Abstract
The Warburg effect (or aerobic glycolysis), which was first described in 1926 by Otto Heinrich Warburg, consists of the change in glucose metabolism in cancer cells. In normal cells, glucose metabolism finalizes in the mitochondria through oxidative phosphorylation (OXPHOS) in the presence of oxygen. However, the Warburg effect describes a change in the glucose metabolism in cancer cells, consuming excess glucose and converting it into lactate independently of the presence of oxygen. During this process, a wide variety of enzymes can modify their expression and activity to contribute to the mechanism of deregulated cancer metabolism. Therefore, the modulation of enzymes regulating aerobic glycolysis is a strategy for cancer treatment. Although numerous enzymes play a role in regulating aerobic glycolysis, hexokinase 2 (HK2), pyruvate dehydrogenase kinase (PDK), pyruvate kinase (PK), and lactate dehydrogenase (LDH) are worth mentioning. Numerous modulators of these enzymes have been described in recent years. This review aims to present and group, according to their chemical structure, the most recent emerging molecules targeting the above-mentioned enzymes involved in the Warburg effect in view of the future development of cancer treatments.
1. Introduction
Cancer is one of the major health problems of the human population and the second cause of death worldwide, only surpassed by cardiovascular diseases [1]. Over the years of intense study on the biology of cancer, some common characteristics have been described in most tumors. These are called “The Hallmarks of Cancer”. Although there were originally six hallmarks [2], nowadays, these features have been refined and novel hallmarks have been identified and included alongside the previous ones, bringing the total number to fourteen [3,4]. Among these key features of cancer cells, deregulated metabolism and reprogrammed cellular energetics are worth mentioning due to their influence on the development of tumor progression, metastasis, and tumor microenvironment (TME). The wide variety of enzymes and transporters implicated in this process and their different localizations into cells make them interesting targets for novel therapeutic approaches [5,6,7].
In normal cells, glucose, the principal carbon source, is transported to the cell by glucose transporter (GLUT) proteins and is catabolized in the glycolytic pathway through the tricarboxylic acid (TCA) cycle. The energy production process is finalized in the mitochondria with OXPHOS in the presence of oxygen, with a balance of 34 generated adenosine triphosphate (ATP) molecules, and the production of intermediates to support the biosynthesis of macromolecules along the process. This process is stimulated by different growth and transcription factors [8]. In hypoxic conditions, the product of glycolysis (pyruvate) is transformed into lactate by lactate dehydrogenase (LDH) to maintain ATP production. This survival mechanism is started by hypoxia-inducible factor 1 (HIF-1), a transcription factor responsible for oxygen homeostasis that is upregulated under hypoxic conditions by different signaling pathways in response to a low oxygen level. Its activation involves the expression of glycolytic and angiogenesis-related enzymes and transporters by the association with the hypoxia response element (HRE) in the promoters of those genes [9,10].
In 1926, Otto Heinrich Warburg described for the first time the change in glucose metabolism in cancer cells, which consumes excess glucose and converts it into lactate independently of the presence of oxygen, naming this process aerobic glycolysis or the Warburg effect [8,11]. This discovery was first associated with damage to the mitochondria and the OXPHOS chain, considering that this mitochondrial damage could be the cause of cancer [12]. However, after years of controversy and research, the correct function of the mitochondria and an adequate level of oxygen was shown in cancer cells. Aerobic glycolysis not only is an adaptation of hypoxia. It is also a metabolic reprogramming in the early stage of carcinogenesis independent of the exposition of oxygen, which results in the loss of function of tumor suppressors, altered signaling pathways, and oncogene activation, generating proliferation and malignant progression [13]. The use of the Warburg effect in non-transformed cells with pro-proliferative signals demonstrated that it is a metabolic strategy for cell proliferation [8]. Aerobic glycolysis provides some advantages to the cell, such as the faster regeneration of ATP than the TCA cycle. Despite a lower ATP production, the increment in the glucose consumption compensates for it in addition to lactate, a key molecule in the progression of tumors [14], whose production and shuttle to the extracellular matrix involves establishing a favorable emerging TME. Additionally, the production of lactate through pyruvate contributes to the regeneration of nicotinamide adenine dinucleotide (NAD) NAD+ to NADH, regulating the redox state of the cell [15]. Furthermore, the capacity to generate intermediates is used as the precursors of macromolecules [8]. These benefits are also possible by the activation of other metabolic pathways, such as pentose phosphate pathways (PPPs) or glutaminolysis [15,16,17,18,19].
The reduction in or disablement of mitochondria produces the alkalinization of the cytoplasm due to a decrease in carbon dioxide (CO2) secretion that reacts with water (H2O) and produces carbonic acid [12]. On the contrary, the acidification of the TME by the excretion of lactate derived in a deregulated pH gradient favors tumor progression, hence reducing the T-cell activity [20], evasion of apoptosis, migration, chemoresistance, etc. [12,20]. Lactate is shuttled through monocarboxylate transporters (MCTs) to the TME with the aim of supplying energy to cancer-associated fibroblasts (CAFs) and stromal cells when the tumor size surpasses the capacity of blood delivery [20,21]. This is a mechanism to compensate for the poor vascularization in the TME and the consequent reduction in nutrients [5,22]. In contrast, there is a certain controversy surrounding the presence of aerobic glycolysis in CAFs and stromal cells that generate a symbiosis between these cells and cancer cells, where the CAFs contribute to the metabolic requirements of cancer cells. This process is known as the “Reverse Warburg Effect” [20,23,24,25,26].
During this process, a wide variety of enzymes, transcription factors, and transporters modify their expression and activity to contribute to the mechanism of deregulated cancer metabolism. Mutations in these genes can be the cause of their deregulated activity. It has been described that the oncogene C-MYC is one of the most relevant genes in the regulation of the gene expression implicated in glucose, glutamine, and fatty acid metabolism. Additionally, C-MYC is implicated in the induction of the expression of glycolytic enzymes, such as LDH and transporters such as GLUT and MCT in cancer cells, in addition to increasing the uptake of glutamine and the activity of the PPPs [5,27,28].
Another signaling pathway that is of special interest is phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), which upregulates glycolytic enzymes as well. Through the PI3K/AKT pathway, hexokinase 2 (HK2), which is the enzyme responsible for phosphorylating glucose in order to retain this molecule inside the cell, is upregulated. A similar behavior occurs with the expression of GLUT. The product of HK2 (glucose-6-phosphate) can initiate the PPP, an essential pathway to maintain redox homeostasis and avoid oxidative stress in cancer cells [5]. AKT signaling favors the expression of HIF-1, also in the presence of oxygen, which acts over the activation of glycolytic components such as the GLUT or LDH, pyruvate kinase M2 (PKM2), etc., or the reduction in the pyruvate dehydrogenase kinase (PDK) activity that is essential for performing aerobic glycolysis [5,29,30,31].
TP53, a tumor suppressor gene, encodes the expression of the protein p53, which in normal cells, reduces aerobic glycolysis and promotes mitochondrial catabolic processes, such as OXPHOS and fatty acid oxidation. Furthermore, this protein diminishes the GLUT expression. However, in cancer cells, this TP53 can be mutated and can consequently favor aerobic glycolysis [5,32,33].
During this process, relevant enzymes participate in essential activities, which can influence the metabolic programming of the cancer cell. Their activity converts these enzymes into a potential target for new drugs. Even though there are many enzymes regulating aerobic glycolysis, four of them are worth mentioning (Figure 1).
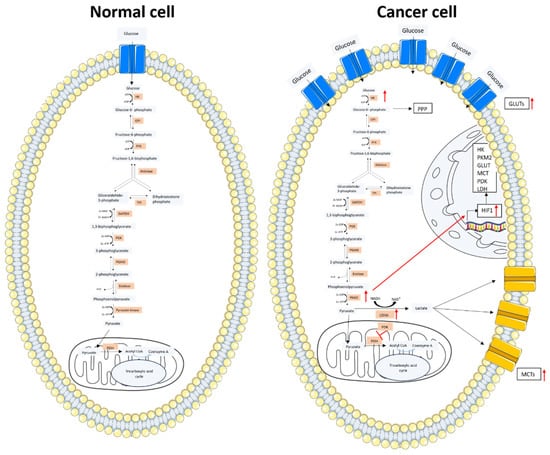
Figure 1.
Glucose metabolism scheme in a normal cell and a cancer cell. In the left image, the glucose metabolism of a normal cell is represented. Glucose enters through the GLUT protein and acts as a substrate of the glycolytic pathway. The produced pyruvate enters into the mitochondria and is converted into Acetyl-CoA, which is incorporated into the TCA cycle. In the right image, the cancer cell metabolism is represented. Glucose enters through the overexpressed GLUT protein with an increased uptake compared to the normal cell. An increased expression of the glycolytic enzymes is observed. The majority of the pyruvate obtained from the glycolytic pathway is converted into lactate by LDH. Lactate is transported outside the cell by the MCTs. The overexpressed PKM2 stimulates the HIF1 transcription factor whose increased expression results in the overexpression of the key elements for glucose metabolism. The figure was partly generated using Servier Medical Art, provided by Servier and licensed under a Creative Commons Attribution 3.0 unported license.
Hexokinase (HK) is the first enzyme implicated in glycolysis and the first rate-limiting step. The activity of this enzyme consists of the phosphorylation of glucose and other hexoses. Phosphorylated glucose is metabolized in different pathways as glycolysis, PPP, glycogenic, etc. The phosphorylation produced by this enzyme in the glucose molecule prevents it from leaving the cell. This enzyme can be found in four isoforms. One of them, HK2, is overexpressed in several cancer cells. HK2 can facilitate chemoresistance due to its interaction with the outer mitochondrial membrane when HK2 interacts with the voltage-dependent anion channel in the outer mitochondrial membrane, provoking an anti-apoptotic and chemoresistance effect. The inhibition of this enzyme allows for the blockage of glucose metabolism, diminishing ATP production and proliferation [34,35,36].
Lactate dehydrogenase (LDH) is the enzyme responsible for catalyzing the reversible transformation of pyruvate into lactate, producing the oxidation of NADH to NAD+. This enzyme is composed of two subunits, LDHA and LDHB. The amount of both subunits is distinguished between each of the five isoforms. Both subunits are overexpressed in cancer tissues. The subunit LDHA, which is specially overexpressed, has a higher affinity to pyruvate, catalyzing its conversion to lactate. The relation of this subunit with tumor progression, invasion, and drug resistance has been shown. The obtention of lactate by this enzyme is essential to the development of aerobic glycolysis, which is the principal source of energy independent of oxygen, and the acidification of the TME. The role of this enzyme converts it into an interesting target because its inhibition can seriously damage cell metabolism without affecting normal tissues where it is not essential [28,37,38,39].
Pyruvate kinase (PK) is a glycolytic enzyme whose function is to transfer a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), obtaining an ATP and a pyruvate molecule. This enzyme possesses two isoforms. One of them, PKM2, is overexpressed in proliferating cells, including cancer cells. The function of PKM2 implies the regulation of glucose oxidation and the progression of the cell cycle. The presence of the dimeric form of PKM2 and its lower catalytic activity involves an increment of anabolic intermediates that form macromolecules in the PPP, glycerol synthesis, and the production of NADPH. On the other hand, the PKM2 activity can trigger the total degradation of the intermediates to produce ATP, sending PEP into pyruvate and OXPHOS. There is controversy about the activity of this enzyme in the regulation of metabolism and whether it should be inhibited or activated. Its inhibition and the consequent accumulation of PEP prohibit cells from obtaining energy. In contrast, its activation implies an increment of OXPHOS to the detriment of aerobic glycolysis. Several drugs in preclinical and clinical phases have shown potent anti-tumor activity supporting both theories [31,40,41,42].
Pyruvate dehydrogenase kinase (PDK) is a serine/threonine kinase which forms a complex in conjunction with pyruvate dehydrogenase (PDH) through the reversible phosphorylation of any of the three serine residues of the alpha-subunit of PDH. PDH is an enzyme that decarboxylates pyruvate into acetyl coenzyme A (CoA), which is the junction between glycolysis and the TCA cycle. This step is critical for cellular metabolism, where the association of both enzymes supposes the inhibition of PDH and the consequent inhibition of the TCA cycle, forcing the cell into the aerobic glycolysis pathway. The inhibition of PDK and the subsequent preservation of the unphosphorylated active form of PDH implies a disadvantageous state for cell metabolism due to the destination of pyruvate into OXPHOS. All the PDK isoforms are overexpressed in diverse types of tumors [43,44].
The objective of this review is to compile the most up-to-date and significant outcome of the research in the development of new modulators of key enzymes implicated in the Warburg effect as a strategy to fight cancer. Notwithstanding, most of the reported results were in vitro and the new modulators were in a very early stage of their development. Hence, this review can shed some light on future advances in molecules targeting the Warburg effect.
2. Target Enzymes and the Reported Inhibitors
2.1. HK2 Modulators
Since HK2 is the first enzyme implicated in the Warburg effect, several molecules have been reported to be HK2 modulators. Within this group, there is a great variety of molecular structures, which have been grouped into six different groups: small molecules, polyphenols, oxygen and sulfur heterocycles, metal complexes, miscellanea, and compounds in combination with other therapies (Table 1).
2.1.1. Small Molecules
Different authors provided evidence that the HK2 inhibitor 3-bromopyruvic acid (compound one in Figure 2) significantly inhibited the viability and growth of colon cancer cells, prompting apoptosis via the mitochondrial apoptosis signaling pathway [45]. The 3-bromopyruvic acid inhibitor has been demonstrated to be a potent HK2 inhibitor in other cell lines, including 8505C (human anaplastic thyroid cancer cells) [46], HCC143 (triple-negative breast cancer), MCF-7 (non-triple-negative breast cancer) [47], and Panc-2 (pancreatic cancer cells) [48]. These findings showed the capacity of 3-bromopyruvic acid for inhibiting HK2, turning it into a strategy to develop new treatments for several types of cancer.
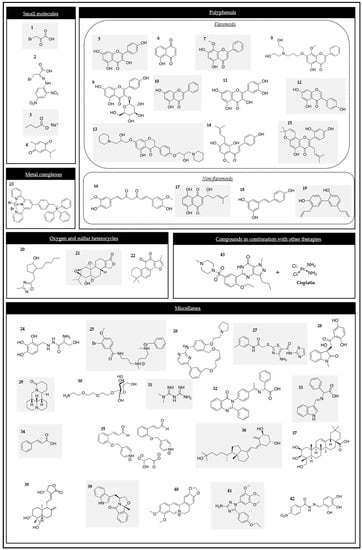
Figure 2.
Chemical structures of the HK2 modulators.
Owing to all the evidence regarding the potent anti-cancer activity of 3-bromopyruvate, some researchers have synthesized and studied the anti-cancer activity of several 3-bromopyruvate derivatives. Specifically, the cytotoxic activity of novel hydrazone derivatives, such as compound two (Figure 2), were assessed on colon, breast, lung, and liver cancer cell lines. The results showed that these derivatives presented a higher HK2 affinity than 3-bromopyruvate, converting them into suitable candidates for developing potent HK2 inhibitors [49].
Sodium butyrate (compound three in Figure 2), a gut microbiota metabolite, inhibited glycolysis in hepatocellular carcinoma (HCC) cells [50]. This outcome suggested that sodium butyrate inhibited the expression of HK2 and subsequently downregulated aerobic glycolysis and the proliferation of HCC cells along with the induction of apoptosis via the C-MYC pathway.
Thymoquinone (compound four in Figure 2), an active component from Nigella sativa, showed antioxidant, antimicrobial, antidiabetic, and anti-inflammatory effects, among others. It has also been extensively studied against cancer. Recent studies have evaluated its role in cancer metabolism. Thymoquinone was demonstrated to inhibit glycolytic metabolism in colorectal cancer cell lines because it works as a HK2 inhibitor via the modulation of the PI3K/AKT axis [51].
2.1.2. Polyphenols
- Flavonoid phenolic compounds
Several polyphenolic compounds have been reported to be potent HK2 inhibitors. Flavone derivatives are widely distributed in nature and have antioxidant, antiangiogenic, and antineoplastic activities. Furthermore, some flavone derivatives have been demonstrated to be HK2 inhibitors. This capacity could convert them into anti-cancer drugs [52,53,54].
Kaempferol (compound five in Figure 2), one of the active ingredients of traditional Chinese medicine, has been demonstrated to interfere with the cell cycle, angiogenesis, and the capacity of metastasis in tumor cells. It also produces apoptosis. Moreover, the studies by Zheng et al., proved that kaempferol inhibited metastasis by blocking aerobic glycolysis in melanoma cells, since the binding of HK2 and a voltage-dependent anion (VDAC1) on the mitochondria was disrupted [55].
Juglone (compound six in Figure 2), another natural compound, has shown an anti-cancer activity. Hu and co-workers proved that juglone suppressed OXPHOS and glycolysis through the inhibition of HK, phosphofructokinase (PFK), and PK activity in prostate cancer [56].
Another example of a flavonoid compound is wogonin (compound seven in Figure 2), which is extracted from Scutellariae radix. It has shown a variety of properties in HT144 melanoma cells, such as an anti-inflammatory effect. This compound also inhibited cell proliferation, colony formation, and tumor growth in the mentioned cells and decreased the activities of HK, PFK, and PK [57].
FV-429 (compound eight in Figure 2), a derivative of wogonin and a glycolysis inhibitor, was considered to be a promising anti-cancer compound. Extensive research into its mechanism of action (MOA) has revealed that it induced glycolysis inhibition and apoptosis in human prostate cancer cells by downregulating the androgen receptor (AR)-AKT-HK2 signaling network. This evidence reinforces that FV-429 is a promising candidate for the treatment of advanced prostate cancer [58].
An extensive number of references have studied the effectiveness of different flavones against HCC, such as astragalin, chrysin, and quercetin. This is one of the most commonly diagnosed malignant types of cancer, whose advanced stages still have no therapeutic options. Under these circumstances, the effects of this class of compounds have been studied in HCC.
Astragalin (compound nine in Figure 2), commonly found in a variety of food components, was previously demonstrated to be cytotoxic on human leukemia cells. Recently, it has also been demonstrated to suppress the proliferation of HCC cells. It suppresses the expression of HK2 through the upregulation of miR-125b [59].
Chrysin (compound 10 in Figure 2), a natural flavone found in plant extracts and widely used in Chinese medicine, has been reported to be a glycolysis inhibitor. It has been proved that when HCC cells are exposed to chrysin, the HK2 expression is decreased, producing cell apoptosis. Chrysin is a promising candidate for novel therapy for HCC [60].
Quercetin (compound 11 in Figure 2), a bio-active flavonoid, has an anti-tumor effect on HCC. Thus, researchers wanted to define the underlying mechanism of this effect. They found that similar to the previous flavones, quercetin lowered the protein levels of HK2 and suppressed the AKT/mTOR pathway within HCC cells [61].
