
Bradykinin-Mediated Angioedema Induced by Commonly Used Cardiovascular Drugs

Abstract
1. Non-Allergic Angioedema
2. Epidemiology
3. Diagnosis
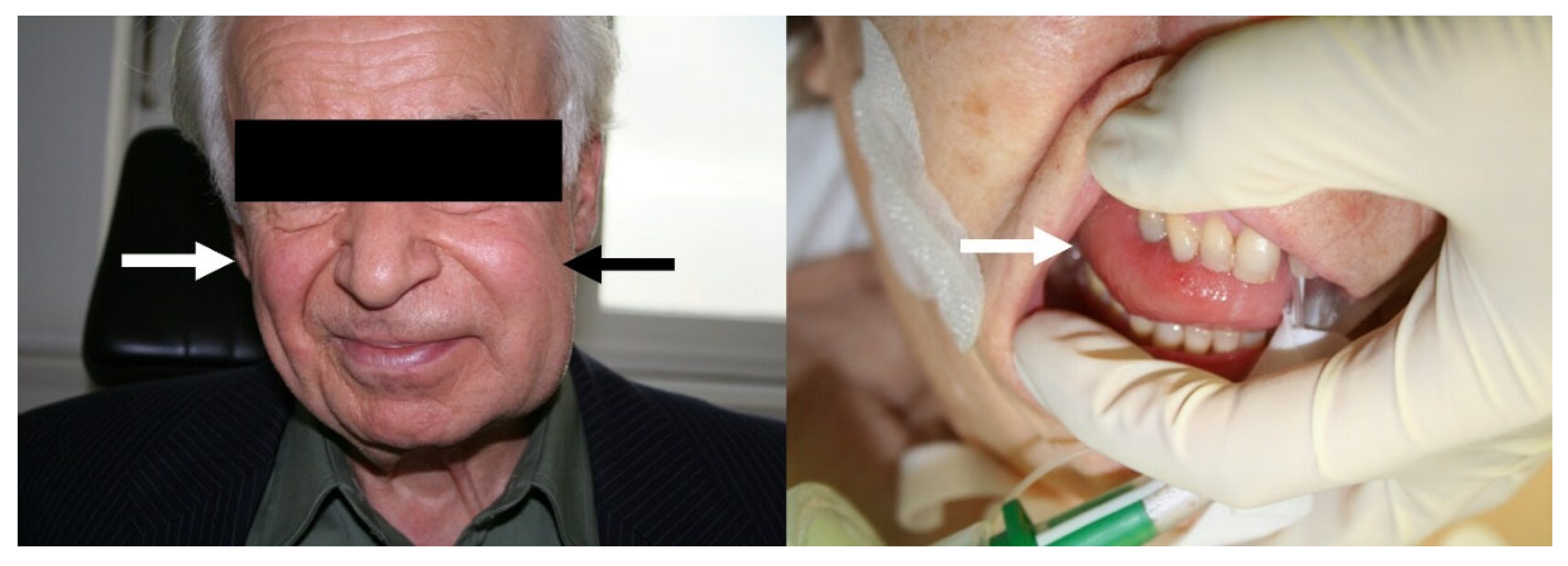
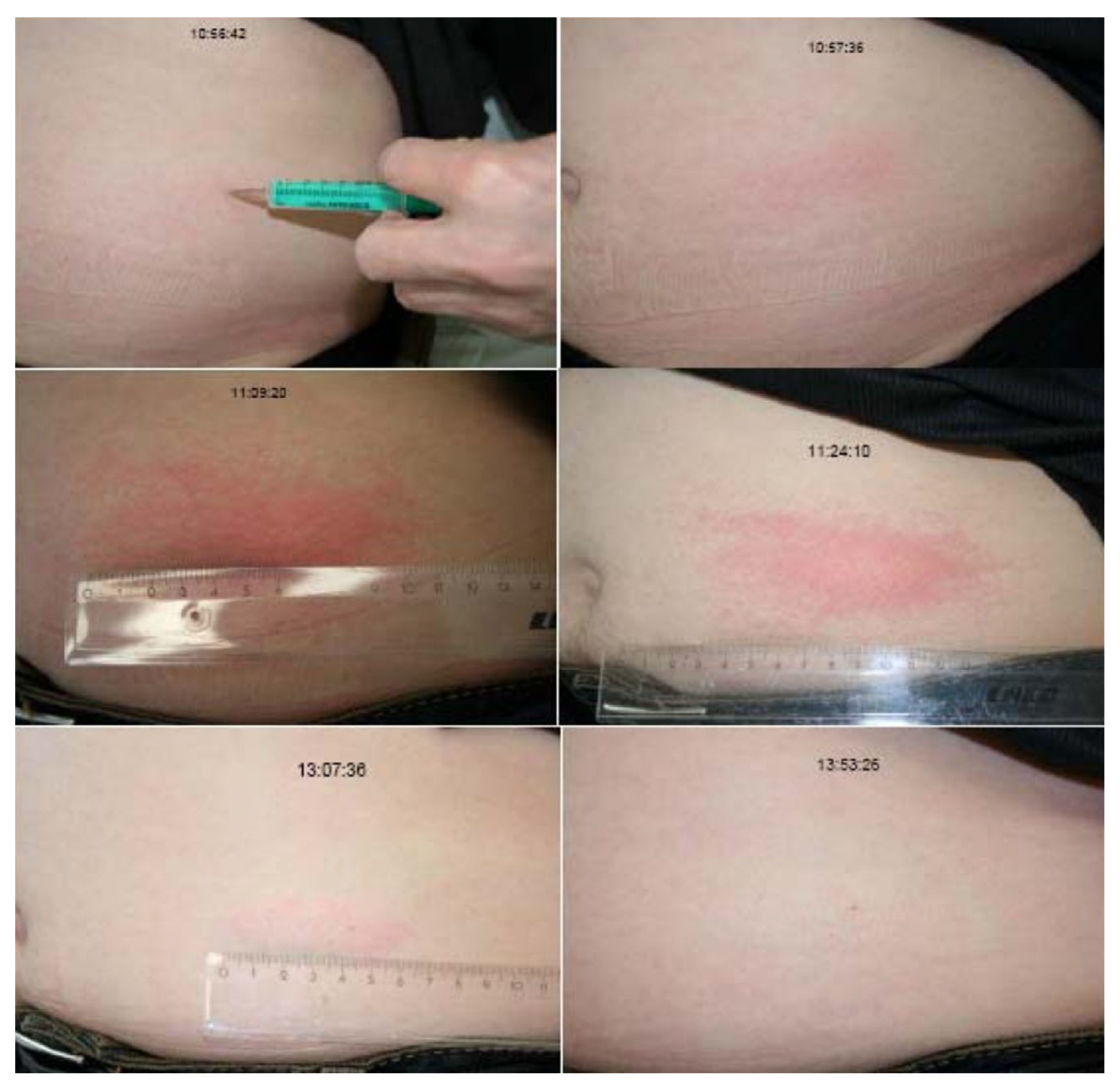
3.1. Drug-Induced-Angioedema Mediated by Bradykinin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medication | Influence on Bradykinin | Indications | Further Remarks |
|---|---|---|---|
| ACE-inhibitors | ACE is involved in the degradation of bradykinin to bradykinin1–7 and bradykinin1–5 | Hypertension, heart failure 1, diabetic nephropathy, event prevention in cardiovascular disease | Event prevention at high cardiovascular risk only, unless required to control hypertension [53] |
| Aliskiren | Direct renin inhibitor; the exact pathophysiology of angioedema is not yet understood | Essential hypertension | No reduction of cardiovascular or renal endpoints |
| Angiotensin II receptor blockers (sartans) | reduced bradykinin metabolism by ACE and Neprilysin, molecular mechanism remains unclear [37] | Hypertension, diabetic nephropathy, heart failure 1, event prevention in cardiovascular disease | Event prevention at high cardiovascular risk only, unless required to control hypertension [53] |
| Direct DPP-4 inhibitors 2 | DPP-4 further degrades bradykinin after cleavage by aminopeptidase P to the ineffective bradykinin2–9 | Type 2 diabetes mellitus | unlikely leading to bradykinin-mediated angioedema alone, only in combination, e.g., with an ACE inhibitor |
| m-TOR inhibitors | The mechanism of angioedema development remains unclear. | Prophylaxis of kidney, heart and liver transplant rejection | Significantly higher incidence of angioedema in combination with an ACE inhibitor |
| Neprilysin inhibitors | Neprilysin is involved in the degradation of bradykinin. | Heart failure 1 | No combination with ACE-inibitors or sartans |
| Tissue plasminogen activator | catalyzes the conversion of plasminogen to plasmin; plasmin has influence on bradykinin production | Fibrinolysis in acute myocardial infarction, acute ischemic stroke and pulmonary embolism | Relatively high number of potential life-threatening orolingual manifestations reported |
3.2. Hereditary Angioedema
3.3. Acute Urticaria
3.4. Allergic Angioedema
4. Symptoms and Course
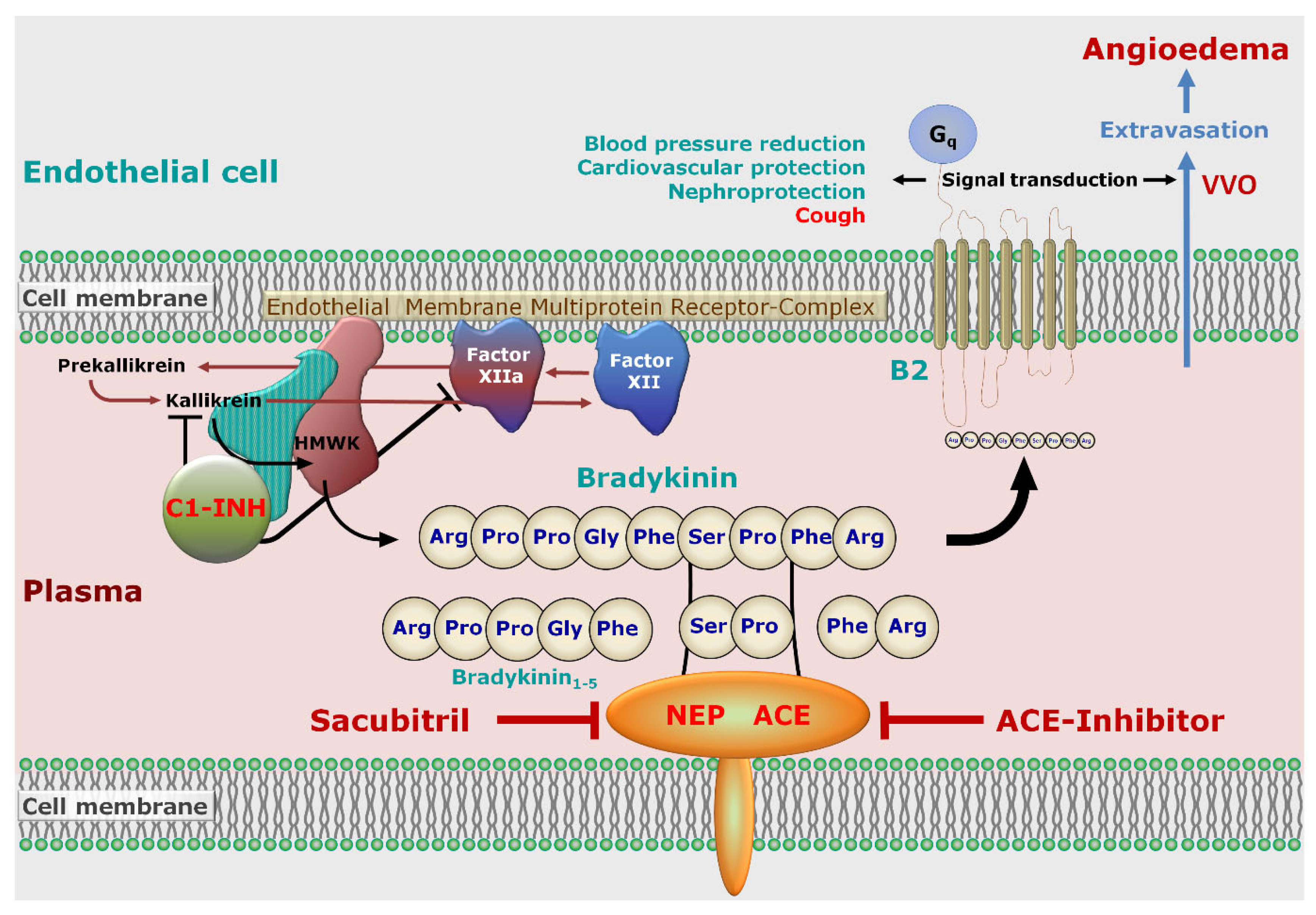
5. Pathophysiology
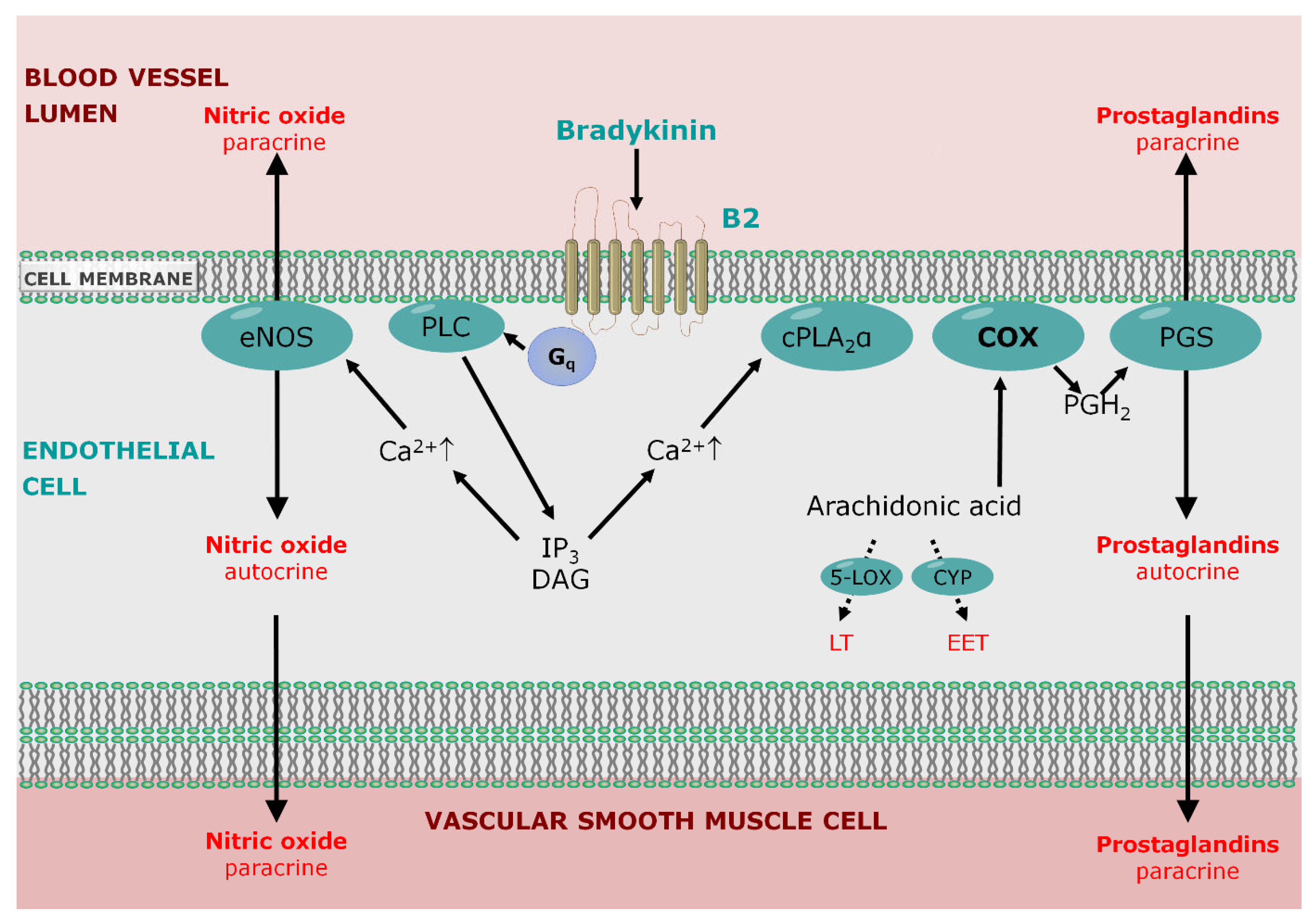
5.1. Endothelial Permeability
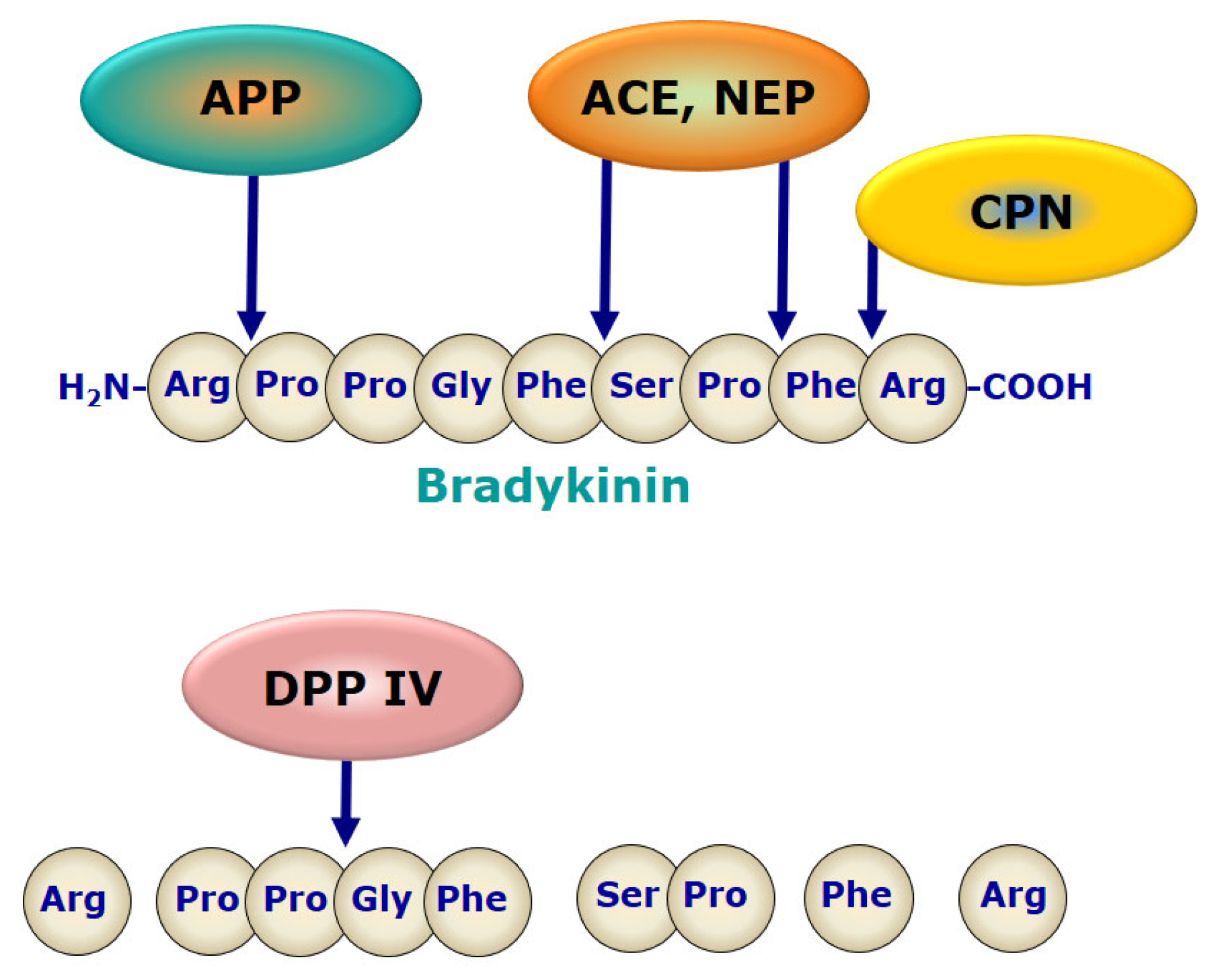
5.2. Role of Bradykinin Signaling
6. Therapy
6.1. Discontinuation of ACE-Inhibitors
6.2. Antifibrinolytics
6.3. Ecallantide
6.4. C1-INH
6.5. Icatibant
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cicardi, M.; Aberer, W.; Banerji, A.; Bas, M.; Bernstein, J.A.; Bork, K.; Caballero, T.; Farkas, H.; Grumach, A.; Kaplan, A.P.; et al. Classification, diagnosis, and approach to treatment for angioedema: Consensus report from the hereditary angioedema international working group. Allergy 2014, 69, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Bas, M.; Adams, V.; Suvorava, T.; Niehues, T.; Hoffmann, T.K.; Kojda, G. Non-allergic angioedema. Role of bradykinin. Allergy 2007, 62, 842–856. [Google Scholar] [CrossRef]
- Mujer, M.T.P.; Rai, M.P.; Nemakayala, D.R.; Yam, J.L. Angioedema of the small bowel caused by lisinopril. Drug Ther. Bull. 2019, 57, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Bas, M.; Greve, J.; Strassen, U.; Khosravani, F.; Hoffmann, T.K.; Kojda, G. Angioedema induced by cardiovascular drugs: New players join old friends. Allergy 2015, 70, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, T.; Maurer, M. Classification and pathophysiology of angioedema. Hautarzt 2019, 70, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Hoffmann, T.K.; Bock, B.; Nordmann-Kleiner, M.; Trainotti, S.; Greve, J. Angioedema. Dtsch. Arztebl. Int. 2017, 114, 489–496. [Google Scholar] [CrossRef][Green Version]
- White, A.A.; Stevenson, D.D. Aspirin-exacerbated respiratory disease. N. Engl. J. Med. 2018, 379, 1060–1070. [Google Scholar] [CrossRef]
- Bas, M.; Greve, J.; Bier, H.; Knopf, A.; Stark, T.; Schuler, P.; Hoffmann, T.K.; Kojda, G. Notfallsituation akutes angioödem [Emergency management of acute angioedema]. Dtsch. Med. Wochenschr. 2010, 135, 1027–1031. [Google Scholar] [CrossRef]
