Abstract
Background/Objectives: Pancreatic cancer is the seventh most lethal type of cancer in the world, and its treatment, which is largely inefficient, is based on surgery and/or non-specific chemotherapy. Its malignant features are characterized by complex cell signaling pathways, which can be used as targets for new drugs. Methods: In this study, glucal-based compounds were synthetized, with substitution based on fluorine, nitrogen and aromatic ring addition. The compounds were tested in the pancreatic cell culture Mia-PaCa-2 and cell viability was assessed, with further IC50 calculation, stability and selectivity. Molecular docking was performed to evaluate the probable molecular target for 5b and in silico physicochemical properties were determined. Results: One molecule, named 5b, with two fluorine atoms inserted in the aromatic ring, exerted potent inhibitory activity on cell growth (IC50 = 1.39 µM), which was selective for pancreatic cells. Through molecular docking studies, the compound was found to be positioned in the active site of JAK3, indicating inhibition of such protein, which has a role in tumoral cell growth. Moreover, 5b was stable for 24 months and had physicochemical properties to permeate cell membranes, good oral absorption, and low potential to cause toxicity. Conclusions: These data suggest that 5b can be druggable and can be considered as a prototype for a new course of treatment in pancreatic cancer.
1. Introduction
Pancreatic cancer is the seventh most lethal type of cancer in the world. It has a poor prognosis and very low survival rate of only 6% [1]. The most prevalent histological variation of pancreatic cancer is pancreatic adenocarcinoma, which appears in the exocrine portion of the pancreas and is responsible for secreting enzymes into the duodenum in order to aid the digestive process [2]. This cancer type has several subtypes, including ductal cell adenocarcinoma, which corresponds to around 85% of the cases [3]. The main characteristic of ductal cell adenocarcinoma is that it is proliferative and invasive, becoming more aggressive and lethal [4].
There are several risk factors associated with this type of cancer, such as obesity, diabetes, tobacco use, and chronic pancreatitis, besides genetic causes [5].
The mechanism underlying this malignant feature is a specific genetic mutation relating to oncogene K-RAS (Kirsten rat sarcoma viral oncogenic homolog), and three tumor suppressor genes: TP53, p16/CDKN2A, and SMAD4 [6].
KRAS is a part of the tumoral proliferative signaling pathway [4]. In a physiological setting, the Ras protein is activated by growth factor through GTP binding. However, specific mutations are capable of inhibiting GTPase, making K-ras constantly active. This mutation seems to be the most common in pancreatic adenocarcinoma, especially at positions G12D and G12V [7]. Secondarily, mutations in tumor suppressor genes (CDKNA2 and TP53) are also present and contribute to tumor growth and invasion [8].
K-ras signaling regulates phosphoinositide 3-kinase activity [7]. This pathway is responsible for cell and matrix proliferation, maintenance of cell cytoskeleton, apoptosis, and protein synthesis. These mechanisms occur through secondary pathways, with participation of the mammalian target of rapamycin (mTOR), nuclear factor kB (NF-kB) and glycogen synthase kinase-3 beta (GSK3β) [4].
Existing studies have focused on these pathways and protein targets, aiming to identify a specific and effective treatment for pancreatic cancer. However, there is no pharmacological agent in the market that acts on these molecules, focusing specifically on pancreatic cancer [4,7].
Among the current pharmacological therapies for this type of tumor, gemcitabine is the most common treatment, but causes up to a 10% response rate. Recently, FOLFIRINOX (fluoruracil, leucovorin, irinotecan, and oxaliplatin) was introduced, and was shown to increase the survival rate, although it has also been associated with increased toxicity [2,9,10].
New compounds have been obtained with the aim of treating pancreatic cancer. One example is 1,3,4-thiadiazole derivatives, which have anticancer effects due to their ability to interfere with DNA replication [11]. Natural sources have provided several molecules which act on this type of cancer, including polyphenols, alkaloids, terpenoids, and flavonoids [12]. Monoterpene indole alkaloid derivatives (BBIT20) also had effects on pancreatic tumors in in vitro and in vivo models, inhibiting the homologous recombination (HR) of DNA repair [13].
Compounds derived from glycosides have been shown to exert activity on tumor cells, including pancreatic ones. Cardiac glycosides and glycosides isolated from natural products, such as stevioside obtained from the leaves of the Stevia rebaudiana plant and from sea cucumbers, inhibit cell growth by inducing apoptosis and modulating the signaling pathways of pancreatic cancer cells [14,15].