Genistein (compound 12 in Figure 2) is a natural isoflavone known for its numerous health advantages, including anti-tumor effects. Its effect on HIF-1α and glycolysis in HCC was still unclear, so it has been studied. Genistein has been demonstrated to inhibit aerobic glycolysis and induce mitochondrial apoptosis in HCC cells by directly downregulating HIF-1α, therefore inactivating GLUT1 and HK2 [62]. One derivative of the mentioned compound, 5-hydroxy-7-(2-hydroxy-3-(piperidin-1-yl)propoxy)-3-(4-(2-hydroxy-3-(piperidin-1-yl)propoxy)phenyl)-4H-chromen-4-one, called gen-27 (compound 13 in Figure 2), is a newly synthesized isoflavone which has been demonstrated to inhibit the proliferation of HCC cells as well as prevent the development of colitis-derived cancer. It has also been demonstrated that gen-27 inhibited glycolysis in human breast cancer cells by decreasing the HK2 expression, leading to the induction of apoptosis [63].
Xanthohumol (2′,4′,4-trihydroxy-6′-methoxy-3′-prenylchalcone) (compound 14 in Figure 2) is an attractive compound due to its multiple pharmacological activities. It has been demonstrated that xanthohumol suppressed colorectal cancer cells via the suppression of HK2 and glycolysis. Thus, it could be considered an interesting new anti-tumor agent [64].
Morusin (compound 15 in Figure 2) is a compound that is known for its antioxidant anti-inflammatory, antiangiogenic, antimigratory, and apoptotic effects. Recently, it has been demonstrated that it has an anti-tumor activity in HCC. Particularly, it has been studied in Huh7 and Hep3B cells. Morusin has been shown to attenuate the expression of p-AKT, p-mTOR, c-Myc, HK2, PKM2, and LDHA in those cancer cell lines. These findings provide evidence that Morusin exhibits an anti-tumor effect in HCC and has the potential to be a potent dietary anti-cancer candidate [65].
- Non-flavonoid phenolic compounds
Several studies have reported that curcumin (compound 16 in Figure 2), a natural polyphenolic pigment extracted from the Curcuma aromatica salisb, has significant pharmacological actions, including a chemo-preventive efficacy. It has been demonstrated that curcumin could be a potent drug for the treatment of colorectal cancers. Curcumin downregulated the expression and activity of HK2 in HTC116 and HT29 cells, and it also induced a dissociation of HK2 from the mitochondria, resulting in mitochondrial-mediated apoptosis [66]. More recent studies have been conducted regarding the promising efficacy of curcumin, especially in thyroid cancer. Curcumin decreased the cell proliferation of PTC B-CPAP and promoted apoptosis by the inhibition of LDHA and HK2 [67].
Other studies have revealed that the polyphenolic compound curcumin promoted apoptosis. The mechanism of curcumin in glycolytic inhibition and apoptotic induction in HCT116 and HT29 of HCC has been investigated. They found that curcumin mainly diminished the activity and expression of HK2, having a minor effect on the PFK, phosphoglucomutase (PGM), and LDH enzymes. They also demonstrated that curcumin was involved in the dissociation of HK2 from the mitochondria, inducing mitochondrial-mediated apoptosis [66].
Shikonin (SK) (compound 17 in Figure 2), an active naphthoquinone that is also present in natural sources in traditional Chinese herbal medicine, is an inhibitor of PKM2 in various cancers. Further studies have been performed with this product. For example, its mechanism has been studied in esophageal squamous cell carcinoma where it has been determined that it not only suppressed the expression of PKM2, but also the expression of HK2 and GLUT1 [68].
Resveratrol (compound 18 in Figure 2) is a small polyphenol that has demonstrated both chemo-preventive and chemotherapeutic effects in cancer. In 2016, the capacity of resveratrol for impairing metabolism by inhibiting the expression of HK2 mediated by the Akt signaling pathway was published for the first time, demonstrating its anti-tumor effect on human non-small cell lung cancer (NSCLC) [69].
Honokiol (HNK) (compound 19 in Figure 2), a naturally occurring phenolic compound derived from Magnolia, is frequently used for its anti-inflammatory and antioxidant properties. Moreover, it has been reported to exhibit inhibitory effects of HNK on the HIF-1α and hypoxia-related signaling pathways. Its effect has been studied in various human breast cancer models, which revealed that the inhibitory effect of HNK on glycolysis was HIF-1α-dependent and that it downregulated the expression of HIF-1α and its downstream regulators, including GLUT1, HK2, and PDK1 [70].
2.1.3. Oxygen and Sulfur Heterocycles
Several oxygen and sulfur heterocycles have been demonstrated to be HK2 inhibitors. The natural product methyl jasmonate is one of the compounds that blocks HK2 activity in cancer cells. Studies have shown that methyl jasmonate suppressed the proliferation of cells and triggered cell death in various human cancer cell lines. In view of these results, the methyl jasmonate derivative, compound 20 (Figure 2), was studied as an effective novel HK2 inhibitor in glioblastoma cells [71].
Triptolide (compound 21 in Figure 2), a natural diterpenoid epoxide derived from a traditional Chinese herb, presents multiple biological activities, including anti-inflammatory, immunologic suppression, and potent anti-cancer effects. Several studies have been carried out to study its mechanism among different cancers. In 2021, it was found that triptolide reduced the expression of C-MYC and mitochondrial HK2 in head and neck cancer cells and activated the BAD/BAX-caspase and 3-GSDME cascade, triggering gasdermin E(GSDME)-mediated pyroptosis [72]. These findings encouraged other researchers to study the role of triptolide in other cancer cells. Particularly, a recent study evaluated its effect in intrahepatic cholangiocarcinoma and revealed that it significantly inhibited the proliferation of those cells [73].
Tanshinone IIA (Tan IIA) (compound 22 in Figure 2), the main component of Salvia miltiorrhiza, is a known active compound that exhibits anti-tumor properties. It was known that the anti-tumor action of Tan IIA in lung cancer involved multiple mechanisms, including the inhibition of cell growth, promotion of cell apoptosis, and modulation of cellular metabolism. It was later confirmed that Tan IIA decreased the sine oculis homeobox homolog 1 (SIX1), PKM2, HK2, and LDHA levels [74].
2.1.4. Metal Complexes
[Cu(ttpy-tpp)Br2]Br], abbreviated as CTB, (compound 23 in Figure 2) is a novel copper II complex containing tri-phenyl-phosphonium that targets the mitochondria. CTB inhibits aerobic glycolysis and tumor acidity by reducing the activity of HK2 in hepatoma cells through the dissociation of HK2 from the mitochondria [75].
2.1.5. Miscellanea
There is significant diversity in the structures of the molecules that can be used as HK2 inhibitors. Due to this heterogeneity, in this section, we have compiled some recent molecules whose structures did not fit in our established classification.
Benserazide (BEN) (compound 24 in Figure 2) was identified as a selective HK2 inhibitor after a structure-based virtual ligand screening of a library of 2924 US Food and Drug Administration (FDA)-approved drugs and a nutraceutical database. The molecular docking results showed that BEN adopted an extended conformation in which the pyrogallol group of the molecule occupied the binding site of the substrate glucose in HK2. BEN was specifically bound to HK2 as a competitive inhibitor. The SW480 colorectal cancer cell line was selected to demonstrate its efficacy. This compound showed antiproliferative effects, decreased the concentration of lactate, and triggered the activation of the cellular energy regulator AMP-activated protein kinase (AMPK). Additionally, BEN also activated p53 and p27 through energetic stress, producing apoptosis in both in vitro and in vivo models [76].
High-throughput virtual screening is a widely used strategy for searching for new anti-cancer molecules. By using this method, several chemical scaffolds have been identified to be promising HK2 inhibitors. Among them, compound 25 (Figure 2) was highlighted due to its outstanding activity in tumor cell suppression and its capacity to be solubilized in aqueous environments. It featured a high efficacy in HK2 inhibition in U87 glioma cells, an effective penetration of the blood–brain barrier, and minimal side effects on normal tissues [77].
Additionally, this high-throughput screening approach led to the discovery of new compounds for treating squamous cell lung cancer. Among all of them, the inhibitors of squamous cell lung cancer that decreased glucose consumption by more than 50% were chosen. For example, pacritinib (compound 26 in Figure 2) decreased glucose consumption in cell cultures and in vivo in tumor tissues. This compound blocked the HK activity and its protein expression [78].
Other HK2 inhibitors have been identified using structured-based virtual screening. The most promising compounds, meaning those with low binding energies, were selected for cytotoxicity tests and evaluated in different cancer cells. Particularly, the most promising compounds acting as HK2 inhibitors were 4244-3659 and K611-0094 (compounds 27 and 28 in Figure 2). The results indicated that the stable binding of the compounds to the receptor was predominantly influenced by hydrogen bonding [79].
Matrine (compound 29 in Figure 2) is a pleiotropic alkaloid from Chinese traditional medicine. It has been demonstrated that this compound could have a possible effect on the suppression of cell proliferation and the induction of apoptosis caused by HK2 depletion and a reduction in C-MYC binding to the HK2 gene in the chronic myeloid leukemia K562 and acute myeloid leukemia HL-60 cell lines. Matrine inhibited cell proliferation and induced apoptosis by reducing the HK2 expression in myeloid leukemia cells [80].
A novel glucose analog with modifications in carbon 2 (C2), called 2-(2-[2-(2-aminoethoxy)ethoxy]ethoxy)-D-glucose (compound 30 in Figure 2), was synthesized as a new strategy for cancer treatment, taking advantage of the increment of glucose consumption in cancer cells. This compound inhibited HK binding in its active site [81].
Metformin (compound 31 in Figure 2), a widely used anti-hyperglycemic drug, has also shown important anti-cancer properties. It has been demonstrated that metformin can affect glucose metabolism. It decreases the 18F-fluorodeoxyglucose (FDG) uptake by the direct inhibition of the enzymatic activity of HK2 and HK1 and mimics glucose-6-phosphate G6P in NSCLC Calu-1 cells. In silico models indicated that this action was due to the capability of metformin to mimic the G6P features because it was bound to the pocket of HK2, which resulted in mitochondrial depolarization and subsequent cell death [82]. Furthermore, it has also been demonstrated that the enzymatic function of HK1 and HK2 was directly Inhibited by metformin in the cancer cell line of triple-negative breast cancer (MDA-MB-231) [83].
New amino acid Schiff bases of quinazolinones and indoles have been studied as anti-cancer agents. In silico studies predicted their ability to inhibit mitochondrial NADH ubiquinone oxidoreductase (complex I) by targeting the AMPK/mTOR signaling pathway and inhibiting HK. Two compounds (32 and 33 in Figure 2) were the most promising ones in accordance with the docking analysis and were selected for the in vitro analyses. They showed a significant activation of the AMPK proteins and oxidative stress, which led to an elevated expression of p53 and Bax, a reduced Bcl-2 expression, and caused cell cycle arrest at the G0/G1 phase. They also showed significant inhibitory effects on the HK enzymes [84].
Cinnamon bark extract (compound 34 in Figure 2) has received special attention due to the wide variety of pharmacological activities reported for this species. It has been used in traditional medicines for diabetes, inflammation, arthritis, and cancer. It has been reported that cinnamon bark extract exerted a suppressive effect on the metastatic dissemination of cancer cells, reducing both cell motility and invasion in MDA-MB-231 human breast cancer cells as it decreased the expression of HK2 [85]. Owing to the demonstrated activity of cinnamon bark extract, other derivatives have been studied against cancer. Among them, the cinnamaldehyde derivative (CB-PIC) (compound 35 in Figure 2), a major component of cinnamon, possesses a well-known anti-cancer activity. Its anti-tumor mechanism, which was studied in HCC, is associated with the Warburg effect. The suppression of the expression of HK2 and PKM2 and the reduction in the production of lactate in HepG2 and Huh7 Cells has been reported [86].
Calcitriol (compound 36 in Figure 2), the biologically active form of vitamin D, has been reported to prevent cancer progression by reducing cell proliferation, increasing cell differentiation, and inhibiting angiogenesis. Mechanistically, calcitriol reduced the expression of cyclin D1, C-MYC, GLUT1, and the key glycolytic enzymes HK2 and LDHA. It suppressed glycolysis and cell growth in human colorectal cells [87].
Centella asiática and Andrographis peniculata are two ethnomedicinal anti-cancer herbs. They have been studied with the aim of improving cancer therapeutics. Particularly, bayogenin (compound 37 in Figure 2) and andrographolide (compound 38 in Figure 2) were extracted from Centella asiática and Andrographis peniculta, respectively. Both compounds were docked against HK2, a drug-likeness prediction was performed, and the docked complexes were subjected to molecular dynamics simulations. The results of the study suggested that both natural compounds have the potential to target HK2, although in vitro studies are necessary to validate these results [88].
The SLMP53-1 (compound 39 in Figure 2) p53 activation by (S)-tryptophanol-derived oxazoloisoindolinone has showed that it regulated glycolysis by particularly downregulating the key glycolytic enzymes HK2, GLUT1, and PFKFB3. Furthermore, it also exhibited a synergistic growth inhibitory activity in combination with the metabolic modulator DCA. These data revealed the promising potential of the p53-activating compound SLMP53-1 in cancer treatment, specifically by targeting the pathways associated with p53-mediated growth control and dissemination [89].
Berberine (compound 40 in Figure 2) is an alkaloid which has a significant anti-tumor effect. However, its mechanism in tumor metabolism was not known. Recently, its molecular mechanism regulating the Warburg effect in ovarian cancer cells has been studied. Those studies demonstrated that berberine inhibited the Warburg effect by inhibiting the expression of HK2 in ovarian cancer cells [90].
A novel tubulin inhibitor, 5-(4-ethoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-1H-1,2,4-triazol-3-amine (YAN) (compound 41 in Figure 2) effectively suppressed glycolysis in both A549 and A549/Taxol cells. The inhibition was achieved by downregulating the expression of HK2, LDHA, and GLUT1 and it was able to block the mitochondrial binding of HK2 in A549 and A549/Taxol cells. Moreover, YAN can promote the production of ROS. These findings provide evidence for considering YAN as a potential candidate for the treatment of multidrug-resistant cancers characterized by a highly glycolytic phenotype [91].
In 2020, benitrobenzerazide (compound 42 in Figure 2) (BZNZ) was reported as a novel selective HK2 inhibitor with EC50 values in the nanomolar range. BZNZ induced cell apoptosis in the SW 1990 cells pancreatic cancer cell line. Its capacity was proved by the authors both for in vitro and in vivo activity. Furthermore, this compound presented low toxicity, so it was considered for development as a novel HK2 small molecule candidate for future cancer therapy [92]. Following the above-mentioned observations and drawing inspiration from this breakthrough, subsequent research teams have undertaken studies on BNBZ and its various derivatives [93], particularly the effect of trihydroxy moiety. Several compounds with different activities were described by this group. Remarkably, the dihydroxy derivatives showed an inhibitory activity inside the cells and some of them showed less toxicity. Based on these results, BNBZ is a promising derivative worthy of being further developed.
2.1.6. Compounds in Combination with Other Therapies
Sildenafil (compound 43 in Figure 2) has been demonstrated to enhance the cytotoxic effect of cisplatin by the induction of apoptosis. Previous studies have reported the promising chemo-sensitizing potential of the phosphodiesterase 5 inhibitor sildenafil in breast, colon, prostate, glioma, and lung tumors. Other studies have highlighted the potential of sildenafil as a promising candidate in combinatorial chemotherapeutic therapies against T-cell lymphoma. Regarding the mechanism, sildenafil deregulates glucose metabolism by lowering the expression of HK2, GLUT1, LDHA, PKM2, and PDK1 via the suppression of the HIF-1α expression. Therefore, sildenafil enhances its capacity to eliminate tumor cells by increasing ROS production through switching the glucose metabolism from aerobic glycolysis to OXPHOS [94].

Table 1.
Efficacy and mode of action of the HK2 inhibitors.
Table 1.
Efficacy and mode of action of the HK2 inhibitors.