- Goring, H.D. In memory of the 160th birthday and the 80th anniversary of the death of heinrich irenaus quincke, as well as of his description of angioedema 120 years ago. Hautarzt 2002, 53, 822–825. [Google Scholar]
- Hofman, Z.; de Maat, S.; Hack, C.E.; Maas, C. Bradykinin: Inflammatory product of the coagulation system. Clin. Rev. Allergy Immunol. 2016, 51, 152–161. [Google Scholar] [CrossRef]
- Hill, M.D.; Barber, P.A.; Takahashi, J.; Demchuk, A.M.; Feasby, T.E.; Buchan, A.M. Anaphylactoid reactions and angioedema during alteplase treatment of acute ischemic stroke. CMAJ 2000, 162, 1281–1284. [Google Scholar]
- Bozkurt, S.; Arslan, E.D.; Köse, A.; Ayrık, C.; Yılmaz, A.; Dündar, G.A. Lingual angioedema after alteplase treatment in a patient with acute ischemic stroke. World J. Emerg. Med. 2015, 6, 74–76. [Google Scholar] [CrossRef][Green Version]
- Dewald, G.; Bork, K. Missense mutations in the coagulation factor xii (Hageman factor) gene in hereditary angioedema with normal c1 inhibitor. Biochem. Biophys. Res. Commun. 2006, 343, 1286–1289. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Steinmüller-Magin, L.; Braenne, I.; Staubach-Renz, P.; Witzke, G.; Hardt, J. Hereditary angioedema with a mutation in the plasminogen gene. Allergy 2018, 73, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Bafunno, V.; Firinu, D.; D’Apolito, M.; Cordisco, G.; Loffredo, S.; Leccese, A.; Bova, M.; Barca, M.P.; Santacroce, R.; Cicardi, M.; et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J. Allergy Clin. Immunol. 2018, 141, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Bork, K.; Wulff, K.; Rossmann, H.; Steinmüller-Magin, L.; Braenne, I.; Witzke, G.; Hardt, J. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the n-terminal cleavage site of bradykinin. Allergy 2019, 74, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Ariano, A.; D’Apolito, M.; Bova, M.; Bellanti, F.; Loffredo, S.; D’Andrea, G.; Intrieri, M.; Petraroli, A.; Maffione, A.B.; Spadaro, G.; et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy 2020, 75, 2989–2992. [Google Scholar] [CrossRef] [PubMed]
- Bork, K.; Wulff, K.; Möhl, B.S.; Steinmüller-Magin, L.; Witzke, G.; Hardt, J.; Meinke, P. Novel hereditary angioedema linked with a heparan sulfate 3-o-sulfotransferase 6 gene mutation. J. Allergy Clin. Immunol. 2021, 148, 1041–1048. [Google Scholar] [CrossRef]
- Zuraw, B.L. Hereditary angioedema with normal c1 inhibitor: Four types and counting. J. Allergy Clin. Immunol. 2018, 141, 884–885. [Google Scholar] [CrossRef]
- Longhurst, H.; Cicardi, M. Hereditary angio-oedema. Lancet 2012, 379, 474–481. [Google Scholar] [CrossRef]
- Bas, M.; Hoffmann, T.K.; Kojda, G. Evaluation and management of angioedema of the head and neck. Curr. Opin. Otolaryngol. Head Neck Surg. 2006, 14, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Agostoni, A.; Cicardi, M. Drug-induced angioedema without urticaria. Drug Saf. 2001, 24, 599–606. [Google Scholar] [CrossRef]
- Kostis, J.B.; Kim, H.J.; Rusnak, J.; Casale, T.; Kaplan, A.; Corren, J.; Levy, E. Incidence and characteristics of angioedema associated with enalapril. Arch. Intern. Med. 2005, 165, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Ray, W.A.; Snowden, M.; Griffin, M.R. Black americans have an increased rate of angiotensin converting enzyme inhibitor-associated angioedema. Clin. Pharmacol. Ther. 1996, 60, 8–13. [Google Scholar] [CrossRef]
- Gainer, J.V.; Nadeau, J.H.; Ryder, D.; Brown, N.J. Increased sensitivity to bradykinin among african americans. J. Allergy Clin. Immunol. 1996, 98, 283–287. [Google Scholar] [CrossRef] [PubMed]
- McDowell, S.E.; Coleman, J.J.; Ferner, R.E. Systematic review and meta-analysis of ethnic differences in risks of adverse reactions to drugs used in cardiovascular medicine. BMJ 2006, 332, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Sleight, P.; Pogue, J.; Bosch, J.; Davies, R.; Dagenais, G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar] [PubMed]
- Pfeffer, M.A.; McMurray, J.J.; Velazquez, E.J.; Rouleau, J.L.; Kober, L.; Maggioni, A.P.; Solomon, S.D.; Swedberg, K.; Van de Werf, F.; White, H.; et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N. Engl. J. Med. 2003, 349, 1893–1906. [Google Scholar] [CrossRef]