The stereogenic center of carbohydrates tends to increase the stability and polarity of molecules, which makes them compatible with biologic fluids, representing a good option for bioactive compounds [16,17].
Therefore, in this work, we present four new synthetic glucal-derived compounds and their activity on pancreatic tumor cells.
2. Results
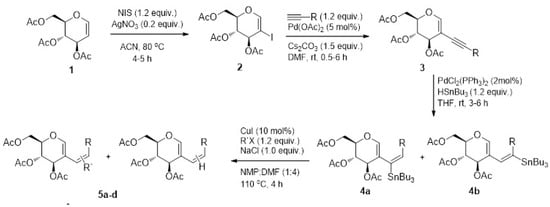
The synthesis is summarized in Figure 1 and the compounds are referred to as 5a, 5b, 5c, and 5d.
Figure 1.
Synthetic scheme of compounds 5a-d from 3,4,6-tri-O-acetyl-D-glucal.
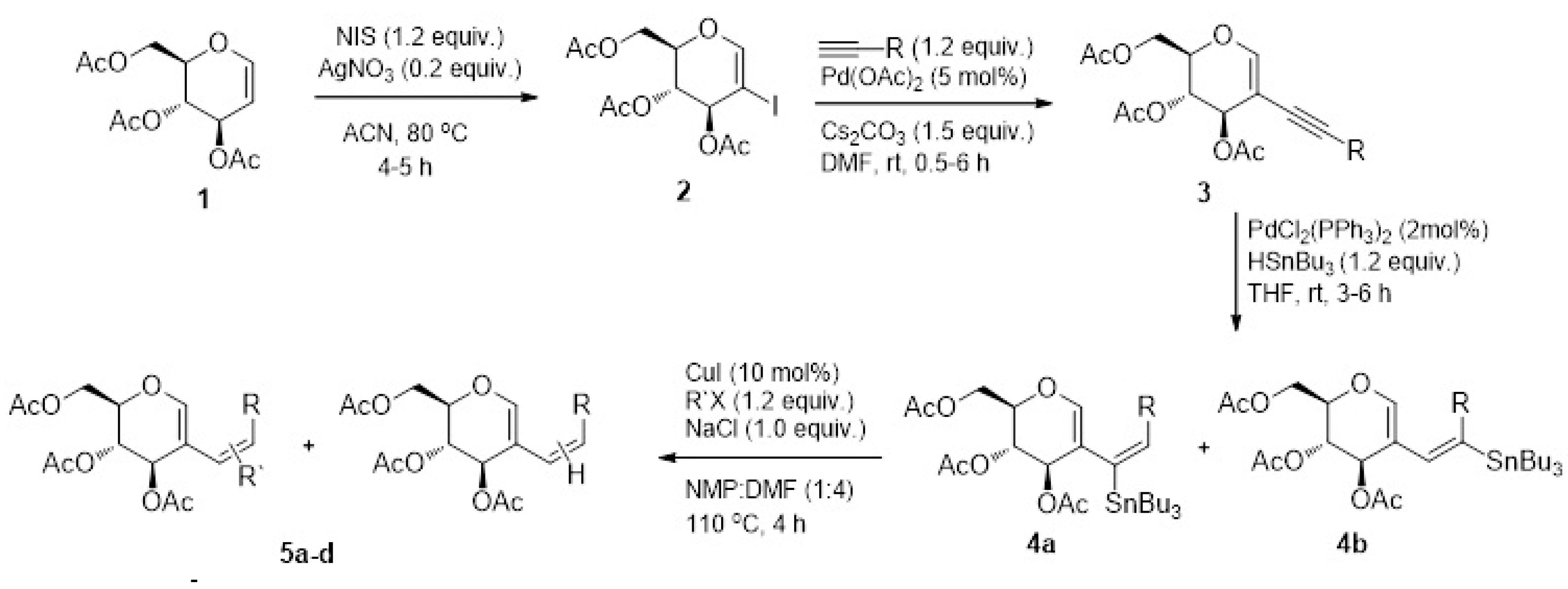
The 2D chemical structures of the synthesized compounds are presented in Figure 2 and include fluorinated aryl rings along with phenyl and pyridyl analogs of acetylated 2-alkenyl-D-glucal.
Figure 2.
Chemical structures of aryl/heteroaryl derivatives of acetylated D-glucal.