| Scaffold | Model | Mode of Action | Refs. |
|---|---|---|---|
| Polyphenols | |||
| Flavonoids | |||
| Astragalin | In vitro | Inhibition of the HK2 expression by the upregulation of miR125b | [59] |
| HCC cells | |||
| Huh-7, HepG2, H22 | |||
| Chrysin | In vitro | Decrease in the expression of HK2 | [60] |
| HCC cells | |||
| HCC-LM3, SMMC-7721, Bel-7402 | |||
| Genistein | In vitro | Inhibition of the HK2 expression | [62] |
| HCC | |||
| HCC-LM3, Bel-7402 | |||
| Gen-27 | In vitro | HK2 release from the mitochondria | [63] |
| Human breast cancer | |||
| MDA-MB-231, MCF-7 | |||
| Xanthohumol | In vitro | Suppression of the HK2 protein expression | [64] |
| Colorectal cancer cells | |||
| HCT-116, HT-29, SW620 | |||
| Morusin | In vitro | Attenuation the activity of HK2 | [65] |
| HCC cells | |||
| Huh7, Hep3B | |||
| FV-429 | In vitro | Downregulation of the HK2 expression | [58] |
| Prostate cancer cells | |||
| LNCaP | |||
| Quercetin | In vitro | Decrease in the levels of HK2 | [61] |
| HCC cells | |||
| SMMG-7721, BEL7402 | |||
| Kaempferol | In vitro | Inhibition of HK2 | [55] |
| Melanoma cells | |||
| A375, B16F10 | |||
| Juglone | In vitro | Inhibition of the HK activity | [56] |
| Prostate cancer cells | |||
| LNCaP, C4-2 | |||
| Wogonin | In vitro | Decrease in the activity of HK | [57] |
| Melanoma cells | |||
| HT-144 | |||
| Non-flavonoids | |||
| Shikonin | Esophageal squamous cell carcinoma | Inhibition of HK2 | [68] |
| Resveratrol | In vivo | Inhibition of the HK2 expression | [69] |
| Non-small cell lung cancer model | |||
| H460 nude mice xenograft | |||
| Curcumin | In vitro | Reduction in the expression of HK2 and its activity | [66] |
| Colorectal cancer cells | |||
| HTC116, HT29 | |||
| In vitro | Reduction in the expression of HK2 | [67] | |
| Papillary thyroid cancer | |||
| B-CPAP | |||
| Honokiol | In vitro | Reduction in the HK2 expression | [70] |
| Breast cancer cells | |||
| MCF-7, MDA-MB-231 | |||
| Small molecules | |||
| 3-bromopyruvic acid | In vitro | Inhibition of the HK2 activity | [45,46,47,48] |
| Breast cancer cells | |||
| HCC1143 | |||
| Pancreatic cancer cells | |||
| Panc-2 | |||
| Compound 2 | In silico | Inhibition of HK2 | [49] |
| (3-bromopyruvate derivative) | |||
| Sodium butyrate | In vitro | Suppression of the HK2 protein expression | [50] |
| HCC | |||
| HCC-LM3, Bel-7402 | |||
| Thymoquinone | In vitro | Inhibition of the HK2 expression | [51] |
| Colorectal cancer cell lines | |||
| HCT-116, SW480 | |||
| Oxygen and sulfur heterocycles | |||
| Triptolide | In vitro | Inhibition of HK2 | [72,73] |
| Neck and head cancer cells | |||
| HK1, FaDu, C666-1 | |||
| Cholangiocarcinoma cells | |||
| HuCCT1, RBE | |||
| Compound 20 | Clinical | Inhibition of the HK2 activity | [71] |
| (Methyl jasmonate derivative) | Glioma cohort data | ||
| Tanshinone IIA | In vivo | Inhibition of the HK2 expression | [74] |
| Lung cancer model | |||
| Nude mouse xenograft | |||
| Metal complexes | |||
| CTB | In vivo | Inhibition of the HK2 expression | [75] |
| HCC xenograft model | |||
| SMMC-7721 cells in nude mice | |||
| Miscellanea | |||
| Benserazide | In vitro | Inhibition of the HK2 activity | [76] |
| Colorectal cancer cells | |||
| SW480 | |||
| Compound 25 | In vitro | Inhibition of the HK2 expression | [77] |
| Glioma cells | |||
| U87 | |||
| Pacritinib | In vitro | Blockage of the HK activity and expression | [78] |
| Squamous cell in lung cancer | |||
| SK-MES-1, H520, H596 | |||
| 4244-3659 | Colorimetric assay | Inhibition of the HK2 activity | [79] |
| K611-0094 | Colorimetric assay | Inhibition of the HK2 activity | [79] |
| Matrine | In vitro | Depletion of the HK2 expression | [80] |
| Myeloid leukemia cells k562, HL-60 | |||
| 2-(2-[2-(2-aminoethoxy)ethoxy]ethoxy)-D-glucose | Kinetic assay | Inhibition of the HK2 activity | [81] |
| Metformin | In vitro | Inhibition of the HK2 activity | [82,83] |
| Lung cancer cells; breast cancer cells | |||
| Calu-1; MDA-MB-231 | |||
| Compounds 32 and 33 (Quizalinone and indole derivatives) | In vitro | Inhibition of the HK activity | [84] |
| Breast cancer cells | |||
| MCF-7, MDA-MB-231 | |||
| Cinnamon bark extract | In vitro | Inhibition of the HK2 expression | [85] |
| Breast cancer cells | |||
| MDA-MB-231 | |||
| CB-PIC | In vitro | Suppression of HK2 | [86] |
| HCC cells | |||
| HepG2 and Huh7 | |||
| Calcitriol | In vivo | Reduction in the expression of HK2 | [87] |
| HT29 xenograft mouse model | |||
| Bayogenin | In silico | Inhibition of the HK2 activity | [88] |
| Andrographolide | In silico | Inhibition of the HK2 activity | [88] |
| SLMP53-1 | In vitro | Decrease in the HK2 expression | [89] |
| Colon cancer cells | |||
| HCT-116 | |||
| Berberine | In vitro | Downregulation of the HK2 expression | [90] |
| Ovarian cancer cells | |||
| SKOV3, 3AO | |||
| 5-(4-ethoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-1H-1,2,4-triazol-3-amine | In vitro | Inhibition of the HK2 expression | [91] |
| Lung cancer cells | |||
| A549 | |||
| Benitrobenzerazide | In vivo | Inhibition of the HK2 activity (non-competitive inhibitor) | [92,93] |
| Colorectal cancer | |||
| SW480 xenograft mouse model | |||
| Compounds in combination with other therapies | |||
| Sildenafil + cisplatin | In vitro | Inhibition of the HK2, PKM2, and LDHA expression | [94] |
| Lymphoma cells | |||
| HuT-78 cells | |||
2.2. PDK Modulators
PDK inhibitors have emerged as a promising class of molecules in the field of cancer research and therapy. In this section, the different classes of PDK inhibitors and their potential applications in cancer treatment have been collected. The molecules have been organized into different groups: small molecules, metal complexes, fused heterocyclic rings, disulfide derivatives, nonsteroidal anti-inflammatory drugs, miscellanea, and compounds in combination with other therapies (Table 2).
2.2.1. Small Molecules
The small molecule dichloroacetate (DCA) (compound 44 in Figure 3) has been proposed as an anti-cancer drug. For several decades, this metabolic modulator has been employed in the treatment of lactic acidosis and inherited mitochondrial diseases. The efficacy of DCA in cancer therapy has been associated with the Warburg effect. The salts of DCA selectively target cancer cells shifting their metabolism from aerobic glycolysis to OXPHOS by the inhibition of PDK. This compound promotes the mitochondrial oxidation of pyruvate and disrupts the metabolic advantage of cancer cells [95,96]. The crystal structure of PDK2 in a complex with DCA revealed that DCA occupied the pyruvate binding site in the N-terminal regulatory (R) domain [97]. Current clinical trials assessed the anti-cancer efficacy of DCA [98].
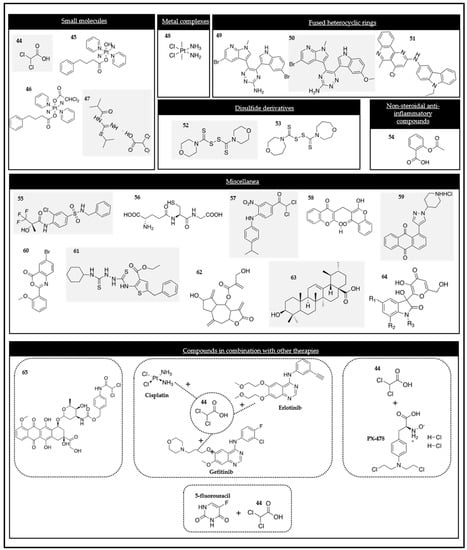
Figure 3.
Chemical structures of the PDK modulators.
The first study of the anti-tumor effect of DCA was performed in an A549 lung adenocarcinoma xenograft model in nude rats. The results showed a significant decrease in tumor growth after being treated with DCA. Furthermore, some experimental groups even showed tumor regression due to the antiproliferative and pro-apoptotic effects of DCA [99].
Some prodrugs have also been described to be PDK inhibitors. These were Pt (IV) prodrugs (compounds 45 and 46 in Figure 3) with bio-active axial ligands that have been designed to selectively target cancer cells and attack several cellular components at the same time, which can result in an enhanced anti-cancer potency. Particularly, the mono-functionalized complex of the PDK inhibitor 4-phenylbutyrate and its di-axial functionalized analog have been synthesized. Both exhibited an increased photocytotoxicity and cellular accumulation in A2780 ovarian cancer cells when compared to A549 lung cancer cells and demonstrated minimal cytotoxicity towards normal MRC-5 cells. They are promising derivatives for cancer treatment [100].
In view of those results, a phase one clinical trial (No. NCT 01111097) in adults with recurrent malignant brain tumors was published in 2014. The study revealed that oral chronic treatment of DCA was feasible and well tolerated in patients with recurrent malignant gliomas and other metastatic tumors to the brain using the dose range established for metabolic diseases [101].
Additional phase one clinical trials (No. NCT 00566410) were performed in order to assess the safety and pharmacokinetic profile of DCA due to the previously mentioned evidence that suggested that DCA could reverse the Warburg effect and inhibit growth in cancer models through the inhibition of PDK [102].
DCA is currently under investigation. The results obtained from several case studies showed that it has several limiting factors in clinical use, in which neurotoxicity and encephalopathy are included. Despite these disadvantages, the data proved that it is a promising treatment option for patients who have no conventional treatment options. Furthermore, it has been demonstrated that it has potential to extend the patient’s life [103].
A recent study on the effect of DCA against melanoma cell lines has been reported. Melanoma is characterized by a high glucose uptake, partially mediated through elevated PDK. For this reason, PDK can be a promising target in melanoma. In this study, the authors concluded that DCA reduced proliferation in melanoma cell lines through reprogramming the cellular metabolism and its the ability to synergize with other drugs applied in metabolic disorders [104].
Another compound, named T1084 (1-isobutanoyl-2-isopropylisothiourea dichloroacetate) (compound 47 in Figure 3), incorporated the nitric oxide synthase (NOS) inhibiting fragment, 1-isobutanoyl-2isopropylisothiourea, and the PDK inhibiting fragment, DCA, into the molecular structure. This multifunctionality enabled T1084 to concurrently exert two distinct pharmacological actions, anti-angiogenesis and hypoxia-oriented cytotoxicity, that could potentially enhance its anti-tumor effects. The methodology employed in this study potentially allowed for the possibility of developing additional multifunctional anti-tumor compounds with diverse molecular mechanisms of action [105].
2.2.2. Metal Complexes
The novel platinum (Pt) (IV) complex c,c,t-[Pt(NH3)2Cl2(C10H15N2O3S)(C2HO2Cl2)] (DPB) (compound 48 in Figure 3), containing DCA as one of the axial ligands in the structure, was synthetized and evaluated as an anti-tumor agent. DCA interfered in the mitochondrial energy metabolism after its reduction, activated PDH due to the inhibition of PDK, depolarized the mitochondrial membrane potential, and promoted ROS. This novel complex induced apoptosis after the inhibition of both glycolysis and glucose oxidation, thus highlighting the interest in these types of drugs [106].
2.2.3. Fused Heterocyclic Rings
To find novel classes of potential PDK inhibitors, 1,2,4-amino triazine derivatives (compounds 49 and 50 in Figure 3) were reported to be promising candidates for inhibiting the PDK expression. Despite the different candidates that were synthetized and evaluated, compounds 49 and 50 must be emphasized [107]. These novel compounds demonstrated a promising ability to inhibit the enzymatic activity of PDK1 and PDK4. Further docking studies also confirmed these results. The novel derivatives exhibited a strong antiproliferative activity against pancreatic cells by targeting the PDK enzyme and consequently reducing tumor growth.
The 2-amino substituted 2,4-dichlorobenzo[h]naphthyridine derivatives, concretely compound 51 (Figure 3), have been proposed to be PDK1 inhibitors. As a first step, they have been studied in silico with docking studies and PDK1 inhibitors. The structure–activity relationship revealed that the compounds holding aminocarbazole moiety and triazole amine moiety increased the activity profile. Afterwards, all of them were also evaluated in vitro against six human cancer cell lines (A549, HCT-15, T47D, C6, HepG2, and Hep-2), showing a potent activity [108].
2.2.4. Disulfide Derivatives
Some disulfide derivatives have demonstrated a capacity to inhibit PDK1. JX06 (compound 52 in Figure 3) has been identified as a selective covalent inhibitor of PDK1. This compound formed a disulfide bond with its thiol group, showing a cysteine residue localized in the hydrophobic pocket of the PDK1 enzyme. This bond induced conformation changes in arginine 286 through Van der Waals forces and hindered access of ATP to this binding pocket, blocking the PDK1 enzymatic activity. As a result, this finding explained the mechanism of inhibition of PDK1 by the JX06 compound and its derivatives [109].
Based on the scaffold of JX06, new disulfide-based PDK1 inhibitors have been synthesized. Among all the new novel compounds, one of them, compound 53 (Figure 3) was shown to inhibit PDK1 more potently than JX06. Furthermore, it was more selective with PDK1 and induced notable alterations in the glucose metabolic pathways within A549 cells by reducing ECAR and increasing the ROS levels. The compound, which has been studied in vivo, showed significant anti-tumor activity without any adverse effect to the weight of the mice. These findings strongly suggested its potential as a promising lead compound for cancer treatment, acting as a selective and covalent inhibitor of PDK1 [110].
2.2.5. Nonsteroidal Anti-Inflammatory Drugs
It was found that aspirin (compound 54 in Figure 3) attenuated glycolysis by suppressing PDK1. The study identified a novel role and regulatory mechanism of PDK1 in breast cancer stem cells (BCSC) reprogramming. It was concluded that PDK1 regulated by the HIF-1 pathway was required for BCSC self-renewal reprogramming, which could be blocked by aspirin. This provides a promising strategy for breast cancer therapy [111].
2.2.6. Miscellanea
Numerous works have highlighted the new therapeutic approach of PDK inhibitors for the treatment of numerous pathologies, such as diabetes, lactic acidosis, cerebro-vascular and cardiovascular diseases, cancer, hypertension, and neurodegenerative diseases. Therefore, many authors have developed novel PDK inhibitors. The N-(4-(N-alkyl/aralkylsulfamoyl)phenyl)-2-methylpropanamides (compound 55 in Figure 3) were synthesized and studied. The results revealed that they had an inhibitory capacity. The structure–activity relationship was interesting for the further design of new PDK inhibitors.
The researchers identified an interesting structure–activity relationship for further study, such as that the ring-opened analogues were more active than the ring-closed ones [112].
α-Lipoic acid (compound 56 in Figure 3) is a natural coenzyme in the mitochondrial PDH complex commonly known as an antioxidant. It has been studied in cancer research. A-lipoic acid has inhibitory effects on the proliferation and migration of cells and demonstrated a proapoptotic effect in the NSCLC A549 and PC9 cell lines. It downregulated the expression of PDK1, resulting in less phospho-PDH phenotype which could decrease the antioxidant activity-associated genes via NRF2-Keap1 pathway [113].
The compound 2,2-Dichloro-1-(4-((4-isopropylphenyl)amino)-3-nitrophenyl)ethan-1-one, namely XB-1 (compound 57 in Figure 3), was discovered as a new PDK inhibitor. This novel compound demonstrated its ability to inhibit PDK activity with an IC50 value of 337 nM and it reduced A549 cell proliferation with an EC50 value of 330 nM. Nevertheless, this compound exhibits serious toxicity issues, such as the formation of a serum albumin complex via hydrogen bond interaction and a non-specific action in normal and cancer cells [114,115].
Dicumarol (compound 58 in Figure 3) is a member of a family of coumarins with various pharmacological properties, including anti-inflammatory and anti-cancer activities. It was demonstrated that dicumarol inhibited the PDK1 activity with an IC50 of 19.42 μM in SKOV3. It also inhibited an A2780 human ovarian cancer cell line and SKOV3 xenografts in vivo through two pair of π–π interactions, van der Waals, and electrostatic interactions between dicumarol and the enzyme. The inhibition of PDK1 provoked an increase in the OXPHOS, ROS production, induced apoptosis, and suppression of xenograft growth, and diminished the lactate production and extracellular acidification [116].
New compounds have been designed and synthesized specifically to be PDK inhibitors. Among them, compound 59 (Figure 3) was a potent novel compound. Particularly, it was predicted and later verified that it binds to the lipoyl-binding site and interrupts intermolecular interactions with the E2-E3bp subunits of PDC. It has been studied in HeLa cells and the results revealed that it displayed a strong cytotoxicity [117,118].
The limited number of reported PDK inhibitors motivated several authors to continue discovering novel small molecules that can serve as PDK inhibitors. High-throughput screening is a commonly used strategy in these cases and has led to the discovery of two novel compounds (compounds 60 and 61 in Figure 3) that inhibited cancer cell proliferation and colony formation in A549 cells. Therefore, they may serve as initial candidates for improving the potency of PDK1 inhibition for anti-cancer treatments [119].
Hemistepsin A (compound 62 in Figure 3) is a sesquiterpene lactone isolated from Hemistepta lyrata Bunge. It has been reported as a PDK1 inhibitor in colorectal cancer cells. Firstly, through computational simulation and biochemical assays, it was demonstrated that Hemistepsin A directly binds to the lipoamide-binding site of PDK1. This binding leads to the inhibition of the interaction between PDK1 with the E2 subunit of the PDH complex. Consequently, the inhibition of PDK1 resulted in a reduction in the lactate production and mitochondrial ROS levels and damage was also increased. This observation revealed that the apoptosis of colorectal cancer cells is promoted by Hemistepsin A. Therefore, it might be a potential candidate for developing a novel anti-cancer drug [120].
Ursolic acid (compound 63 in Figure 3), a triterpenoid widely found in food, has been investigated on adult T-cell leukemia cells. It was demonstrated that it inhibits the proliferation of adult cells due to the inhibition of PDK, AKT, RSK1, and TOR [121].
A group of synthetic compounds formed by the aldolic reaction of isatins and kojic acid have been studied as inhibitors of PDK. This new family of compounds, 3-hydroxy-3-[3-hydroxy-6-(hydroxymethyl)-4-oxo-4H-pyran-2-yl] indolin-2-ones (compound 64 in Figure 3), have been proven to have different biomedical applications, such as anticonvulsant, anti-inflammatory, and anti-cancer effects. The docking studies revealed that these compounds were susceptible to occupying the binding site in PDK, thus inhibiting the binding of ATP. Therefore, they are promising ligands for inhibiting PDK in the Warburg effect [122].
2.2.7. Compounds in Combination with Other Therapies
As mentioned before, the well-known PDK inhibitor DCA has been reported to have anti-cancer effects through its reversal of tumor-associated glycolysis. To explore the anti-cancer potential of DCA, studies of DCA in combination with other anti-cancer drugs have been done.
Particularly, a binary prodrug named PDOX (compound 65 in Figure 3) was designed and investigated. It consisted of a combination of DCA (PDK inhibitor) and the anti-cancer drug doxorubicin (DOX). The prodrug was activated selectively by cancer-associated esterase to deliver DCA and DOX.
DCA and DOX induced synergistic effects. This synergistic effect was evaluated in HepG2 cells. PDOX showed a higher anti-cancer activity than DOX and could serve as an alternative strategy to diminish toxicity [123].
The PDK inhibitor DCA (compound 44) has also been explored in lung cancer in combination with commercial therapeutic drugs, particularly A549 and LNM35. Al-Azawi and co-workers demonstrated that DCA significantly enhanced the anti-cancer effect of cisplatin, gefitinib, and erlotinib. The additive effects on the inhibition of the mentioned cell lines due to the synergistic effect were shown. This study reinforced DCA as a promising therapeutic agent for lung cancer [124].
Alongside the drug combination approach, new evidence for the synergism between the PDK inhibitor DCA and the HIF-1α inhibitor PX-478 was found. The group demonstrated that the combination of DCA and PX-478 produced synergistic effects in colorectal, lung, breast, cervical, liver, and brain cancer cell lines. Mechanistically, while DCA suppressed PDK1, HIF-1α increased the PDK1 expression. Thus, PX-478 reinforced the primary effect of DCA indirectly, thereby synergistically increasing ROS production when combined with DCA [125].
The synergistic anti-tumor effect of DCA in combination with the chemotherapeutic agent 5-fluorouracil has also been studied, particularly in four colorectal cancer cells. The results of this study showed that DCA had a synergistic antiproliferative effect in combination with 5-fluorouracil. The apoptosis of colorectal cancer cells treated with both compounds was enhanced, showing changes in the Bcl-2, Bax, and caspase-3 proteins [126]. Furthermore, an investigation was conducted to assess the potential resensitization of gastric cancer cells, which have developed a resistance to 5-fluorouracil due to hypoxia. The study concluded that low doses of DCA successfully restored sensitivity in these gastric cancer cells with a hypoxia-induced resistance to 5-fluorouracil by modifying the glucose metabolism [127].
Table 2.
Efficacy and modes of action of the PDK inhibitors.
Table 2.