- Yusuf, S.; Teo, K.K.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P.; Anderson, C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N. Engl. J. Med. 2008, 358, 1547–1559. [Google Scholar]
- Makani, H.; Messerli, F.H.; Romero, J.; Wever-Pinzon, O.; Korniyenko, A.; Berrios, R.S.; Bangalore, S. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am. J. Cardiol. 2012, 110, 383–391. [Google Scholar] [CrossRef]
- Brown, N.J.; Byiers, S.; Carr, D.; Maldonado, M.; Warner, B.A. Dipeptidyl peptidase-iv inhibitor use associated with increased risk of ace inhibitor-associated angioedema. Hypertension 2009, 54, 516–523. [Google Scholar] [CrossRef]
- Byrd, J.B.; Shreevatsa, A.; Putlur, P.; Foretia, D.; McAlexander, L.; Sinha, T.; Does, M.D.; Brown, N.J. Dipeptidyl peptidase iv deficiency increases susceptibility to angiotensin-converting enzyme inhibitor-induced peritracheal edema. J. Allergy Clin. Immunol. 2007, 120, 403–408. [Google Scholar] [CrossRef]
- Yusuf, S.; Teo, K.; Anderson, C.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet 2008, 372, 1174–1183. [Google Scholar]
- Rasmussen, E.R.; Pottegård, A.; Bygum, A.; von Buchwald, C.; Homøe, P.; Hallas, J. Angiotensin ii receptor blockers are safe in patients with prior angioedema related to angiotensin-converting enzyme inhibitors—A nationwide registry-based cohort study. J. Intern. Med. 2019, 285, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Hellebrand, M.C.; Kojda, G.; Hoffmann, T.K.; Bas, M. Angioedema due to ace inhibitors and AT1 receptor antagonists. Hautarzt 2005, 57, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.K.; Visconti, J.A.; Tschampel, M.M. Angiotensin ii receptor blockers in patients with ace inhibitor-induced angioedema. Ann. Pharmacother. 2000, 34, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Krum, H.; Esler, M.D. Losartan increases bradykinin levels in hypertensive humans. Circulation 2005, 111, 315–320. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Kostis, J.B.; Packer, M.; Black, H.R.; Schmieder, R.; Henry, D.; Levy, E. Omapatrilat and enalapril in patients with hypertension: The omapatrilat cardiovascular treatment vs. Enalapril (octave) trial. Am. J. Hypertens. 2004, 17, 103–111. [Google Scholar] [CrossRef]
- Kostis, W.J.; Shetty, M.; Chowdhury, Y.S.; Kostis, J.B. Ace inhibitor-induced angioedema: A review. Curr. Hypertens. Rep. 2018, 20, 55. [Google Scholar] [CrossRef]
- Hurford, R.; Rezvani, S.; Kreimei, M.; Herbert, A.; Vail, A.; Parry-Jones, A.R.; Douglass, C.; Molloy, J.; Alachkar, H.; Tyrrell, P.J.; et al. Incidence, predictors and clinical characteristics of orolingual angio-oedema complicating thrombolysis with tissue plasminogen activator for ischaemic stroke. J. Neurol. Neurosurg. Psychiatry 2015, 86, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Lapostolle, A.; Weisenburger-Lile, D.; Yger, M.; Alamowitch, S.; Fain, O. Bradykinin-mediated angioedema following tenecteplase administration in an acute ischemic stroke. Stroke 2022, 53, e446–e447. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Kanaa, M.; Alkawaleet, Y.; Ayub, M.T. Angioedema: A life-threatening complication of tissue plasminogen activator. Cureus 2018, 10, e2392. [Google Scholar] [CrossRef]
- White, W.B.; Bresalier, R.; Kaplan, A.P.; Palmer, B.F.; Riddell, R.H.; Lesogor, A.; Chang, W.; Keefe, D.L. Safety and tolerability of the direct renin inhibitor aliskiren: A pooled analysis of clinical experience in more than 12,000 patients with hypertension. J. Clin. Hypertens. 2010, 12, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.B.; Touzin, K.; Sile, S.; Gainer, J.V.; Yu, C.; Nadeau, J.; Adam, A.; Brown, N.J. Dipeptidyl peptidase iv in angiotensin-converting enzyme inhibitor associated angioedema. Hypertension 2008, 51, 141–147. [Google Scholar] [CrossRef]
- Duerr, M.; Glander, P.; Diekmann, F.; Dragun, D.; Neumayer, H.H.; Budde, K. Increased incidence of angioedema with ace inhibitors in combination with mtor inhibitors in kidney transplant recipients. Clin. J. Am. Soc. Nephrol. 2010, 5, 703–708. [Google Scholar] [CrossRef]
- Lenschow, M.; Bas, M.; Johnson, F.; Wirth, M.; Strassen, U. A score for the differential diagnosis of bradykinin- and histamine-induced head and neck swellings. Eur. Arch. Otorhinolaryngol. 2018, 275, 1767–1773. [Google Scholar] [CrossRef]