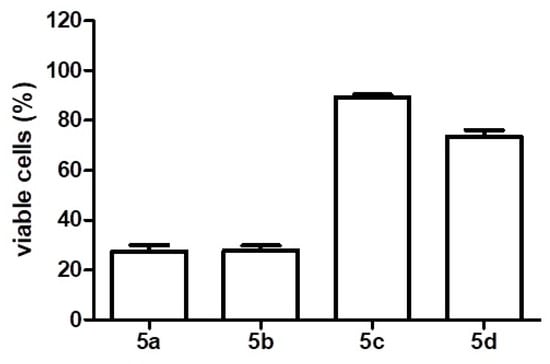
The four analogues were tested in Mia-Paca-2 pancreatic cancer cells, which were analyzed to verify their viability. Compounds 5a and 5b caused significant cell death (about 80%) when incubated in cells for 48 h, according to the MTT assay, with 2 µM. Compounds 5c and 5d had little effects on cell viability within the same incubation time (Figure 3).
Figure 3.
Viable Mia-Paca-2 cells (percent), determined by MTT method, after the treatment for 48 h with 2 µM glucal-based molecules.
In order to check if compounds 5a and 5b were selective for pancreatic cancer cells, they were tested in a panel of cancer cell lines: glioblastoma (U251), breast (MCF-7), ovary (OVCAR-3), leukemia (K562) and non-tumor fibroblast (HCAT), with a 48 h incubation time, and in a higher concentration range, in order to detect any effects of these cell lines. It was possible to determine that 5a caused a reduction in cell viability for MCF-7, OVCAR-3, and HCAT, showing IC50 values below 1 M. On the other hand, AS-5b did not have any effect on any of the tested cell lines, even at a very high concentration, indicating a targeted and exclusive effect on pancreas cancer cells (Table 1).
Table 1.
IC50 of tumor cell panel after treatment with glucal-based compounds, expressed in molar.
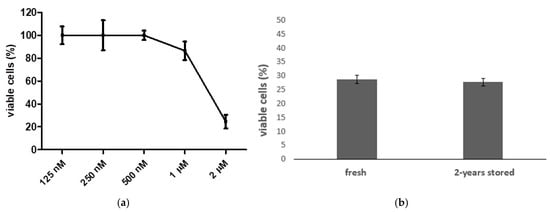
Considering the highly selective, potent effects on Mia-Paca-2, we elected compound 5b as the best option for further studies. Thus, the IC50 value was estimated at 1.39 ± 0.006 µM in this cell line, as shown in Figure 4a. This compound was stable in an aqueous solution. A cell viability MTT assay was used to verify the activity of 5b stored in 2–8 °C for 24 months in 0.9% sterile saline solution, in comparison to a sample that was synthesized, diluted and immediately used. No significant differences were noticed in the cell viability of Mia-Paca-2: 27.8% of cells were viable after 2 years of storage compared to the 28.78% viability of the fresh cells (Figure 4b).
Figure 4.
Mia-Paca2 cells viability by MTT method after treatment with 5b. (a) Percentage of viable cells in increasing concentrations of 5b for determination of IC50 value. (b) Percentage of viable cells after treatment with 2 µM 5b fresh or stored by 2 years at 2–8 °C.
The characteristics of the molecules are reflected in their important physicochemical parameters: 5b has logP 3.17, TPSA 88.13, and is moderately soluble in water (4.54−0.3 mg/mL). Regarding pharmacokinetics, it has an elevated gastrointestinal absorption rate and is not permeable to the brain–blood barrier, according to the Brain Or IntestinaL EstimateD permeation method (BOILED-Egg), a predictive model based on the lipophilicity and polarity of small molecules [18]. These data indicate that the molecule can be administered orally, which is an easy and convenient pathway, and it has a moderate capacity to permeate membranes, which allows it to enter the gastrointestinal barrier, but not the brain–blood barrier, and thus has no effect on the central nervous system.
It also has no effect on the P-gp substrate, according to the Support Vector Machines (SVMs) model, used to predict the pharmacokinetic properties of P-gp substrate based on molecular descriptors and comparison with known molecules [19]. This parameter indicates that this chemotherapy has no resistance profile.
This compound is a weak inhibitor of hERG (pIC50 ≤ 6.0 mol/L), and it was considered nontoxic based on AMES and acute oral toxicity (Class III = 500 mg/kg–5000 mg/kg). The toxicity was estimated by a lethal dose 50% (LD50), according to categories established by EPA-USA.
