
Heat Shock Proteins Mediate Intercellular Communications within the Tumor Microenvironment through Extracellular Vesicles

 , and
, and
Abstract
1. Introduction
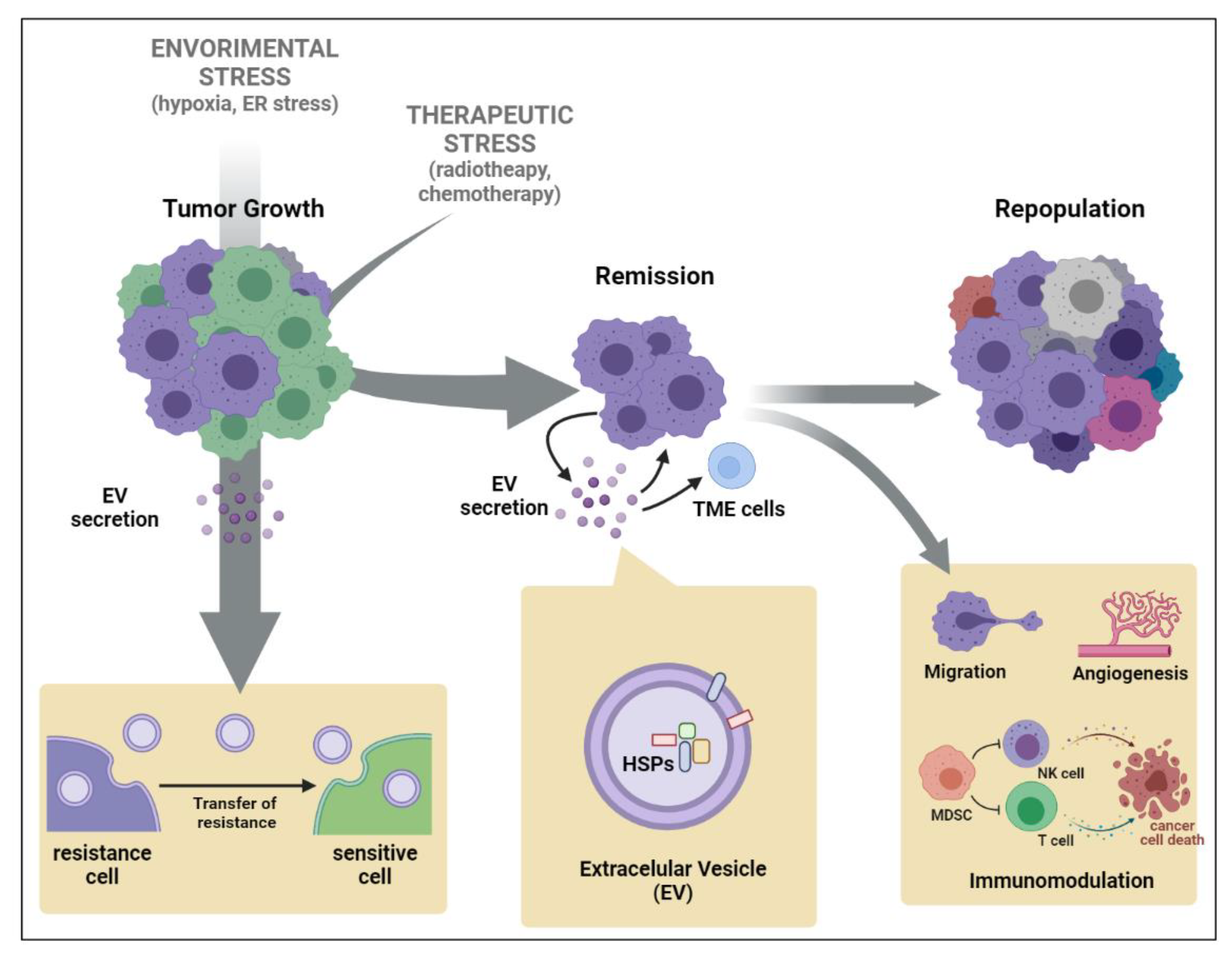
2. Extracellular Vesicle (EV)-Mediated Stress Propagation during Tumor Development and Progression
3. Heat Shock Protein (HSP) Secretion by EVs Triggered by Chemo- or Radiotherapy
4. Transfer of Therapy Resistance Mediated by HSP-EVs’ Release by Tumor Cells
5. Cancer Cell Intrinsic Mechanism Modulated by HSP-EVs That Impact Therapy Response
6. Immunological Roles of Cancer HSP-EVs That Impact Therapy Response
7. Future Perspectives
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kampinga, H.H.; Mayer, M.P.; Mogk, A. Protein quality control: From mechanism to disease: EMBO Workshop, Costa de la Calma (Mallorca), Spain, April 28–May 03, 2019. Cell Stress Chaperones 2019, 24, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C.; Barral, J.M.; Hartl, F.U. More than folding: Localized functions of cytosolic chaperones. Trends Biochem. Sci. 2003, 28, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Pagliara, V.; Moltedo, O.; Remondelli, P. Structural and Functional Significance of the Endoplasmic Reticulum Unfolded Protein Response Transducers and Chaperones at the Mitochondria-ER Contacts: A Cancer Perspective. Front. Cell Dev. Biol. 2021, 9, 641194. [Google Scholar] [CrossRef]
- Lindquist, S. Protein Folding Sculpting Evolutionary Change. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 103–108. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm 2022, 3, e161. [Google Scholar] [CrossRef]
- Ferrarini, M.; Heltai, S.; Zocchi, M.R.; Rugarli, C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 1992, 51, 613–619. [Google Scholar] [CrossRef]
- Jäättelä, M. Over-expression of hsp70 confers tumorigenicity to mouse fibrosarcoma cells. Int. J. Cancer 1995, 60, 689–693. [Google Scholar] [CrossRef]
- Vargas-Roig, L.M.; Gago, F.E.; Tello, O.; Aznar, J.C.; Ciocca, D.R. Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int. J. Cancer 1998, 79, 468–475. [Google Scholar] [CrossRef]
- Tao, S.-C.; Guo, S.-C. Role of extracellular vesicles in tumour microenvironment. Cell Commun. Signal. 2020, 18, 163. [Google Scholar] [CrossRef]
- Dixson, A.C.; Dawson, T.R.; Di Vizio, D.; Weaver, A.M. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat. Rev. Mol. Cell Biol. 2023, 24, 454–476. [Google Scholar] [CrossRef]
- Théry, C. Exosomes: Secreted vesicles and intercellular communications. F1000 Biol. Rep. 2011, 3, 15. [Google Scholar] [CrossRef]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef]
- Andrade, L.N.S.; Otake, A.H.; Cardim, S.G.B.; da Silva, F.I.; Ikoma Sakamoto, M.M.; Furuya, T.K.; Uno, M.; Pasini, F.S.; Chammas, R. Extracellular Vesicles Shedding Promotes Melanoma Growth in Response to Chemotherapy. Sci. Rep. 2019, 9, 14482. [Google Scholar] [CrossRef]
