; Li, H.; Chen, X.; Sun, D.; Yu, D.; Lu, J.; Xie, Y.; Zhang, Q.; Xu, J.; et al. Danshensu Inhibits SARS-CoV-2 by Targeting Its Main Protease as a Specific Covalent Inhibitor and Discovery of Bifunctional Compounds Eliciting Antiviral and Anti-Inflammatory Activity. Int. J. Biol. Macromol. 2024, 257, 128623. [Google Scholar] [CrossRef]
- Hicks, E.G.; Kandel, S.E.; Lampe, J.N. Identification of Aloe-Derived Natural Products as Prospective Lead Scaffolds for SARS-CoV-2 Main Protease (Mpro) Inhibitors. Bioorg Med. Chem. Lett. 2022, 66, 128732. [Google Scholar] [CrossRef]
- Chen, Z.; Cui, Q.; Cooper, L.; Zhang, P.; Lee, H.; Chen, Z.; Wang, Y.; Liu, X.; Rong, L.; Du, R. Ginkgolic Acid and Anacardic Acid Are Specific Covalent Inhibitors of SARS-CoV-2 Cysteine Proteases. Cell Biosci. 2021, 11, 45. [Google Scholar] [CrossRef]
- Lee, H.; Mittal, A.; Patel, K.; Gatuz, J.L.; Truong, L.; Torres, J.; Mulhearn, D.C.; Johnson, M.E. Identification of Novel Drug Scaffolds for Inhibition of SARS-CoV 3-Chymotrypsin-like Protease Using Virtual and High-Throughput Screenings. Bioorg. Med. Chem. 2014, 22, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Soledade, M.; Pedras, C.; Zheng, Q. The Chemistry of Arabidopsis thaliana. In Comprehensive Natural Products II; Elsevier: Amsterdam, The Netherlands, 2010; pp. 1297–1315. ISBN 978-0-08-045382-8. [Google Scholar]
- Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.M.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; Elfaky, M.A. Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals 2021, 14, 213. [Google Scholar] [CrossRef]
- Srinivasan, V.; Brognaro, H.; Prabhu, P.R.; De Souza, E.E.; Günther, S.; Reinke, P.Y.A.; Lane, T.J.; Ginn, H.; Han, H.; Ewert, W.; et al. Antiviral Activity of Natural Phenolic Compounds in Complex at an Allosteric Site of SARS-CoV-2 Papain-like Protease. Commun. Biol. 2022, 5, 805. [Google Scholar] [CrossRef]
- Elkhalifa, D.; Al-Hashimi, I.; Al Moustafa, A.-E.; Khalil, A. A Comprehensive Review on the Antiviral Activities of Chalcones. J. Drug Target. 2021, 29, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khan, J.; Dukhyil, A.A.B.; Alarousy, R.M.I.I.; Attah, E.I.; Sharma, T.; Khairnar, S.J.; Bendale, A.R. Chalcone Scaffolds, Bioprecursors of Flavonoids: Chemistry, Bioactivities, and Pharmacokinetics. Molecules 2021, 26, 7177. [Google Scholar] [CrossRef]
- Li, Z.; Xie, H.; Tang, C.; Feng, L.; Ke, C.; Xu, Y.; Su, H.; Yao, S.; Ye, Y. Flavonoids from the Roots and Rhizomes of Sophora Tonkinensis and Their in Vitro Anti-SARS-CoV-2 Activity. Chin. J. Integr. Med. 2023, 21, 65–80. [Google Scholar] [CrossRef]
- Herzog, A.-M.; Göbel, K.; Marongiu, L.; Ruetalo, N.; Alonso, M.C.; Leischner, C.; Busch, C.; Burkard, M.; Lauer, U.M.; Geurink, P.P.; et al. Compounds Derived from Humulus Lupulus Inhibit SARS-CoV-2 Papain-like Protease and Virus Replication. Phytomedicine 2024, 123, 155176. [Google Scholar] [CrossRef]
- Khamto, N.; Utama, K.; Tateing, S.; Sangthong, P.; Rithchumpon, P.; Cheechana, N.; Saiai, A.; Semakul, N.; Punyodom, W.; Meepowpan, P. Discovery of Natural Bisbenzylisoquinoline Analogs from the Library of Thai Traditional Plants as SARS-CoV-2 3CLPro Inhibitors: In Silico Molecular Docking, Molecular Dynamics, and In Vitro Enzymatic Activity. J. Chem. Inf. Model. 2023, 63, 2104–2121. [Google Scholar] [CrossRef] [PubMed]
- Kanjanasirirat, P.; Suksatu, A.; Manopwisedjaroen, S.; Munyoo, B.; Tuchinda, P.; Jearawuttanakul, K.; Seemakhan, S.; Charoensutthivarakul, S.; Wongtrakoongate, P.; Rangkasenee, N.; et al. High-Content Screening of Thai Medicinal Plants Reveals Boesenbergia Rotunda Extract and Its Component Panduratin A as Anti-SARS-CoV-2 Agents. Sci. Rep. 2020, 10, 19963. [Google Scholar] [CrossRef]
- Ma, M.; Luan, X.; Zheng, H.; Wang, X.; Wang, S.; Shen, T.; Ren, D. A Mulberry Diels-Alder-Type Adduct, Kuwanon M, Triggers Apoptosis and Paraptosis of Lung Cancer Cells through Inducing Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2023, 24, 1015. [Google Scholar] [CrossRef]
- Lin, Y.; Zang, R.; Ma, Y.; Wang, Z.; Li, L.; Ding, S.; Zhang, R.; Wei, Z.; Yang, J.; Wang, X. Xanthohumol Is a Potent Pan-Inhibitor of Coronaviruses Targeting Main Protease. Int. J. Mol. Sci. 2021, 22, 12134. [Google Scholar] [CrossRef] [PubMed]
- Badshah, S.L.; Faisal, S.; Muhammad, A.; Poulson, B.G.; Emwas, A.H.; Jaremko, M. Antiviral Activities of Flavonoids. Biomed. Pharmacother. 2021, 140, 111596. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Hu, H.; Gao, M.; et al. Anti-SARS-CoV-2 Activities in Vitro of Shuanghuanglian Preparations and Bioactive Ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Hua, Y.; Zhang, W.; Zhou, X.; He, J.; Chen, D. Tannic Acid as a Natural Ferroptosis Inhibitor: Mechanisms and Beneficial Role of 3’-O-Galloylation. ChemistrySelect 2021, 6, 1562–1569. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhu, G.-H.; Wang, H.-N.; Hu, Q.; Chen, L.-L.; Guan, X.-Q.; Li, H.-L.; Chen, H.-Z.; Tang, H.; Ge, G.-B. Discovery of Naturally Occurring Inhibitors against SARS-CoV-2 3CLpro from Ginkgo biloba Leaves via Large-Scale Screening. Fitoterapia 2021, 152, 104909. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kwon, E.-B.; Kim, B.; Chung, H.-S.; Choi, G.; Kim, Y.-H.; Choi, J.-G. Mulberry Component Kuwanon C Exerts Potent Therapeutic Efficacy In Vitro against COVID-19 by Blocking the SARS-CoV-2 Spike S1 RBD:ACE2 Receptor Interaction. Int. J. Mol. Sci. 2022, 23, 12516. [Google Scholar] [CrossRef]
- Yi, Y.; Zhang, M.; Xue, H.; Yu, R.; Bao, Y.-O.; Kuang, Y.; Chai, Y.; Ma, W.; Wang, J.; Shi, X.; et al. Schaftoside Inhibits 3CLpro and PLpro of SARS-CoV-2 Virus and Regulates Immune Response and Inflammation of Host Cells for the Treatment of COVID-19. Acta Pharm. Sin. B 2022, 12, 4154–4164. [Google Scholar] [CrossRef]