- Bas, M.; Greve, J.; Stelter, K.; Havel, M.; Strassen, U.; Rotter, N.; Veit, J.; Schossow, B.; Hapfelmeier, A.; Kehl, V.; et al. A randomized trial of icatibant in ace-inhibitor-induced angioedema. N. Engl. J. Med. 2015, 372, 418–425. [Google Scholar] [CrossRef]
- Balla, Z.; Zsilinszky, Z.; Pólai, Z.; Andrási, N.; Kőhalmi, K.V.; Csuka, D.; Varga, L.; Farkas, H. The importance of complement testing in acquired angioedema related to angiotensin-converting enzyme inhibitors. J. Allergy Clin. Immunol. Pract. 2021, 9, 947–955. [Google Scholar] [CrossRef]
- Hahn, J.; Bock, B.; Muth, C.M.; Pfaue, A.; Friedrich, D.; Hoffmann, T.K.; Greve, J. The ulm emergency algorithm for the acute treatment of drug-induced, bradykinin-mediated angioedema. Med. Klin. Intensivmed. Notfmed. 2019, 114, 708–716. [Google Scholar] [CrossRef]
- Gangnus, T.; Burckhardt, B.B. Targeted lc-ms/ms platform for the comprehensive determination of peptides in the kallikrein-kinin system. Anal. Bioanal. Chem. 2021, 413, 2971–2984. [Google Scholar] [CrossRef]
- Marceau, F.; Rivard, G.E.; Hébert, J.; Gauthier, J.; Bachelard, H.; Gangnus, T.; Burckhardt, B.B. Picomolar sensitivity analysis of multiple bradykinin-related peptides in the blood plasma of patients with hereditary angioedema in remission: A pilot study. Front. Allergy 2022, 3, 837463. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 esc guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international wao/eaaci guideline for the management of hereditary angioedema-the 2021 revision and update. Allergy 2022, 77, 1961–1990. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Witzke, G.; Staubach, P.; Hardt, J.; Meinke, P. Gene mutations linked to hereditary angioedema in solitary angioedema patients with normal c1 inhibitor. J. Allergy Clin. Immunol. Pract. 2023, 11, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Zuberbier, T.; Abdul Latiff, A.H.; Abuzakouk, M.; Aquilina, S.; Asero, R.; Baker, D.; Ballmer-Weber, B.; Bangert, C.; Ben-Shoshan, M.; Bernstein, J.A.; et al. The international EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis, and management of urticaria. Allergy 2022, 77, 734–766. [Google Scholar] [CrossRef] [PubMed]
- Muraro, A.; Worm, M.; Alviani, C.; Cardona, V.; DunnGalvin, A.; Garvey, L.H.; Riggioni, C.; de Silva, D.; Angier, E.; Arasi, S.; et al. Eaaci guidelines: Anaphylaxis (2021 update). Allergy 2022, 77, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Palmquist, S.; Mathews, B. Isolated intestinal type angioedema due to ace-inhibitor therapy. Clin. Case Rep. 2017, 5, 707–710. [Google Scholar] [CrossRef]
- Ichikawa, K.; Matsuoka, D.; Murase, T.; Nakazawa, Y. Angiotensin-converting enzyme inhibitor-induced angioedema following posterior reversible encephalopathy syndrome in a child: A case report. Cureus 2022, 14, e31568. [Google Scholar] [CrossRef]
- Pfaue, A.; Schuler, P.J.; Mayer, B.; Hoffmann, T.K.; Greve, J.; Hahn, J. Clinical features of angioedema induced by renin-angiotensin-aldosterone system inhibition: A retrospective analysis of 84 patients. J. Community Hosp. Intern. Med. Perspect. 2019, 9, 453–459. [Google Scholar] [CrossRef]
- Dean, D.E.; Schultz, D.L.; Powers, R.H. Asphyxia due to angiotensin converting enzyme (ACE) inhibitor mediated angioedema of the tongue during the treatment of hypertensive heart disease. J. Forensic Sci. 2001, 46, 1239–1243. [Google Scholar] [CrossRef]
- Atalay, E.; Ozdemir, M.T.; Cigsar, G.; Omurca, F.; Aslan, N.; Yildiz, M.; Gey, Z.B. Angiotensin converting enzyme inhibitor-related angioedema: A case of an unexpected death. Iran. J. Allergy Asthma Immunol. 2015, 14, 642–645. [Google Scholar] [PubMed]
- Betschel, S.; Badiou, J.; Binkley, K.; Borici-Mazi, R.; Hebert, J.; Kanani, A.; Keith, P.; Lacuesta, G.; Waserman, S.; Yang, B.; et al. The international/canadian hereditary angioedema guideline. Allergy Asthma Clin. Immunol. 2019, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Magerl, M.; Ansotegui, I.; Aygoren-Pursun, E.; Betschel, S.; Bork, K.; Bowen, T.; Balle Boysen, H.; Farkas, H.; Grumach, A.S.; et al. The international wao/eaaci guideline for the management of hereditary angioedema-the 2017 revision and update. Allergy 2018, 73, 1575–1596. [Google Scholar] [CrossRef]