- Otake, A.H.; Saito, R.D.F.; Duarte, A.P.M.; Ramos, A.F.; Chammas, R. GD3 ganglioside-enriched extracellular vesicles stimulate melanocyte migration. Biochim. et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2019, 1864, 422–432. [Google Scholar] [CrossRef]
- Gregory, C.D.; Rimmer, M.P. Extracellular vesicles arising from apoptosis: Forms, functions, and applications. J. Pathol. 2023. [Google Scholar] [CrossRef]
- Dai, C.; Dai, S.; Cao, J. Proteotoxic stress of cancer: Implication of the heat-shock response in oncogenesis. J. Cell. Physiol. 2011, 227, 2982–2987. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Bister, N.; Pistono, C.; Huremagic, B.; Jolkkonen, J.; Giugno, R.; Malm, T. Hypoxia and extracellular vesicles: A review on methods, vesicular cargo and functions. J. Extracell. Vesicles 2020, 10, e12002. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic re-ticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, G.; Huang, L.; Yuan, Y.; Wu, C.; Li, Y. Transmissible Endoplasmic Reticulum Stress: A Novel Perspective on Tumor Immunity. Front. Cell Dev. Biol. 2020, 8, 846. [Google Scholar] [CrossRef] [PubMed]
- Collett, G.P.; Redman, C.W.; Sargent, I.L.; Vatish, M. Endoplasmic reticulum stress stimulates the release of ex-tracellular vesicles carrying danger-associated molecular pattern (DAMP) molecules. Oncotarget 2018, 9, 6707–6717. [Google Scholar] [CrossRef]
- Kanemoto, S.; Nitani, R.; Murakami, T.; Kaneko, M.; Asada, R.; Matsuhisa, K.; Saito, A.; Imaizumi, K. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2016, 480, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Regnault, A.; Garin, J.; Wolfers, J.; Zitvogel, L.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 1999, 147, 599–610. [Google Scholar] [CrossRef]
- Wubbolts, R.; Leckie, R.S.; Veenhuizen, P.T.; Schwarzmann, G.; Möbius, W.; Hoernschemeyer, J.; Slot, J.W.; Geuze, H.J.; Stoorvogel, W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 2003, 278, 10963–10972. [Google Scholar] [CrossRef]
- Géminard, C.; Nault, F.; Johnstone, R.M.; Vidal, M. Characteristics of the Interaction between Hsc70 and the Transferrin Receptor in Exosomes Released during Reticulocyte Maturation. J. Biol. Chem. 2001, 276, 9910–9916. [Google Scholar] [CrossRef]
- Van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome–like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef]
- Clayton, A.; Turkes, A.; Navabi, H.; Mason, M.D.; Tabi, Z. Induction of heat shock proteins in B-cell exosomes. J. Cell Sci. 2005, 118 Pt 16, 3631–3638. [Google Scholar] [CrossRef]
- Ono, K.; Eguchi, T.; Sogawa, C.; Calderwood, S.K.; Futagawa, J.; Kasai, T.; Seno, M.; Okamoto, K.; Sasaki, A.; Kozaki, K. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J. Cell. Biochem. 2018, 119, 7350–7362. [Google Scholar] [CrossRef]