To understand its mechanism of action, 5b was submitted to the Swiss Target Prediction webtool. Proteases/enzymes and the family A/G protein-coupled receptor and kinases were identified as the most probable macromolecular targets (43.3, 33.3 and 26.7%, respectively). Among kinases pathway, MAP2K1, MAPK8, and EGFR were the most probable.
Thus, these kinases and other related kinases were selected from the Protein Data Bank (PDB) and used for molecular docking evaluation to verify a possible inhibition caused by 5b. We considered proteins from different pathways related to cell proliferation and apoptosis, which had already been described as important targets for pancreatic cancer. The results of the docking are presented in Table 2, where it is possible to see all of the evaluated targets and binding profiles.
Table 2.
Binding parameters between protein targets for pancreatic cancer and compound 5b.
Of all the analyzed proteins, only PI3K, JNK1, JAK3, and TAK 1 had 5b positioned on the same site of a known inhibitor, where the proteins elicit their pharmacological effects, which demonstrated this compound’s potential application as a drug. From that, PI3K had low affinity to 5b, so although it was in a good site for pharmacological activity, it was deemed unlikely to bind to this protein.
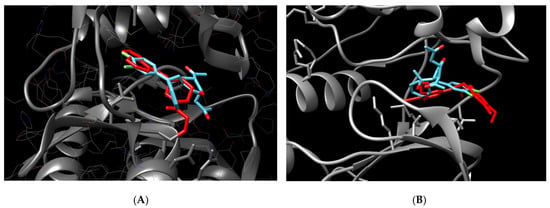
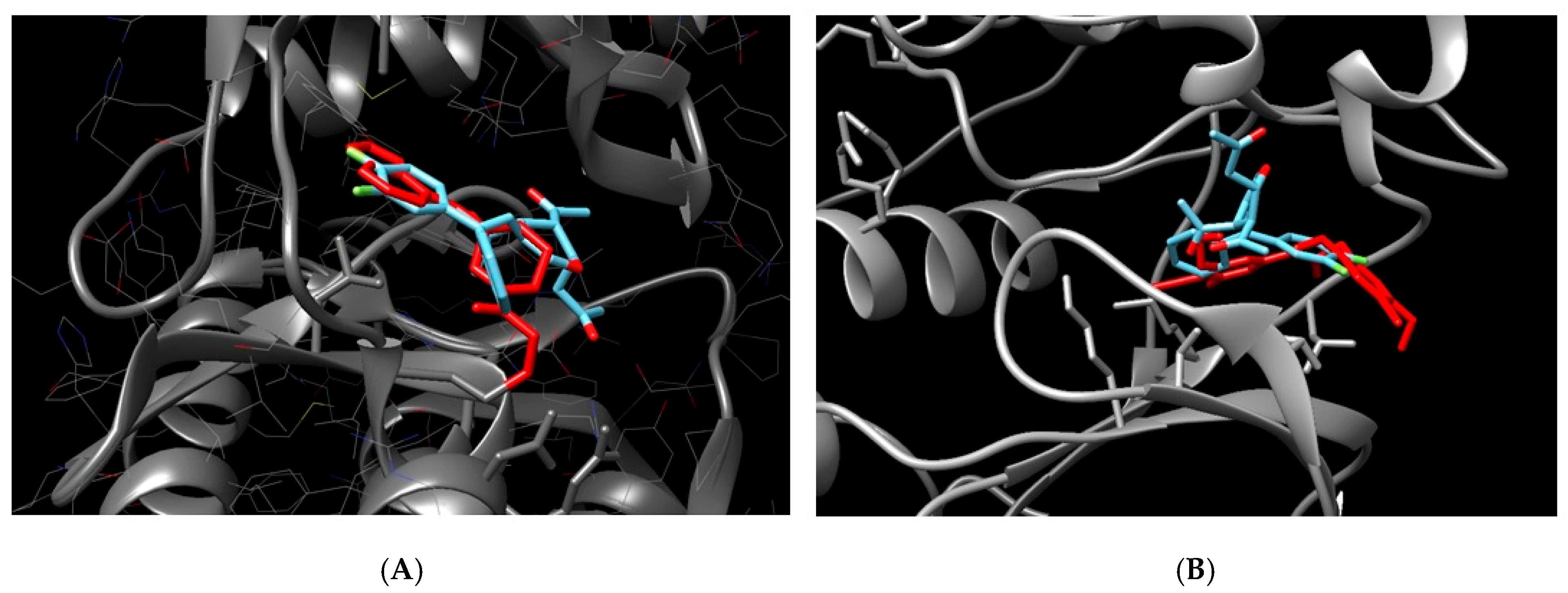
On the other hand, JNK1, JAK3, and TAK were placed in adequate protein sites, with low binding energy, of which JAK3 had the best affinity (−9.4 kcal/mol), as shown in Figure 5A. In the figure, it is possible to see that 5b is superposed with a specific inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one
Figure 5.