- Raheja, H.; Kumar, V.; Kamholz, S.; Hollander, G.; Shani, J. Life threatening angioedema due to valsartan/sacubitril with previously well-tolerated ace inhibitor. Am. J. Ther. 2017, 10, e508–e509. [Google Scholar] [CrossRef] [PubMed]
- Bas, M.; Hoffmann, T.K.; Bier, H.; Kojda, G. Increased c-reactive protein in ace-inhibitor-induced angioedema. Br. J. Clin. Pharmacol. 2005, 59, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.; Gonzalez, J.; Monteleone, C. Angiotensin-converting enzyme inhibitor-induced angioedema: A review of the literature. J. Clin. Hypertens 2017, 19, 1377–1382. [Google Scholar] [CrossRef]
- Banerji, A.; Clark, S.; Blanda, M.; LoVecchio, F.; Snyder, B.; Camargo, C.A., Jr. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann. Allergy Asthma Immunol. 2008, 100, 327–332. [Google Scholar] [CrossRef]
- Sondhi, D.; Lippmann, M.; Murali, G. Airway compromise due to angiotensin-converting enzyme inhibitor-induced angioedema: Clinical experience at a large community teaching hospital. Chest 2004, 126, 400–404. [Google Scholar] [CrossRef]
- Kim, S.J.; Brooks, J.C.; Sheikh, J.; Kaplan, M.S.; Goldberg, B.J. Angioedema deaths in the united states, 1979–2010. Ann. Allergy Asthma Immunol. 2014, 113, 630–634. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Quay, S.C.; Orenstein, N.S.; Dvorak, A.M.; Hahn, P.; Bitzer, A.M.; Carvalho, A.C. Tumor shedding and coagulation. Science 1981, 212, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Gewaltig, M.T.; Kojda, G. Vasoprotection by nitric oxide: Mechanisms and therapeutic potential. Cardiovasc. Res. 2002, 55, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Curry, F.R.; Adamson, R.H. Vascular permeability modulation at the cell, microvessel, or whole organ level: Towards closing gaps in our knowledge. Cardiovasc. Res. 2010, 87, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef]
- Breslin, J.W. Mechanical forces and lymphatic transport. Microvasc. Res. 2014, 96, 46–54. [Google Scholar] [CrossRef]
- Takada, M.; Hattori, S. Presence of fenestrated capillaries in the skin. Anat. Rec. 1972, 173, 213–219. [Google Scholar] [CrossRef]
- Weibel, E.R.; Palade, G.E. New cytoplasmatic components in arterial endothelia. J. Cell Biol. 1964, 23, 101–112. [Google Scholar] [CrossRef]
- Majno, G.; Palade, G.E. Studies on inflammation. I. The effect of histamine and serotonin on vascular permeability: An electron microscopic study. J. Biophys. Biochem. Cytol. 1961, 11, 571–605. [Google Scholar] [CrossRef]
- Kohn, S.; Nagy, J.A.; Dvorak, H.F.; Dvorak, A.M. Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumor blood vessels. Lab. Investig. 1992, 67, 596–607. [Google Scholar]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the vascular barrier: Endothelial signaling processes controlling extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Majno, G.; Palade, G.E.; Schoefl, G.I. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: A topographic study. J. Biophys. Biochem. Cytol. 1961, 11, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Borgono, C.A.; Diamandis, E.P. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer 2004, 4, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.P.; Joseph, K. Pathogenic mechanisms of bradykinin mediated diseases: Dysregulation of an innate inflammatory pathway. Adv. Immunol. 2014, 121, 41–89. [Google Scholar]
- Fuller, R.W.; Dixon, C.M.; Cuss, F.M.; Barnes, P.J. Bradykinin-induced bronchoconstriction in humans. Mode of action. Am. Rev. Respir. Dis. 1987, 135, 176–180. [Google Scholar]
- Fox, A.J.; Lalloo, U.G.; Belvisi, M.G.; Bernareggi, M.; Chung, K.F.; Barnes, P.J. Bradykinin-evoked sensitization of airway sensory nerves: A mechanism for ace-inhibitor cough. Nat. Med. 1996, 2, 814–817. [Google Scholar] [CrossRef]
- Hulsmann, A.R.; Raatgeep, H.R.; Saxena, P.R.; Kerrebijn, K.F.; de Jongste, J.C. Bradykinin-induced contraction of human peripheral airways mediated by both bradykinin beta 2 and thromboxane prostanoid receptors. Am. J. Respir. Crit. Care Med. 1994, 150, 1012–1018. [Google Scholar] [CrossRef]
- Jaspard, E.; Wei, L.; Alhenc-Gelas, F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin i-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J. Biol. Chem. 1993, 268, 9496–9503. [Google Scholar] [CrossRef]