- Merendino, A.M.; Bucchieri, F.; Campanella, C.; Marcianò, V.; Ribbene, A.; David, S.; Zummo, G.; Burgio, G.; Corona, D.F.V.; de Macario, E.C.; et al. Hsp60 is actively secreted by human tumor cells. PLoS ONE 2010, 5, e9247. [Google Scholar] [CrossRef]
- Huang, M.-B.; Wu, J.Y.; Lillard, J.; Bond, V.C. SMR peptide antagonizes mortalin promoted release of extracellular vesicles and affects mortalin protection from complement-dependent cytotoxicity in breast cancer cells and leukemia cells. Oncotarget 2019, 10, 5419–5438. [Google Scholar] [CrossRef]
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular Vesicles and Exosomes: Insights From Exercise Science. Front. Physiol. 2021, 11, 604274. [Google Scholar] [CrossRef]
- O’neill, C.P.; Gilligan, K.E.; Dwyer, R.M. Role of Extracellular Vesicles (EVs) in Cell Stress Response and Resistance to Cancer Therapy. Cancers 2019, 11, 136. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Heinrich, J.C.; Donakonda, S.; Haupt, V.J.; Lennig, P.; Zhang, Y.; Schroeder, M. New HSP27 inhibitors efficiently suppress drug resistance development in cancer cells. Oncotarget 2016, 7, 68156–68169. [Google Scholar] [CrossRef]
- Boudesco, C.; Cause, S.; Jego, G.; Garrido, C. Hsp70: A Cancer Target Inside and Outside the Cell. Methods Mol. Biol. 2017, 1709, 371–396. [Google Scholar] [CrossRef]
- Kumar, P.; Siripini, S.; Sreedhar, A.S. The matrix metalloproteinase 7 (MMP7) links Hsp90 chaperone with acquired drug resistance and tumor metastasis. Cancer Rep. 2020, 5, e1261. [Google Scholar] [CrossRef]
- Dempsey, N.C.; Ireland, H.E.; Smith, C.M.; Hoyle, C.F.; Williams, J.H. Heat Shock Protein translocation induced by membrane fluidization increases tumor-cell sensitivity to chemotherapeutic drugs. Cancer Lett. 2010, 296, 257–267. [Google Scholar] [CrossRef]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar] [CrossRef]
- Garrido, C.; Schmitt, E.; Candé, C.; Vahsen, N.; Parcellier, A.; Kroemer, G. HSP27 and HSP70: Potentially oncogenic apoptosis inhibitors. Cell Cycle 2003, 2, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.G.; Martins, V.R.; Hajj, G.N.M. Unconventional Secretion of Heat Shock Proteins in Cancer. Int. J. Mol. Sci. 2017, 18, 946. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Hunter-Lavin, C.; Davies, E.L.; Bacelar, M.M.; Marshall, M.J.; Andrew, S.M.; Williams, J.H. Hsp70 release from peripheral blood mononuclear cells. Biochem. Biophys. Res. Commun. 2004, 324, 511–517. [Google Scholar] [CrossRef]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef]
- Tamura, Y.; Torigoe, T.; Kutomi, G.; Hirata, K.; Sato, N. New paradigm for intrinsic function of heat shock proteins as endogenous ligands in inflammation and innate immunity. Curr. Mol. Med. 2012, 12, 1198–1206. [Google Scholar] [CrossRef]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: A role for plasminogen activation. BMC Cancer 2010, 10, 294. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Lin, Y.-S.; Pan, Y.-C.; Tsai, P.-H.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Wyciszkiewicz, A.; Kalinowska-Łyszczarz, A.; Nowakowski, B.; Kaźmierczak, K.; Osztynowicz, K.; Michalak, S. Expression of small heat shock proteins in exosomes from patients with gynecologic cancers. Sci. Rep. 2019, 9, 9817. [Google Scholar] [CrossRef]