Efficacy and modes of action of the PDK inhibitors.
| Scaffold | Model | Mode of Action | Refs. |
|---|---|---|---|
| Small molecules | |||
| DCA | In vitro | Inhibition of the PDK1 and PDK2 activity | [95,96,97,98,99,100,101,102,103,104] |
| Diverse tumor types | |||
| HCT-116, SiHa, A549, A375, MeWo | |||
| In vivo | |||
| Human patients | |||
| 1-isobutanoyl-2isopropylisothioureadichloroacetate | In vivo | Inhibition of PDK | [105] |
| (T1084) | Mouse model | ||
| Metal complexes | |||
| DPB | In vitro | Inhibition of the PDK expression | [106] |
| Cervix cancer cells | |||
| HeLa cells | |||
| Fused heterocyclic rings | |||
| Compound 49 and 50 | In vivo | Inhibition of the PDK activity | [107] |
| (1,2,4-amino triazine derivative) | Murine Lewis Lung Carcinoma tumor model | ||
| Compound 51 | In silico | Inhibition of the PDK activity | [108] |
| (2-amino substituted 2,4-dichlorobenzo[h]naphthyridine) | |||
| Sulfide and disulfide derivatives | |||
| JX06 | In vitro | Inhibition of the PDK1 activity | [109] |
| (disulfide derivative) | Lung cancer cells | ||
| A549 | |||
| Compound 53 | Kinetic assay | Inhibition of the PDK activity | [110] |
| Nonsteroidal anti-inflammatory compounds | |||
| Aspirin | In vitro | Suppression of the PDK1 expression | [111] |
| Breast cancer cells | |||
| MDA-MB-231, MCF-7 | |||
| Miscellanea | |||
| N-(4-(N-alkyl/aralkylsulfamoyl)phenyl)-2-methylpropanamides | Kinetic assay | Inhibitor of the PDK activity | [112] |
| α-lipoic acid | In vivo | Downregulation of the expression of PDK1 | [113] |
| Mouse model | |||
| XB-1 | Kinetic assay | Inhibition of the PDK1 activity | [114,115] |
| Dicumarol | In vitro | Inhibition of the PDK1 activity | [116] |
| Ovarian cancer cells | |||
| SKOV3, A2780 | |||
| Compound 59 | In silico | Inhibition of the PDK activity | [117,118] |
| Hemistepsin A | In vitro | Decreases the PDK activity | [120] |
| Colon cancer | |||
| DLD-1 | |||
| Ursolic acid | In vitro | Inhibition the PDK activity | [121] |
| Leukemia cells | |||
| MT-4 | |||
| 3-hydroxy-3-[3-hydroxy-6-(hydroxymethyl)-4-oxo-4H-pyran-2-yl] indolin-2-ones | In silico | Inhibition of the PDK activity | [122] |
| Compounds in combination with other therapies | |||
| DCA + DOX (PDOX) | No data | Inhibition of PDK | [123] |
| DCA + cisplatin/erlotinib/gefitinib | No data | Inhibition of PDK | [124] |
| DCA + PX-478 | No data | Suppression of PDK1 (DCA) and an increase in the PDK1 expression | [125] |
| DCA + 5-fluorouracil | No data | Inhibition of PDK | [126,127] |
2.3. PK Modulators
When it comes to studying and analyzing the available molecules for targeting and interfering in the MOA of PKM2 by the direct interaction with enzymes, not only compounds with capacity to inhibit, but also other molecules with the ability to activate this enzyme are described. As a result, both types of modulation can be a strategy for fighting against cell proliferation and growth. This section collects different PK modulators that have been organized into the following groups: small molecules, polyphenolic compounds, quinoline derivatives, nonsteroidal anti-inflammatory drugs, and miscellanea (Table 3).
2.3.1. Small Molecules
Small molecules include molecules that activate the enzyme, along with others that can trigger PKM2 and promote different mechanisms that inhibit tumor growth.
TEPP-46 and DASA-58 (compounds 66 and 67 in Figure 4), two allosteric activators, were studied in five breast cancer cell lines to investigate the potential effect of PKM2 when it is activated. The studies showed that the molecules were able to increase the activity of the enzyme in those cancer cells without affecting the overall cell survival and that they were also able to reduce an intracellular glucose sensor (TXNIP levels) [128].
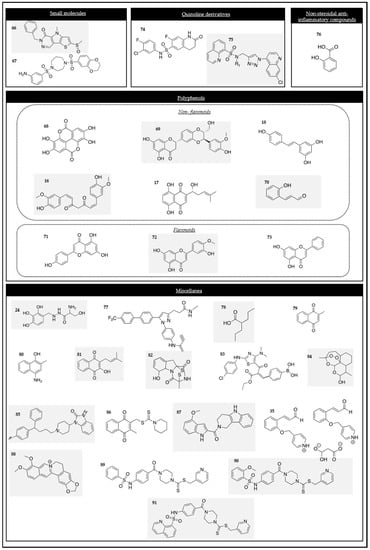
Figure 4.
Chemical structures of the PK modulators.
Other studies were carried out with those molecules to prove the relevance of the enzyme in LPS-induced macrophages infected with Mycobacterium tuberculosis. They provided important evidence, showing how DASA-58 and TEPP-46 could promote the formation of active PKM2 tetramers. In this way, they could boost the PKM2 enzymatic activity and limit tumor growth in vivo. The study helped clear up how Warburg-like metabolic changes can affect macrophages and how they are important in chronic, inflammatory, and oxidative processes [129].
2.3.2. Polyphenolic Compounds
- Non-flavonoids
The evaluation of these phenolic compounds against three binding sites of PKM2 provides insight into how molecule–enzyme binding occurs and functionally how amino residues of PKM2 are important. These compounds have different IC50 values and can exert anti-tumor effects in different cancer cell lines.
Ellagic acid (compound 68 in Figure 4) is an organic heterotetracyclic compound and is found in numerous fruits and vegetables [130]. Ellagic acid can induce apoptosis in MCF-7 and MDA-MB-231 cancer cells and can induce a reduction in the key glycolytic enzymes, such as PKM2, HK2, and GLUT1, which were measured in MCF-7 and MDA-MB-231 cells. It induced apoptosis in those cancer cell lines with IC50 values of 23 µM and 27 µM, respectively [131].
Silibinin (compound 69 in Figure 4) is the principal component of silymarin, a standardized extract of silybum marianum. It is well known due to its chemo-preventive, anti-angiogenic, and apoptosis inducer properties. It has been identified as a potent PKM2 inhibitor with an IC50 value of 0.91 µM. It can reduce PKM2 and the protein in TNBC cells (MDA-MB-231 and BT549 cells) and induce a reduction in the key glycolytic enzymes, such as PKM2, HK2, and GLUT1. Silibinin can inhibit the HK2 activity in TNBC cells, and when it is combined with paclitaxel, it can promote antineoplastic drug toxicity in gastric cancerous cells [131,132,133,134].
Resveratrol (compound 18 in Figure 4) is a natural stilbene and non-flavonoid polyphenol that can be found in cereals, fruits, and vegetables. It has been demonstrated that it possesses antioxidant, anti-inflammatory, anti-tumoral, and cardioprotective effects. According to reports, resveratrol exhibited the potential to reverse multidrug resistance in some cancer cells. Moreover, its combination with clinically approved drugs has been found to enhance the sensitivity of cancer cells to standard chemotherapeutic agents. Resveratrol downregulated the PKM2 expression by inhibiting mTOR signaling and suppressed cancer metabolism in the HeLa, MCF-7, and HepG2 cell lines [131,135,136,137].
Curcumin (compound 16 in Figure 4) has been studied as an anti-cancer compound. It has been demonstrated that curcumin inhibited the glucose uptake and lactate production in the H1299 (lung), MCF-7 (breast), HeLa (cervical), and PC-3 (prostate) cancer cell lines by downregulating the PKM2 expression via the inhibition of the mTOR-HIF-1α axis [138].
Shikonin (compound 17 in Figure 4), the active naphthoquinone presented in Section 2.1.2, is also a PKM2 inhibitor. It suppressed the expression of PKM2, HK, and GLUT1 [68].
HCA (compound 70 in Figure 4) is an active component isolated from cinnamon bark. It inhibits the PKM2 and STAT3 signaling pathways. Its biochemical methods (affinity chromatography, drug affinity, responsive stability assay, etc.) demonstrated that HCA bound directly to PKM2 and decreased the protein kinase activity of the enzyme by decreasing the phosphorylation at the tyrosine 105 residue [139]. STAT3 phosphorylation at this residue was suppressed, which ultimately led to a downregulation of the genes, such as MEK5 and cyclin 5, that are implicated in cancer growth when they are altered [140].
- Flavonoids
This section highlights the various flavonoids derived from vegetables and fruits, which have demonstrated antiproliferative effects on human cancer cells.
Apigenin (compound 71 in Figure 4) inhibits cancer cell proliferation by triggering cell apoptosis and modulating the cell cycle. It interferes with multiple signaling pathways and protein kinases (PI3K/AKT, MAPK/ERK, JAK/STAT, NF-κB and Wnt/β-catenin) [141]. Apigenin targeted and reduced the PKM2 enzyme expression and activity in colon cancer cells (HCT-116, HT-29, and DLD1) [142].
Other flavones, such as diosmetin and chrysin (compounds 72 and 73 in Figure 4), have been studied due to their structural features, showing their capacity to inhibit the PKM2 enzyme. This interruption of glycolysis resulted in a reduction in tumor growth [143].
2.3.3. Quinoline Derivatives
Quinoline derivatives (NZT) have been the subject of syntheses, structure–activity relationships, selectivity, and other properties research. Certain studies focused on the 2-oxo-N-aryl-1,2,3,4-tetrahydroquinoline-6-sulfonamide scaffold (compound 74 in Figure 4). These studies revealed an EC50 value of 90 nM with a significant selectivity over other PK isoforms [144].
In silico studies confirmed that the 8-quinolinesulfonamide derivatives, concretely compound 79 (compound 75 in Figure 4), were potential PKM2 modulators. In vitro research studies showed that this compound could reduce the intracellular pyruvate level in A549 lung cancer cells, reduce cell viability, and induce apoptosis [145].
2.3.4. Nonsteroidal Anti-Inflammatory Drugs
Salicylic acid (SA) (compound 76 in Figure 4) is the major and active metabolite of aspirin. Both compounds are well-described drugs that can be used without the need of a prescription for lowering fever, reducing inflammation, and relieving low to moderate pain. Studies were done to report and identify the potential SA-binding proteins (SABPs) in human HEK-293 cells. One of the proteins that resulted in binding with SA was PKM2. A genome-wide high-throughput screening study was conducted to identify the potential binding interactions between the SA-binding proteins (SABPs) in human HEK293 cells. The screening strategy utilized an adaptation of a previously established method used for identifying SABPs in plants [146]. Salicylic acid (SA) inhibited the activity of PKM2 in a dose-dependent manner, with 400μM of SA inhibiting up to 40% of its activity. Unfortunately, the inhibition at higher concentrations of SA could not be tested. Importantly, the inhibition of PKM2 was selective, as SA binding did not affect the enzymatic activity of PKM1. In addition, other studies have demonstrated that SA derivatives can reduce the activity of PKM2, PFK, HK2, as well as the tumor angiogenesis-related vascular endothelial growth factor. Furthermore, the lactate level in the hepatoma cell line SMMC-7721 was significantly reduced [146].
2.3.5. Miscellanea
Benserazide (BEN) (compound 24 in Figure 4) is currently in clinical use as a co-adjuvant treatment in Parkinson’s disease with L-DOPA. It has been demonstrated, first by a structure-based virtual ligand screening in an FDA-approved drug database and then by in vitro and in vivo studies, that BEN was able to direct PKM2 binding, hence blocking its activity. In this way, the aerobic glycolysis concurrent upregulation of OXPHOS can be inhibited in a dose-dependent manner. It has been verified as a novel inhibitor for targeting PKM2 in melanoma cells, in which the enzyme expression is higher, converting BEN into a potential therapeutic candidate for melanoma [147].
N-(4-(3-(3-(methylamino)-3-oxopropyl)-5-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-1H-pyrazol-1-yl)phenyl)propiolamide (compound 77 in Figure 4) is a novel irreversible PKM2 inhibitor that has demonstrated tumor suppressive effects in different cell lines. This compound can inhibit PKM2 by interacting with CYS326 and CYS317 of the enzyme and the propiolamide electrophile terminus of compound 77. The inhibition of the enzyme and, as result, the growth suppression of the tumor was tested in PC9 cells where the PKM2 expression was shown to be decreased. Moreover, there was substantial evidence indicating that the remarkable in vitro and in vivo anti-tumor effectiveness of compound one resulted from its capacity to target both metabolic and nonmetabolic functions within cancer cells [148].
Valproic acid (compound 78 in Figure 4) has been found to reduce the PKM2 expression in breast cancer cells, specifically in MCF-7 and MDA-MB-231 cells [149].
Vitamin K compounds, known as VKs, are fat-soluble compounds. VK3 (compound 79 in Figure 4) and VK5 (compound 80 in Figure 4) have been reported to be adjuvant agents for cancer therapy. Chen et al., demonstrated that these vitamins acted as selective PKM2 inhibitors in Hela cells [150,151].
Lapachol (compound 81 in Figure 4), a compound derived from Tabebuia impetiginosa, has been shown to have a greater affinity compared to other related natural products, such as shikonin, which was demonstrated using computer-aided design. In melanoma cells, lapachol requires a lower concentration to inhibit 50% of the PKM2 activity. The results indicated that lapachol inhibited glycolysis in melanoma cells by interacting with PKM2, a key regulator of glycolysis. This inhibition of glycolysis by lapachol sensitized the melanoma cells to apoptosis. The in silico studies revealed that the lapachol structure interacted with 19 amino acid residues through Van der Waals interactions, hydrogen bonds, alkyl interactions, and pi–alkyl interactions. These molecular interactions contributed to the lapachol binding and inhibitory effects on PKM2 and glycolysis in melanoma cells [150,152].
Glyotoxin (compound 82 in Figure 4) bound directly and strongly to PKM2 and suppressed its activity. Moreover, it exhibited a dose-dependent inhibition of glycolytic activity, resulting in a reduced glucose consumption and lactate production within the U87 human glioma cell line. The inhibition of PKM2 activity further led to the suppression of STAT3 phosphorylation. These studies demonstrated the potential of gliotoxin as a treatment for glioma by targeting cancer metabolism [153].
Boronic acid has the ability to activate the PKM2 enzyme, leading to redox metabolism in oral cancer cells. Consequently, a novel class of boronic acid derivatives was developed. Particularly, the PKM2 activity of compound 83 (Figure 4) was tested in CAL-27 cells. It was found that compound 83 activated PKM2 with an AC50 of 25 nM [154].
Dihydroartemisinin (DHA) (compound 84 in Figure 4), also known as artenimol, is an active metabolite of the anti-malarial drug artemisinin. DHA induces a pro-inflammatory cell death called pyroptosis. It has been shown to downregulate the PKM2 expression and induce cell death in the esophageal squamous cell carcinoma (ESCC) cells Eca109 and EC9706 at 24 h. DHA has the potential to be a therapeutic agent for ESCC by downregulating the PKM2 expression, activating the caspase-8/3-GSDME axis, and mediating tumor cell pyroptosis [155].
The antipsychotic medication Pimozide (compound 85 in Figure 4) acts on the signaling pathway and promotes the effect of p53, leading to a decrease in the expression of PKM2. Pimozide has demonstrated strong anti-breast cancer effects both in vitro and in vivo. However, it remains unclear whether Pimozide exerts inhibitory effects on aerobic glycolysis. The in vitro and in vivo studies have shown that Pimozide inhibited the survival of breast cancer cells by blocking the PI3K/Akt/MDM2 signaling pathway. This inhibition enhanced the p53 expression, resulting in the downregulation of the PKM2 enzyme and alleviating the Warburg effect. To further validate the effects of Pimozide, the in vivo experiments were conducted using mice transplanted with MCF-7 breast cancer cells. The tumor size was significantly reduced in the Pimozide-treated group compared to the control group, and the tumor weight was inhibited by 44%. These findings support the potential therapeutic use of Pimozide in breast cancer treatment [156].
Compound 86 (Figure 4) is a specific inhibitor of the PKM2 enzyme. Investigations on the anti-cancer effects of this inhibitor were conducted using MTT and colony formation assays in SK-OV-3 cells. Compound 86 was found to induce AMPK activation, which is associated with suppressing the tumor progression. The inhibition of PKM2 by compound 86 affected the Warburg effect and led to autophagic cell death. This mechanism highlighted the potential of specific PKM2 inhibitors for targeting cancer cell metabolism by blocking the glycolytic pathway [157].
NPD10084 (compound 87 in Figure 4) has demonstrated an antiproliferative activity against colorectal cancer cells both in vitro and in vivo. In this study, PKM2 was identified as a potential target protein for this compound. Interestingly, NPD10084 disrupted the protein–protein interactions between PKM2 and β-catenin or STAT3, leading to the suppression of the downstream signaling pathways [158].
The cinnamaldehyde derivative (CB-PIC) (compound 35 in Figure 4), the major component of cinnamon, has been found to inhibit the migration of H1299 lung tumor cells in scratch and transwell assays. This compound was directly and specifically bound to recombinant PKM2, resulting in a reduction in its catalytic activity. The cells with PKM2 knockdown demonstrated a significantly reduced migration compared to the control cells when subjected to glucose and oxygen deprivation. The anti-cancer mechanism of compound 35 was investigated in human HCC cells in relation to STAT3 signaling. Compound 35 suppressed the phosphorylation of STAT3 in HepG2 and Huh7 cells. Conversely, the depletion of STAT3 enhanced the capacity of this derivative to suppress the expression of HK2, PKM2, and pro-caspase3, and reduced cell viability in Huh7 cells [86].
In addition, a literature-based phytochemical analysis of Mangifera indica identified a total of 94 compounds, which were docked against three binding sites of PKM2 to identify the potential PKM2 inhibitors. Among these compounds, berberine (compound 88 in Figure 4), isolated from Coptis and Hydrastis canadensis, was found to inhibit the PKM2 activity, showing antiproliferative effects in HCT116 and HeLa cells. However, further studies are needed to validate the PKM2 inhibitory potential of the identified compounds through in vitro biochemical assays [159].
A novel sulfonamide compound with a pyridin-3-ylmethyl 4-(benzoyl)piperazine-1-carbodithioate moiety has been identified as a potent activator of PKM2. A series of analogs based on the lead compound 89 (Figure 4) were designed and synthetized. Among these analogs, compounds 90 and 91 (Figure 4) demonstrated higher PKM2 activation activities. Furthermore, they exhibited more significant antiproliferative effects against human tumor cell lines. Notably, compound 91 specifically inhibited the proliferation of multiple cancer cell types while showing minimal toxicity towards normal cells. Additionally, it was observed that compound 91 effectively inhibited the colony formation in MCF7 cells. These findings suggest that compound 91 and its analogs have the potential to act as potent PKM2 activators, displaying enhanced antiproliferative activities against cancer cells. Further research is needed to explore the therapeutic applications of these compounds and elucidate their mechanisms of action [160].