- Qadri, F.; Bader, M. Kinin b1 receptors as a therapeutic target for inflammation. Expert Opin. Ther. Targets 2018, 22, 31–44. [Google Scholar] [CrossRef]
- Kato, T.; Nagatsu, T.; Fukasawa, K.; Harada, M.; Nagatsu, I.; Sakakibara, S. Successive cleavage of n-terminal arg1-pro2 and lys3-pro4 from substance p but no release of arg1-pro2 from bradykinin, by X-pro dipeptidyl-aminopeptidase. Biochim. Biophys. Acta 1978, 525, 417–422. [Google Scholar] [CrossRef]
- Blaukat, A. Structure and signalling pathways of kinin receptors. Andrologia 2003, 35, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Leeb-Lundberg, L.M.; Marceau, F.; Muller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, D.; Fu, Y.; Chen, A.; Yang, X.; Zhang, H. Cryo-EM structures of human bradykinin receptor-Gq proteins complexes. Nat. Commun. 2022, 13, 714. [Google Scholar] [CrossRef]
- Ghouse, J.; Ahlberg, G.; Andreasen, L.; Banasik, K.; Brunak, S.; Schwinn, M.; Larsen, I.H.; Petersen, O.; Sørensen, E.; Ullum, H.; et al. Association of variants near the bradykinin receptor B2 gene with angioedema in patients taking ace inhibitors. J. Am. Coll. Cardiol. 2021, 78, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Nicoletti, P. Novel pharmacogenomic locus implicated in angiotensin-converting enzyme inhibitor-induced angioedema. J. Am. Coll. Cardiol. 2021, 78, 710–712. [Google Scholar] [CrossRef]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [CrossRef]
- Busse, R.; Fleming, I. Regulation of endothelium-derived vasoactive autacoid production by hemodynamic forces. Trends Pharmacol. Sci. 2003, 24, 24–29. [Google Scholar] [CrossRef]
- Gholamreza-Fahimi, E.; Bisha, M.; Hahn, J.; Strassen, U.; Krybus, M.; Khosravani, F.; Hoffmann, T.K.; Hohlfeld, T.; Greve, J.; Bas, M.; et al. Cyclooxygenase activity in bradykinin-induced dermal extravasation. A study in mice and humans. Biomed. Pharmacother. 2020, 123, 109797. [Google Scholar] [CrossRef]
- Krybus, M.; Sieradzki, M.; Fahimi, E.; Metry, S.; Nüsing, R.; Geisslinger, G.; Steiner, I.; Daldrup, T.; Lehr, M.; Kojda, G. Contribution of cyclooxygenase-1-dependent prostacyclin synthesis to bradykinin-induced dermal extravasation. Biomed. Pharmacother. 2022, 148, 112786. [Google Scholar] [CrossRef]
- Suvorava, T.; Metry, S.; Pick, S.; Kojda, G. Alterations in endothelial nitric oxide synthase activity and their relevance to blood pressure. Biochem. Pharmacol. 2022, 205, 115256. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef]
- Han, E.D.; MacFarlane, R.C.; Mulligan, A.N.; Scafidi, J.; Davis, A.E., III. Increased vascular permeability in c1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J. Clin. Investig. 2002, 109, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, P.; Emanueli, C.; Gaspa, L.; Salis, B.; Milia, A.F.; Chao, L.; Chao, J. Role of the bradykinin b2 receptor in the maturation of blood pressure phenotype: Lesson from transgenic and knockout mice. Immunopharmacology 1999, 44, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Oschatz, C.; Maas, C.; Lecher, B.; Jansen, T.; Bjorkqvist, J.; Tradler, T.; Sedlmeier, R.; Burfeind, P.; Cichon, S.; Hammerschmidt, S.; et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 2011, 34, 258–268. [Google Scholar] [CrossRef]
- Bisha, M.; Dao, V.T.; Gholamreza-Fahimi, E.; Vogt, M.; van Zandvoort, M.; Weber, S.; Bas, M.; Khosravani, F.; Kojda, G.; Suvorava, T. The role of bradykinin receptor type 2 in spontaneous extravasation in mice skin: Implications for non-allergic angio-oedema. Br. J. Pharmacol. 2018, 175, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Veronez, C.L.; Maghsodi, S.; Todiras, M.; Popova, E.; Rodrigues, A.F.; Qadri, F.; Pesquero, J.B.; Bader, M. Endothelial B2-receptor overexpression as an alternative animal model for hereditary angioedema. Allergy 2019, 74, 1998–2002. [Google Scholar] [CrossRef]
- Hornig, B.; Kohler, C.; Drexler, H. Role of bradykinin in mediating vascular effects of angiotensin-converting enzyme inhibitors in humans. Circulation 1997, 95, 1115–1118. [Google Scholar] [CrossRef]
- Gainer, J.V.; Morrow, J.D.; Loveland, A.; King, D.J.; Brown, N.J. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N. Engl. J. Med. 1998, 339, 1285–1292. [Google Scholar] [CrossRef]