Molecular docking of 5b compound and potential protein targets related to pancreatic cancer inhibition. (A) = docking between JAK3 and 5b. (B) = docking between JNK1 and 5b. Protein in gray, 5b in blue, and a known inhibitor in red (1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one for JAK 1 and N-(4-Amino-5-cyano-6-ethoxypyridin-2-yl)-2-(4-bromo-2, 5-dimethoxyphenyl)acetamide for JNK1.
Although JAK1 and 2 have low affinity as well, the binding was identified in the amino acids far from the inhibition pocket, with no possible inhibition characterized, as well as STAT3. Tyrosine kinase 2 (TYK2), belonging to the JAK family, did not bind with the ligand.
C-Jun N-terminal kinase 1 (JNK1 or MAPK8) also was identified as an important target due to low binding energy (−8.17 kcal/mol) and correct place for the protein inhibition, superposed to the known inhibitor N-(4-Amino-5-cyano-6-ethoxypyridin-2-yl)-2-(4-bromo-2, 5-dimethoxyphenyl)acetamide (Figure 5B).
3. Discussion
Pancreatic cancer, the seventh leading cause of cancer-related deaths worldwide, has a poor prognosis with a survival rate of only 6%. Although there is pharmacological therapy for this tumor, the efficacy is poor, and the adverse effects are intense. Thus, pharmacological alternatives are necessary. In this paper, we presented new synthetic compounds able to reduce the cell viability of pancreatic cells, in in vitro and in silico approaches.
MIA PaCa-2 is derived from pancreas tumor tissue obtained from a 65-year-old Caucasian male (ATCC). It is a cell line that is widely used in the in vitro studies of adenocarcinomas.
Here, we showed that one molecule, named 5b, reduced the viability of MIA PaCa-2, without having any effect on other cell lines, which represents a high selectivity for pancreatic cells. These results may represent that, in a living organism, the potential of presenting side effects is reduced, making this a good drug prototype. Moreover, the molecule is highly stable, maintaining its activity for up to 2 years, which is another important factor when considering a drug. In addition to the selectivity, the potency of the molecule was high, as de IC50 was calculated as only 1.39 µM.
In a tentative attempt to elucidate the mechanism of action, molecular docking was performed with the most studied proteins relating to pancreatic tumors. Proteins JAK3 and JNK1 were the most promising ones, with low energy (−9.4 and−8.17 kcal/mol, respectively), in the same inhibitor site as a known inhibitor. Both parameters indicate that 5b has high potential to interact with the proteins, in an important region for their biological activity. Thus, 5b’s potential to inhibit them is high, which explains the inhibition of MIA PaCa-2 growth.
The Janus kinase/signal transducers and activators of the transcription (JAK-STAT) pathway are activated by the action of several types of cytokines and growth factors, and have been associated with pancreatic cancer [20,21].
A study showed that gemcitabine and erlotinib could inhibit pancreatic tumor growth through the downregulation of JAKs and STATs, and induced apoptosis of pancreatic cancer cells [22]. Another study showed that the inhibition of JAK3 and STAT3 inhibited the growth, invasion, and migration of human pancreatic cancer cells [23].
Activator protein-1 (AP-1) is a group of proteins that comprise the Jun, Fos, Maf and ATF families, related to several types of cancer, including pancreatic cancer, responsible for cell proliferation and apoptosis control [24]. The proto-oncogene c-jun is overexpressed in human pancreatic cancer cells, especially c-Jun, regulated by JNK, and results in cancer cell proliferation. Thus, specific inhibitors have been studied for such target [25,26].