- Zhu, D.; Su, H.; Ke, C.; Tang, C.; Witt, M.; Quinn, R.J.; Xu, Y.; Liu, J.; Ye, Y. Efficient Discovery of Potential Inhibitors for SARS-CoV-2 3C-like Protease from Herbal Extracts Using a Native MS-Based Affinity-Selection Method. J. Pharm. Biomed. Anal. 2022, 209, 114538. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, M.; Fu, B.; Li, M.; Yang, J.; Zhang, Z.; Li, C.; Zhang, H.; Wu, H.; Xue, W.; et al. Structure-Based Discovery of the SARS-CoV-2 Main Protease Noncovalent Inhibitors from Traditional Chinese Medicine. J. Chem. Inf. Model. 2024, 64, 1319–1330. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, W.; Hu, Y.; Ding, T.; Zafar, M.M.; Jia, X.; Zhang, L.; Ren, M.; Li, F.; Wang, W. Cotton Flower Metabolites Inhibit SARS-CoV-2 Main Protease. FEBS Open Bio 2022, 12, 1886–1895. [Google Scholar] [CrossRef]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of Pyrogallol as a Warhead in Design of Covalent Inhibitors for the SARS-CoV-2 3CL Protease. Nat. Commun. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, D.-Y.; Scartelli, C.; Xie, H.; Merrill-Skoloff, G.; Yang, M.; Sun, L.; Saeed, M.; Flaumenhaft, R. Plant Flavonoid Inhibition of SARS-CoV-2 Main Protease and Viral Replication. iScience 2023, 26, 107602. [Google Scholar] [CrossRef]
- Li, H.; Sun, M.; Lei, F.; Liu, J.; Chen, X.; Li, Y.; Wang, Y.; Lu, J.; Yu, D.; Gao, Y.; et al. Methyl Rosmarinate Is an Allosteric Inhibitor of SARS-CoV-2 3 CL Protease as a Potential Candidate against SARS-Cov-2 Infection. Antiviral Res. 2024, 224, 105841. [Google Scholar] [CrossRef] [PubMed]
- Dayem, A.A.; Choi, H.Y.; Kim, Y.B.; Cho, S.-G. Antiviral Effect of Methylated Flavonol Isorhamnetin against Influenza. PLoS ONE 2015, 10, e0121610. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Sayed, A.M.; Hassan, H.M.; Youssif, K.A.; Gaber, Y.; Moatasim, Y.; Kutkat, O.; Mostafa, A.; Ali, M.A.; Rateb, M.E.; et al. Cnicin as an Anti-SARS-CoV-2: An Integrated In Silico and In Vitro Approach for the Rapid Identification of Potential COVID-19 Therapeutics. Antibiotics 2021, 10, 542. [Google Scholar] [CrossRef]
- Hu, X.; Wang, M.; Pan, Y.; Xie, Y.; Han, J.; Zhang, X.; Niayale, R.; He, H.; Li, Q.; Zhao, T.; et al. Anti-Inflammatory Effect of Astragalin and Chlorogenic Acid on Escherichia Coli-Induced Inflammation of Sheep Endometrial Epithelium Cells. Front. Vet. Sci. 2020, 7, 201. [Google Scholar] [CrossRef]
- Du, A.; Zheng, R.; Disoma, C.; Li, S.; Chen, Z.; Li, S.; Liu, P.; Zhou, Y.; Shen, Y.; Liu, S.; et al. Epigallocatechin-3-Gallate, an Active Ingredient of Traditional Chinese Medicines, Inhibits the 3CLpro Activity of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 176, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.-H.; Rao, Y.K.; Tzeng, Y.-M. Inhibitory Effects of Flavonol Glycosides from Cinnamomum osmophloeum on Inflammatory Mediators in LPS/IFN-γ-Activated Murine Macrophages. Bioorg. Med. Chem. 2005, 13, 2381–2388. [Google Scholar] [CrossRef]
- Choi, J.-G.; Kim, Y.S.; Kim, J.H.; Chung, H.-S. Antiviral Activity of Ethanol Extract of Geranii Herba and Its Components against Influenza Viruses via Neuraminidase Inhibition. Sci. Rep. 2019, 9, 12132. [Google Scholar] [CrossRef]
- Liao, Q.; Chen, Z.; Tao, Y.; Zhang, B.; Wu, X.; Yang, L.; Wang, Q.; Wang, Z. An Integrated Method for Optimized Identification of Effective Natural Inhibitors against SARS-CoV-2 3CLpro. Sci. Rep. 2021, 11, 22796. [Google Scholar] [CrossRef]
- Schwarz, S.; Sauter, D.; Wang, K.; Zhang, R.; Sun, B.; Karioti, A.; Bilia, A.; Efferth, T.; Schwarz, W. Kaempferol Derivatives as Antiviral Drugs against the 3a Channel Protein of Coronavirus. Planta Med. 2014, 80, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Smeriglio, A.; Iraci, N.; Denaro, M.; Mandalari, G.; Giofrè, S.V.; Trombetta, D. Synergistic Combination of Citrus Flavanones as Strong Antioxidant and COX-Inhibitor Agent. Antioxidants 2023, 12, 972. [Google Scholar] [CrossRef]
- Tutunchi, H.; Naeini, F.; Ostadrahimi, A.; Hosseinzadeh-Attar, M.J. Naringenin, a Flavanone with Antiviral and Anti-inflammatory Effects: A Promising Treatment Strategy against COVID-19. Phytother. Res. 2020, 34, 3137–3147. [Google Scholar] [CrossRef] [PubMed]
- Khamto, N.; Utama, K.; Boontawee, P.; Janthong, A.; Tatieng, S.; Arthan, S.; Choommongkol, V.; Sangthong, P.; Yenjai, C.; Suree, N.; et al. Inhibitory Activity of Flavonoid Scaffolds on SARS-CoV-2 3CLpro: Insights from the Computational and Experimental Investigations. J. Chem. Inf. Model. 2024, 64, 874–891. [Google Scholar] [CrossRef]
- Luo, Y.; Jian, Y.; Liu, Y.; Jiang, S.; Muhammad, D.; Wang, W. Flavanols from Nature: A Phytochemistry and Biological Activity Review. Molecules 2022, 27, 719. [Google Scholar] [CrossRef]
- Liga, S.; Paul, C.; Péter, F. Flavonoids: Overview of Biosynthesis, Biological Activity, and Current Extraction Techniques. Plants 2023, 12, 2732. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Martínez, J.; Jiménez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcón, D.; Vega, S.; Calvo, C.; Benítez, C.; Abian, O.; Velázquez-Campoy, A.; Thomson, T.M.; et al. Discovery of Diverse Natural Products as Inhibitors of SARS-CoV-2 Mpro Protease through Virtual Screening. J. Chem. Inf. Model. 2021, 61, 6094–6106. [Google Scholar] [CrossRef]
- Tun, M.M.N.; Luvai, E.; Nwe, K.M.; Toume, K.; Mizukami, S.; Hirayama, K.; Komatsu, K.; Morita, K. Anti-SARS-CoV-2 Activity of Various PET-Bottled Japanese Green Teas and Tea Compounds in Vitro. Arch. Virol. 2022, 167, 1547–1557. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Wang, W.; Ke, J.-P.; Zhang, P.; Chu, G.-X.; Bao, G.-H. Discovery of Camellia Sinensis Catechins as SARS-CoV-2 3CL Protease Inhibitors through Molecular Docking, Intra and Extra Cellular Assays. Phytomedicine 2022, 96, 153853. [Google Scholar] [CrossRef]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2009. [Google Scholar]
- Křížová, L.; Dadáková, K.; Kašparovská, J.; Kašparovský, T. Isoflavones. Molecules 2019, 24, 1076. [Google Scholar] [CrossRef]
- Bo, S.; Chang, S.K.; Chen, Y.; Sheng, Z.; Jiang, Y.; Yang, B. The Structure Characteristics, Biosynthesis and Health Benefits of Naturally Occurring Rare Flavonoids. Crit. Rev. Food Sci. Nutr. 2024, 64, 2490–2512. [Google Scholar] [CrossRef] [PubMed]