Table 3.
Efficacy and modes of action of the PK modulators.
Table 3.
Efficacy and modes of action of the PK modulators.
| Scaffold | Model | Mode of Action | Refs. |
|---|---|---|---|
| Polyphenols | |||
| Flavonoids | |||
| Chrysin | Kinetic assay | Inhibition of PKM2 | [143] |
| Apigenin | In vitro | Reduction in the PKM2 activity | [141,142] |
| Colon cancer cells | |||
| HCT116, HT29, DLD1 | |||
| Morusin | In vitro | Attenuation of the activity of PKM2 | [65] |
| HCC cells | |||
| Huh7, Hep3B | |||
| Diosmetin | Kinetic assays | Inhibition of PKM2 | [143] |
| Non-flavonoids | |||
| Shikonin | Esophageal squamous cell carcinoma | Inhibition of the PKM2 expression | [68] |
| Resveratrol | In vitro | Inhibition of the PKM2 expression | [131,135,136,137] |
| Diverse tumor types | |||
| HeLa, MCF-7, HepG2 | |||
| Curcumin | In vitro | Downregulation of the PKM2 expression | [138] |
| Diverse tumor types | |||
| H1299, PC-3, MCF-7, HeLa | |||
| Ellagic acid | Kinetic assay | Inhibition of the PKM2 activity | [130,131] |
| Silibinin | SAR studies | Inhibition of the PKM2 activity | [131,132,133,134] |
| HCA | In vivo | Inhibition of the PKM2 expression | [139,140] |
| Nude mice model | |||
| DU-145 tumor xenografts | |||
| Small molecules | |||
| TEPP-46 | In vitro | PKM2 activator | [128,129] |
| Breast cancer cells | |||
| MCF-7, MDA-MB-231, T47-D, MDA-MB-468, HCC1443 | |||
| DASA-58 | In vitro | PKM2 activator | [128,129] |
| Breast cancer cells | |||
| MCF-7, MDA-MB-231, T47-D, MDA-MB-468, HCC1443 | |||
| Oxygen and sulfur heterocycles | |||
| Triptolide | In vitro | Inhibition of the PKM2 expression | [72,73] |
| Neck and head cancer cells | |||
| HK1, FaDu, C666-1 | |||
| Cholangiocarcinoma cells | |||
| HuCCT1, RBE | |||
| Tanshinone IIA | In vivo | Inhibition of the PKM2 expression | [74] |
| Lung cancer model | |||
| Nude mouse xenograft | |||
| Nonsteroidal anti-inflammatory compounds | |||
| Salicylic acid | In vitro | Inhibition of PKM2 | [146] |
| HEK293 cells | |||
| Quinoline derivatives | |||
| Compound 105 | In vitro | PKM2 activation | [161] |
| (Quinoline 3-sulfonamide derivative) | HCC | ||
| Snu398 | |||
| Compound 78 | In silico | Modulation of the PKM2 activity | [144] |
| (2-oxo-N-aryl-1,2,3,4-tetrahydroquinoline-6-sulfonamide derivative) | |||
| Compound 79 | In vitro | Inhibition of the PKM2 activity | [145] |
| (8-quinolinesulfonamide derivative) | Lung cancer cells | ||
| A549 | |||
| Miscellanea | |||
| Benserazide | In vitro | Inhibition of the PKM2 activity | [147] |
| Melanoma cells | |||
| SK-MEL-5, SK-MEL-28 | |||
| CB-PIC | In vitro | Suppression of the PKM2 expression | [86] |
| HCC cells | |||
| HepG2 and Huh7 | |||
| N-(4-(3-(3-(methylamino)-3-oxopropyl)-5-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-1H-pyrazol-1-yl)phenyl)propiolamide | In vivo | Irreversible inhibition of PKM2 and the PKM2 expression | [148] |
| Athymic nude mice | |||
| PC9 (lung cancer) xenograft tumor model | |||
| Valproic acid | In vitro | Reduction in the PKM2 expression | [149] |
| Breast cancer cells | |||
| MCF-7 | |||
| MDA-MB-231 | |||
| Vitamin K | Kinetic assay | PKM2 selective inhibitors | [150,151] |
| Lapachol | In vitro | Inhibition of the PKM2 activity | [150,152] |
| Melanoma cells | |||
| SK-MEL103 | |||
| Glyotoxin | In vitro | Suppression of the PKM2 activity | [153] |
| Glioma cells | |||
| U87 | |||
| Compound 88 | In vitro | Activation of PKM2 | [154] |
| (Boronic acid derivative) | Oral squamous carcinoma cells | ||
| CAL-27 | |||
| Dihydroartemisinin | In vitro | Downregulation of the PKM2 expression | [155] |
| Esophageal squamous cell carcinoma | |||
| Eca109, Ec9706 | |||
| Pimozide | In vivo | Decrease in the PKM2 expression | [156] |
| Nude mice model | |||
| MCF-7 tumor xenograft model | |||
| Compound 91 | In vitro | Inhibition of the PKM2 expression | [157] |
| Ovarian cancer cells | |||
| SK-OV-3 | |||
| NPD10084 | In vitro | Inhibition of the PKM2 activity | [158] |
| Colorectal cancer cells | |||
| HCT-116, LoVo | |||
| Compound 95, 96, 97 | In vitro | Activation of PKM2 | [160] |
| (Pyridine-3-yl-methyl-4-(benzoyl)piperazine-1-carbodithioate) | Breast cancer cells | ||
| MCF-7 | |||
| Berberine | In vitro | Decrease in the PKM2 activity | [159] |
| Diverse tumor cells | |||
| HCT-116, HeLa | |||
| Compounds in combination with other therapies | |||
| Sildenafil + cisplatin | In vitro | Inhibition of PKM2 | [94] |
| Lymphoma cells | |||
| HuT-78 cells | |||
2.4. LDH Enzyme Modulators
The enzymatic activity of LDH is focused on lowering the extracellular pH, thus avoiding tumor invasion and metastasis. Therefore, the inhibition of its activity will put up a protective barrier against tumor growth. In this section, different LDH modulators were collected. They have been organized into the following groups: small molecules, pyridazines and piperazines, polyphenols, quinoline-based derivatives, sulfide and disulfide derivatives, nonsteroidal anti-inflammatory compounds, compounds in combination with other therapies, and miscellanea (Table 4).
2.4.1. Small Molecules
Sodium oxamate (compound 92 in Figure 5) exerted a competitive inhibition of human LDHA in vitro with a Ki of 136.3 uM in two NPC cancer cell lines. However, this derivative presented several problems related to a low and limited selectivity, weak potency, and poor cellular uptake [162]. The studies showing the MOA [163] stated that compound 92 could promote cell apoptosis via the downregulation of the CDK1/cyclin B1 pathway, which ultimately provoked the inhibition of LDHA.
Figure 5.
Chemical structures of the LDH modulators.
2.4.2. Pyridazine and Piperazine
Trimetazidine (TMZ) (compound 93 in Figure 5) is a drug indicated exclusively in adults as additional therapy for the symptomatic treatment of patients with stable angina pectoris. Silica-induced pulmonary fibrosis studies in rats were done to explore the effects of this drug over the LDH activity. The results showed that the derivative of compound 93 was able to reduce and normalize the levels of the enzyme in bronchoalveolar lavage fluid (BALF) [164]. TMZ can be used alone or in combination with antineoplastic drugs, such as gemcitabine or abraxane, to reduce cell viability and induce apoptosis in human pancreatic cancer cells [165]. Additionally, the derivative of compound 93 also induced cell death in breast cancer cells [166].
Pyridazine derivatives exhibited potent anti-cancer activity and were synthesized due to structure-based virtual screening studies. Compound 94 (Figure 5) exerted its effect on multiple cancer lines: Med1-MB (a cell line obtained from Sonic Hedgehog medulloblastoma), CRC HCT116, SW620, lung cancer A549, and pancreatic PANC-1 cancer cell lines. This derivative has also been tested using an enzymatic assay, measuring the rate of NADH consumption by spectrophotometry. It displayed a significant inhibitory activity with an IC50 of 12 μM. When compared to sodium oxamate (IC50 = 16.47 μM), a reference inhibitor, compound 94 exerted a lower dose dependency over LDH [167].
2.4.3. Polyphenols
- Flavonoids
Epigallocatechin (EGCG) (compound 95 in Figure 5) is one of the main compounds of green tea and a compound found in the medical plant Spatholobus suberectus, which is commonly used in China for the treatment of cancer-related blood stasis. EGCG can inhibit the LDHA enzyme, targeting the metabolism of human pancreatic adenocarcinoma (MIA PaCa-2 cells). This derivative can significantly reduce the lactate production, anaerobic glycolysis, and glucose consumption [168]. Other studies have demonstrated its ability for inhibiting the LDHA activity in both estrogen-dependent human MCF-7 cells and estrogen-independent MDA-MB-231 cells. The mechanism of action is believed to be related to the potential acceleration of HIF-1α proteasome degradation [169,170].
- Non-flavonoids
FX11 (3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid) (compound 96 in Figure 5) is a synthetic molecule that was discovered using an HITS assay. FX11 is structurally related to gossypol and is well known to be a sensitizer in chemotherapy-resistant tumor cells [171,172,173]. It is an NADH competitive, small, reversible, and selective LDHA inhibitor and it has demonstrated preclinical success in the progression of multiple adult cancers (lymphoma, pancreas, osteosarcoma, and prostate) [174].
Gossypol (compound 97 in Figure 5) is a natural aldehyde derived from cotton seed and it can inhibit several cancer cell lines, such as melanoma, lung, breast, cervix, and leukemia. This compound is a non-selective LDH inhibitor with Ki values of 1.9 and 1.4 µM in LDHA and LDHB, respectively. The derivative of compound 97 is well tolerated but it presents a poor response in metastatic adrenal cancer, malignant glioma, and anthracycline and taxane-resistant metastatic breast cancers [175,176].
Galloflavin (compound 98 in Figure 5), a synthetic derivative of gallic acid, binds to LDHA, but it is also able to block LDHB without competing with substrates and cofactors. The cytotoxic effects were demonstrated by this analog in various human breast cancer cells (MCF-7, triple-negative MDA-MB-321, and tamoxifen-resistant MCF-7tam cell lines) [171,177].
2.4.4. Quinoline-Based Derivatives
Compound 99 (Figure 5) a quinoline 3-sulfonamide derivative, is a selective and competitive LDHA inhibitor, competing with NADH. This derivative can exert its effect in multiple cell lines, including hepatocellular and breast carcinomas [161].
2.4.5. Sulfide and Disulfide Derivatives
GNE-140 (compound 100 in Figure 5), a synthetic molecule discovered using an HITS assay, was tested in MDA-MB-231 and HCC143 cells in vitro. GNE-140 has been demonstrated to block LDH [178].
Several substituted 3-hydroxy-mercaptocyclo-2-enone compounds, especially compound 101 (Figure 5), have been identified as potent novel class LDH inhibitors using the HITS approach. Compound 101 can bind to the active site of LDH where pyruvate usually binds, hence limiting and competing with the substrate binding. Furthermore, biochemical and surface plasmon resonance experiments supported the fact that these derivatives required an NADH cofactor [179].
2.4.6. Nonsteroidal Anti-Inflammatory Compounds
Diclofenac (compound 102 in Figure 5) is commonly used to reduce inflammation and alleviate pain. Apart from its classical role as a cyclooxygenase (COX) inhibitor, diclofenac has exhibited potent anti-cancer effects. Studies were conducted to investigate whether diclofenac could induce cancer cell death in HeLa cells through the production of ROS. It was also examined at non-toxic concentrations to determine whether it could decrease lactate production in murine glioma cells and inhibit the expression of LDHA in vitro using GL281 cells [180]. Other studies have revealed its ability to disrupt the Warburg effect through the targeting of glucose transport and the inhibition of LDH and MCT1, leading to a reduced glucose uptake and lactate secretion [181,182].
2.4.7. Compounds in Combination with Other Therapies
Different concentrations of METABLOC (compound 103 in Figure 5), a combination of hydroxycitrate and lipoic acid, was used in combination with diclofenac and metformin in LL/2 lung carcinoma cells. Cisplatin was used as a positive control. The effects of METABLOC appeared to be enhanced when high-dose metformin was used. In a lung cancer model in C57BL/6 mice, high doses of metformin and diclofenac were administered, and METABLOC was used as the positive controls. The combination of metformin and diclofenac slightly delayed tumor growth compared to the control groups. METABLOC, when used in combination with metformin and diclofenac, was also tested to observe the progression of LL/2 carcinoma, and it successfully delayed tumor growth [181,183].
In another study, the combination of diclofenac (102 in Figure 5) and ibuprofen was examined. Both drugs are NSAIDs associated with anti-tumoral effects in glioma cells. However, ibuprofen showed a stronger inhibition of cell growth compared to diclofenac. Both drugs were able to decrease STAT3 phosphorylation. Interestingly, diclofenac led to a decreased C-MYC expression and a subsequent reduction in the LDHA activity. On the other hand, the treatment with higher doses of ibuprofen induced the C-MYC expression and had less impact on LDHA alteration. These findings highlighted the different MOAs of diclofenac and ibuprofen. Diclofenac acted by inhibiting STAT3 signaling and the downstream modulation of glycolysis. Ibuprofen, on the other hand, inhibited the COX and lactate mechanisms independently [184].
2.4.8. Miscellanea
Crocetin (compound 104 in Figure 5) is a 20-carbon dicarboxylic acid diterpenoid that was obtained using chemical synthesis. Its LDH inhibition properties have been tested against two cancer cell lines (A549 and HeLa) [185,186].
Machillin A (compound 105 in Figure 5), a natural product found mainly in Machilus thunbergii, Iryanthera lancifolia, and Magnolia sinica, has been demonstrated to reduce tumor growth by inhibiting LDHA. It is a competitive inhibitor, which blocks the NAD binding site [185,187].
Nifurtimox (compound 106 in Figure 5), a drug used for treating parasitic protozoan Trypanosoma cruzi infections, has also been reported to present cytotoxic properties in the neuroblastoma cell lines LA-N-1, IMR-32, LS and SK-N-SH. Nifurtimox can achieve a reduction in the LDH enzyme activity [188].
N-hydroxyndole-based inhibitors have been demonstrated to be small, competitive, and LDHA isoform-selective inhibitors. The derivative of compound 107 (Figure 5) can compete against pyruvate and NADH. SAR studies confirmed the importance of the OH-COOH pharmacophore, since the enzyme active site will usually have a gap for lactate (hydroxyl group) and another for pyruvate (alpha-keto acid group) [189].
Selenobenzenes, such as (1-(phenylseleno)-4-(trifluoromethyl)benzene (PSTMB) (compound 108 in Figure 5), demonstrated their ability to decrease tumor growth in several tumor cell lines (NCI-H460, MCF-7, Hep3B, A375, HT-29, and LLC). Thus, in HT-29 human colon cancer cells, PSTMB exerted a dose-dependent inhibition of the cell viability and of the activity of LDHA. Biochemical assays showed that compound 108 did not affect the tetramer formation of LDHA. The conversion of pyruvate to lactate was inhibited because this derivative inhibited the NADH binding to LDHA [190].
Oxaloacetate (OAA) (compound 109 in Figure 5) is a competitive inhibitor of human LDHA and has been found to be regulated by PKM2 activity. An elevated PKM2 activity can promote the de novo synthesis of OAA through glutaminolysis, leading to the inhibition of LDHA in cancer cells. A high pyruvate kinase activity can result in increased cytosolic OAA levels, which act as a competitive inhibitor of LDHA at concentrations that are sufficient for inhibiting its activity in cancer cells. Additionally, the activation of PKM2 can enhance the OAA levels by increasing the de novo synthesis of OAA [191].
N-acylhydrazone derivatives have been studied as LDHA inhibitors. For this purpose, a virtual screening procedure was conducted. Afterwards, chemical modifications were made to the selected compounds to enhance their inhibitory activity. The new molecules demonstrated activity in the micro-molar range. Specifically, compound 110 (Figure 5) exhibited a significant effect on lactate production in the Raji human cell line. A total of 67 molecules were chosen, and a structure–activity relationship (SAR) study was carried out to investigate their inhibitory effects on purified LDHA. Compound 110 of the study showed pronounced effects, and the N-acylhydrazone scaffold was identified as an important structural feature for exerting the inhibitory effect [192].
Table 4.
Efficacy and modes of action of the LDH inhibitors.
Table 4.