- Campanella, C.; Bucchieri, F.; Merendino, A.M.; Fucarino, A.; Burgio, G.; Corona, D.F.V.; Barbieri, G.; David, S.; Farina, F.; Zummo, G.; et al. The odyssey of Hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and ex-osomal protein-trafficking modalities. PLoS ONE 2012, 7, e42008. [Google Scholar] [CrossRef]
- Lv, L.-H.; Wan, Y.-L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.-L.; Lin, H.-M.; Shang, C.-Z.; Chen, Y.-J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Febbraio, M.A. Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef]
- Bausero, M.A.; Gastpar, R.; Multhoff, G.; Asea, A. Alternative mechanism by which IFN-gamma enhances tumor recognition: Active release of heat shock protein 72. J. Immunol. 2005, 175, 2900–2912. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2013, 54, 554–565. [Google Scholar] [CrossRef]
- Li, Z.; Zhuang, M.; Zhang, L.; Zheng, X.; Yang, P.; Li, Z. Acetylation modification regulates GRP78 secretion in colon cancer cells. Sci. Rep. 2016, 6, 30406. [Google Scholar] [CrossRef]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef]
- Namee, N.M.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.-C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anticancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Shedden, K.; Xie, X.T.; Chandaroy, P.; Chang, Y.T.; Rosania, G.R. Expulsion of small molecules in vesicles shed by cancer cells: Association with gene expression and chemosensitivity profiles. Cancer Res. 2003, 63, 4331–4337. [Google Scholar] [PubMed]
- Dai, S.; Wan, T.; Wang, B.; Zhou, X.; Xiu, F.; Chen, T.; Wu, Y.; Cao, X. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immun-ization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin. Cancer Res. 2005, 11, 7554–7563. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, M.D.; Kaur, P.; Nagaraja, G.M.; Bausero, M.A.; Manola, J.; Asea, A. Radiation therapy induces circulating serum Hsp72 in patients with prostate cancer. Radiother. Oncol. 2010, 95, 350–358. [Google Scholar] [CrossRef]
- Arscott, W.T.; Tandle, A.T.; Zhao, S.; Shabason, J.E.; Gordon, I.K.; Schlaff, C.D.; Zhang, G.; Tofilon, P.J.; Camphausen, K.A. Ionizing radiation and glioblastoma exosomes: Implications in tumor biology and cell migration. Transl. Oncol. 2013, 6, 638–648. [Google Scholar] [CrossRef]
- Al-Mayah, A.; Bright, S.; Chapman, K.; Irons, S.; Luo, P.; Carter, D.; Goodwin, E.; Kadhim, M. The non-targeted effects of radiation are perpetuated by exosomes. Mutat. Res. Mol. Mech. Mutagen. 2015, 772, 38–45. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nature 2018, 21, 190–202. [Google Scholar] [CrossRef]