When its physicochemical properties were analyzed using in silico tools, 5b proved to be permeable to cell membranes, as its logP is 3.17 and its TPSA is 88.13. It was found to be moderately soluble in water, compatible with biological fluids, and able to be absorbed and distributed and to reach the molecular target. The pharmacokinetics data confirmed that its elevated gastrointestinal absorption rate indicates that it is suitable for oral administration, the optimal choice for ministering medication. Moreover, the lack of permeability to the brain–blood barrier indicates no action on the central nervous system, and, most importantly for a drug used in chemotherapy, no P-gp substrate, indicating a low resistance profile.
One factor which characterizes 5b from this series of compounds is the presence of a fluorine atom, which has high electronegativity, but a small size, changing the electronic properties, steric interactions, and stability of the molecule, in addition to facilitating cell permeation, which contributes to better pharmacokinetics and interaction with biological targets. This could explain the better effect observed by compound 5b compared to other compounds. It is important to mention that the replacement of hydrogen with fluorine is commonly used in medicinal chemistry and drug design and development [27].
In addition to its high potency, selectivity, and stability, 5b is a poor inhibitor of hERG, which indicates that it does not inhibit potassium channels during cardiac repolarization, as it is not cardiotoxic. Furthermore, it was considered nontoxic based on AMES and acute oral toxicity; therefore, it has shown low potential to cause DNA mutation and toxic reaction, respectively.
According to all these features, we can determine that 5b does not violate any of Lipinski’s rules or the PAINS alert for medicinal chemistry, and this can be considered as a druggable compound [28].
In conclusion, we obtained a new compound that is able to inhibit the growth of a human tumor pancreatic cell, selectively and with high potency, and thus is considered druggable. JAK3 was identified as a probable target of this molecule, which represents a therapeutic alternative for this lethal cancer type.
4. Materials and Methods
4.1. Synthesis
Four glucal-derived compounds were synthesized using the synthetic procedures already communicated by our group [17,29] and others [30,31]. The characterization by 1H NMR, 13C NMR, and mass spectrometry of these compounds has also been communicated by our group in the above-mentioned publications.
4.2. Cell Viability Assay
Cancer cell lines of pancreas (Mia-Paca-28), glioblastoma (U251), breast (MCF-7), ovary (OVCAR-3), leukemia (K562) and non-tumor fibroblast (HCAT) were obtained from the American Type Culture Collection—ATCC.
Cells were cultured in DMEM media containing 5% fetal bovine serum (Nutricell, South Tangerang, Indonesia) and 1% penicillin–streptomycin (Nutricell), in 37 °C and 5% CO2 atmosphere and humidity. Cells (3 × 104) were grown in a 96-well plate and then treated with compounds (125 nM–2 µM for pancreatic cells and 10 µM–1 M for other cell lines) diluted in sterile saline for 48 h, in triplicate. After that, MTT (3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide) (Sigma, St. Louis, MO, USA) was incubated for 4 h. The absorbance was read at 540 nm and the media from the triplicates were obtained. The IC50 (half-maximal inhibitory concentration) value was calculated using Prism Graph Software v 5.0.
For the stability evaluation, the compound was immediately diluted in 0.9% sterile saline after synthesis or maintained at 2–8 °C for 24 months, diluted in the same buffer. The MTT assay was used for biological activity determination, as described above.
4.3. Target Prediction
The macromolecular targets for the compound were estimated by the webtool Swiss TargetPrediction. The 3D structure of the ligand was submitted and searched in a Homo sapiens database and founded according to the similarity with a library of several compounds [32].
4.4. Molecular Docking
The ligand was drawn in OpenBabel Cheminformatics for a mol2 file obtention, with 3D structure, protonated at pH 7, and no changes were made in terms of adding or deleting hydrogens.
The protein targets were selected in the Protein Data Bank (PDB) by their resolution (up to 3 Å) and binding to an inhibitor. Missing atoms, chain breaks, and water molecules were removed using Chimera 1.15 software.
Ligands and proteins were subjected to analysis by the Swiss Dock webserver docking https://www.swissdock.ch/, accessed on September 2024, with parameters set to automatic search, considering the CHARMM method [33]. Binding energy (kcal/mol) values were obtained, and the docking was analyzed by Chimera 1.15 software to verify the ligand positioning in the protein.
4.5. ADMET
The physicochemical properties of compounds and ADME parameters (gastrointestinal absorption, blood–brain barrier permeation, and liver enzymes inhibition) were predicted using the Swiss ADME server from the Molecular Modeling Group of the Swiss Institute of Bioinformatics [19].