- Aly, S.H.; Elissawy, A.M.; Eldahshan, O.A.; Elshanawany, M.A.; Efferth, T.; Singab, A.N.B. The Pharmacology of the Genus Sophora (Fabaceae): An Updated Review. Phytomedicine 2019, 64, 153070. [Google Scholar] [CrossRef] [PubMed]
- Abd-Alla, H.I.; Kutkat, O.; Sweelam, H.M.; Eldehna, W.M.; Mostafa, M.A.; Ibrahim, M.T.; Moatasim, Y.; GabAllah, M.; Al-Karmalawy, A.A. Investigating the Potential Anti-SARS-CoV-2 and Anti-MERS-CoV Activities of Yellow Necklacepod among Three Selected Medicinal Plants: Extraction, Isolation, Identification, In Vitro, Modes of Action, and Molecular Docking Studies. Metabolites 2022, 12, 1109. [Google Scholar] [CrossRef]
- Püntener, A.G.; Schlesinger, U. Natural Dyes. In Colorants for Non-Textile Applications; Elsevier: Amsterdam, The Netherlands, 2000; pp. 382–455. ISBN 978-0-444-82888-0. [Google Scholar]
- Li, J.; Liao, W.; Huang, D.; Ou, M.; Chen, T.; Wang, X.; Zhao, R.; Zhang, L.; Mei, L.; Liu, J.; et al. Current Strategies of Detecting Aβ Species and Inhibiting Aβ Aggregation: Status and Prospects. Coord. Chem. Rev. 2023, 495, 215375. [Google Scholar] [CrossRef]
- Low, K.J.Y.; Venkatraman, A.; Mehta, J.S.; Pervushin, K. Molecular Mechanisms of Amyloid Disaggregation. J. Adv. Res. 2022, 36, 113–132. [Google Scholar] [CrossRef]
- Li, J.; Zhao, R.; Miao, P.; Xu, F.; Chen, J.; Jiang, X.; Hui, Z.; Wang, L.; Bai, R. Discovery of Anti-Inflammatory Natural Flavonoids: Diverse Scaffolds and Promising Leads for Drug Discovery. Eur. J. Med. Chem. 2023, 260, 115791. [Google Scholar] [CrossRef]
- Drouet, S.; Leclerc, E.A.; Garros, L.; Tungmunnithum, D.; Kabra, A.; Abbasi, B.H.; Lainé, É.; Hano, C. A Green Ultrasound-Assisted Extraction Optimization of the Natural Antioxidant and Anti-Aging Flavonolignans from Milk Thistle Silybum marianum (L.) Gaertn. Fruits for Cosmetic Applications. Antioxidants 2019, 8, 304. [Google Scholar] [CrossRef]
- Xiao, T.; Wei, Y.; Cui, M.; Li, X.; Ruan, H.; Zhang, L.; Bao, J.; Ren, S.; Gao, D.; Wang, M.; et al. Effect of Dihydromyricetin on SARS-CoV-2 Viral Replication and Pulmonary Inflammation and Fibrosis. Phytomedicine 2021, 91, 153704. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Leung, P.S. Pancreatic Cancer, Pancreatitis, and Oxidative Stress. In Gastrointestinal Tissue; Elsevier: Amsterdam, The Netherlands, 2017; pp. 173–186. ISBN 978-0-12-805377-5. [Google Scholar]
- Sun, Y.; Liu, S.; Yang, S.; Chen, C.; Yang, Y.; Lin, M.; Liu, C.; Wang, W.; Zhou, X.; Ai, Q.; et al. Mechanism of Dihydromyricetin on Inflammatory Diseases. Front. Pharmacol. 2022, 12, 794563. [Google Scholar] [CrossRef]
- Selim, S.; Albqmi, M.; Alanazi, A.; Alruwaili, Y.; Al-Sanea, M.M.; Alnusaire, T.S.; Almuhayawi, M.S.; Al Jaouni, S.K.; Hussein, S.; Warrad, M.; et al. Antiviral Activities of Olive Oil Apigenin and Taxifolin against SARS-CoV-2 RNA-Dependent RNA Polymerase (RdRP): In Silico, Pharmacokinetic, ADMET, and in-Vitro Approaches. Cogent Food Agric. 2023, 9, 2236828. [Google Scholar] [CrossRef]
- Rudrapal, M.; Issahaku, A.R.; Agoni, C.; Bendale, A.R.; Nagar, A.; Soliman, M.E.S.; Lokwani, D. In Silico Screening of Phytopolyphenolics for the Identification of Bioactive Compounds as Novel Protease Inhibitors Effective against SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 10437–10453. [Google Scholar] [CrossRef]
- Hamdy, R.; Mostafa, A.; Abo Shama, N.M.; Soliman, S.S.M.; Fayed, B. Comparative Evaluation of Flavonoids Reveals the Superiority and Promising Inhibition Activity of Silibinin against SARS-CoV-2. Phytother. Res. 2022, 36, 2921–2939. [Google Scholar] [CrossRef]
- Pillai, J.U.; Cherian, L.; Taunk, K.; Iype, E.; Dutta, M. Identification of Antiviral Phytochemicals from Cranberry as Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro). Int. J. Biol. Macromol. 2024, 261, 129655. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K.; Islam, N.; Faruk, O.; Ashaduzzaman, M.; Dungani, R. Review on Tannins: Extraction Processes, Applications and Possibilities. S. Afr. J. Bot. 2020, 135, 58–70. [Google Scholar] [CrossRef]
- Sharma, K.; Kumar, V.; Kaur, J.; Tanwar, B.; Goyal, A.; Sharma, R.; Gat, Y.; Kumar, A. Health Effects, Sources, Utilization and Safety of Tannins: A Critical Review. Toxin Rev. 2019, 40, 432–444. [Google Scholar] [CrossRef]
- Alharbi, Y.T.M.; Abdel-Mageed, W.M.; Basudan, O.A.; Mothana, R.A.; Tabish Rehman, M.; ElGamal, A.A.; Alqahtani, A.S.; Fantoukh, O.I.; AlAjmi, M.F. Investigation of Phytochemicals Isolated from Selected Saudi Medicinal Plants as Natural Inhibitors of SARS CoV-2 Main Protease: In Vitro, Molecular Docking and Simulation Analysis. Saudi Pharm. J. 2024, 32, 102023. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, U.R.; Albohy, A.; Abdulrazik, B.S.; Bayoumi, S.A.L.; Malak, L.G.; Khallaf, I.S.A.; Bringmann, G.; Farag, S.F. Natural Coumarins as Potential Anti-SARS-CoV-2 Agents Supported by Docking Analysis. RSC Adv. 2021, 11, 16970–16979. [Google Scholar] [CrossRef]
- Barot, K.P.; Jain, S.V.; Kremer, L.; Singh, S.; Ghate, M.D. Recent Advances and Therapeutic Journey of Coumarins: Current Status and Perspectives. Med. Chem. Res. 2015, 24, 2771–2798. [Google Scholar] [CrossRef]
- Chidambaram, S.K.; Ali, D.; Alarifi, S.; Radhakrishnan, S.; Akbar, I. In Silico Molecular Docking: Evaluation of Coumarin Based Derivatives against SARS-CoV-2. J. Infect. Public Health 2020, 13, 1671–1677. [Google Scholar] [CrossRef]
- Salgado-Moran, G.; Cardona, V.W.; Gerli-Candia, L.; Mendoza-Huizar, L.H.; Abdizadeh, T. Identification of Novel Coumarin Based Compounds as Potential Inhibitors of the 3-Chymotrypsin-like Main Protease of SARS-CoV-2 Using DFT, Molecular Docking and Molecular Dynamics Simulation Studies. J. Chil. Chem. Soc. 2022, 67, 5521–5536. [Google Scholar] [CrossRef]
- Mir, S.A.; Meher, R.K.; Nayak, B. Molecular Modeling and Simulations of Some Antiviral Drugs, Benzylisoquinoline Alkaloid, and Coumarin Molecules to Investigate the Effects on Mpro Main Viral Protease Inhibition. Biochem. Biophys. Rep. 2023, 34, 101459. [Google Scholar] [CrossRef] [PubMed]