- Emam, S.E.; Ando, H.; Abu Lila, A.S.; Kobayashi, S.; Shimizu, T.; Okuhira, K.; Ishima, Y.; Ishida, T. Doxorubicin Expands in Vivo Secretion of Circulating Exosome in Mice. Biol. Pharm. Bull. 2018, 41, 1078–1083. [Google Scholar] [CrossRef]
- König, L.; Kasimir-Bauer, S.; Bittner, A.-K.; Hoffmann, O.; Wagner, B.; Manvailer, L.F.S.; Kimmig, R.; Horn, P.A.; Rebmann, V. Elevated levels of extracellular vesicles are associated with therapy failure and disease progression in breast cancer patients undergoing neoadjuvant chemotherapy. Oncoimmunology 2017, 7, e1376153. [Google Scholar] [CrossRef]
- Bandari, S.K.; Purushothaman, A.; Ramani, V.C.; Brinkley, G.J.; Chandrashekar, D.S.; Varambally, S.; Mobley, J.A.; Zhang, Y.; Brown, E.E.; Vlodavsky, I.; et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2017, 65, 104–118. [Google Scholar] [CrossRef]
- Campanella, C.; D’Anneo, A.; Gammazza, A.M.; Bavisotto, C.C.; Barone, R.; Emanuele, S.; Cascio, F.L.; Mocciaro, E.; Fais, S.; De Macario, E.C.; et al. The histone deacetylase inhibitor SAHA induces HSP60 nitration and its extracellular release by exosomal vesicles in human lung-derived carcinoma cells. Oncotarget 2015, 7, 28849–28867. [Google Scholar] [CrossRef]
- Shao, H.; Chung, J.; Balaj, L.; Charest, A.; Bigner, D.D.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Weissleder, R.; Lee, H. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med. 2012, 18, 1835–1840. [Google Scholar] [CrossRef]
- Kıyga, E.; Adıgüzel, Z.; Uçar, E. Temozolomide increases heat shock proteins in extracellular vesicles released from glioblastoma cells. Mol. Biol. Rep. 2022, 49, 8701–8713. [Google Scholar] [CrossRef] [PubMed]
- Vinik, Y.; Ortega, F.G.; Mills, G.B.; Lu, Y.; Jurkowicz, M.; Halperin, S.; Aharoni, M.; Gutman, M.; Lev, S. Proteomic analysis of circulating extracellular vesicles identifies potential markers of breast cancer progression, recurrence, and response. Sci. Adv. 2020, 6, eaba5714. [Google Scholar] [CrossRef]
- Rothammer, A.; Sage, E.K.; Werner, C.; Combs, S.E.; Multhoff, G. Increased heat shock protein 70 (Hsp70) serum levels and low NK cell counts after radiotherapy—Potential markers for predicting breast cancer recurrence? Radiat. Oncol. 2019, 14, 78. [Google Scholar] [CrossRef]
- Tsen, F.; Bhatia, A.; O’Brien, K.; Cheng, C.F.; Chen, M.; Hay, N.; Stiles, B.; Woodley, D.T.; Li, W. Extracellular heat shock protein 90 signals through subdomain II and the NPVY motif of LRP-1 receptor to Akt1 and Akt2: A circuit essential for promoting skin cell migration in vitro and wound healing in vivo. Mol. Cell. Biol. 2013, 33, 4947–4959. [Google Scholar] [CrossRef]
- Ono, K.; Sogawa, C.; Kawai, H.; Tran, M.T.; Taha, E.A.; Lu, Y.; Oo, M.W.; Okusha, Y.; Okamura, H.; Ibaragi, S.; et al. Triple knockdown of CDC37, HSP90-alpha and HSP90-beta diminishes extracellular vesicles-driven malignancy events and macrophage M2 polarization in oral cancer. J. Extracell. Vesicles 2020, 9, 1769373. [Google Scholar] [CrossRef]