The Lipinski rules were used to determine the drug-likeness of the compound, considering molecular mass, membrane permeability, TPSA, and water solubility [28]. Moreover, values for gastrointestinal absorption rate and p-gp substrate were obtained. These values were obtained by the Swiss ADME server, as well as the drug-likeness.
Toxicity was predicted by admetSAR 2.0 server [34]. The ligand was drawn and values of hERG inhibition, AMES, and acute oral toxicity were retrieved for carcinogenicity and acute oral toxicity evaluation, respectively.
Author Contributions
P.A., H.S., A.S., D.G.P. and K.C.Q.B.: Investigation, Methodology, Formal analysis. H.A.S.: Formal analysis, Funding acquisition, Supervision. J.M.S.: Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing—original draft. All authors: Writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. JMS is the National Council for Scientific and Technological Development (CNPq) fellow (313402/2023-0).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding authors.
Acknowledgments
The authors thank Robert for encouraging this research and publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- New Global Cancer Data: GLOBOCAN. Available online: https://gco.iarc.fr/en (accessed on 1 July 2024).
- Goess, R.; Friess, H. A Look at the Progress of Treating Pancreatic Cancer over the Past 20 Years. Expert Rev. Anticancer Ther. 2018, 18, 295–304. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic Cancer: A Review of Clinical Diagnosis, Epidemiology, Treatment and Outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Polireddy, K.; Chen, Q. Cancer of the Pancreas: Molecular Pathways and Current Advancement in Treatment. J. Cancer 2016, 7, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Brody, J.R.; Abrams, R.A.; Lewis, N.L.; Yeo, C.J. Cancer of the Pancreas. In DeVita, Hellman and Rosenberg’s Cancer: Principles and Practice of Oncology; Wolters Kluwer Health: Philadelphia, PA, USA, 2015. [Google Scholar]
- Iacobuzio-Donahue, C.A.; Velculescu, V.E.; Wolfgang, C.L.; Hruban, R.H. Genetic Basis of Pancreas Cancer Development and Progression: Insights from Whole-Exome and Whole-Genome Sequencing. Clin. Cancer Res. 2012, 18, 4257–4265. [Google Scholar] [CrossRef]
- Murthy, D.; Attri, K.S.; Singh, P.K. Phosphoinositide 3-Kinase Signaling Pathway in Pancreatic Ductal Adenocarcinoma Progression, Pathogenesis, and Therapeutics. Front. Physiol. 2018, 9, 335. [Google Scholar] [CrossRef]
- Bernard, V.; Fleming, J.; Maitra, A. Molecular and Genetic Basis of Pancreatic Carcinogenesis. Surg. Oncol. Clin. N. Am. 2016, 25, 227–238. [Google Scholar] [CrossRef]
- von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Żurawska, K.; Stokowy, M.; Kapica, P.; Olesiejuk, M.; Kudelko, A.; Papaj, K.; Skonieczna, M.; Szeja, W.; Walczak, K.; Kasprzycka, A. Synthesis and Preliminary Anticancer Activity Assessment of N-Glycosides of 2-Amino-1,3,4-Thiadiazoles. Molecules 2021, 26, 7245. [Google Scholar] [CrossRef]
- Indelicato, S.; Bongiorno, D.; Mauro, M.; Cascioferro, S. Recent Developments of 1,3,4-Thiadiazole Compounds as Anticancer Agents. Pharmaceuticals 2025, 18, 580. [Google Scholar] [CrossRef]
- Sahu, S.K.; Prabhakar, P.K.; Vyas, M. Therapeutical potential of natural products in treatment of pancreatic cancer: A review. Mol. Biol. Rep. 2025, 52, 179. [Google Scholar] [CrossRef]
- Calheiros, J.; Silva, R.; Barbosa, F.; Morais, J.; Moura, S.R.; Almeida, S.; Fiorini, E.; Mulhovo, S.; Aguiar, T.Q.; Wang, T.; et al. A first-in-class inhibitor of homologous recombination DNA repair counteracts tumour growth, metastasis and therapeutic resistance in pancreatic cancer. J. Exp. Clin. Cancer Res. 2025, 44, 129. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Sikander, M.; Malik, S.; Tripathi, M.K.; Hafeez, B.B.; Yallapu, M.M.; Chauhan, S.C.; Khan, S.; Jaggi, M. Steviol Represses Glucose Metabolism and Translation Initiation in Pancreatic Cancer Cells. Biomedicines 2021, 9, 1814. [Google Scholar] [CrossRef]