- Amen, Y.; Elsbaey, M.; Othman, A.; Sallam, M.; Shimizu, K. Naturally Occurring Chromone Glycosides: Sources, Bioactivities, and Spectroscopic Features. Molecules 2021, 26, 7646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, W.; Chen, Q.; Huang, W.; Yang, Z.; Li, X.; Wang, X. Inhibition Viral RNP and Anti-Inflammatory Activity of Coumarins against Influenza Virus. Biomed. Pharmacother. 2017, 87, 583–588. [Google Scholar] [CrossRef]
- Su, X.; Zhou, D.; Li, N. Bioactive Stilbenes from Plants. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2022; Volume 73, pp. 265–403. ISBN 978-0-323-91097-2. [Google Scholar]
- Teka, T.; Zhang, L.; Ge, X.; Li, Y.; Han, L.; Yan, X. Stilbenes: Source Plants, Chemistry, Biosynthesis, Pharmacology, Application and Problems Related to Their Clinical Application-A Comprehensive Review. Phytochemistry 2022, 197, 113128. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.-C.; Wu, Y.-C.; Chen, Z.-W.; Yang, W.-C. Naturally Occurring Anthraquinones: Chemistry and Therapeutic Potential in Autoimmune Diabetes. Evid. Based Complement. Alternat Med. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Carvalho, N.K.G.; Wellisson Da Silva Mendes, J.; Martins Da Costa, J.G. Quinones: Biosynthesis, Characterization of 13C Spectroscopical Data and Pharmacological Activities. Chem. Biodivers. 2023, 20, e202301365. [Google Scholar] [CrossRef]
- Ntemafack, A.; Singh, R.V.; Ali, S.; Kuiate, J.-R.; Hassan, Q.P. Antiviral Potential of Anthraquinones from Polygonaceae, Rubiaceae and Asphodelaceae: Potent Candidates in the Treatment of SARS-COVID-19, A Comprehensive Review. S. Afr. J. Bot. 2022, 151, 146–155. [Google Scholar] [CrossRef]
- Abdullah; Hussain, Y. Anthraquinones and SARS-CoV-2. In Application of Natural Products in SARS-CoV-2; Elsevier: Amsterdam, The Netherlands, 2023; pp. 171–184. ISBN 978-0-323-95047-3. [Google Scholar]
- Rodríguez-García, C.; Sánchez-Quesada, C.; Toledo, E.; Delgado-Rodríguez, M.; Gaforio, J.J. Naturally Lignan-Rich Foods: A Dietary Tool for Health Promotion? Molecules 2019, 24, 917. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Wang, D.-Y.; Li, Y.-P.; Deyrup, S.T.; Zhang, H.-J. Plant-Derived Lignans as Potential Antiviral Agents: A Systematic Review. Phytochem. Rev. 2022, 21, 239–289. [Google Scholar] [CrossRef]
- Cui, Q.; Du, R.; Liu, M.; Rong, L. Lignans and Their Derivatives from Plants as Antivirals. Molecules 2020, 25, 183. [Google Scholar] [CrossRef]
- Sureja, D.K.; Shah, A.P.; Gajjar, N.D.; Jadeja, S.B.; Bodiwala, K.B.; Dhameliya, T.M. In Silico Computational Investigations of AntiViral Lignan Derivatives as Potent Inhibitors of SARS-CoV-2. ChemistrySelect 2022, 7, e202202069. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Horváti, K.; Kraszni, M.; Ausbüttel, T.; Pályi, B.; Kis, Z.; Mucsi, Z.; Kovács, G.M.; Bősze, S.; Boldizsár, I. Arylnaphthalene Lignans with Anti-SARS-CoV-2 and Antiproliferative Activities from the Underground Organs of Linum austriacum and Linum perenne. J. Nat. Prod. 2023, 86, 672–682. [Google Scholar] [CrossRef]
- Li, B.; Qiao, L.; Xiao, Q.; Zhang, J.; Liu, J.; Zhang, B.; Liu, H. Effects of Diarylbutane Lignans from Schisandra Chinensis Fruit on SARS-CoV-2 3CLpro and PLpro and Their in Vitro Anti-Inflammatory Properties. Phytomedicine Plus 2023, 3, 100432. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiao, L.; Zhang, J.; Xiao, Q.; Liu, J.; Zhang, B.; Liu, H. Natural 7,8-Secolignans from Schisandra sphenanthera Fruit Potently Inhibit SARS-CoV-2 3CLpro and Inflammation. J. Tradit. Complement. Med. 2024, 14, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Omar, F.; Tareq, A.M.; Alqahtani, A.M.; Dhama, K.; Sayeed, M.A.; Emran, T.B.; Simal-Gandara, J. Plant-Based Indole Alkaloids: A Comprehensive Overview from a Pharmacological Perspective. Molecules 2021, 26, 2297. [Google Scholar] [CrossRef]
- Sayed, A.M.; Ibrahim, A.H.; Tajuddeen, N.; Seibel, J.; Bodem, J.; Geiger, N.; Striffler, K.; Bringmann, G.; Abdelmohsen, U.R. Korupensamine A, but Not Its Atropisomer, Korupensamine B, Inhibits SARS-CoV-2 in Vitro by Targeting Its Main Protease (Mpro). Eur. J. Med. Chem. 2023, 251, 115226. [Google Scholar] [CrossRef]
- Ikram, N.K.B.K.; Zhan, X.; Pan, X.-W.; King, B.C.; Simonsen, H.T. Stable Heterologous Expression of Biologically Active Terpenoids in Green Plant Cells. Front. Plant Sci. 2015, 6, 129. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, Z.; Gan, Y.; Xie, J.; Xia, Z.; Liu, T.; Chen, X.; Li, X.; Zhou, H.; Sun, P.; et al. From Ancient Remedy to Modern Medicine: Artemisia Argyi Sesquiterpenoids as a Promising Natural Treatment for COVID-19. Arab. J. Chem. 2023, 16, 105298. [Google Scholar] [CrossRef]
- Fan, Y.-W.; Ao, Z.-Y.; Zhang, W.-J.; Chen, J.-Y.; Lian, X.; Chen Pan, Y.; Chen, L.-P.; Wu, J.-W.; Yuan, J. The Sesquiterpenes with the COVID-19 Mpro Inhibitory Activity from the Carpesium abrotanoides L. Nat. Prod. Res. 2023, 38, 1909–1917. [Google Scholar] [CrossRef]
- Waqas, M.; Ullah, S.; Halim, S.A.; Rehman, N.U.; Ali, A.; Jan, A.; Muhsinah, A.B.; Khan, A.; Al-Harrasi, A. Targeting Papain-like Protease by Natural Products as Novel Therapeutic Potential SARS-CoV-2. Int. J. Biol. Macromol. 2024, 258, 128812. [Google Scholar] [CrossRef]
- Djouonzo, P.T.; Mukim, M.S.I.; Kemda, P.N.; Kowa, T.K.; Tchinda, A.T.; Agbor Agbor, G.; Pan, C.-H.; Song, D.-G. SARS-CoV-2 Main Protease Inhibitors from the Stem Barks of Discoglypremna caloneura (Pax) Prain (Euphorbiaceae) and Pterocarpus erinaceus Poir (Fabaceae) and Their Molecular Docking Investigation. Appl. Biol. Chem. 2023, 66, 76. [Google Scholar] [CrossRef]