- Tang, X.; Chang, C.; Guo, J.; Lincoln, V.; Liang, C.; Chen, M.; Woodley, D.T.; Li, W. Tumour-Secreted Hsp90α on External Surface of Exosomes Mediates Tumour—Stromal Cell Communication via Autocrine and Paracrine Mechanisms. Sci. Rep. 2019, 9, 15108. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Mao, R.; Zhang, Y.; Wen, J.; Liu, Q.; Liu, Y.; Zhang, T. DNAJB8 in small extracellular vesicles promotes Oxaliplatin resistance through TP53/MDR1 pathway in colon cancer. Cell. Death Dis. 2022, 13, 151. [Google Scholar] [CrossRef]
- Yukawa, H.; Suzuki, K.; Aoki, K.; Arimoto, T.; Yasui, T.; Kaji, N.; Ishikawa, T.; Ochiya, T.; Baba, Y. Imaging of angi-ogenesis of human umbilical vein endothelial cells by uptake of exosomes secreted from hepatocellular carcinoma cells. Sci. Rep. 2018, 8, 6765. [Google Scholar] [CrossRef]
- Bohonowych, J.E.; Gopal, U.; Isaacs, J.S. Hsp90 as a gatekeeper of tumor angiogenesis: Clinical promise and po-tential pitfalls. J. Oncol. 2010, 2010, 412985. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Zhang, C.; Lum, D.; Druso, J.E.; Blank, B.; Wilson, K.F.; Welm, A.; Antonyak, M.A.; Cerione, R.A. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat. Commun. 2017, 8, 14450. [Google Scholar] [CrossRef] [PubMed]
- Campanella, C.; Rappa, F.; Sciumè, C.; Gammazza, A.M.; Barone, R.; Bucchieri, F.; David, S.; Curcurù, G.; Bavisotto, C.C.; Pitruzzella, A.; et al. Heat shock protein 60 levels in tissue and circulating exosomes in human large bowel cancer before and after ablative surgery. Cancer 2015, 121, 3230–3239. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Cipolla, C.; Graceffa, G.; Barone, R.; Bucchieri, F.; Bulone, D.; Cabibi, D.; Campanella, C.; Marino Gammazza, A.; Pitruzzella, A.; et al. Immunomorphological Pattern of Molecular Chaperones in Normal and Pathological Thyroid Tissues and Circulating Exosomes: Potential Use in Clinics. Int. J. Mol. Sci. 2019, 20, 4496. [Google Scholar] [CrossRef]
- Diao, J.; Yang, X.; Song, X.; Chen, S.; He, Y.; Wang, Q.; Chen, G.; Luo, C.; Wu, X.; Zhang, Y. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med. Oncol. 2015, 32, 35–453. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. J. Natl. Cancer Inst. 2016, 108, djv330. [Google Scholar] [CrossRef]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano, J.; Malzahn, D.; Bickeböller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef]
- Xie, Y.; Bai, O.; Zhang, H.; Yuan, J.; Zong, S.; Chibbar, R.; Slattery, K.; Qureshi, M.; Wei, Y.; Deng, Y.; et al. Mem-brane-bound HSP70-engineered myeloma cell-derived exosomes stimulate more efficient CD8(+) CTL- and NK-mediated an-titumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic HSP70. J. Cell. Mol. Med. 2010, 14, 2655–2666. [Google Scholar] [CrossRef]