- Lindholm, H.; Ejeskär, K.; Szekeres, F. Digitoxin Affects Metabolism, ROS Production and Proliferation in Pancreatic Cancer Cells Differently Depending on the Cell Phenotype. Int. J. Mol. Sci. 2022, 23, 8237. [Google Scholar] [CrossRef]
- Vieira Veloso, R.; Shamim, A.; Lamarrey, Y.; Stefani, H.A.; Mozer Sciani, J. Antioxidant and Anti-Sickling Activity of Glucal-Based Triazoles Compounds—An in Vitro and in Silico Study. Bioorg. Chem. 2021, 109, 104709. [Google Scholar] [CrossRef]
- Shamim, A.; Barbeiro, C.S.; Ali, B.; Stefani, H.A. Synthesis of Stannyl-Substituted D-Glucal Derivatives via Palladium-Catalyzed Regioselective Hydrostannation and Their Synthetic Applications. ChemistrySelect 2016, 1, 5653–5659. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A Boiled-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT Signaling Pathway. J. Cell. Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Mace, T.A.; Shakya, R.; Elnaggar, O.; Wilson, K.; Komar, H.M.; Yang, J.; Pitarresi, J.R.; Young, G.S.; Ostrowski, M.C.; Ludwig, T.; et al. Single Agent BMS-911543 Jak2 Inhibitor Has Distinct Inhibitory Effects on STAT5 Signaling in Genetically Engineered Mice with Pancreatic Cancer. Oncotarget 2015, 6, 44509–44522. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, D.; Liu, Z.; Huang, X.; Liu, Q.; Kang, Y.; Chen, Z.; Guo, Y.; Zhu, H.; Sun, C. Combination of Gemcitabine and Erlotinib Inhibits Recurrent Pancreatic Cancer Growth in Mice via the JAK-STAT Pathway. Oncol. Rep. 2018, 39, 1081–1089. [Google Scholar] [CrossRef]
- Tang, S.-N.; Fu, J.; Shankar, S.; Srivastava, R.K. EGCG Enhances the Therapeutic Potential of Gemcitabine and CP690550 by Inhibiting STAT3 Signaling Pathway in Human Pancreatic Cancer. PLoS ONE 2012, 7, e31067. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a Regulator of Cell Life and Death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Tessari, G.; Ferrara, C.; Poletti, A.; Dubrovich, A.; Corsini, A.; del Favero, G.; Naccarato, R. The Expression of Proto-Oncogene c-Jun in Human Pancreatic Cancer. Anticancer Res. 1999, 19, 863–867. [Google Scholar]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The Specificities of Protein Kinase Inhibitors: An Update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef]
- Chandra, G.; Singh, D.V.; Mahato, G.K.; Patel, S. Fluorine-a small magic bullet atom in the drug development: Perspective to FDA approved and COVID-19 recommended drugs. Chem. Pap. 2023, 13, 4085–4106. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug. Deliv. Rev. 2001, 46, 3. [Google Scholar] [CrossRef]
- Shamim, A.; Vasconcelos, S.N.S.; Ali, B.; Madureira, L.S.; Zukerman-Schpector, J.; Stefani, H.A. Ligand and Copper Free Sonogashira Coupling to Achieve 2-Alkynyl d-Glucal Derivatives: Regioselective Electrophile Promoted Nucleophilic 5-Endo-Dig Cyclization. Tetrahedron Lett. 2015, 56, 5836–5842. [Google Scholar] [CrossRef]
- Betzer, J.-F.; Delaloge, F.; Muller, B.; Pancrazi, A.; Prunet, J. Radical Hydrostannylation, Pd(0)-Catalyzed Hydrostannylation, Stannylcupration of Propargyl Alcohols and Enynols: Regio- and Stereoselectivities. J. Org. Chem. 1997, 62, 7768–7780. [Google Scholar] [CrossRef]
- Dharuman, S.; Vankar, Y.D. N-Halosuccinimide/AgNO3-Efficient Reagent Systems for One-Step Synthesis of 2-Haloglycals from Glycals: Application in the Synthesis of 2C-Branched Sugars via Heck Coupling Reactions. Org. Lett. 2014, 16, 1172–1175. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).