- Zhao, L.; Qin, X.; Lin, T.; Xie, F.; Yao, L.; Li, Y.; Xiong, B.; Xu, Z.; Ye, Y.; Chen, H.; et al. Multi-Target Mechanisms against Coronaviruses of Constituents from Chinese Dagang Tea Revealed by Experimental and Docking Studies. J. Ethnopharmacol. 2022, 297, 115528. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Wan, X.; Li, B.; Guan, M.; Ning, X.; Hu, X.; Li, S.; Liu, S.; Song, G. Discovery and Structural Optimization of 3-O-β-Chacotriosyl Betulonic Acid Saponins as Potent Fusion Inhibitors of Omicron Virus Infections. Bioorg. Chem. 2023, 131, 106316. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Zhang, Y.; Zhang, Q.; Liu, Q.; Hu, Y.; Chen, X.; Wang, J.; Shi, Y.; Deng, C.; et al. Oridonin Inhibits SARS-CoV-2 Replication by Targeting Viral Proteinase and Polymerase. Virologica Sinica 2023, 38, 470–479. [Google Scholar] [CrossRef]
- Wadhwa, G.; Kumar, S.; Chhabra, L.; Mahant, S.; Rao, R. Essential Oil–Cyclodextrin Complexes: An Updated Review. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 39–58. [Google Scholar] [CrossRef]
- Oleszek, M.; Oleszek, W. Saponins in Food. In Handbook of Dietary Phytochemicals; Xiao, J., Sarker, S.D., Asakawa, Y., Eds.; Springer: Singapore, 2021; pp. 1501–1540. ISBN 978-981-15-4147-6. [Google Scholar]
- Dinda, B.; Debnath, S.; Mohanta, B.C.; Harigaya, Y. Naturally Occurring Triterpenoid Saponins. Chem. Biodivers. 2010, 7, 2327–2580. [Google Scholar] [CrossRef]
- Van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O.; et al. Glycyrrhizin Effectively Inhibits SARS-CoV-2 Replication by Inhibiting the Viral Main Protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Deng, W.; Ding, L.; Ye, X.; Yin, S.; Huang, W. Glycyrrhetinic Acid and Its Derivatives as Potential Alternative Medicine to Relieve Symptoms in Nonhospitalized COVID-19 Patients. J. Med. Virol. 2020, 92, 2200–2204. [Google Scholar] [CrossRef]
- Yi, Y.; Li, J.; Lai, X.; Zhang, M.; Kuang, Y.; Bao, Y.-O.; Yu, R.; Hong, W.; Muturi, E.; Xue, H.; et al. Natural Triterpenoids from Licorice Potently Inhibit SARS-CoV-2 Infection. J. Adv. Res. 2022, 36, 201–210. [Google Scholar] [CrossRef]
- Güçlü-Üstündağ, Ö.; Mazza, G. Saponins: Properties, Applications and Processing. Crit. Rev. Food Sci. Nutr. 2007, 47, 231–258. [Google Scholar] [CrossRef]
- Yekeen, N.; Malik, A.A.; Idris, A.K.; Reepei, N.I.; Ganie, K. Foaming Properties, Wettability Alteration and Interfacial Tension Reduction by Saponin Extracted from Soapnut (Sapindus mukorossi) at Room and Reservoir Conditions. J. Pet. Sci. Eng. 2020, 195, 107591. [Google Scholar] [CrossRef]
- Nicoliche, T.; Bartolomeo, C.S.; Lemes, R.M.R.; Pereira, G.C.; Nunes, T.A.; Oliveira, R.B.; Nicastro, A.L.M.; Soares, É.N.; Da Cunha Lima, B.F.; Rodrigues, B.M.; et al. Antiviral, Anti-Inflammatory and Antioxidant Effects of Curcumin and Curcuminoids in SH-SY5Y Cells Infected by SARS-CoV-2. Sci. Rep. 2024, 14, 10696. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.; Muller, F.L.; Van Remmen, H. An Objective Appraisal of the Free Radical Theory of Aging. In Handbook of the Biology of Aging; Elsevier: Amsterdam, The Netherlands, 2011; pp. 177–202. ISBN 978-0-12-378638-8. [Google Scholar]
- Kao, Y.-W.; Hsu, S.-K.; Chen, J.Y.-F.; Lin, I.-L.; Chen, K.-J.; Lee, P.-Y.; Ng, H.-S.; Chiu, C.-C.; Cheng, K.-C. Curcumin Metabolite Tetrahydrocurcumin in the Treatment of Eye Diseases. Int. J. Mol. Sci. 2020, 22, 212. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S. In Silico Studies to Identify Potential Natural Antiviral Agents to Treat and Control SARS-CoV-2 (COVID-19). J. Glob. Biosci. 2021, 10, 8720–8743. [Google Scholar]
- Dembitsky, V.M. Biological Activity and Structural Diversity of Steroids Containing Aromatic Rings, Phosphate Groups, or Halogen Atoms. Molecules 2023, 28, 5549. [Google Scholar] [CrossRef]
- Castilla, V.; Ramirez, J.; Coto, C.E. Plant and Animal Steroids a New Hope to Search for Antiviral Agents. CMC 2010, 17, 1858–1873. [Google Scholar] [CrossRef]
- Yerlikaya, P.O.; Arısan, E.; Mehdizadehtapeh, L.; Uysal Onganer, P.; Çoker Gürkan, A. The Use of Plant Steroids in Viral Disease Treatments: Current Status and Future Perspectives. Eur. J. Biol. 2023, 82, 86–94. [Google Scholar] [CrossRef]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchianò, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 Receptor Interaction by Natural and Semi-Synthetic Steroidal Agents Acting on Functional Pockets on the Receptor Binding Domain. Front. Chem. 2020, 8, 572885. [Google Scholar] [CrossRef]
- Sirotkin, A.V.; Kolesarova, A. Plant Molecules and Their Influence on Health and Female Reproduction. In Environmental Contaminants and Medicinal Plants Action on Female Reproduction; Elsevier: Amsterdam, The Netherlands, 2022; pp. 245–399. ISBN 978-0-12-824292-6. [Google Scholar]
- Giacalone, M.; Forfori, F.; Giunta, F. Chili Pepper Compounds in the Management of Neuropathic Pain. In Bioactive Nutraceuticals and Dietary Supplements in Neurological and Brain Disease; Elsevier: Amsterdam, The Netherlands, 2015; pp. 187–195. ISBN 978-0-12-411462-3. [Google Scholar]
- Guldiken, B.; Catalkaya, G.; Ozkan, G.; Ceylan, F.D.; Capanoglu, E. Toxicological Effects of Commonly Used Herbs and Spices. In Toxicology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 201–213. ISBN 978-0-12-819092-0. [Google Scholar]
- Nag, A.; Paul, S.; Banerjee, R.; Kundu, R. In Silico Study of Some Selective Phytochemicals against a Hypothetical SARS-CoV-2 Spike RBD Using Molecular Docking Tools. Comput. Methods Programs Biomed. 2021, 137, 104818. [Google Scholar] [CrossRef]
- Rahmattullah, N.; Arumingtyas, E.; Widyananda, M.; Ahyar, A.; Tabroni, I. Bioinformatics Analysis of Bioactive Compounds of Four Capsicum Species against SARS-CoV-2 Infection. Int. J. Adv. Biol. Biomed. Res. 2021, 9, 298–319. [Google Scholar] [CrossRef]
- Pandey, A.K.; Verma, S. An in Silico Evaluation of Dietary Components for Structural Inhibition of SARS-Cov-2 Main Protease. J. Biomol. Struct. Dyn. 2022, 40, 136–142. [Google Scholar] [CrossRef]
- Badea, G.I.; Radu, G.L. Introductory Chapter: Carboxylic Acids-Key Role in Life Sciences. In Carboxylic Acid-Key Role in Life Sciences; Badea, G.I., Radu, G.L., Eds.; InTech: London, UK, 2018; ISBN 978-1-78923-278-3. [Google Scholar]