Efficacy and modes of action of the LDH inhibitors.
| Scaffold | Model | Mode of Action | Refs. |
|---|---|---|---|
| Polyphenols | |||
| Flavonoids | |||
| Morusin | In vitro | Attenuation of the activity of LDHA | [65] |
| HCC cells | |||
| Huh7, Hep3B | |||
| Epigallocatechin | In vitro | Inhibition of LDHA | [168,169,170] |
| Human pancreatic adenocarcinoma | |||
| (MIA PaCa-2cells) | |||
| Breast cancer cells | |||
| MDA-MB-231, MCF-7 | |||
| Non-flavonoids | |||
| Galloflavin | HCC | Inhibition of the LDHA activity | [171,177] |
| In vitro | |||
| Breast cancer cells | |||
| MCF-7, MCF-Tam, MDA-MB-231 | |||
| FX11 | In vitro | Competitive inhibition of the LDHA activity | [171,173,174] |
| Gallbladder cancer cells | |||
| NOZ, GBC-SD | |||
| Prostate cancer cells | |||
| PC-3 | |||
| Gossypol | Kinetic assay | Non-selective inhibition of LDH | [175,176] |
| Curcumin | In vitro | Reduction in the expression of LDHA and its activity | [67] |
| Papillary thyroid cancer | |||
| B-CPAP | |||
| Small molecules | |||
| Sodium oxamate | In vitro | Inhibition of the LDHA activity | [162,163] |
| Gastric cancer cells | |||
| SGC-7901, BGC823 | |||
| Nasopharyngeal carcinoma cells | |||
| CNE-1, CNE-2 | |||
| Oxygen and sulfur heterocycles | |||
| Tanshinone IIA | In vivo | Inhibition of the LDHA expression | [74] |
| Lung cancer model | |||
| Nude mouse xenograft | |||
| Sulfide and disulfide derivatives | |||
| GNE-140 | In vivo | Inhibition of the LDH activity | [178] |
| Immunocompromised mice | |||
| MiaPaCa-2 tumor xenograft | |||
| Compound 107 | In silico | Inhibition of the LDH activity | [179] |
| (Substituted 3-hydroxy-mercaptocyclo-2-enone compound) | |||
| Nonsteroidal anti-inflammatory compounds | |||
| Diclofenac | In vitro | Inhibition of the LDHA expression | [180,181,182] |
| Melanoma cells | |||
| MeIIm | |||
| Metformin + Diclofenac (METABLOC) | In silico | Inhibition of the LDHA activity | [181,183] |
| Ibuprofen + diclofenac | In vitro | Reduction in the LDHA activity | [184] |
| Mouse glioma cells | |||
| GL261 | |||
| Quinoline derivatives | |||
| Compound 105 | In vitro | Inhibition of the LDHA activity | [161] |
| (Quinoline 3-sulfonamide derivative) | Multiple tumor cell lines | ||
| Pyridazine and piperazine | |||
| TMZ | No data | Inhibition of the LDH activity | [164] |
| Compound 100 (pyridazine derivative) | In vitro | Inhibition of the LDH activity | [167] |
| Medulloblastoma cells | |||
| Med1-MB | |||
| Miscellanea | |||
| Calcitriol | In vivo | Reduction in the expression of LDHA | [87] |
| HT29 xenograft mouse model | |||
| Crocetin | Kinetic assay | Inhibition of LDHA | [185,186] |
| Machilin A | Kinetic assay | Inhibition of the LDHA activity | [185,187] |
| Nifurtimox | In vitro | Reduction in the LDH enzyme activity | [188] |
| Neuroblastoma cells | |||
| LA-N-1, IMR-32, SK-N-SH, LS | |||
| N-hydroxyndole | Kinetic assay | Inhibition of the LDHA activity | [189] |
| PSTMB | In vitro | Inhibition of the LDHA activity | [190] |
| Different cancer types | |||
| NCI-H1299, MCF-7, Hep3B, HT29, LLC | |||
| Compound 82 | SAR studies | LDHA inhibitors | [192] |
| (N-acylhydrazone derivative) | |||
| Oxaloacetate | In vitro | Inhibition of the LDHA activity | [191] |
| Lung cancer cells | |||
| H1299 | |||
| Compounds in combination with other therapies | |||
| Sildenafil + cisplatin | In vitro | Inhibition of the LDHA expression | [94] |
| Lymphoma cells | |||
| HuT-78 cells | |||
| TMZ + gemcitabine or abraxane | No data | Inhibition of LDH | [165,166] |
3. Conclusions
During the last years, the research of cancer has allowed us to extend our knowledge about this malignant disease, which damages the life of those who suffer from it. This new knowledge has enabled us to identify some features about this pathology, which provide an opportunity for the scientific community to delve into the mechanism of action of the disease, favoring the research of novel treatments. One of the features described in the last years is the deregulated metabolism and reprogrammed cellular energetics, considered a new hallmark of cancer. Changes in the glucose metabolism and the metabolic pathways implied in the deregulation of glycolysis to lactate production in aerobic conditions, called the Warburg effect or aerobic glycolysis, and the enzymes or transporters that participate in the deregulated cancer metabolism can be used as a target in the research of novel treatment due to the necessity to improve the actual treatments. In this review, the most representative molecules synthetized since 2010 were collected to compile the latest data for their modulation activity towards four enzymes that participate in aerobic glycolysis, namely HK2, PKM2, PDK, and LDH. Small molecules, flavonoids and non-flavonoids polyphenolic compounds, metal complexes, and other structures, a total of more than 100 compounds were described as novel molecules for inhibiting one or more of the target enzymes. The chemical versality of new molecules creates a wide range of possibilities for expanding cancer treatment options. Although most of the compounds have been evaluated in silico, in vitro, or in animal models, this is a promising starting point in the process of approving new drugs. Hence, this review can shed some light on future advances in molecules targeting the Warburg effect for potential new cancer treatments.
Author Contributions
Conceptualization, E.A.-E., S.R.-I. and D.P.; writing—original draft preparation, E.A.-E., L.G.-S., I.Z. and S.R.-I.; writing—review and editing, C.S. and D.P.; supervision, C.S. and D.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADP | Adenosin diphosphate |
| AKT | Protein kinase B |
| AML | Acute myelogenous leukemia |
| AMPK AMP | Activated protein kinase |
| ATP | Adenosine triphosphate |
| BCSC | Breast cancer stem cells |
| C | Carbon |
| CAF | Cancer-associated fibroblast |
| CoA | Coenzyme A |
| COX | Cyclooxygenase |
| EC50 | Efficacy concentration 50 |
| ESCC | Esophageal squamous cell carcinoma |
| FDG | 18F-fluorodesoxyglucose |
| G6P | Glucose-6-phosphate |
| GLUT | Glucose transporter |
| HCC | Hepatocellular carcinoma |
| HIF-1 | Hipoxia-inducible factor 1 |
| HK | Hexokinase |
| HRE | Hypoxia response element |
| IC50 | Inhibitory concentration 50 |
| JAK2 | Janus kinase 2 |
| LDH | Lactate dehydrogenase |
| Lys | Lysine |
| MCT | Monocarboxylate transporter |
| MOA | Mechanism of action |
| MPC | Mitochondrial pyruvate carrier |
| NAD | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOS | Nitric oxide synthase |
| NSAID | Nonsteroidal anti-inflammatory drug |
| NSCLC | Non-small cell lung cancer |
| OXPHOS | Oxidative phosphorylation |
| PDH | Pyruvate dehydrogenase |
| PDK | Pyruvate dehydrogenase kinase |
| PEP | Phosphoenolpyruvate |
| PFK | Phosphofructokinase |
| PGM | Phosphoglucomutase |
| PI3K | Phosphatidylinositol 3-kinase |
| PK | Pyruvate kinase |
| PPP | Pentose phosphate pathway |
| Pt | Platinum |
| ROS | Reactive oxygen species |
| SABP | SA-binding protein |
| SAR | Structure activity relationship |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCA | Tricarboxylic acid |
| TME | Tumor microenvironment |
| VDAC1 | Voltage-dependent anion channel 1 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef]
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell Biosci. 2017, 7, 62. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef]
- Li, C.; Zhang, G.; Zhao, L.; Ma, Z.; Chen, H. Metabolic reprogramming in cancer cells: Glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J. Surg. Oncol. 2016, 14, 15. [Google Scholar] [CrossRef]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T.P. From Warburg effect to Reverse Warburg effect; the new horizons of anti-cancer therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, W.; Garcia-Prieto, C.; Huang, P. The Warburg effect and its cancer therapeutic implications. J. Bioenerg. Biomembr. 2007, 39, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Li, W.; Li, X.; Jin, X.; Liao, Q.; Li, Y.; Zhou, Y. ‘Reverse Warburg effect’ of cancer-associated fibroblasts (Review). Int. J. Oncol. 2022, 60, 67. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Update 2018, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Perkins, N.D. Nuclear factor-κB, p53, and mitochondria: Regulation of cellular metabolism and the Warburg effect. Trends Biochem. Sci. 2012, 37, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Zhou, Y.; Tam, K.Y. The development of small-molecule inhibitors targeting hexokinase 2. Drug. Discov. Today 2022, 27, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The multiple roles of LDH in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin. Cancer Biol. 2022, 87, 184–195. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Pyruvate kinase M2 at a glance. J. Cell Sci. 2015, 128, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Lu, S.; Jia, X.; Li, Q.; Liu, L.; Xie, P.; Wang, G.; Lu, M.; Gao, W.; Zhao, T.; et al. Role of endoplasmic reticulum stress in apoptosis induced by HK2 inhibitor and its potential as a new drug combination strategy. Cell Stress Chaperones 2022, 27, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Aggarwal, A.; Marshall, J.A.; Barletta, J.A.; Kijewski, M.F.; Lorch, J.H.; Nehs, M.A. Glycolytic inhibition with 3-bromopyruvate suppresses tumor growth and improves survival in a murine model of anaplastic thyroid cancer. Surgery 2022, 171, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, J.; Liu, Y.; Luo, X.; Yang, C.; Xiao, W.; Li, Q.; Yang, L.; Zhang, X. 3-Bromopyruvic acid regulates glucose metabolism by targeting the c-Myc/TXNIP axis and induces mitochondria-mediated apoptosis in TNBC cells. Exp. Ther. Med. 2022, 24, 520. [Google Scholar] [CrossRef]
- Roy, S.; Dukic, T.; Bhandary, B.; Tu, K.J.; Molitoris, J.; Ko, Y.H.; Shukla, H.D. 3-Bromopyruvate inhibits pancreatic tumor growth by stalling glycolysis, and dismantling mitochondria in a syngeneic mouse model. Am. J. Cancer Res. 2022, 12, 4977–4987. [Google Scholar]
- Beygi, F.; Mostoufi, A.; Mojaddami, A. Novel Hydrazone Derivatives of 3-Bromopyruvate: Synthesis, Evaluation of the Cytotoxic Effects, Molecular Docking and ADME Studies. Chem. Biodivers. 2022, 19, e202100754. [Google Scholar] [CrossRef]
- Yu, Q.; Dai, W.; Ji, J.; Wu, L.; Feng, J.; Li, J.; Zheng, Y.; Li, Y.; Cheng, Z.; Zhang, J.; et al. Sodium butyrate inhibits aerobic glycolysis of hepatocellular carcinoma cells via the c-myc/hexokinase 2 pathway. J. Cell. Mol. Med. 2022, 26, 3031–3045. [Google Scholar] [CrossRef]
- Karim, S.; Burzangi, A.S.; Ahmad, A.; Siddiqui, N.A.; Ibrahim, I.M.; Sharma, P.; Abualsunun, W.A.; Gabr, G.A. PI3K-AKT Pathway Modulation by Thymoquinone Limits Tumor Growth and Glycolytic Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 2305. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zheng, J.; Li, Y.; Xu, D.P.; Li, S.; Chen, Y.M.; Li, H.B. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients 2016, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Carresi, C.; Musolino, V.; Oppedisano, F.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Macri, R.; et al. Nutraceuticals and Cancer: Potential for Natural Polyphenols. Nutrients 2021, 13, 3834. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, S.; Fichna, J.; Lewandowska, U. Polyphenols as mitochondria-targeted anticancer drugs. Cancer Lett. 2015, 366, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Pan, Y.; Yang, G.; Liu, Y.; Zou, J.; Zhao, H.; Yin, G.; Wu, Y.; Li, X.; Wei, Z.; et al. Kaempferol impairs aerobic glycolysis against melanoma metastasis via inhibiting the mitochondrial binding of HK2 and VDAC1. Eur. J. Pharmacol. 2022, 931, 175226. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Xu, H.; Li, Z.; Liu, D.; Zhang, S.; Fang, F.; Wang, L. Juglone promotes antitumor activity against prostate cancer via suppressing glycolysis and oxidative phosphorylation. Phytother. Res. 2023, 37, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ji, Y.; Zhang, L.; Cai, H.; Ji, Z.; Gu, L.; Yang, S. Wogonin inhibits the growth of HT144 melanoma via regulating hedgehog signaling-mediated inflammation and glycolysis. Int. Immunopharmacol. 2021, 101, 108222. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, L.; Yang, L.; Guo, W.; Guo, Q.; Zhou, Y. Glycolysis inhibition and apoptosis induction in human prostate cancer cells by FV-429-mediated regulation of AR-AKT-HK2 signaling network. Food Chem. Toxicol. 2020, 143, 111517. [Google Scholar] [CrossRef]
- Li, W.; Hao, J.; Zhang, L.; Cheng, Z.; Deng, X.; Shu, G. Astragalin Reduces Hexokinase 2 through Increasing miR-125b to Inhibit the Proliferation of Hepatocellular Carcinoma Cells in Vitro and in Vivo. J. Agric. Food Chem. 2017, 65, 5961–5972. [Google Scholar] [CrossRef]
- Xu, D.; Jin, J.; Yu, H.; Zhao, Z.; Ma, D.; Zhang, C.; Jiang, H. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J. Exp. Clin. Cancer Res. 2017, 36, 44. [Google Scholar] [CrossRef]
- Wu, H.; Pan, L.; Gao, C.; Xu, H.; Li, Y.; Zhang, L.; Ma, L.; Meng, L.; Sun, X.; Qin, H. Quercetin Inhibits the Proliferation of Glycolysis-Addicted HCC Cells by Reducing Hexokinase 2 and Akt-mTOR Pathway. Molecules 2019, 24, 1993. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Dai, W.; Zhang, Q.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer 2017, 117, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wei, L.; Liu, Y.; Ding, Y.; Liu, X.; Zhang, X.; Wang, X.; Yao, Y.; Lu, J.; Wang, Q.; et al. Gen-27, a newly synthesized flavonoid, inhibits glycolysis and induces cell apoptosis via suppression of hexokinase II in human breast cancer cells. Biochem. Pharmacol. 2017, 125, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, W.; Liu, H.; Yu, X. Xanthohumol inhibits colorectal cancer cells via downregulation of Hexokinases II-mediated glycolysis. Int. J. Biol. Sci. 2019, 15, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.R.; Park, W.Y.; Lee, H.J.; Sim, D.Y.; Im, E.; Park, J.E.; Ahn, C.H.; Shim, B.S.; Kim, S.H. Antitumor Effect of Morusin via G1 Arrest and Antiglycolysis by AMPK Activation in Hepatocellular Cancer. Int. J. Mol. Sci. 2021, 22, 10619. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Fan, H.; Chen, Q.; Ma, G.; Zhu, M.; Zhang, X.; Zhang, Y.; Yu, J. Curcumin inhibits aerobic glycolysis and induces mitochondrial-mediated apoptosis through hexokinase II in human colorectal cancer cells in vitro. Anticancer Drugs 2015, 26, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gao, Y.; Li, L.; Xie, S. Curcumin Inhibits Papillary Thyroid Cancer Cell Proliferation by Regulating lncRNA LINC00691. Anal. Cell Pathol. 2022, 2022, 5946670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, Q.; Zheng, S.; Liu, T.; Yang, L.; Han, X.; Lu, X. Shikonin Inhibits Tumor Growth of ESCC by suppressing PKM2 mediated Aerobic Glycolysis and STAT3 Phosphorylation. J. Cancer 2021, 12, 4830–4840. [Google Scholar] [CrossRef]