- Cho, J.-A.; Lee, Y.-S.; Kim, S.-H.; Ko, J.-K.; Kim, C.-W. MHC independent anti-tumor immune responses induced by Hsp70-enriched exosomes generate tumor regression in murine models. Cancer Lett. 2009, 275, 256–265. [Google Scholar] [CrossRef]
- Menay, F.; Herschlik, L.; De Toro, J.; Cocozza, F.; Tsacalian, R.; Gravisaco, M.J.; Di Sciullo, M.P.; Vendrell, A.; Waldner, C.I.; Mongini, C. Exosomes Isolated from Ascites of T-Cell Lymphoma-Bearing Mice Expressing Surface CD24 and HSP-90 Induce a Tumor-Specific Immune Response. Front. Immunol. 2017, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Sen, K.; Sheppe, A.E.F.; Singh, I.; Hui, W.W.; Edelmann, M.J.; Rinaldi, C. Exosomes released by breast cancer cells under mild hyperthermic stress possess immunogenic potential and modulate polarization. Int. J. Hyperth. 2020, 37, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a mem-brane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef]
- Takayama, S.; Reed, J.C.; Homma, S. Heat-shock proteins as regulators of apoptosis. Oncogene 2003, 22, 9041–9047. [Google Scholar] [CrossRef]
- Beere, H.M. The stress of dying’: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117 Pt 13, 2641–2651. [Google Scholar] [CrossRef]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Meriin, A.B.; Sherman, M.Y.; Morimoto, R.I.; Massie, B. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol. 2000, 20, 7146–7159. [Google Scholar] [CrossRef]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar] [CrossRef]
- Cesa, L.C.; Shao, H.; Srinivasan, S.R.; Tse, E.; Jain, C.; Zuiderweg, E.R.P.; Southworth, D.R.; Mapp, A.K.; Gestwicki, J.E. X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhi-bition. J. Biol. Chem. 2018, 293, 2370–2380. [Google Scholar] [CrossRef]
- Hunter, A.M.; LaCasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef]
- Petersen, J.D.; Mekhedov, E.; Kaur, S.; David, D.; Roberts, D.D.; Zimmerberg, J. Endothelial cells release microvesicles that harbour multivesicular bodies and secrete exosomes. J. Extracell. Biol. 2023, 2, e79. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Lettini, G.; Lepore, S.; Crispo, F.; Sisinni, L.; Esposito, F.; Landriscina, M. Heat shock proteins in cancer stem cell maintenance: A potential therapeutic target? Histol. Histopathol. 2020, 35, 25–37. [Google Scholar]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Dong, L.; Wang, X. Emerging role of exosome signalling in maintaining cancer stem cell dynamic equilibrium. J. Cell. Mol. Med. 2018, 22, 3719–3728. [Google Scholar] [CrossRef]
- Nakano, I.; Garnier, D.; Minata, M.; Rak, J. Extracellular vesicles in the biology of brain tumour stem cells—Implications for inter-cellular communication, therapy and biomarker development. Semin. Cell. Dev. Biol. 2015, 40, 17–26. [Google Scholar] [CrossRef]
- Al-Sowayan, B.S.; Al-Shareeda, A.T.; Alrfaei, B.M. Cancer Stem Cell-Exosomes, Unexposed Player in Tumorigenicity. Front. Pharmacol. 2020, 11, 384. [Google Scholar] [CrossRef]