- Pedreira, A.; Taşkın, Y.; García, M.R. A Critical Review of Disinfection Processes to Control SARS-CoV-2 Transmission in the Food Industry. Foods 2021, 10, 283. [Google Scholar] [CrossRef]
- Shahab, S.; Kaviani, S.; Sheikhi, M.; Almodarresiyeh, H.A.; Al Saud, S. DFT Calculations and In Silico Study of Chlorogenic, Ellagic and Quisqualic Acids as Potential Inhibitors of SARS-CoV-2 Main Protease Mpro. Biointerface Res. Appl. Chem. 2021, 12, 61–73. [Google Scholar] [CrossRef]
- Opgrande, J.L.; Dobratz, C.J.; Brown, E.; Liang, J.; Conn, G.S.; Shelton, F.J.; With, J. Benzaldehyde. In Kirk-Othmer Encyclopedia of Chemical Technology; Kirk-Othmer, Ed.; Wiley Online Library: Hoboken, NJ, USA, 2000; ISBN 978-0-471-48494-3. [Google Scholar]
- Khairan, K.; Idroes, R.; Tumilaar, S.G.; Tallei, T.E.; Idroes, G.M.; Rahmadhany, F.; Futri, M.U.; Dinura, N.M.; Mauliza, S.; Diana, M.; et al. Molecular Docking Study of Fatty Acids from Pliek U Oil in the Inhibition of SARS-CoV-2 Protein and Enzymes. IOP Conf. Ser: Mater. Sci. Eng. 2021, 1087, 012058. [Google Scholar] [CrossRef]
- Kang, N.; Heo, S.-Y.; Cha, S.-H.; Ahn, G.; Heo, S.-J. In Silico Virtual Screening of Marine Aldehyde Derivatives from Seaweeds against SARS-CoV-2. Mar. Drugs 2022, 20, 399. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-B.; Long, C.; Kennelly, E.J. Structural Diversity and Bioactivities of Natural Benzophenones. Nat. Prod. Rep. 2014, 31, 1158–1174. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Ramu, R.; Jayanthi, M.K.; Ranganatha, L.V.; Khanum, S.A.; Silina, E.; Stupin, V.; Achar, R.R. Discovery of Novel Benzophenone Integrated Derivatives as Anti-Alzheimer’s Agents Targeting Presenilin-1 and Presenilin-2 Inhibition: A Computational Approach. PLoS ONE 2022, 17, e0265022. [Google Scholar] [CrossRef] [PubMed]
- Mustieles, V.; Balogh, R.K.; Axelstad, M.; Montazeri, P.; Márquez, S.; Vrijheid, M.; Draskau, M.K.; Taxvig, C.; Peinado, F.M.; Berman, T.; et al. Benzophenone-3: Comprehensive Review of the Toxicological and Human Evidence with Meta-Analysis of Human Biomonitoring Studies. Environ. Int. 2023, 173, 107739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Q.; Liu, M.; Ruan, J.; Yu, H.; Li, J.; Wang, T. Effects of Benzophenones from Mango Leaves on Lipid Metabolism. Chem. Pharm. Bull. 2019, 67, 634–639. [Google Scholar] [CrossRef]
- Coelho, C.; Gallo, G.; Campos, C.B.; Hardy, L.; Würtele, M. Biochemical Screening for SARS-CoV-2 Main Protease Inhibitors. PLoS ONE 2020, 15, e0240079. [Google Scholar] [CrossRef]
- Malik, H.N.; Jabeen, A.; Ashraf, S.; Haq, Z.U.; Salar, U.; Arshia; Khan, K.M. Benzophenone and Coumarin Derivatives as 3-CLPro Inhibitors: Targeting Cytokine Storm through in Silico and in Vitro Approaches. J. Biomol. Struct. Dyn. 2022, 1265, 133478. [Google Scholar] [CrossRef]
- Costa, M.A.M.; De Sousa, N.F.; Mansur Pontes, C.L.; Scotti, M.T.; De Assis, F.F.; Braga, A.L.; Sandjo, L.P. Inhibitory Effects against SARSCoV-2 Main Protease (Mpro) of Biflavonoids and Benzophenones from the Fruit of Platonia insignis. Fitoterapia 2024, 173, 105784. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Yang, Y.; Li, Y.; He, C. Naturally Occurring Coumestans from Plants, Their Biological Activities and Therapeutic Effects on Human Diseases. Pharmacol. Res. 2021, 169, 105615. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.M.; Hop, N.Q.; Son, N.T. Wedelolactone: A Molecule of Interests. Fitoterapia 2023, 164, 105355. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, X.; Fu, L.; Xu, H. Natural Product-Based Screening for Lead Compounds Targeting SARS CoV-2 Mpro. Pharmaceuticals 2023, 16, 767. [Google Scholar] [CrossRef]
- Kapoor, N.; Ghorai, S.M.; Khuswaha, P.K.; Bandichhor, R.; Brogi, S. Butein as a Potential Binder of Human ACE2 Receptor for Interfering with SARS-CoV-2 Entry: A Computer-Aided Analysis. J. Mol. Model. 2022, 28, 270. [Google Scholar] [CrossRef]
- Mazziotti, I.; Petrarolo, G.; La Motta, C. Aurones: A Golden Resource for Active Compounds. Molecules 2022, 27, 2. [Google Scholar] [CrossRef]
- Caleffi, G.S.; Rosa, A.S.; De Souza, L.G.; Avelar, J.L.S.; Nascimento, S.M.R.; De Almeida, V.M.; Tucci, A.R.; Ferreira, V.N.; Da Silva, A.J.M.; Santos-Filho, O.A.; et al. Aurones: A Promising Scaffold to Inhibit SARS-CoV-2 Replication. J. Nat. Prod. 2023, 86, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The Application of in Silico Drug-Likeness Predictions in Pharmaceutical Research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET Modeling Approaches in Drug Discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Kar, S.; Leszczynski, J. Open Access in Silico Tools to Predict the ADMET Profiling of Drug Candidates. Expert. Opin. Drug Deliv. 2020, 15, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Żołek, T.; Maciejewska, D. Theoretical Evaluation of ADMET Properties for Coumarin Derivatives as Compounds with Therapeutic Potential. Eur. J. Pharm. Med. Res. 2017, 109, 486–502. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Saldívar-González, F.I. Cheminformatics to Characterize Pharmacologically Active Natural Products. Biomolecules 2020, 10, 1566. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Gupta, P.; Sharma, S.; Sharma, A.; Agarwal, S.M. ADMET Profiling of Geographically Diverse Phytochemical Using Chemoinformatic Tools. Future Med. Chem. 2019, 12, 69–87. [Google Scholar] [CrossRef]
- Falade, V.A.; Adelusi, T.I.; Adedotun, I.O.; Abdul-Hammed, M.; Lawal, T.A.; Agboluaje, S.A. In Silico Investigation of Saponins and Tannins as Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro). In Silico Pharmacol. 2021, 9, 9. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Wimmerová, M.; Wimmer, Z. Saponins of Selected Triterpenoids as Potential Therapeutic Agents: A Review. Pharmaceuticals 2023, 16, 386. [Google Scholar] [CrossRef]
- Rajasekar, N.; Sivanantham, A.; Ravikumar, V.; Rajasekaran, S. An Overview on the Role of Plant-Derived Tannins for the Treatment of Lung Cancer. Phytochemistry 2021, 188, 112799. [Google Scholar] [CrossRef]