- Li, W.; Ma, X.; Li, N.; Liu, H.; Dong, Q.; Zhang, J.; Yang, C.; Liu, Y.; Liang, Q.; Zhang, S.; et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp. Cell Res. 2016, 349, 320–327. [Google Scholar] [CrossRef]
- Yi, X.; Qi, M.; Huang, M.; Zhou, S.; Xiong, J. Honokiol Inhibits HIF-1α-Mediated Glycolysis to Halt Breast Cancer Growth. Front. Pharmacol. 2022, 13, 796763. [Google Scholar] [CrossRef]
- Uludağ, D.; Bay, S.; Sucu, B.O.; Şavluğ İpek, Ö.; Mohr, T.; Güzel, M.; Karakaş, N. Potential of Novel Methyl Jasmonate Analogs as Anticancer Agents to Metabolically Target HK-2 Activity in Glioblastoma Cells. Front. Pharmacol. 2022, 13, 828400. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yi, M.; Tan, Y.; Li, X.; Li, G.; Zeng, Z.; Xiong, W.; Xiang, B. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-ΙΙ. J. Exp. Clin. Cancer Res. 2021, 40, 190. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, C.; Qiu, Z.; Deng, D.; Chen, X.; Wang, Q.; Meng, Y.; Zhang, B.; Zheng, G.; Hu, J. Triptolide inhibits intrahepatic cholangiocarcinoma growth by suppressing glycolysis via the AKT/mTOR pathway. Phytomedicine 2023, 109, 154575. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Chen, Z.; Qin, Y.; Wang, X.; Zhang, Z.; Li, Y. Tanshinone IIA inhibits cell growth by suppressing SIX1-induced aerobic glycolysis in non-small cell lung cancer cells. Oncol. Lett. 2022, 23, 184. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shao, J.; Guo, Z.; Jin, C.; Wang, L.; Wang, F.; Jia, Y.; Zhu, Z.; Zhang, Z.; Zhang, F.; et al. Novel mitochondrion-targeting copper(II) complex induces HK2 malfunction and inhibits glycolysis via Drp1-mediating mitophagy in HCC. J. Cell Mol. Med. 2020, 24, 3091–3107. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zheng, M.; Wu, S.; Gao, S.; Yang, M.; Li, Z.; Min, Q.; Sun, W.; Chen, L.; Xiang, G.; et al. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 2017, 36, 58. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Pan, P.; Lv, R.; Ma, C.; Wu, E.; Guo, R.; Zhao, Z.; Song, H.; Zhou, J.; Liu, Y.; et al. High-throughput glycolytic inhibitor discovery targeting glioblastoma by graphite dots-assisted LDI mass spectrometry. Sci. Adv. 2022, 8, eabl4923. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, C.; Chen, B.Y.; Damoiseaux, R.; Clark, P.M. Pacritinib inhibits glucose consumption in squamous cell lung cancer cells by targeting FLT3. Sci. Rep. 2023, 13, 1442. [Google Scholar] [CrossRef]
- Liu, R.; Liu, X. Virtual Screening and Biological Activity Evaluation of New Potent Inhibitors Targeting Hexokinase-II. Molecules 2022, 27, 7555. [Google Scholar] [CrossRef]
- Lin, G.; Wu, Y.; Cai, F.; Li, Z.; Su, S.; Wang, J.; Cao, J.; Ma, L. Matrine Promotes Human Myeloid Leukemia Cells Apoptosis Through Warburg Effect Mediated by Hexokinase 2. Front. Pharmacol. 2019, 10, 1069. [Google Scholar] [CrossRef]
- Glenister, A.; Simone, M.I.; Hambley, T.W. A Warburg effect targeting vector designed to increase the uptake of compounds by cancer cells demonstrates glucose and hypoxia dependent uptake. PLoS ONE 2019, 14, e0217712. [Google Scholar] [CrossRef] [PubMed]
- Salani, B.; Marini, C.; Rio, A.D.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci. Rep. 2013, 3, 2070. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Salani, B.; Massollo, M.; Amaro, A.; Esposito, A.I.; Orengo, A.M.; Capitanio, S.; Emionite, L.; Riondato, M.; Bottoni, G.; et al. Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle 2013, 12, 3490–3499. [Google Scholar] [CrossRef]
- Noser, A.A.; Abdelmonsef, A.H.; El-Naggar, M.; Salem, M.M. New Amino Acid Schiff Bases as Anticancer Agents via Potential Mitochondrial Complex I-Associated Hexokinase Inhibition and Targeting AMP-Protein Kinases/mTOR Signaling Pathway. Molecules 2021, 26, 5332. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Konno, Y.; Maruyama, A.; Tomita, M.; Makinoshima, H. Cinnamon bark extract suppresses metastatic dissemination of cancer cells through inhibition of glycolytic metabolism. J. Nat. Med. 2022, 76, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, H.J.; Sim, D.Y.; Park, J.E.; Ahn, C.H.; Park, S.Y.; Jang, E.; Kim, B.; Kim, S.H. The Antitumor Effect of Cinnamaldehyde Derivative CB-PIC in Hepatocellular Carcinoma Cells via Inhibition of Pyruvate and STAT3 Signaling. Int. J. Mol. Sci. 2022, 23, 6461. [Google Scholar] [CrossRef]
- Huang, C.Y.; Weng, Y.T.; Li, P.C.; Hsieh, N.T.; Li, C.I.; Liu, H.S.; Lee, M.F. Calcitriol Suppresses Warburg Effect and Cell Growth in Human Colorectal Cancer Cells. Life 2021, 11, 963. [Google Scholar] [CrossRef]
- Swargiary, G.; Mani, S. Molecular docking and simulation studies of phytocompounds derived from Centella asiatica and Andrographis paniculata against hexokinase II as mitocan agents. Mitochondrion 2021, 61, 138–146. [Google Scholar] [CrossRef]
- Ramos, H.; Calheiros, J.; Almeida, J.; Barcherini, V.; Santos, S.; Carvalho, A.T.P.; Santos, M.M.M.; Saraiva, L. SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner. Int. J. Mol. Sci. 2020, 21, 596. [Google Scholar] [CrossRef]
- Li, J.; Zou, Y.; Pei, M.; Zhang, Y.; Jiang, Y. Berberine inhibits the Warburg effect through TET3/miR-145/HK2 pathways in ovarian cancer cells. J. Cancer 2021, 12, 207–216. [Google Scholar] [CrossRef]
- Gao, M.; Yang, Y.; Gao, Y.; Liu, T.; Guan, Q.; Zhou, T.; Shi, Y.; Hao, M.; Li, Z.; Zuo, D.; et al. The anti-MDR efficacy of YAN against A549/Taxol cells is associated with its inhibition on glycolysis and is further enhanced by 2-deoxy-d-glucose. Chem. Biol. Interact. 2022, 354, 109843. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wu, C.; Yang, K.; Yang, Y.; Liu, Y.; Gao, S.; Wang, Q.; Li, C.; Chen, L.; Li, H. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol. Res. 2021, 164, 105367. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, K.; Kubicka, A.; Kitel, R.; Dzido, G.; Łabieniec-Watała, M.; Zawadzki, S.; Marczak, A.; Walczak, K.; Matczak, K.; Tomczyk, M.D. Hexokinase 2 Inhibition and Biological Effects of BNBZ and Its Derivatives: The Influence of the Number and Arrangement of Hydroxyl Groups. Int. J. Mol. Sci. 2022, 23, 2616. [Google Scholar] [CrossRef]
- Rawat, S.G.; Tiwari, R.K.; Jaiswara, P.K.; Gupta, V.K.; Sonker, P.; Vishvakarma, N.K.; Kumar, S.; Pathak, C.; Gautam, V.; Kumar, A. Phosphodiesterase 5 inhibitor sildenafil potentiates the antitumor activity of cisplatin by ROS-mediated apoptosis: A role of deregulated glucose metabolism. Apoptosis 2022, 27, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxid. Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, C.A.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-induced metabolic reprogramming in tumor cells enhances the anti-proliferative activity of the PDK inhibitor dichloroacetate. Cancer Lett. 2020, 470, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Goliasova, T.; Denko, N.C. Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int. J. Cancer 2011, 128, 1001–1008. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Shi, H.; Imberti, C.; Clarkson, G.J.; Sadler, P.J. Axial functionalisation of photoactive diazido platinum(iv) anticancer complexes. Inorg. Chem. Front. 2020, 7, 3533–3540. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.S.; Sangha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.; Venner, P.; Michelakis, E.D. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Marier, D.; Marsden, E.; Andrews, D.; Eliaz, I. A novel form of dichloroacetate therapy for patients with advanced cancer: A report of 3 cases. Altern. Ther. Health Med. 2014, 20 (Suppl. S2), 21–28. [Google Scholar] [PubMed]
- Tiersma, J.F.; Evers, B.; Bakker, B.M.; Jalving, M.; de Jong, S. Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities. Int. J. Mol. Sci. 2022, 23, 3745. [Google Scholar] [CrossRef] [PubMed]
- Filimonova, M.; Shitova, A.; Soldatova, O.; Shevchenko, L.; Saburova, A.; Podosinnikova, T.; Surinova, V.; Shegay, P.; Kaprin, A.; Ivanov, S.; et al. Combination of NOS- and PDK-Inhibitory Activity: Possible Way to Enhance Antitumor Effects. Int. J. Mol. Sci. 2022, 23, 730. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guo, Y.; Song, D.; Zhu, Z.; Zhang, Z.; Sun, Y.; Yang, T.; Guo, Z.; Wang, X. Targeting Energy Metabolism by a Platinum(IV) Prodrug as an Alternative Pathway for Cancer Suppression. Inorg. Chem. 2019, 58, 6507–6516. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, C.; De Franco, M.; Carbone, D.; Bassani, D.; Pavan, M.; Cascioferro, S.; Parrino, B.; Cirrincione, G.; Dall’Acqua, S.; Moro, S.; et al. 1,2,4-Amino-triazine derivatives as pyruvate dehydrogenase kinase inhibitors: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2023, 249, 115134. [Google Scholar] [CrossRef] [PubMed]
- Prabha, K.; Satheeshkumar, R.; Nasif, V.; Saranya, J.; Sayin, K.; Natarajan, J.; Chandrasekar, C.; Rajendra Prasad, K.J. Synthesis, In Vitro Cytotoxicity, and DFT Studies of Novel 2-Amino Substituted Benzonaphthyridines as PDK1 Inhibitors. ChemistrySelect 2022, 7, e202200288. [Google Scholar] [CrossRef]
- Sun, W.; Xie, Z.; Liu, Y.; Zhao, D.; Wu, Z.; Zhang, D.; Lv, H.; Tang, S.; Jin, N.; Jiang, H.; et al. JX06 Selectively Inhibits Pyruvate Dehydrogenase Kinase PDK1 by a Covalent Cysteine Modification. Cancer Res. 2015, 75, 4923–4936. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, Z.; Zhao, D.; Zhu, J.; Mao, F.; Tang, S.; Xu, H.; Luo, C.; Geng, M.; Huang, M.; et al. Development of the First Generation of Disulfide-Based Subtype-Selective and Potent Covalent Pyruvate Dehydrogenase Kinase 1 (PDK1) Inhibitors. J. Med. Chem. 2017, 60, 2227–2244. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Schoumacher, M.; Dilly, S.; Elmoualij, B.; Zorzi, D.; Quatresooz, P.; Lambert, V.; Noël, A.; Pirotte, B.; de Tullio, P. Design, Synthesis, and Evaluation of Novel Pyruvate Dehydrogenase Kinase Inhibitors. Med. Chem. 2023, 19, 276–296. [Google Scholar] [PubMed]
- Yue, L.; Ren, Y.; Yue, Q.; Ding, Z.; Wang, K.; Zheng, T.; Chen, G.; Chen, X.; Li, M.; Fan, L. α-Lipoic Acid Targeting PDK1/NRF2 Axis Contributes to the Apoptosis Effect of Lung Cancer Cells. Oxid. Med. Cell Longev. 2021, 2021, 6633419. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhu, C.; Huang, D.; Wen, X.; Zhang, S.L.; Shen, Y. Profiling the interaction of a novel toxic pyruvate dehydrogenase kinase inhibitor with human serum albumin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 256, 119733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yang, Z.; Hu, X.; Chakravarty, H.; Tam, K.Y. Anticancer effects of some novel dichloroacetophenones through the inhibition of pyruvate dehydrogenase kinase 1. Eur. J. Pharm. Sci. 2018, 123, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Su, J.; Xu, H.; Yu, S.; Liu, Y.; Zhang, Y.; Sun, L.; Yue, Y.; Zhou, X. Dicumarol inhibits PDK1 and targets multiple malignant behaviors of ovarian cancer cells. PLoS ONE 2017, 12, e0179672. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Pagire, H.S.; Kang, D.; Song, Y.H.; Lee, I.K.; Lee, K.T.; Park, C.J.; Ahn, J.H.; Kim, J. Structural basis for the inhibition of PDK2 by novel ATP- and lipoyl-binding site targeting compounds. Biochem. Biophys. Res. Commun. 2020, 527, 778–784. [Google Scholar] [CrossRef]
- Lee, D.; Pagire, H.S.; Pagire, S.H.; Bae, E.J.; Dighe, M.; Kim, M.; Lee, K.M.; Jang, Y.K.; Jaladi, A.K.; Jung, K.Y.; et al. Discovery of Novel Pyruvate Dehydrogenase Kinase 4 Inhibitors for Potential Oral Treatment of Metabolic Diseases. J. Med. Chem. 2019, 62, 575–588. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, X.; Chakravarty, H.; Yang, Z.; Tam, K.Y. Identification of Novel Pyruvate Dehydrogenase Kinase 1 (PDK1) Inhibitors by Kinase Activity-Based High-Throughput Screening for Anticancer Therapeutics. ACS Comb. Sci. 2018, 20, 660–671. [Google Scholar] [CrossRef]
- Jin, L.; Kim, E.Y.; Chung, T.W.; Han, C.W.; Park, S.Y.; Han, J.H.; Bae, S.J.; Lee, J.R.; Kim, Y.W.; Jang, S.B.; et al. Hemistepsin A suppresses colorectal cancer growth through inhibiting pyruvate dehydrogenase kinase activity. Sci. Rep. 2020, 10, 21940. [Google Scholar] [CrossRef]
- Shen, M.; Wang, D.; Sennari, Y.; Zeng, Z.; Baba, R.; Morimoto, H.; Kitamura, N.; Nakanishi, T.; Tsukada, J.; Ueno, M.; et al. Pentacyclic triterpenoid ursolic acid induces apoptosis with mitochondrial dysfunction in adult T-cell leukemia MT-4 cells to promote surrounding cell growth. Med. Oncol. 2022, 39, 118. [Google Scholar] [CrossRef] [PubMed]
- Elinson, M.N.; Ryzhkova, Y.E.; Ryzhkov, F.V.; Fakhrutdinov, A.N. Kojic acid aldol adduct with isatin as inhibitors of pyruvate dehydrogenase kinase. J. Heterocycl. Chem. 2022, 59, 760–770. [Google Scholar] [CrossRef]
- Sharma, A.; Chun, J.; Ji, M.S.; Lee, S.; Kang, C.; Kim, J.S. Binary Prodrug of Dichloroacetic Acid and Doxorubicin with Enhanced Anticancer Activity. ACS Appl. Bio Mater. 2021, 4, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Al-Azawi, A.; Sulaiman, S.; Arafat, K.; Yasin, J.; Nemmar, A.; Attoub, S. Impact of Sodium Dichloroacetate Alone and in Combination Therapies on Lung Tumor Growth and Metastasis. Int. J. Mol. Sci. 2021, 22, 12553. [Google Scholar] [CrossRef] [PubMed]