- Cicardi, M.; Banerji, A.; Bracho, F.; Malbran, A.; Rosenkranz, B.; Riedl, M.; Bork, K.; Lumry, W.; Aberer, W.; Bier, H.; et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N. Engl. J. Med. 2010, 363, 532–541. [Google Scholar] [CrossRef]
- Zuraw, B.L. Clinical practice. Hereditary angioedema. N. Engl. J. Med. 2008, 359, 1027–1036. [Google Scholar] [CrossRef]
- Lesage, A.; Marceau, F.; Gibson, C.; Loenders, B.; Katzer, W.; Ambrosi, H.D.; Saupe, J.; Faussner, A.; Pardali, E.; Knolle, J. In vitro pharmacological profile of PHA-022121, a small molecule bradykinin B2 receptor antagonist in clinical development. Int. Immunopharmacol. 2022, 105, 108523. [Google Scholar] [CrossRef] [PubMed]
- Riha, H.M.; Summers, B.B.; Rivera, J.V.; Van Berkel, M.A. Novel therapies for angiotensin-converting enzyme inhibitor-induced angioedema: A systematic review of current evidence. J. Emerg. Med. 2017, 53, 662–679. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.; Levy, R.J.; McNeil, D.L.; Li, H.H.; Sheffer, A.L.; Campion, M.; Horn, P.T.; Pullman, W.E. Ecallantide for the treatment of acute attacks in hereditary angioedema. N. Engl. J. Med. 2010, 363, 523–531. [Google Scholar] [CrossRef]
- Banerji, A.; Busse, P.; Shennak, M.; Lumry, W.; Davis-Lorton, M.; Wedner, H.J.; Jacobs, J.; Baker, J.; Bernstein, J.A.; Lockey, R.; et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N. Engl. J. Med. 2017, 376, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Aygoren-Pursun, E.; Bygum, A.; Grivcheva-Panovska, V.; Magerl, M.; Graff, J.; Steiner, U.C.; Fain, O.; Huissoon, A.; Kinaciyan, T.; Farkas, H.; et al. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N. Engl. J. Med. 2018, 379, 352–362. [Google Scholar] [CrossRef]
- Zuraw, B.; Lumry, W.R.; Johnston, D.T.; Aygören-Pürsün, E.; Banerji, A.; Bernstein, J.A.; Christiansen, S.C.; Jacobs, J.S.; Sitz, K.V.; Gower, R.G.; et al. Oral once-daily berotralstat for the prevention of hereditary angioedema attacks: A randomized, double-blind, placebo-controlled phase 3 trial. J. Allergy Clin. Immunol. 2021, 148, 164–172.e9. [Google Scholar] [CrossRef]
- Brown, N.J.; Snowden, M.; Griffin, M.R. Recurrent angiotensin-converting enzyme inhibitor—Associated angioedema. JAMA 1997, 278, 232–233. [Google Scholar] [CrossRef]
- Mahmoudpour, S.H.; Asselbergs, F.W.; Terreehorst, I.; Souverein, P.C.; de Boer, A.; der Zee, A.H.M.-V. Continuation of angiotensin converting enzyme inhibitor therapy, in spite of occurrence of angioedema. Int. J. Cardiol. 2015, 201, 644–645. [Google Scholar] [CrossRef]
- Cicardi, M.; Zingale, L.C.; Bergamaschini, L.; Agostoni, A. Angioedema associated with angiotensin-converting enzyme inhibitor use: Outcome after switching to a different treatment. Arch. Intern. Med. 2004, 164, 910–913. [Google Scholar] [CrossRef]
- Beltrami, L.; Zanichelli, A.; Zingale, L.; Vacchini, R.; Carugo, S.; Cicardi, M. Long-term follow-up of 111 patients with angiotensin-converting enzyme inhibitor-related angioedema. J. Hypertens. 2011, 29, 2273–2277. [Google Scholar] [CrossRef]
- Kainat, A.; Phang, C.R.; Ain, N.U.; Agarwal, B. The first occurrence of angioedema after discontinuation of angiotensin-converting enzyme inhibitor. Cureus 2021, 13, e19553. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; O’Donnell, P.S. The therapeutic potential of a kallikrein inhibitor for treating hereditary angioedema. Expert. Opin. Investig. Drugs 2006, 15, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.M.; Graffeo, C.; Crosley, P.; Klausner, H.A.; Clark, C.L.; Frank, A.; Miner, J.; Iarrobino, R.; Chyung, Y. Ecallantide for the acute treatment of angiotensin-converting enzyme inhibitor-induced angioedema: A multicenter, randomized, controlled trial. Ann. Emerg. Med. 2015, 65, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.A.; Moellman, J.J.; Collins, S.P.; Hart, K.W.; Lindsell, C.J. Effectiveness of ecallantide in treating angiotensin-converting enzyme inhibitor-induced angioedema in the emergency department. Ann. Allergy Asthma Immunol. 2015, 114, 245–249. [Google Scholar] [CrossRef]