- Duran, N.; Polat, M.F.; Aktas, D.A.; Alagoz, M.A.; Ay, E.; Cimen, F.; Tek, E.; Anil, B.; Burmaoglu, S.; Algul, O. New Chalcone Derivatives as Effective against SARS-CoV-2 Agent. Int. J. Clin. Pract. 2021, 75, e14846. [Google Scholar] [CrossRef]
- Prabakaran, G.; Manivarman, S.; Bharanidharan, M. Catalytic Synthesis, ADMET, QSAR and Molecular Modeling Studies of Novel Chalcone Derivatives as Highly Potent Antioxidant Agents. Mater. Today Proc. 2022, 48, 400–408. [Google Scholar] [CrossRef]
- Norisham, M.; Husin, F. Elucidation of Stilbene-Derivatives as Potential Inhibitors of SARS-CoV-2 Mpro Binding Pocket: A Molecular Docking, and ADMET Prediction Studies. IJF 2024, 1, 56–68. [Google Scholar] [CrossRef]
- Mortuza, M.G.; Roni, M.A.H.; Kumer, A.; Biswas, S.; Saleh, M.A.; Islam, S.; Sadaf, S.; Akther, F. A Computational Study on Selected Alkaloids as SARS-CoV-2 Inhibitors: PASS Prediction, Molecular Docking, ADMET Analysis, DFT, and Molecular Dynamics Simulations. Biochem. Res. Int. 2023, 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wanat, K. Biological Barriers, and the Influence of Protein Binding on the Passage of Drugs across Them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Mei, L.; Quach, T.; Porter, C.; Trevaskis, N. Lipophilic Conjugates of Drugs: A Tool to Improve Drug Pharmacokinetic and Therapeutic Profiles. Pharm. Res. 2021, 38, 1497–1518. [Google Scholar] [CrossRef]
- Pangal, A.; Ahmed, K. Synthesis and Biological Evaluation of Coumarin-Quinone Hybrids as Multifunctional Bioactive Agents. ADMET DMPK 2022, 11, 81–96. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Chrobak, E.; Bębenek, E. Lipophilicity and ADMET Analysis of Quinoline-1,4-Quinone Hybrids. Pharmaceutics 2022, 15, 34. [Google Scholar] [CrossRef]
- Zuo, H.-L.; Huang, H.-Y.; Lin, Y.-C.-D.; Cai, X.-X.; Kong, X.-J.; Luo, D.-L.; Zhou, Y.-H.; Huang, H.-D. Enzyme Activity of Natural Products on Cytochrome P450. Molecules 2022, 27, 515. [Google Scholar] [CrossRef]
- Maia, M.D.S.; Raimundo E Silva, J.P.; De Lima Nunes, T.A.; Saraiva De Sousa, J.M.; Soares Rodrigues, G.C.; Messias Monteiro, A.F.; Fechine Tavares, J.; Da Franca Rodrigues, K.A.B.; Mendonça-Junior, F.J.; Scotti, L.; et al. Virtual Screening and the In Vitro Assessment of the Antileishmanial Activity of Lignans. Molecules 2020, 25, 2281. [Google Scholar] [CrossRef]
- Ye, L.; Fan, S.; Zhao, P.; Wu, C.; Liu, M.; Hu, S.; Wang, P.; Wang, H.; Bi, H. Potential Herb–Drug Interactions between Anti-COVID-19 Drugs and Traditional Chinese Medicine. Acta Pharm. Sin. B 2023, 13, 3598–3637. [Google Scholar] [CrossRef]
- Klomp, F.; Wenzel, C.; Drozdzik, M.; Oswald, S. Drug–Drug Interactions Involving Intestinal and Hepatic CYP1A Enzymes. Pharmaceutics 2020, 12, 1201. [Google Scholar] [CrossRef]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Relevance of Half-Life in Drug Design: Miniperspective. J. Med. Chem. 2018, 61, 4273–4282. [Google Scholar] [CrossRef] [PubMed]
- Dobrek, L. A Synopsis of Current Theories on Drug-Induced Nephrotoxicity. Life 2023, 13, 325. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.A.; Mercês, É.A.B.; Portela, F.S.; Malheiro, L.F.L.; Silva, H.B.L.; De Benedictis, L.M.; De Benedictis, J.M.; Silva, C.C.; d’Ávilla, E.; Santos, A.C.L.; et al. An Integrated View of Cisplatin-Induced Nephrotoxicity, Hepatotoxicity, and Cardiotoxicity: Characteristics, Common Molecular Mechanisms, and Current Clinical Management. Clin. Exp. Nephrol. 2024, 28, 711–727. [Google Scholar] [CrossRef]
- Zhang, D.; Wei, C.; Hop, C.E.C.A.; Wright, M.R.; Hu, M.; Lai, Y.; Khojasteh, S.C.; Humphreys, W.G. Intestinal Excretion, Intestinal Recirculation, and Renal Tubule Reabsorption Are Underappreciated Mechanisms That Drive the Distribution and Pharmacokinetic Behavior of Small Molecule Drugs. J. Med. Chem. 2021, 64, 7045–7059. [Google Scholar] [CrossRef] [PubMed]
- Adibah, K.Z.M.; Azzreena, M.A. Plant Toxins: Alkaloids and Their Toxicities. GSC Biol. Pharm. Sci. 2018, 6, 21–29. [Google Scholar] [CrossRef]
- Rajput, A.; Sharma, R.; Bharti, R. Pharmacological Activities and Toxicities of Alkaloids on Human Health. Mater. Today Proc. 2022, 48, 1407–1415. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Zhang, B.-Y.; Qiu, L.-P.; Guan, R.-F.; Ye, Z.-H.; Yu, X.-P. Structure Properties and Mechanisms of Action of Naturally Originated Phenolic Acids and Their Derivatives against Human Viral Infections. CMC 2017, 24, 4279–4302. [Google Scholar] [CrossRef]
- Kiokias, S.; Oreopoulou, V. A Review of the Health Protective Effects of Phenolic Acids against a Range of Severe Pathologic Conditions (Including Coronavirus-Based Infections). Molecules 2021, 26, 5405. [Google Scholar] [CrossRef]
- Schefer, S.; Oest, M.; Rohn, S. Interactions between Phenolic Acids, Proteins, and Carbohydrates—Influence on Dough and Bread Properties. Foods 2021, 10, 2798. [Google Scholar] [CrossRef]
- Gonçalves, C.P.; Marcucci, M.C.; Rocha, O.V.; Utuari, C.L.C.; Fortunato, C.R.M.; Souza, G.F.D.; Farias, A. Computational Studies of Caffeoylquinic Acids from Brazilian Green Propolis and Its Anti-Viral Potential against SARS-CoV-2 Mpro. RSD 2022, 11, e359111536917. [Google Scholar] [CrossRef]
- Gao, K.; Wang, R.; Chen, J.; Tepe, J.J.; Huang, F.; Wei, G.-W. Perspectives on SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2021, 64, 16922–16955. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanouil, C.; Chatziathanasiadou, M.V.; Chatzigiannis, C.; Chontzopoulou, E.; Mavromoustakos, T.; Grdadolnik, S.G.; Tzakos, A.G. Unveiling the Interaction Profile of Rosmarinic Acid and Its Bioactive Substructures with Serum Albumin. J. Enzym. Inhib. Med. Chem. 2020, 35, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhai, G.; Li, Y.; Wang, M.; Chen, X.; Wang, R.; Xie, H.; Zhang, W.; Ge, G.; Zhang, Q.; et al. Ginkgolic Acids Inhibit SARS-CoV-2 and Its Variants by Blocking the Spike Protein/ACE2 Interplay. Int. J. Biol. Macromol. 2023, 226, 780–792. [Google Scholar] [CrossRef]