- Nolan, K.D.; Kaur, J.; Isaacs, J.S. Secreted heat shock protein 90 promotes prostate cancer stem cell heterogeneity. Oncotarget 2017, 8, 19323–19341. [Google Scholar] [CrossRef]
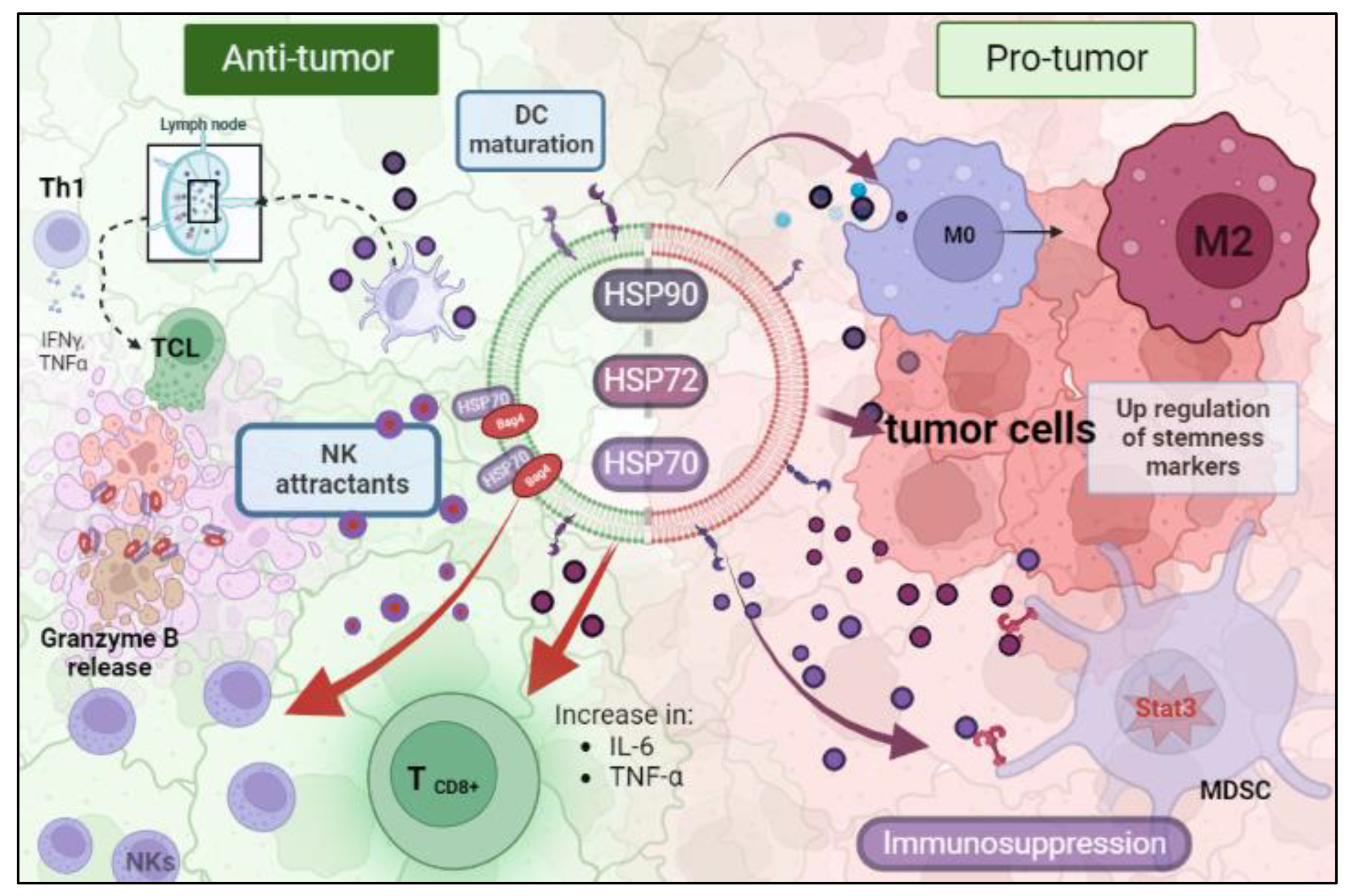

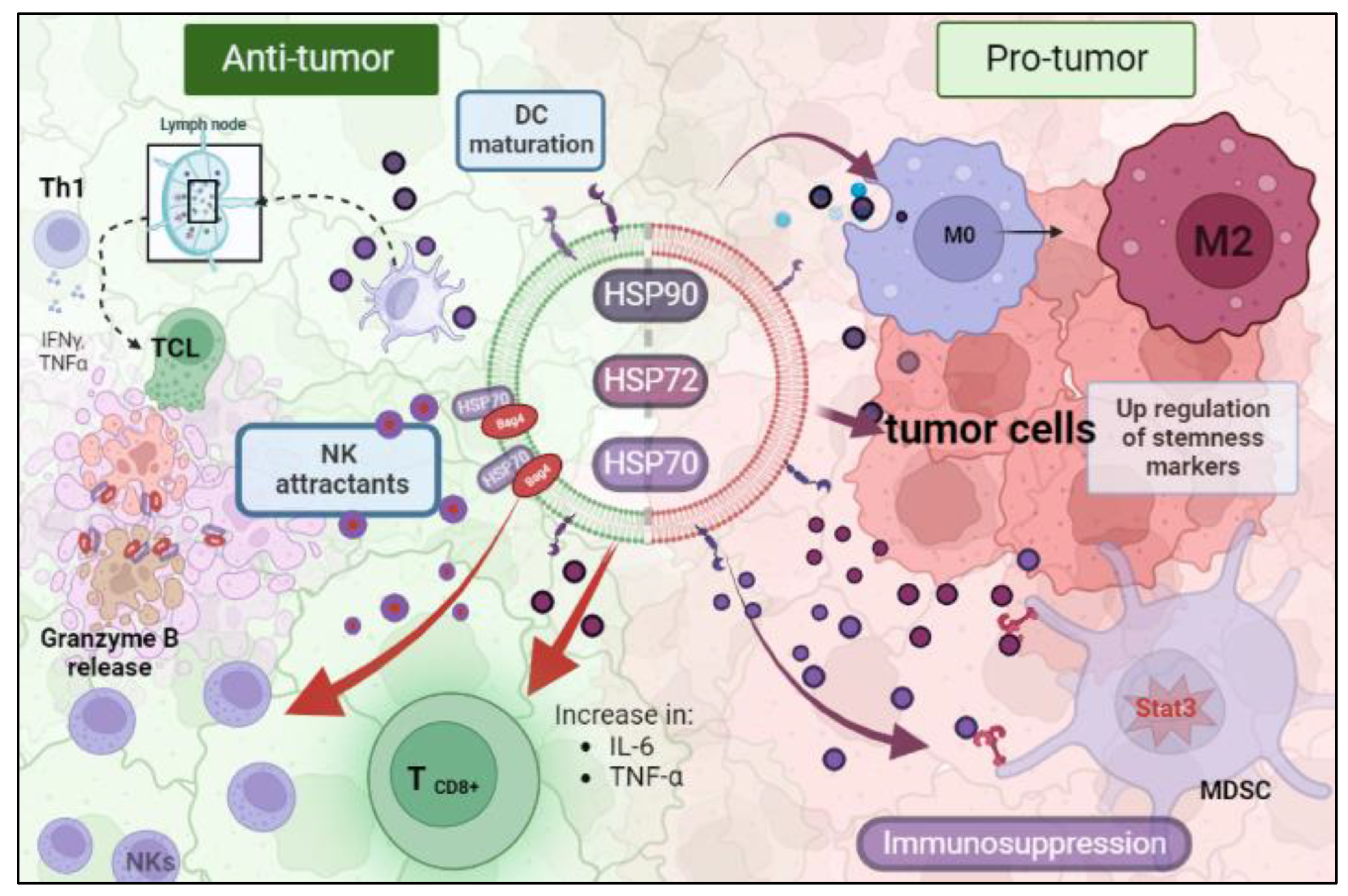{kind=link}
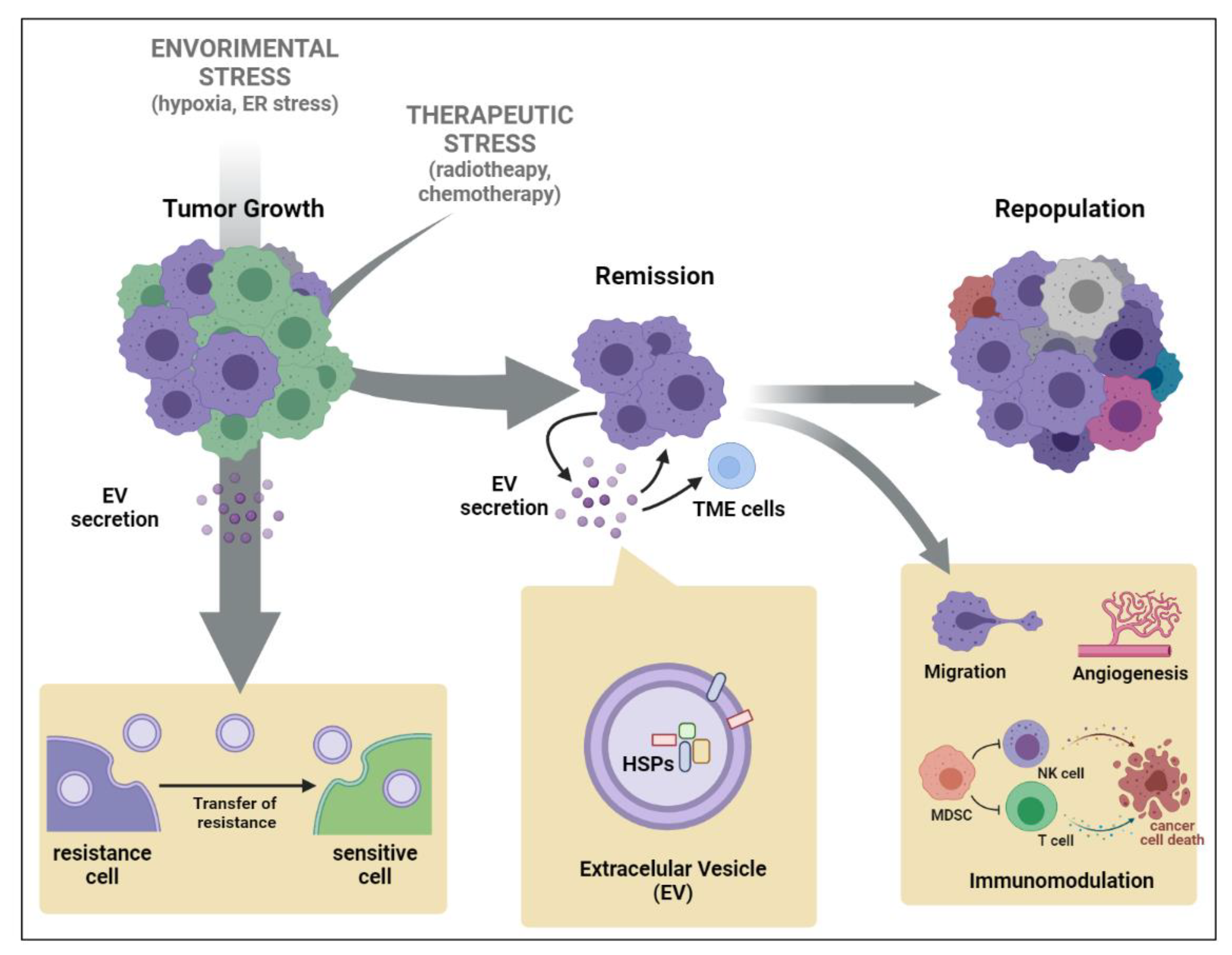{kind=link}
| HSP | Cell Type and Reference |
|---|---|
| HSP20 | Gynecologic cancer cells [51] |
| HSP27 HSC70 HSP70 HSP90 | B cells [29] |
| HSP60 | Human lung carcinoma cells [52] |
| HSP60 HSP70 | H292, A549 and K562 tumor cell lines [31] |
| HSP60 HSP70 HSP90 | Hepatocellular carcinoma cells [53] |
| HSP70 | Human peripheral blood mononuclear cells [54] |
| HSP70 | Natural killer cells [46] |
| HSP70 | Choriocarcinoma cells [23] |
| HSP72 | Breast adenocarcinoma cells Erythroleukemic cells [55] |
| HSC73 | Dendritic cells [25] |
| HSP70 HSP90 | Prostate cancer cell [56] |
| mt-HSP70 | Breast cancer cells [32] |
| GRP78 | Colon cancer cells [57] |
| HSP90 | Cancer stem cell-like [58] |
| Chaperone | Activity | |
|---|---|---|
| Pro-Tumor | Anti-Tumor | |
| HSP70 | Promote cell-survival, protect againist oxidative stress and others, promote protein folding and degradation, and promote cell migration and invasion. | Induce tumor cells’ apoptotic death and sensitize them to chemo- or radiotherapies. |
| HSP72 | Promote angiogenesis, protect cancer cells from oxidative stress. Supress apoptosis and promote cell invation, and migration. | Favor arresting of tumor growth, promote apoptosis, sensitize to chemotherapy. |
| HSP90 | Promote protein folding and stabilization of multiple proteins, promote cell survival, and suppress apoptosis. | Promote apoptosis, sensitize to chemo- and radiotherapy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, R.F.; Machado, C.M.L.; Lomba, A.L.O.; Otake, A.H.; Rangel, M.C. Heat Shock Proteins Mediate Intercellular Communications within the Tumor Microenvironment through Extracellular Vesicles. Appl. Biosci. 2024, 3, 45-58. https://doi.org/10.3390/applbiosci3010003
Saito RF, Machado CML, Lomba ALO, Otake AH, Rangel MC. Heat Shock Proteins Mediate Intercellular Communications within the Tumor Microenvironment through Extracellular Vesicles. Applied Biosciences. 2024; 3(1):45-58. https://doi.org/10.3390/applbiosci3010003
Chicago/Turabian StyleSaito, Renata F., Camila Maria Longo Machado, Ana Luiza Oliveira Lomba, Andréia Hanada Otake, and Maria Cristina Rangel. 2024. "Heat Shock Proteins Mediate Intercellular Communications within the Tumor Microenvironment through Extracellular Vesicles" Applied Biosciences 3, no. 1: 45-58. https://doi.org/10.3390/applbiosci3010003
APA StyleSaito, R. F., Machado, C. M. L., Lomba, A. L. O., Otake, A. H., & Rangel, M. C. (2024). Heat Shock Proteins Mediate Intercellular Communications within the Tumor Microenvironment through Extracellular Vesicles. Applied Biosciences, 3(1), 45-58. https://doi.org/10.3390/applbiosci3010003