- Parczyk, J.; Ruhnau, J.; Pelz, C.; Schilling, M.; Wu, H.; Piaskowski, N.N.; Eickholt, B.; Kühn, H.; Danker, K.; Klein, A. Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer 2021, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Xie, G.; He, J.; Li, J.; Pan, F.; Liang, H. Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J. Biomed. Biotechnol. 2011, 2011, 740564. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Hur, H.; Ham, I.H.; Yun, J.; Lee, J.Y.; Shim, W.; Kim, Y.B.; Lee, G.; Han, S.U.; Cho, Y.K. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp. Cell Res. 2014, 321, 219–230. [Google Scholar] [CrossRef]
- Almouhanna, F.; Blagojevic, B.; Can, S.; Ghanem, A.; Wölfl, S. Pharmacological activation of pyruvate kinase M2 reprograms glycolysis leading to TXNIP depletion and AMPK activation in breast cancer cells. Cancer Metab. 2021, 9, 5. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Battisti, U.M.; Gao, C.; Akladios, F.; Kim, W.; Yang, H.; Bayram, C.; Bolat, I.; Kiliclioglu, M.; Yuksel, N.; Tozlu, O.O.; et al. Ellagic Acid and Its Metabolites as Potent and Selective Allosteric Inhibitors of Liver Pyruvate Kinase. Nutrients 2023, 15, 577. [Google Scholar] [CrossRef]
- Sarfraz, I.; Rasul, A.; Jabeen, F.; Sultana, T.; Adem, Ş. Identification of Natural Compounds as Inhibitors of Pyruvate Kinase M2 for Cancer Treatment. Molecules 2022, 27, 7113. [Google Scholar] [CrossRef] [PubMed]
- Mashhadi Akbar Boojar, M.; Mashhadi Akbar Boojar, M.; Golmohammad, S. Overview of Silibinin anti-tumor effects. J. Herb. Med. 2020, 23, 100375. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Chattopadhyay, S.; Siddiqui, F.A.; Ur Rehman, A.; Siddiqui, S.; Prakasam, G.; Khan, A.; Sultana, S.; Bamezai, R.N. Silibinin induces metabolic crisis in triple-negative breast cancer cells by modulating EGFR-MYC-TXNIP axis: Potential therapeutic implications. FEBS J. 2021, 288, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, Y.; Ping, X.; Yu, M.; Lou, D.; Shi, W. Synergistic apoptotic effects of silibinin in enhancing paclitaxel toxicity in human gastric cancer cell lines. Mol. Med. Rep. 2018, 18, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Im, E.; Kim, N.D. Mechanism of Resveratrol-Induced Programmed Cell Death and New Drug Discovery against Cancer: A Review. Int. J. Mol. Sci. 2022, 23, 13689. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Bamezai, R.N. Resveratrol inhibits cancer cell metabolism by down regulating pyruvate kinase M2 via inhibition of mammalian target of rapamycin. PLoS ONE 2012, 7, e36764. [Google Scholar] [CrossRef]
- Siddiqui, F.A.; Prakasam, G.; Chattopadhyay, S.; Rehman, A.U.; Padder, R.A.; Ansari, M.A.; Irshad, R.; Mangalhara, K.; Bamezai, R.N.K.; Husain, M.; et al. Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF1α inhibition. Sci. Rep. 2018, 8, 8323. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kim, Y.H.; Jin, Y.; Chi, S.W.; Moon, J.H.; Han, D.C.; Kwon, B.M. 2′-hydroxycinnamaldehyde inhibits cancer cell proliferation and tumor growth by targeting the pyruvate kinase M2. Cancer Lett. 2018, 434, 42–55. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Shi, J.; Yang, P.; Jia, B.; Wu, H.; Zhang, X.; Li, Z. Apigenin Restrains Colon Cancer Cell Proliferation via Targeted Blocking of Pyruvate Kinase M2-Dependent Glycolysis. J. Agric. Food Chem. 2017, 65, 8136–8144. [Google Scholar] [CrossRef] [PubMed]
- Aslan, E.; Adem, S. In vitro effects of some flavones on human pyruvate kinase isoenzyme M2. J. Biochem. Mol. Toxicol. 2015, 29, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.J.; Brimacombe, K.R.; Veith, H.; Bougie, J.M.; Daniel, T.; Leister, W.; Cantley, L.C.; Israelsen, W.J.; Vander Heiden, M.G.; Shen, M.; et al. 2-Oxo-N-aryl-1,2,3,4-tetrahydroquinoline-6-sulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg. Med. Chem. Lett. 2011, 21, 6322–6327. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Rzepka, Z.; Chrobak, E.; Boryczka, S.; Latocha, M.; Wrześniok, D.; Beberok, A. Design, Synthesis and Biological Evaluation of Quinoline-8-Sulfonamides as Inhibitors of the Tumor Cell-Specific M2 Isoform of Pyruvate Kinase: Preliminary Study. Molecules 2023, 28, 2509. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Wang, L.; Powell, A.F.; Strickler, S.R.; Wang, D.; Dempsey, D.A.; Schroeder, F.C.; Klessig, D.F. A genome-wide screen for human salicylic acid (SA)-binding proteins reveals targets through which SA may influence development of various diseases. Sci. Rep. 2019, 9, 13084. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.S.; Gopula, B.; Chou, C.C.; Wu, H.Y.; Chang, G.D.; Wu, W.J.; Chang, C.S.; Chu, P.C.; Chen, C.S. Development of Novel Irreversible Pyruvate Kinase M2 Inhibitors. J. Med. Chem. 2019, 62, 8497–8510. [Google Scholar] [CrossRef]
- Li, Z.; Yang, L.; Zhang, S.; Song, J.; Sun, H.; Shan, C.; Wang, D.; Liu, S. Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect. Cell Transpl. 2021, 30, 9636897211027524. [Google Scholar] [CrossRef]
- Sun, X.; Peng, Y.; Zhao, J.; Xie, Z.; Lei, X.; Tang, G. Discovery and development of tumor glycolysis rate-limiting enzyme inhibitors. Bioorg. Chem. 2021, 112, 104891. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Vitamin K(3) and K(5) are inhibitors of tumor pyruvate kinase M2. Cancer Lett. 2012, 316, 204–210. [Google Scholar] [CrossRef]
- Shankar Babu, M.; Mahanta, S.; Lakhter, A.J.; Hato, T.; Paul, S.; Naidu, S.R. Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE 2018, 13, e0191419. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Liu, Z.L.; Mai, X.Y.; Qi, X.; Li, D.H.; Gu, Q.Q.; Li, J. Identification of Gliotoxin isolated from marine fungus as a new pyruvate kinase M2 inhibitor. Biochem. Biophys. Res. Commun. 2020, 528, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Rihan, M.; Vineela Nalla, L.; Dharavath, A.; Patel, S.; Shard, A.; Khairnar, A. Boronic acid derivative activates pyruvate kinase M2 indispensable for redox metabolism in oral cancer cells. Bioorg. Med. Chem. Lett. 2022, 59, 128539. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, Y.; Qi, L.; Li, L.; Song, D.; Gan, J.; Li, Y.; Ling, X.; Song, C. Dihydroartemisinin mediating PKM2-caspase-8/3-GSDME axis for pyroptosis in esophageal squamous cell carcinoma. Chem. Biol. Interact. 2021, 350, 109704. [Google Scholar] [CrossRef]
- Li, J.; Qu, P.; Zhou, X.Z.; Ji, Y.X.; Yuan, S.; Liu, S.P.; Zhang, Q.G. Pimozide inhibits the growth of breast cancer cells by alleviating the Warburg effect through the P53 signaling pathway. Biomed. Pharmacother. 2022, 150, 113063. [Google Scholar] [CrossRef]
- Park, J.H.; Kundu, A.; Lee, S.H.; Jiang, C.; Lee, S.H.; Kim, Y.S.; Kyung, S.Y.; Park, S.H.; Kim, H.S. Specific Pyruvate Kinase M2 Inhibitor, Compound 3K, Induces Autophagic Cell Death through Disruption of the Glycolysis Pathway in Ovarian Cancer Cells. Int. J. Biol. Sci. 2021, 17, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, I.; Muroi, M.; Kawatani, M.; Ohishi, T.; Ohba, S.I.; Kawada, M.; Osada, H. Identification of a Small Compound Targeting PKM2-Regulated Signaling Using 2D Gel Electrophoresis-Based Proteome-wide CETSA. Cell Chem. Biol. 2020, 27, 186–196.e184. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Lu, Y.; Yang, P.; Li, Z. Berberine inhibited the proliferation of cancer cells by suppressing the activity of tumor pyruvate kinase M2. Nat. Prod. Commun. 2017, 12, 1415–1418. [Google Scholar] [CrossRef]
- Li, R.; Ning, X.; He, J.; Lin, Z.; Su, Y.; Li, R.; Yin, Y. Synthesis of novel sulfonamide derivatives containing pyridin-3-ylmethyl 4-(benzoyl)piperazine-1-carbodithioate moiety as potent PKM2 activators. Bioorg. Chem. 2021, 108, 104653. [Google Scholar] [CrossRef]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colón, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Han, F.; Yang, S.; Wu, J.; Zhan, W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: Involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015, 358, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, R.S.; Shawky, N.M. Trimetazidine, a metabolic modulator, attenuates silica-induced pulmonary fibrosis and decreases lactate levels and LDH activity in rats. J. Biochem. Mol. Toxicol. 2022, 36, e23071. [Google Scholar] [CrossRef] [PubMed]
- Atlı Şekeroğlu, Z.; Şekeroğlu, V.; Işık, S.; Aydın, B. Trimetazidine alone or in combination with gemcitabine and/or abraxane decreased cell viability, migration and ATP levels and induced apoptosis of human pancreatic cells. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101632. [Google Scholar] [CrossRef] [PubMed]
- Atlı Şekeroğlu, Z.; Kendigelen, E.; Şekeroğlu, V.; Küçük, N. Effects of trimetazidine on anticancer activity and toxicity of abraxane in MCF-7 breast cancer cells. Rend. Lincei 2022, 33, 879–888. [Google Scholar] [CrossRef]
- Di Magno, L.; Coluccia, A.; Bufano, M.; Ripa, S.; La Regina, G.; Nalli, M.; Di Pastena, F.; Canettieri, G.; Silvestri, R.; Frati, L. Discovery of novel human lactate dehydrogenase inhibitors: Structure-based virtual screening studies and biological assessment. Eur. J. Med. Chem. 2022, 240, 114605. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.Y.; Zhang, L.; Yee, J.K.; Go, V.W.; Lee, W.N. Metabolic Consequences of LDHA inhibition by Epigallocatechin Gallate and Oxamate in MIA PaCa-2 Pancreatic Cancer Cells. Metabolomics 2015, 11, 71–80. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.; Han, S.; Wang, N.; Mo, F.; Loo, T.Y.; Shen, J.; Huang, H.; Chen, J. Bioactivity-guided identification and cell signaling technology to delineate the lactate dehydrogenase A inhibition effects of Spatholobus suberectus on breast cancer. PLoS ONE 2013, 8, e56631. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Guan, R.; Chen, J.; Yang, D.; Zhao, Z.; Wang, D. Chemical characterization of procyanidins from Spatholobus suberectus and their antioxidative and anticancer activities. J. Funct. Foods 2015, 12, 468–477. [Google Scholar] [CrossRef]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, X.; Yu, Y.; Li, J.; Hu, Q.; Xue, C.; Chen, J.; Shen, S.; Luo, Y.; Ren, F.; et al. LDHA is a direct target of miR-30d-5p and contributes to aggressive progression of gallbladder carcinoma. Mol. Carcinog. 2018, 57, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Xian, Z.Y.; Liu, J.M.; Chen, Q.K.; Chen, H.Z.; Ye, C.J.; Xue, J.; Yang, H.Q.; Li, J.L.; Liu, X.F.; Kuang, S.J. Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumour Biol. 2015, 36, 8093–8100. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Deck, J.A.; Hunsaker, L.A.; Deck, L.M.; Royer, R.E.; Goldberg, E.; Vander Jagt, D.L. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem. Pharmacol. 2001, 62, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Bushunow, P.; Reidenberg, M.M.; Wasenko, J.; Winfield, J.; Lorenzo, B.; Lemke, S.; Himpler, B.; Corona, R.; Coyle, T. Gossypol treatment of recurrent adult malignant gliomas. J. Neurooncol. 1999, 43, 79–86. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Fauber, B.P.; Boggs, J.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Giannetti, A.M.; Hunsaker, T.; et al. Identification of substituted 3-hydroxy-2-mercaptocyclohex-2-enones as potent inhibitors of human lactate dehydrogenase. Bioorg. Med. Chem. Lett. 2014, 24, 3764–3771. [Google Scholar] [CrossRef]
- Choi, S.; Kim, S.; Park, J.; Lee, S.E.; Kim, C.; Kang, D. Diclofenac: A Nonsteroidal Anti-Inflammatory Drug Inducing Cancer Cell Death by Inhibiting Microtubule Polymerization and Autophagy Flux. Antioxidants 2022, 11, 1009. [Google Scholar] [CrossRef]
- da Veiga Moreira, J.; Hamraz, M.; Abolhassani, M.; Schwartz, L.; Jolicœur, M.; Peres, S. Metabolic therapies inhibit tumor growth in vivo and in silico. Sci. Rep. 2019, 9, 3153. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New aspects of an old drug--diclofenac targets MYC and glucose metabolism in tumor cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef] [PubMed]
- Gerthofer, V.; Kreutz, M.; Renner, K.; Jachnik, B.; Dettmer, K.; Oefner, P.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma. Int. J. Mol. Sci. 2018, 19, 2586. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Seliger, C.; Jachnik, B.; Welz, T.; Leukel, P.; Vollmann-Zwerenz, A.; Bogdahn, U.; Kreutz, M.; Grauer, O.M.; Hau, P. Ibuprofen and Diclofenac Restrict Migration and Proliferation of Human Glioma Cells by Distinct Molecular Mechanisms. PLoS ONE 2015, 10, e0140613. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, M.; Kim, H.J.; Jang, S.B.; Bae, S.J.; Lee, I.K.; Ryu, D.; Ha, K.T. Targeting Lactate Dehydrogenase A with Catechin Resensitizes SNU620/5FU Gastric Cancer Cells to 5-Fluorouracil. Int. J. Mol. Sci. 2021, 22, 5406. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Fortunato, S.; Meini, S.; Rizzolio, F.; Caligiuri, I.; Tuccinardi, T.; Lee, H.Y.; Hergenrother, P.J.; Minutolo, F. Characterization of the Saffron Derivative Crocetin as an Inhibitor of Human Lactate Dehydrogenase 5 in the Antiglycolytic Approach against Cancer. J. Agric. Food Chem. 2017, 65, 5639–5649. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Kim, E.Y.; Han, C.W.; Park, S.Y.; Jeong, M.S.; Yoon, D.; Choi, H.J.; Jin, L.; Park, M.J.; Kwon, Y.J.; et al. Machilin A Inhibits Tumor Growth and Macrophage M2 Polarization Through the Reduction of Lactic Acid. Cancers 2019, 11, 963. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas Stanchi, K.M.; Bruchelt, G.; Handgretinger, R.; Holzer, U. Nifurtimox reduces N-Myc expression and aerobic glycolysis in neuroblastoma. Cancer Biol. Ther. 2015, 16, 1353–1363. [Google Scholar] [CrossRef]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Kim, E.Y.; Chung, T.W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef]
- Wiese, E.K.; Hitosugi, S.; Loa, S.T.; Sreedhar, A.; Andres-Beck, L.G.; Kurmi, K.; Pang, Y.P.; Karnitz, L.M.; Gonsalves, W.I.; Hitosugi, T. Enzymatic activation of pyruvate kinase increases cytosolic oxaloacetate to inhibit the Warburg effect. Nat. Metab. 2021, 3, 954–968. [Google Scholar] [CrossRef]
- Rupiani, S.; Buonfiglio, R.; Manerba, M.; Di Ianni, L.; Vettraino, M.; Giacomini, E.; Masetti, M.; Falchi, F.; Di Stefano, G.; Roberti, M.; et al. Identification of N-acylhydrazone derivatives as novel lactate dehydrogenase A inhibitors. Eur. J. Med. Chem. 2015, 101, 63–70. [Google Scholar] [CrossRef]
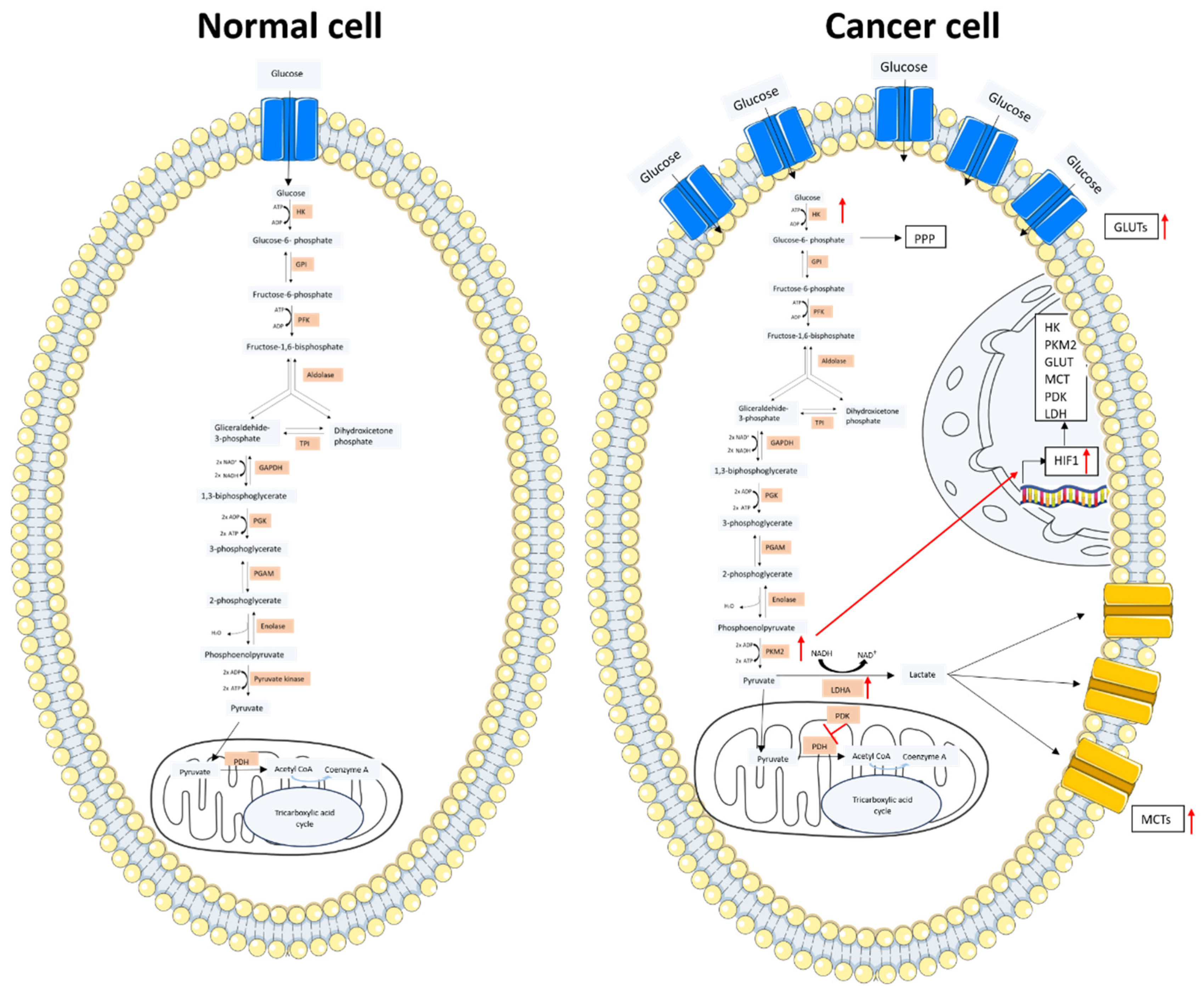
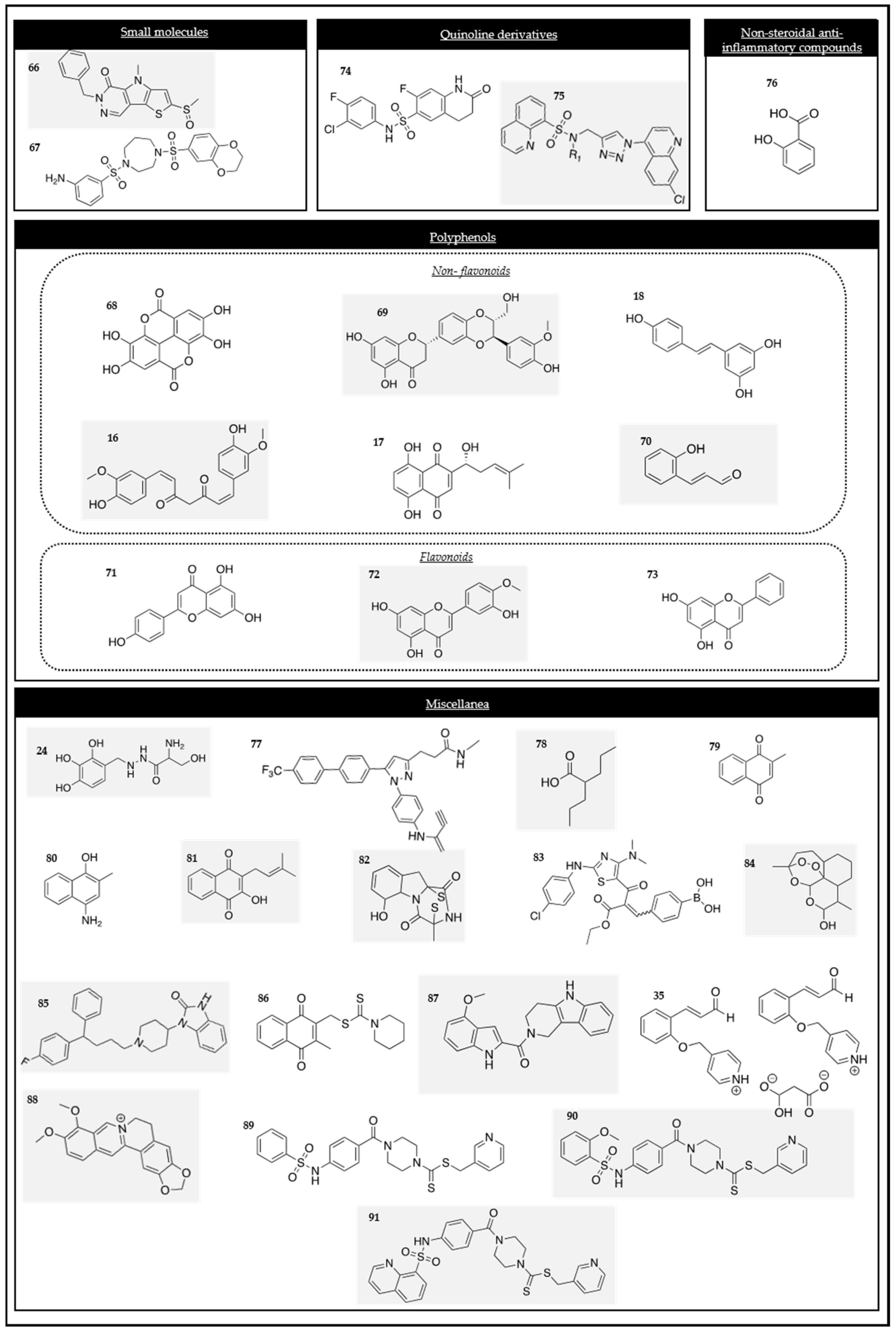
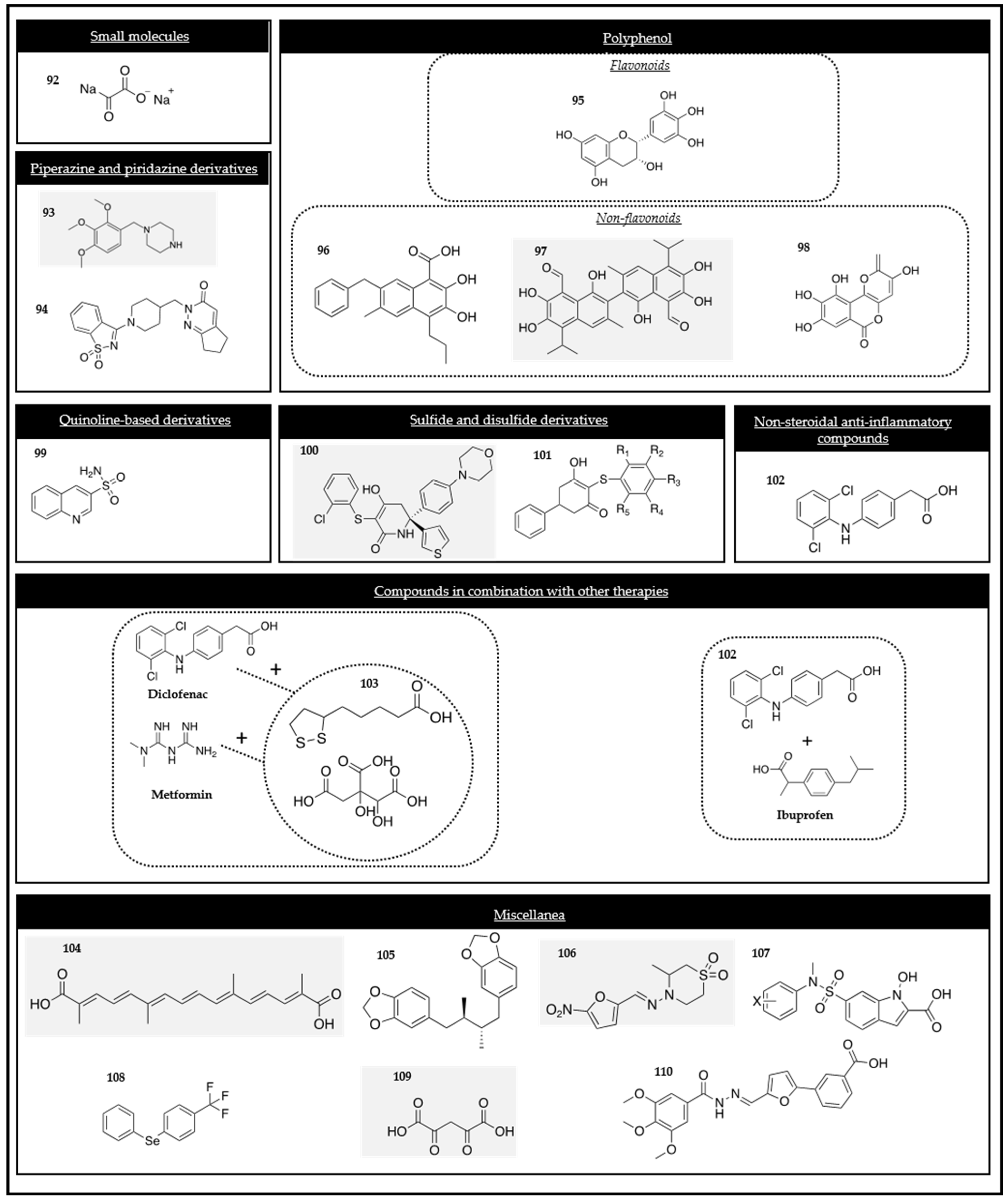
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).