- Geiger, N.; König, E.-M.; Oberwinkler, H.; Roll, V.; Diesendorf, V.; Fähr, S.; Obernolte, H.; Sewald, K.; Wronski, S.; Steinke, M.; et al. Acetylsalicylic Acid and Salicylic Acid Inhibit SARS-CoV-2 Replication in Precision-Cut Lung Slices. Vaccines 2022, 10, 1619. [Google Scholar] [CrossRef]
- Macario, A.; López, J.C.; Blanco, S. Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study. Int. J. Mol. Sci. 2024, 25, 4074. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Xu, X.; Yang, C.; Niu, X.; Yin, S.; Pan, X.; Xu, W.; Hu, G.; Wang, C.; et al. Danshensu Alleviates Pseudo-Typed SARS-CoV-2 Induced Mouse Acute Lung Inflammation. Acta Pharmacol. Sin. 2022, 43, 771–780. [Google Scholar] [CrossRef]
- Reis, B.; Martins, M.; Barreto, B.; Milhazes, N.; Garrido, E.M.; Silva, P.; Garrido, J.; Borges, F. Structure−Property−Activity Relationship of Phenolic Acids and Derivatives. Protocatechuic Acid Alkyl Esters. J. Agric. Food Chem. 2010, 58, 6986–6993. [Google Scholar] [CrossRef] [PubMed]
- Merkl, R.; Hrádková, I.; Filip, V.; Šmidrkal, J. Antimicrobial and Antioxidant Properties of Phenolic Acids Alkyl Esters. Czech J. Food Sci. 2010, 28, 275–279. [Google Scholar] [CrossRef]
- Kakkar, S.; Bais, S. A Review on Protocatechuic Acid and Its Pharmacological Potential. ISRN Pharmacol. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Krzysztoforska, K.; Mirowska-Guzel, D.; Widy-Tyszkiewicz, E. Pharmacological Effects of Protocatechuic Acid and Its Therapeutic Potential in Neurodegenerative Diseases: Review on the Basis of in Vitro and in Vivo Studies in Rodents and Humans. Nutr. Neurosci. 2017, 22, 72–82. [Google Scholar] [CrossRef]
- Song, J.; He, Y.; Luo, C.; Feng, B.; Ran, F.; Xu, H.; Ci, Z.; Xu, R.; Han, L.; Zhang, D. New Progress in the Pharmacology of Protocatechuic Acid: A Compound Ingested in Daily Foods and Herbs Frequently and Heavily. Pharmacol. Res. 2020, 161, 105109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ren, X.; Wu, J.; Li, H.; Yang, L.; Zhang, Y.; Wang, X.; Li, Z. Protocatechuic Acid Protects Mice from Influenza A Virus Infection. Eur. J. Clin. Microbiol. Infect. Dis. 2022, 41, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Chen, Y.; Zhuang, A.-Q.; Chen, S.-M. Natural Phenolic Acids and Their Derivatives against Human Viral Infections. In Infectious Diseases; Aditya Parikesit, A., Ed.; IntechOpen: London, UK, 2023; Volume 27, ISBN 978-1-80356-783-9. [Google Scholar]
- Kumar, N.; Goel, N. Phenolic Acids: Natural Versatile Molecules with Promising Therapeutic Applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]

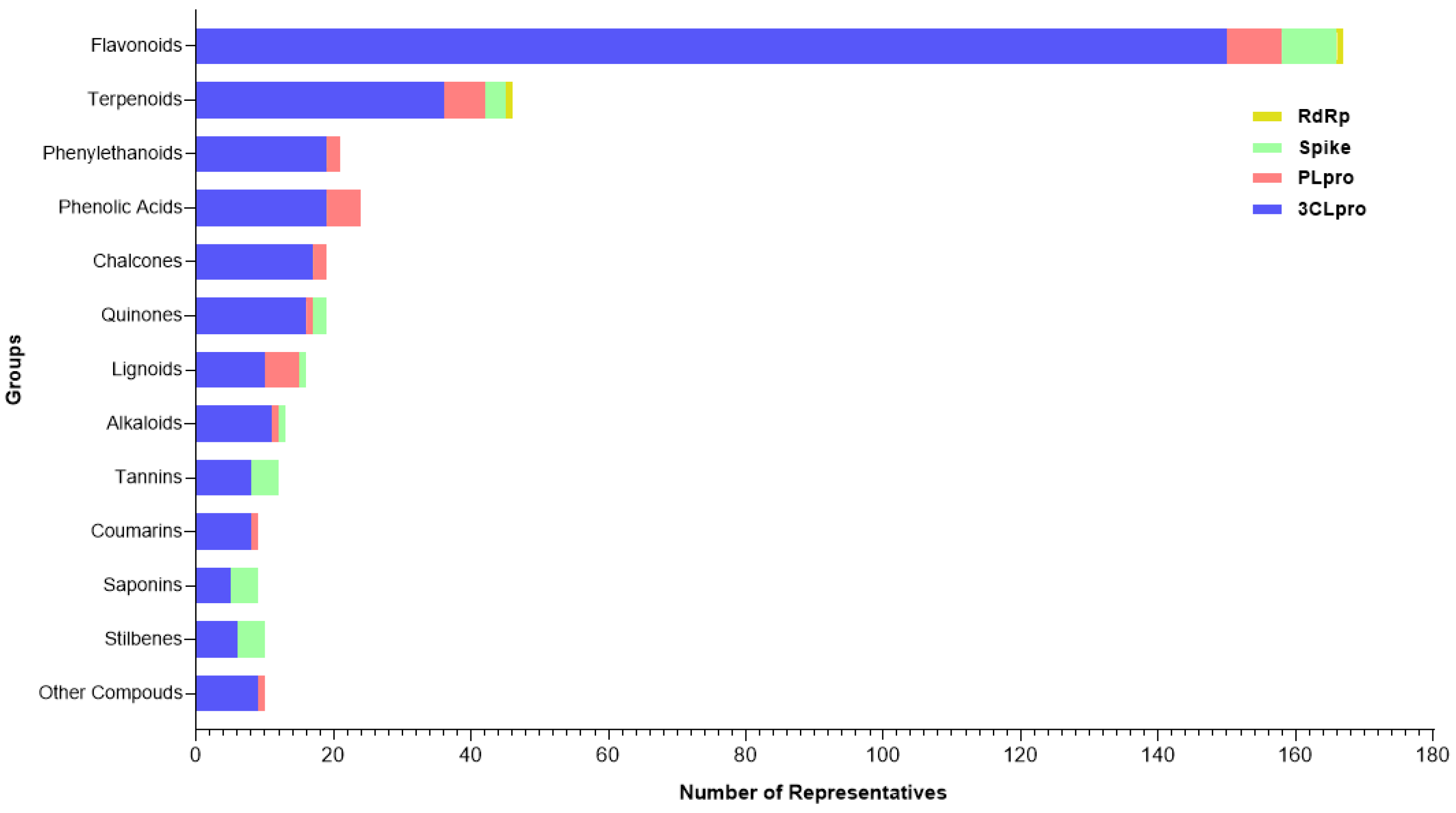


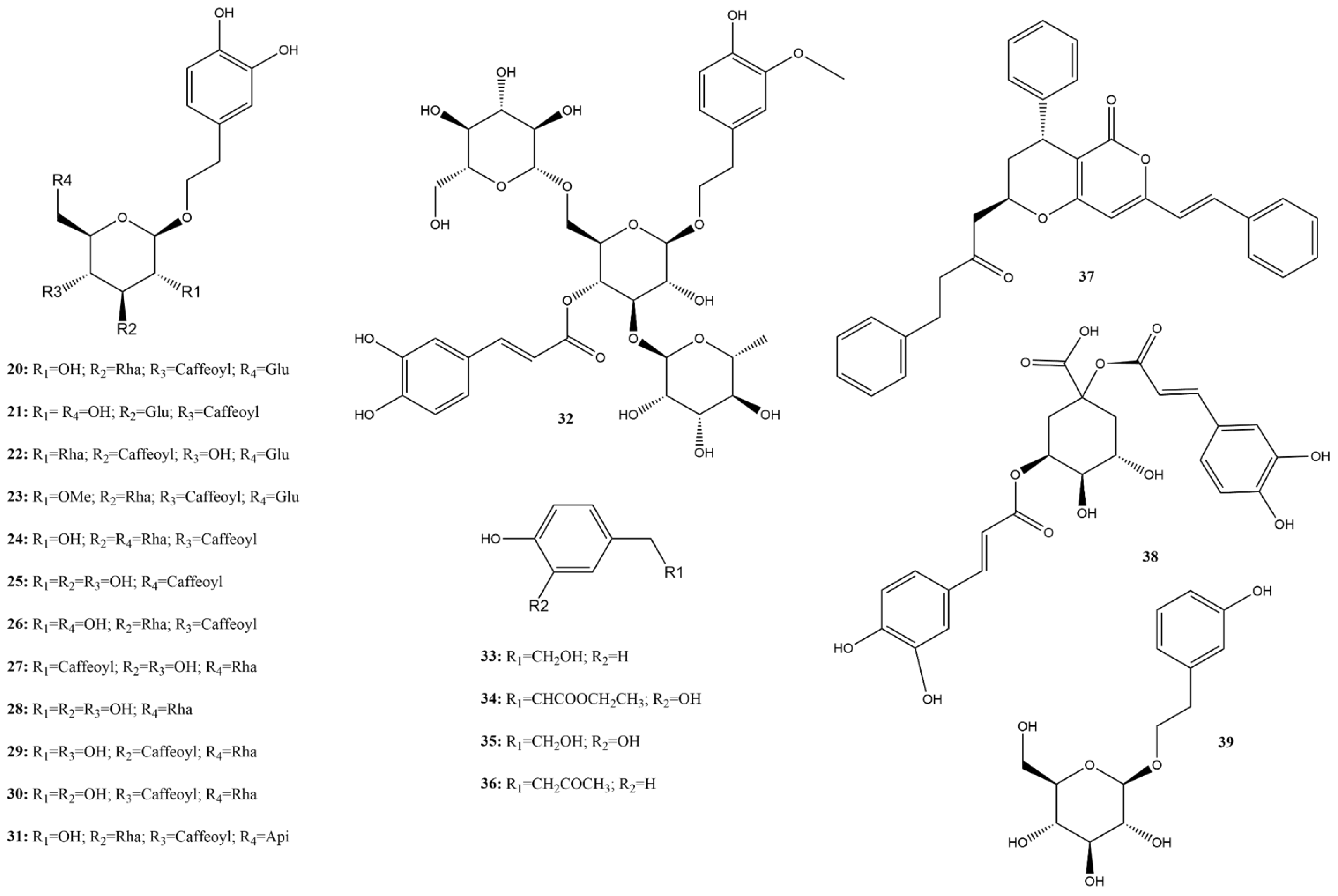
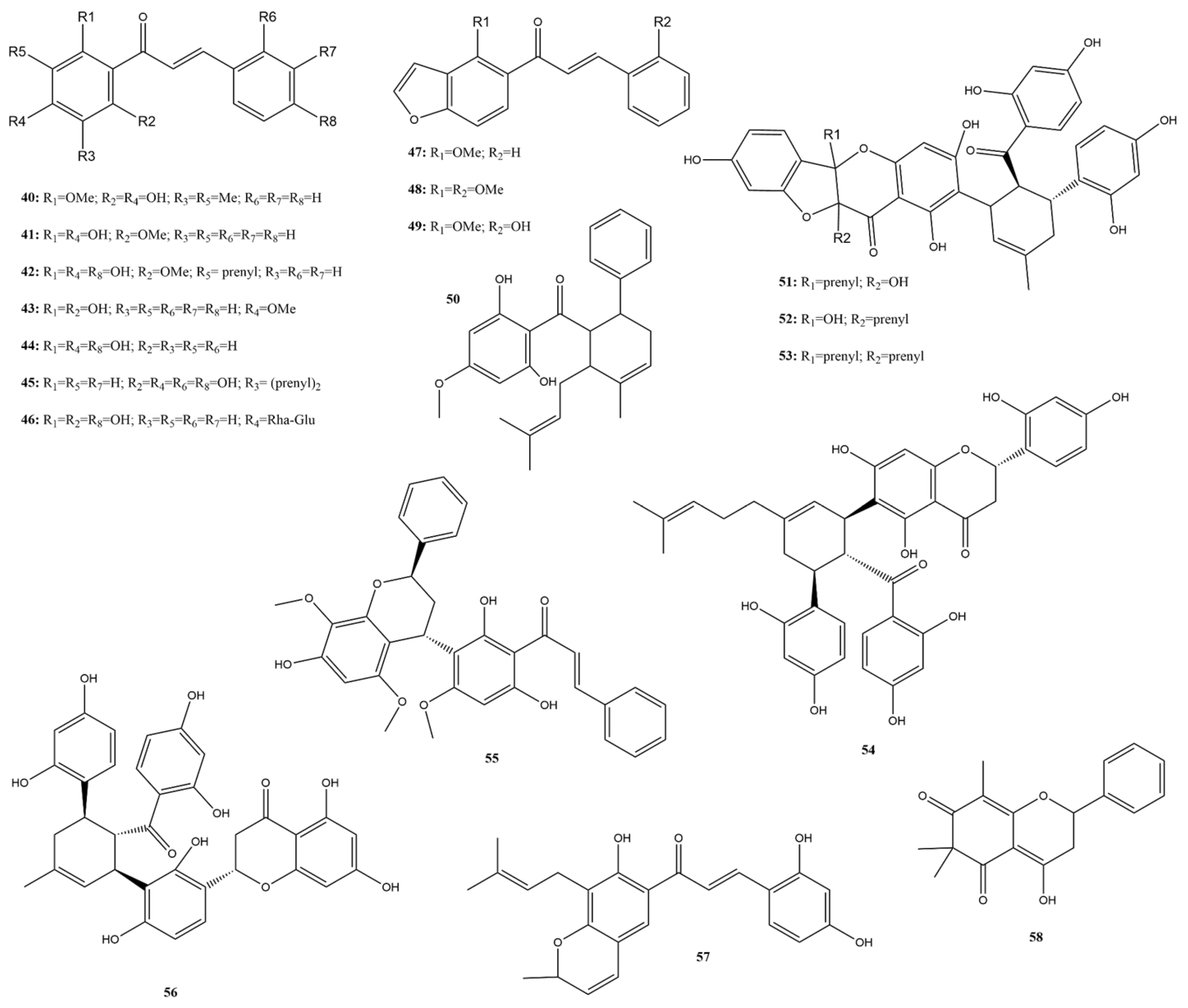
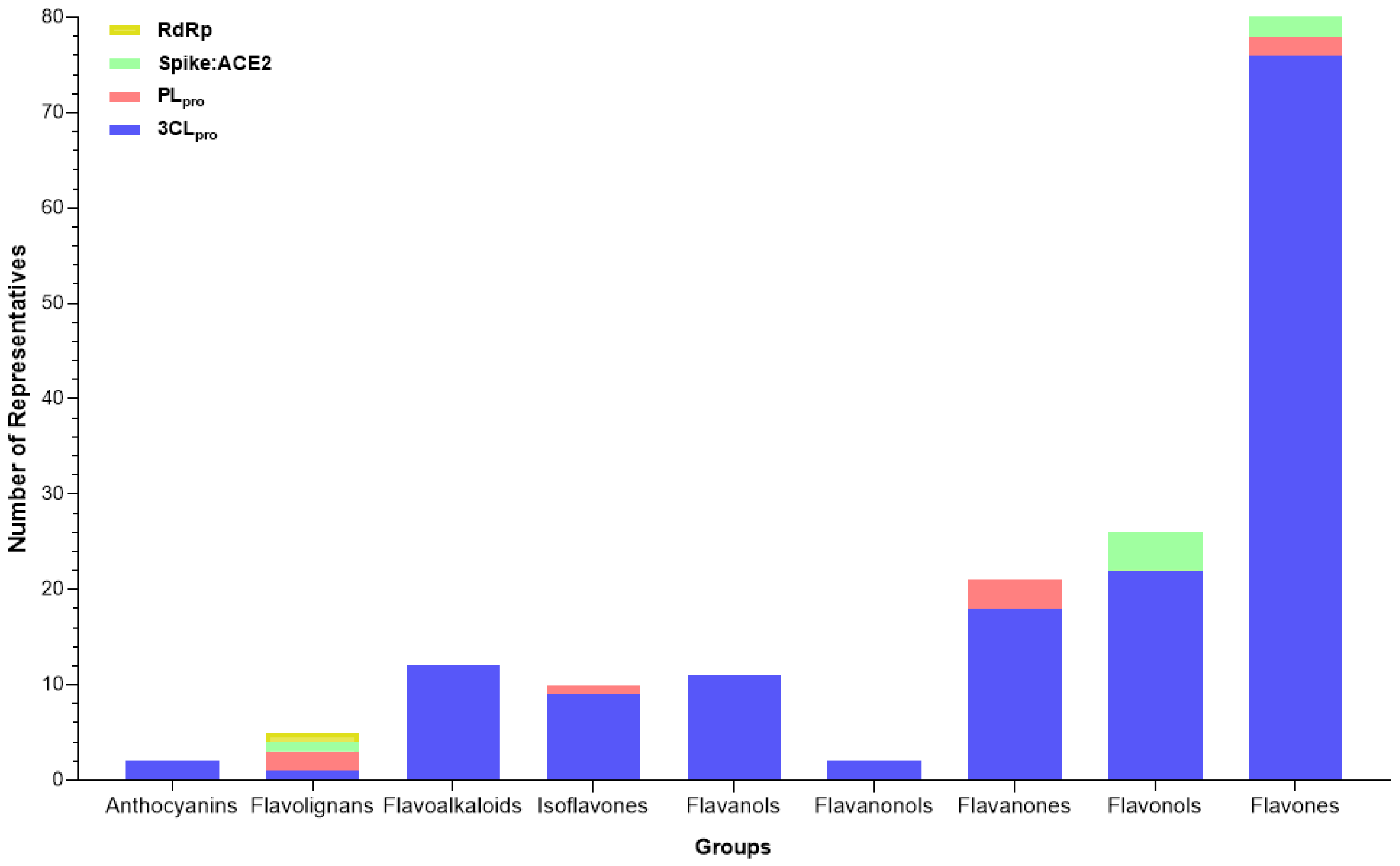


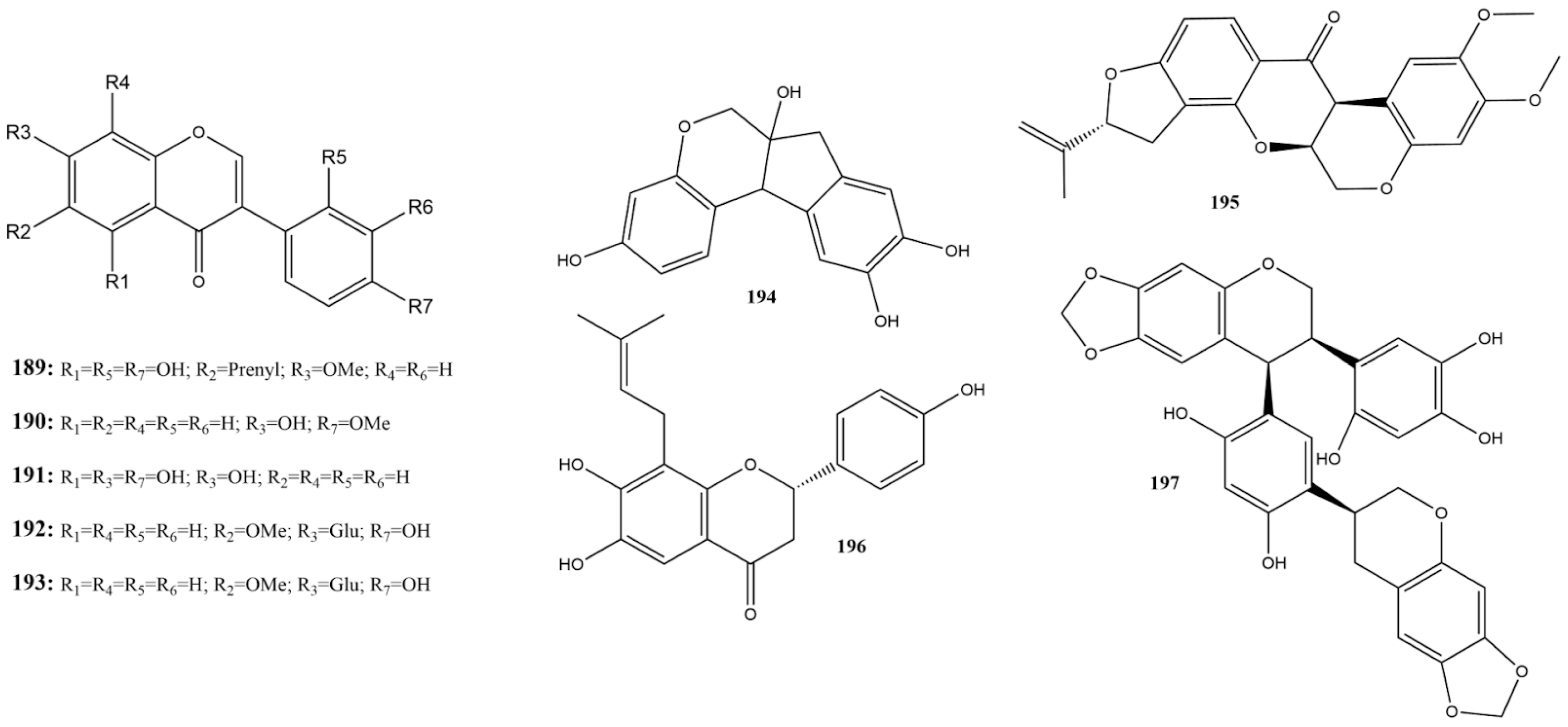
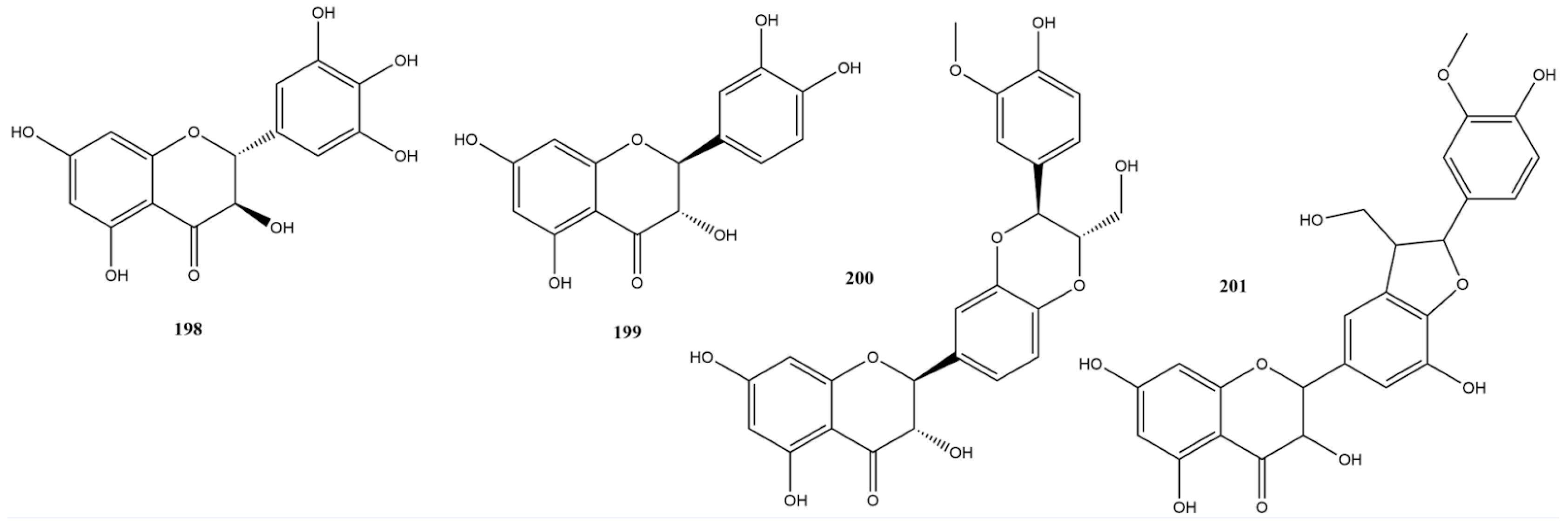
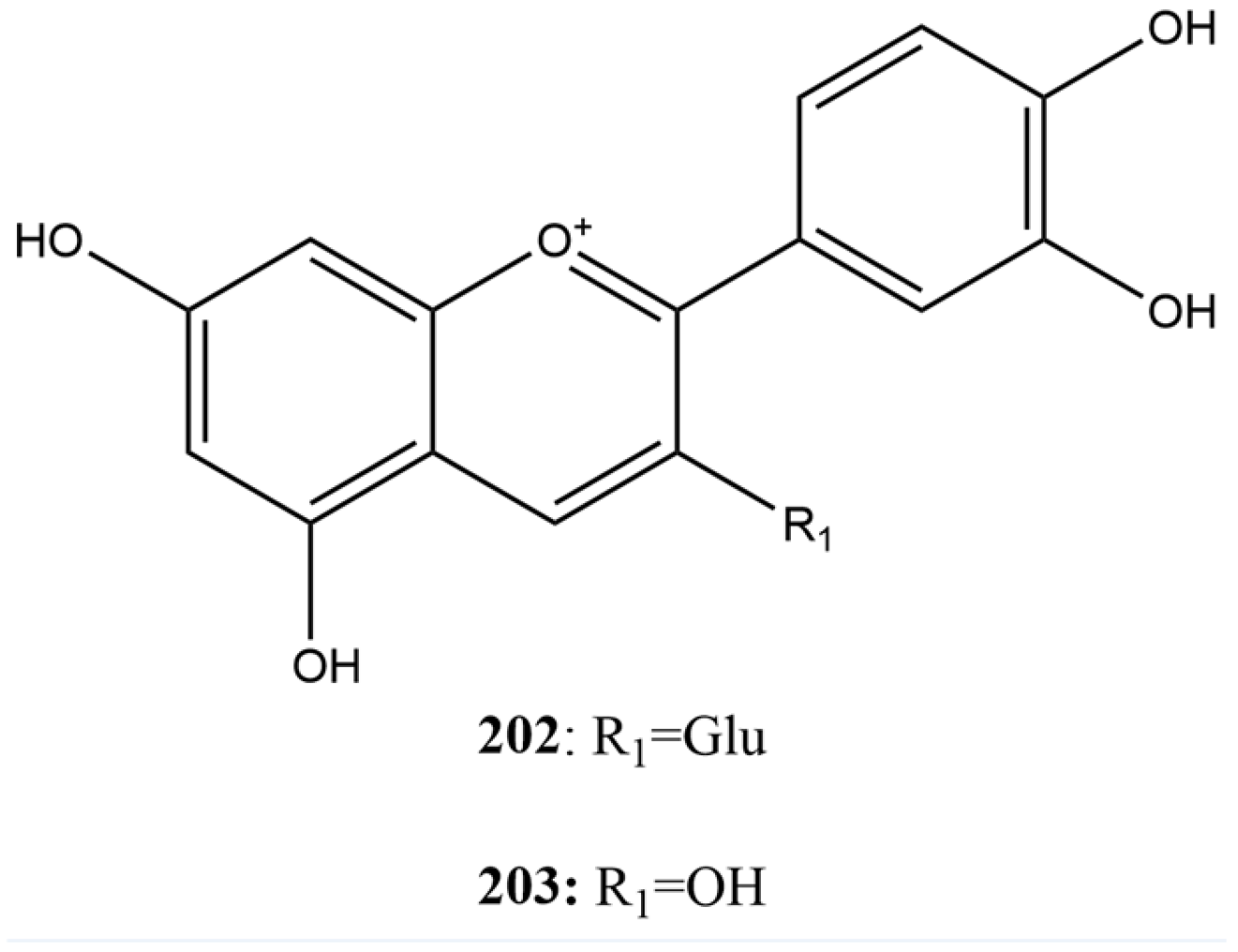
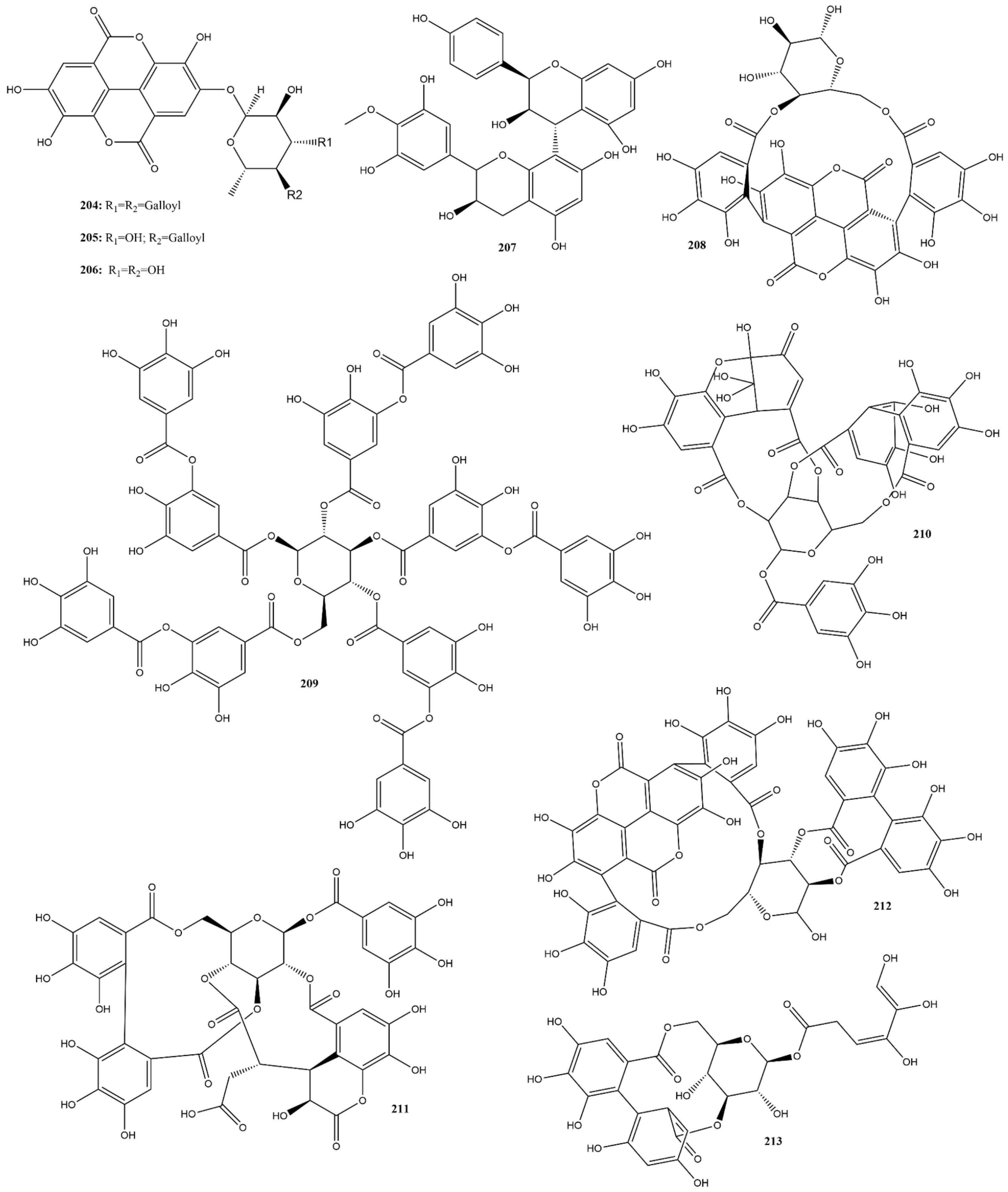

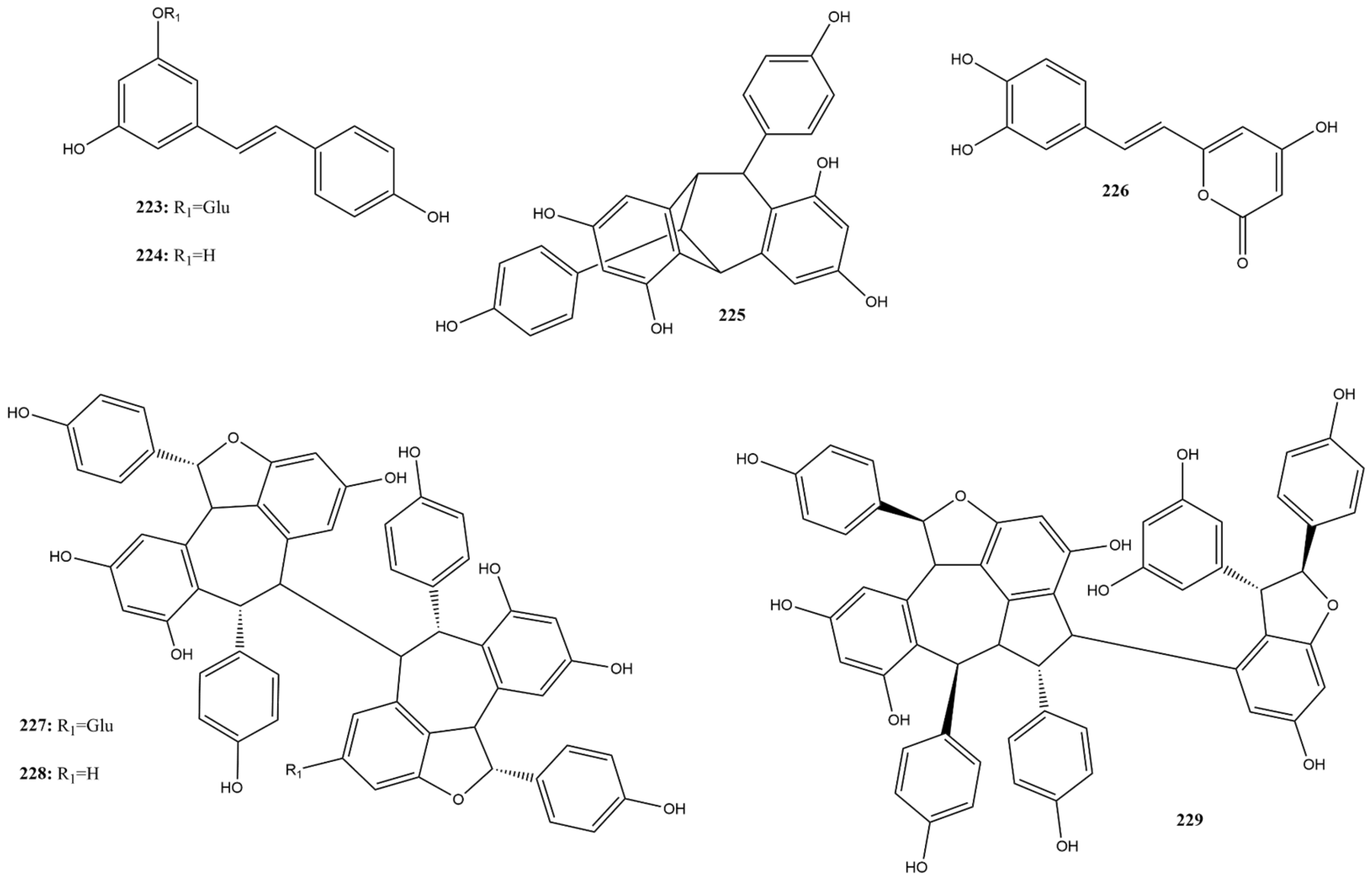
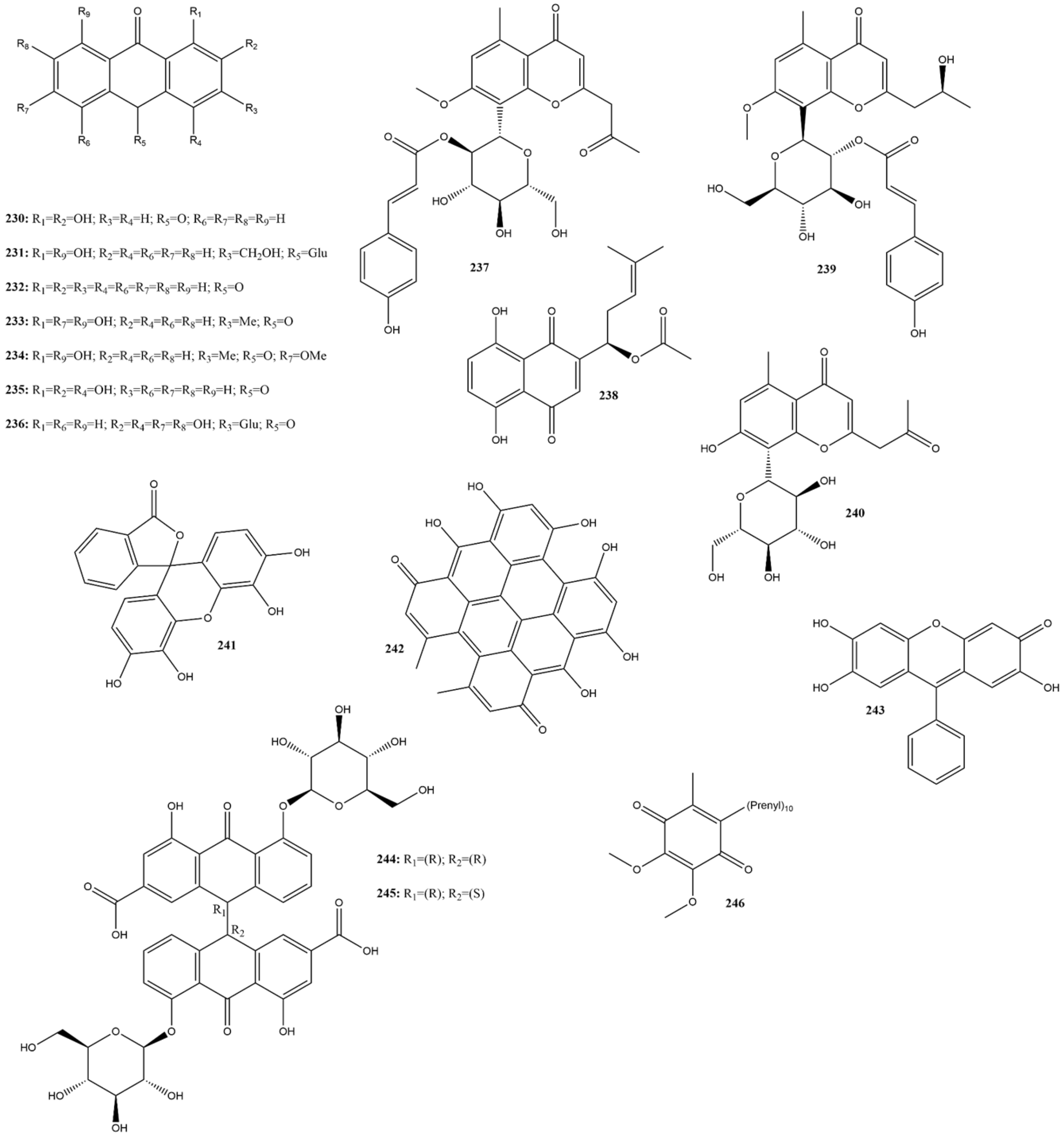


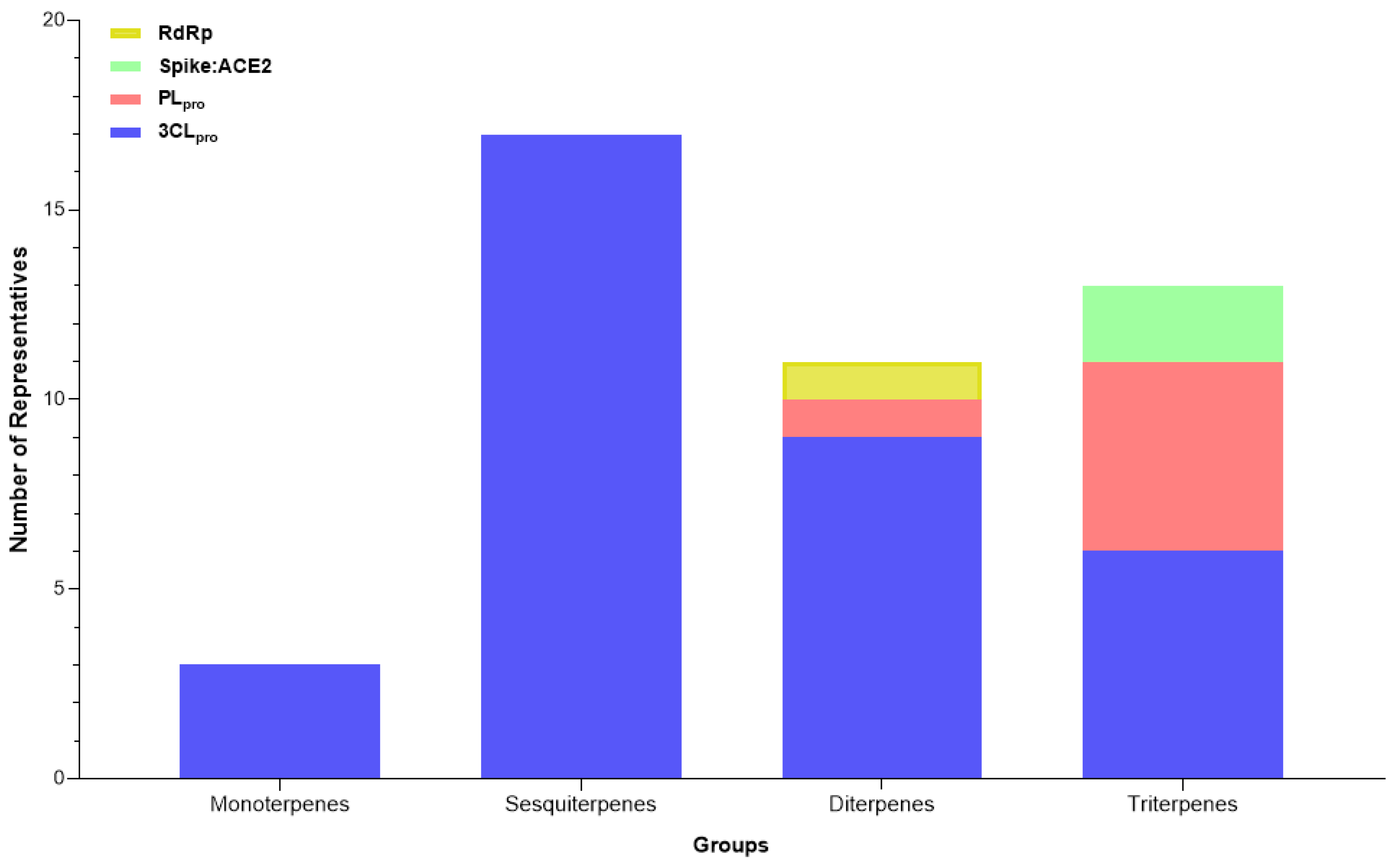
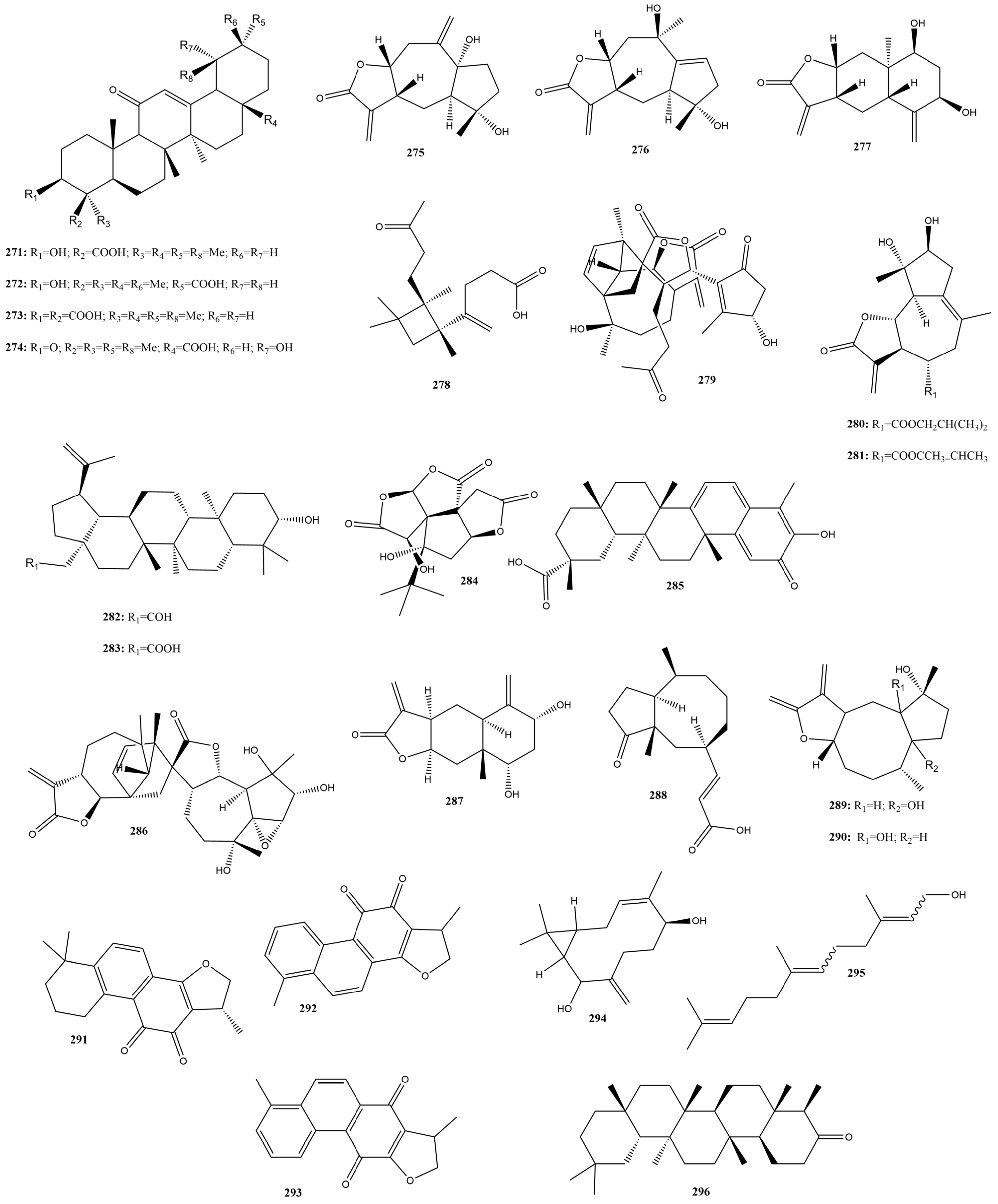
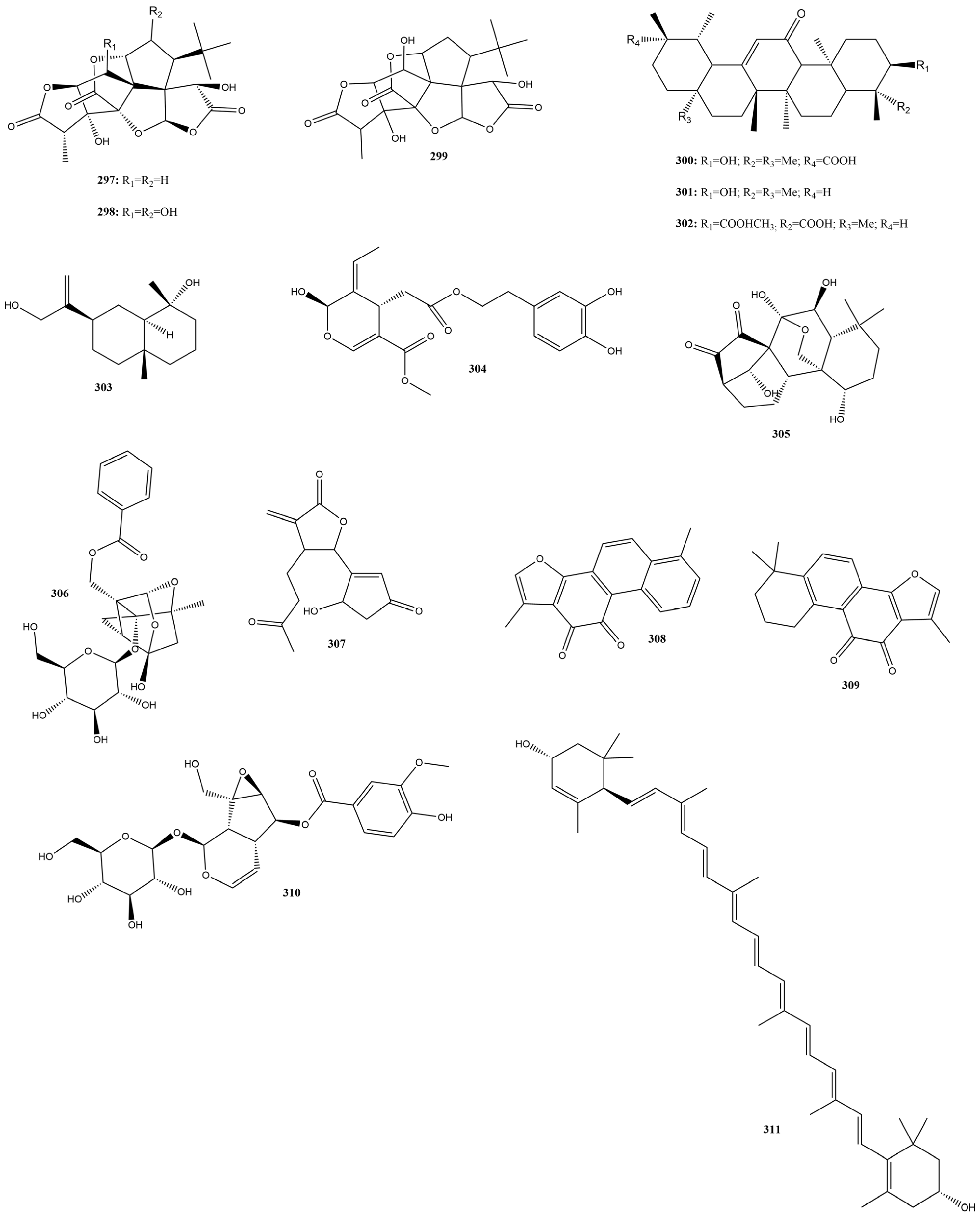
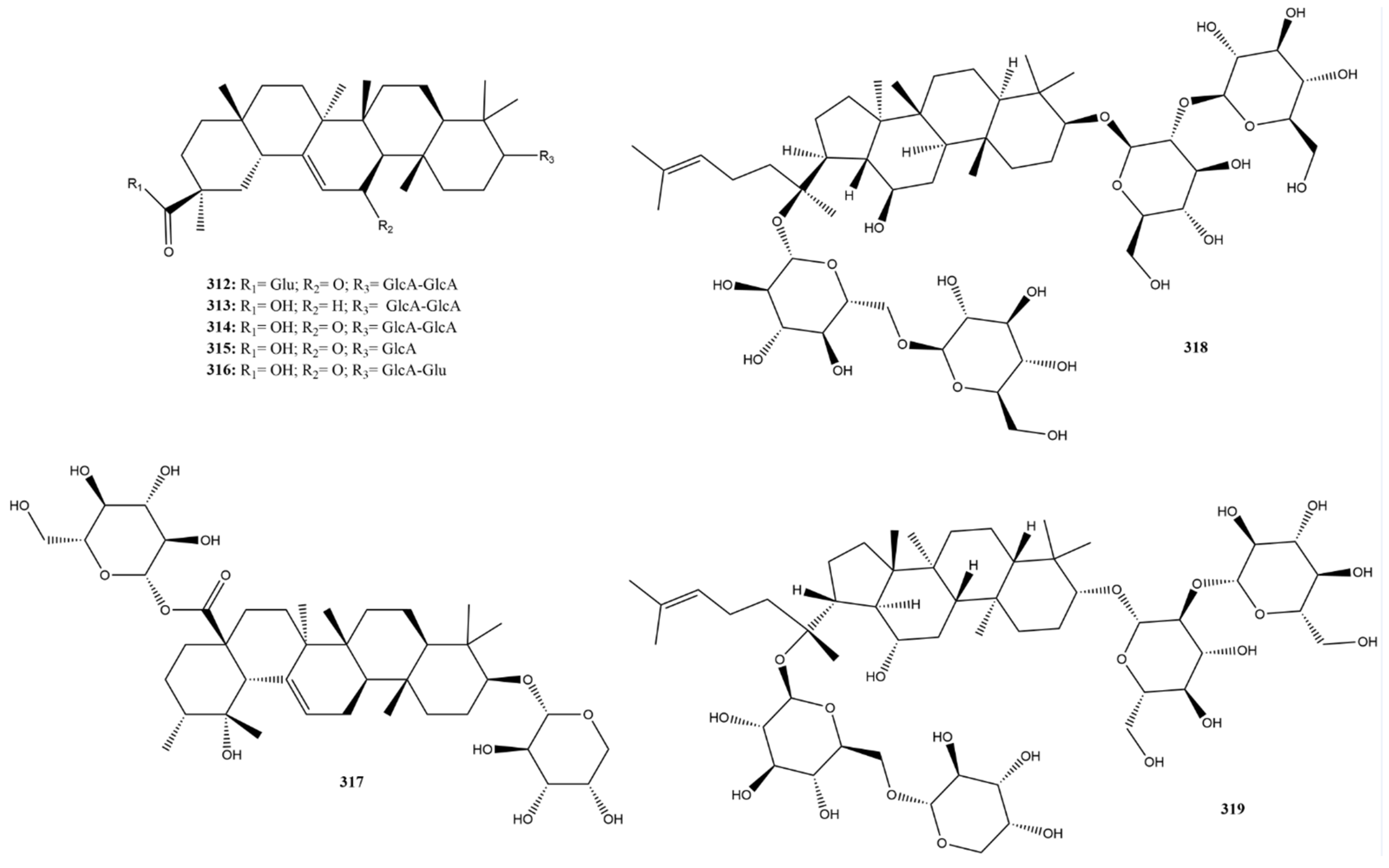

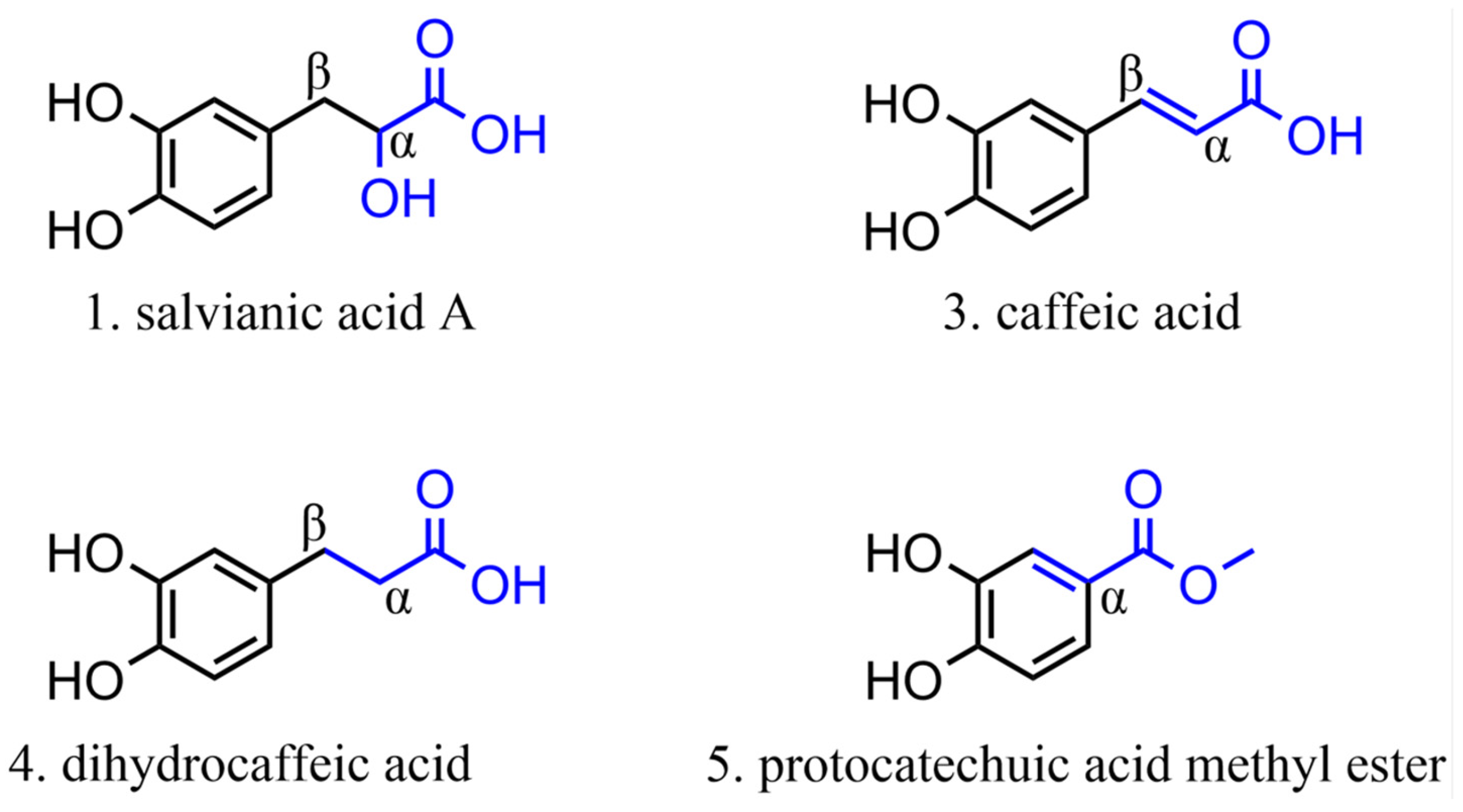
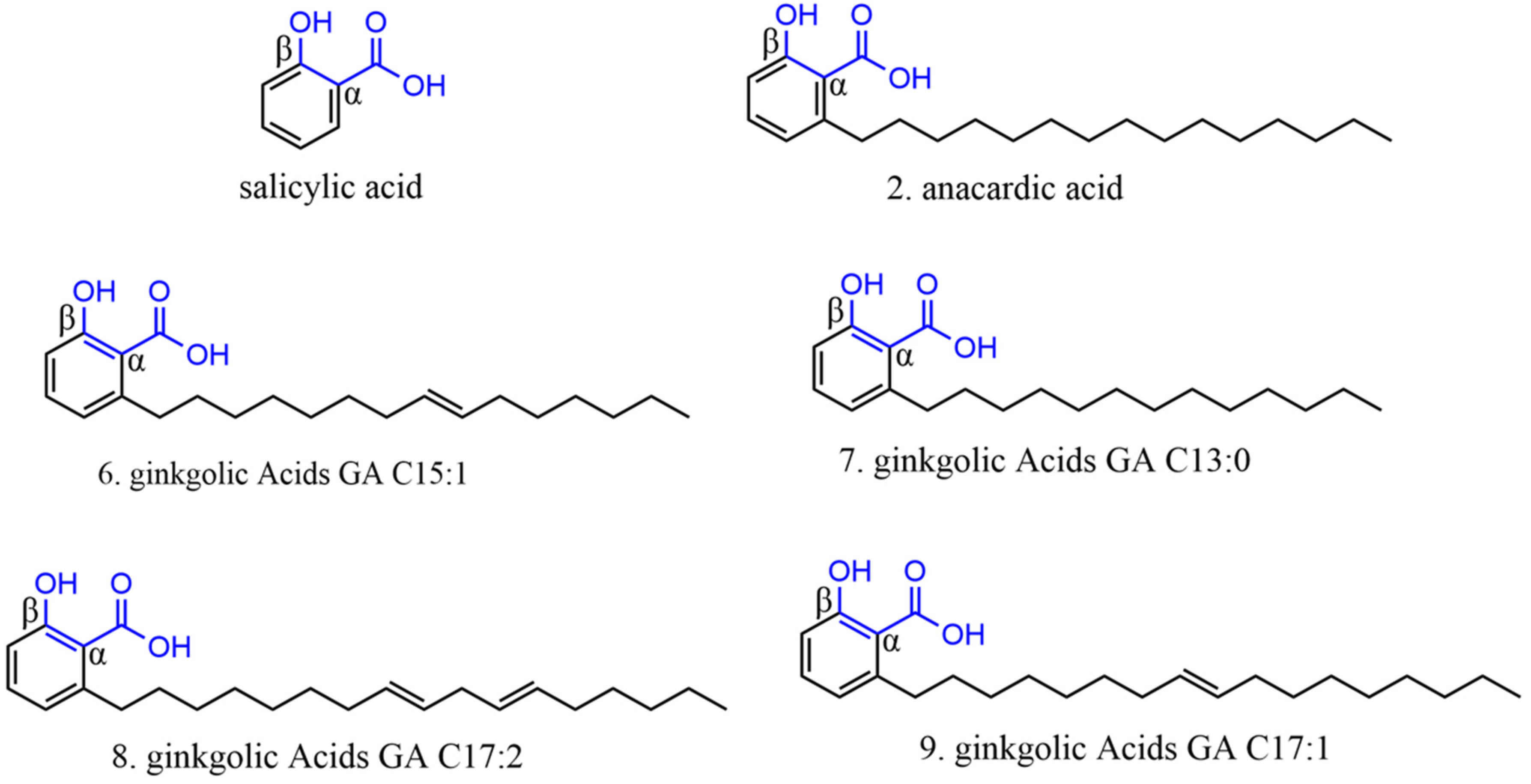

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Classes | DL | A | D | M | E | T | MC | BA |
|---|---|---|---|---|---|---|---|---|
| N° Compounds Retained/Passed | ||||||||
| Phenolic acids (19) | 14/5 | 0/5 | 1/4 | 0/4 | 1/3 | 0/3 | 0/3 | 1/2 |
| Phenylethanoids (20) | 16/4 | 0/4 | 1/3 | 0/3 | 2/1 | 0/1 | 0/1 | # |
| Chalcones (19) | 8/11 | 4/7 | # | - | - | - | - | - |
| Flavonoids (145) | 86/59 | 15/44 | 43/1 | 0/1 | # | - | - | - |
| Tannins (10) | # | - | - | - | - | - | - | - |
| Stilbenes (7) | 4/3 | 1/2 | 1/1 | # | - | - | - | - |
| Coumarins (9) | 0/9 | 0/9 | 1/8 | 0/8 | 2/6 | 3/3 | 1/2 | # |
| Quinones (17) | 9/8 | 1/7 | # | - | - | - | - | - |
| Lignoids (12) | 2/10 | 1/9 | 7/2 | # | - | - | - | - |
| Alkaloids (13) | 7/6 | 2/4 | 0/4 | 0/4 | 3/1 | # | - | - |
| Terpenoids (41) | 19/22 | 2/20 | 5/15 | 0/15 | 11/4 | 3/1 | 0/1 | # |
| Saponins (8) | # | - | - | - | - | - | - | - |
| Other compounds (11) | 2/9 | 0/9 | # | - | - | - | - | - |
| Total compounds (330) | 149 | 120 | 38 | 35 | 15 | 8 | 7 | 2 Best compounds |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, B.A.; Fernandes, D.A.; da Fonseca, T.S.; Campos, M.F.; Jural, P.A.; Toledo e Silva, M.V.; Constant, L.E.C.; da Veiga, A.A.S.; Ferreira, B.R.; Magalhães, E.S.; et al. Plants Metabolites as In Vitro Inhibitors of SARS-CoV-2 Targets: A Systematic Review and Computational Analysis. Drugs Drug Candidates 2025, 4, 27. https://doi.org/10.3390/ddc4020027
Gomes BA, Fernandes DA, da Fonseca TS, Campos MF, Jural PA, Toledo e Silva MV, Constant LEC, da Veiga AAS, Ferreira BR, Magalhães ES, et al. Plants Metabolites as In Vitro Inhibitors of SARS-CoV-2 Targets: A Systematic Review and Computational Analysis. Drugs and Drug Candidates. 2025; 4(2):27. https://doi.org/10.3390/ddc4020027
Chicago/Turabian StyleGomes, Brendo Araujo, Diégina Araújo Fernandes, Thamirys Silva da Fonseca, Mariana Freire Campos, Patrícia Alves Jural, Marcos Vinicius Toledo e Silva, Larissa Esteves Carvalho Constant, Andrex Augusto Silva da Veiga, Beatriz Ribeiro Ferreira, Ellen Santos Magalhães, and et al. 2025. "Plants Metabolites as In Vitro Inhibitors of SARS-CoV-2 Targets: A Systematic Review and Computational Analysis" Drugs and Drug Candidates 4, no. 2: 27. https://doi.org/10.3390/ddc4020027
APA StyleGomes, B. A., Fernandes, D. A., da Fonseca, T. S., Campos, M. F., Jural, P. A., Toledo e Silva, M. V., Constant, L. E. C., da Veiga, A. A. S., Ferreira, B. R., Magalhães, E. S., Pereira, H. B. M., de Mattos, B. G. M., de Oliveira, B. A. C., da Silva Costa, S., do Amaral, F. M. M., de Oliveira, D. R., Leal, I. C. R., Martins, G. R., Leitão, G. G., ... Leitão, S. G. (2025). Plants Metabolites as In Vitro Inhibitors of SARS-CoV-2 Targets: A Systematic Review and Computational Analysis. Drugs and Drug Candidates, 4(2), 27. https://doi.org/10.3390/ddc4020027