- Strassen, U.; Bas, M.; Wirth, M.; Wirth, M.; Gröger, M.; Stelter, K.; Volkenstein, S.; Kehl, V.; Kojda, G.; Hoffmann, T.K.; et al. Efficacy of human c1 esterase inhibitor concentrate for treatment of ace-inhibitor induced angioedema. Am. J. Emerg. Med. 2023, 64, 121–128. [Google Scholar] [CrossRef]
- Bas, M.; Greve, J.; Stelter, K.; Bier, H.; Stark, T.; Hoffmann, T.K.; Kojda, G. Therapeutic efficacy of icatibant in angioedema induced by angiotensin-converting enzyme inhibitors: A case series. Ann. Emerg. Med. 2010, 56, 278–282. [Google Scholar] [CrossRef]
- Le, T.A.; Smith, W.; Hissaria, P. Real-world off-label use of icatibant for acute management of non-hereditary angioedema. Intern. Med. J. 2021, 51, 419–423. [Google Scholar] [CrossRef]
- Straka, B.T.; Ramirez, C.E.; Byrd, J.B.; Stone, E.; Woodard-Grice, A.; Nian, H.; Yu, C.; Banerji, A.; Brown, N.J. Effect of bradykinin receptor antagonism on ace inhibitor-associated angioedema. J. Allergy Clin. Immunol. 2017, 140, 242–248.e2. [Google Scholar] [CrossRef]
- Sinert, R.; Levy, P.; Bernstein, J.A.; Body, R.; Sivilotti, M.L.A.; Moellman, J.; Schranz, J.; Baptista, J.; Kimura, A.; Nothaft, W. Randomized trial of icatibant for angiotensin-converting enzyme inhibitor-induced upper airway angioedema. J. Allergy Clin. Immunol. Pract. 2017, 5, 1402–1409.e3. [Google Scholar] [CrossRef]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef]
- Hubers, S.A.; Kohm, K.; Wei, S.; Yu, C.; Nian, H.; Grabert, R.; Sexton, D.J.; Brown, N.J. Endogenous bradykinin and b1-b5 during angiotensin-converting enzyme inhibitor-associated angioedema. J. Allergy Clin. Immunol. 2018, 142, 1636–1639.e5. [Google Scholar] [CrossRef] [PubMed]
- Nussberger, J.; Cugno, M.; Cicardi, M. Bradykinin-mediated angioedema. N. Engl. J. Med. 2002, 347, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Agostoni, A.; Cicardi, M.; Cugno, M.; Zingale, L.C.; Gioffre, D.; Nussberger, J. Angioedema due to angiotensin-converting enzyme inhibitors. Immunopharmacology 1999, 44, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Morand, E.; Leech, M. Annexin a1: Potential for glucocorticoid sparing in RA. Nat. Rev. Rheumatol. 2013, 9, 595–603. [Google Scholar] [CrossRef]
- Fishel, R.S.; Eisenberg, S.; Shai, S.Y.; Redden, R.A.; Bernstein, K.E.; Berk, B.C. Glucocorticoids induce angiotensin-converting enzyme expression in vascular smooth muscle. Hypertension 1995, 25, 343–349. [Google Scholar] [CrossRef]
- Shiber, J.R. Angioedema of the arytenoids. N. Engl. J. Med. 2005, 353, e15. [Google Scholar] [CrossRef]





| Subtypes of non-allergic Angioedema |
|---|
| Hereditary Angioedema (HAE) |
| Increased generation of bradykinin caused by mutations of the C1-Esterase-Inhibitor (C1-INH) gene SERPING1 inducing a loss of C1-INH (HAE type 1, 85% of the cases), or dysfunction of C1-INH (HAE type 2) |
| Increased generation of bradykinin despite normal C1-INH caused by missense mutations in the Factor 12 gene [13], the plasminogen gene [14], the angiopoietin-1 gene [15], the kininogen 1 gene [16], the myoferlin gene [17], the HS3ST6 gene [18] or of unknown cause [19] |
| Acquired Angioedema |
| Decreased degradation of bradykinin caused by drugs such as ACE-inhibitors, sartans #, plasminogen activators, or the Neprilysin inhibitor sacubitril |
| Increased generation of bradykinin caused by a loss of C1-INH due to autoantibodies and/or underlying (malignant) conditions |
| Angioedema of an unknown cause not responding to antihistamines |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, J.; Greve, J.; Bas, M.; Kojda, G. Bradykinin-Mediated Angioedema Induced by Commonly Used Cardiovascular Drugs. Drugs Drug Candidates 2023, 2, 708-727. https://doi.org/10.3390/ddc2030036
Hahn J, Greve J, Bas M, Kojda G. Bradykinin-Mediated Angioedema Induced by Commonly Used Cardiovascular Drugs. Drugs and Drug Candidates. 2023; 2(3):708-727. https://doi.org/10.3390/ddc2030036
Chicago/Turabian StyleHahn, Janina, Jens Greve, Murat Bas, and Georg Kojda. 2023. "Bradykinin-Mediated Angioedema Induced by Commonly Used Cardiovascular Drugs" Drugs and Drug Candidates 2, no. 3: 708-727. https://doi.org/10.3390/ddc2030036
APA StyleHahn, J., Greve, J., Bas, M., & Kojda, G. (2023). Bradykinin-Mediated Angioedema Induced by Commonly Used Cardiovascular Drugs. Drugs and Drug Candidates, 2(3), 708-727. https://doi.org/10.3390/ddc2030036