
Unlocking Insights into Folding, Structure, and Function of Proteins through Circular Dichroism Spectroscopy—A Short Review



Abstract
:1. Circular Dichroism (CD) and Proteins
1.1. CD
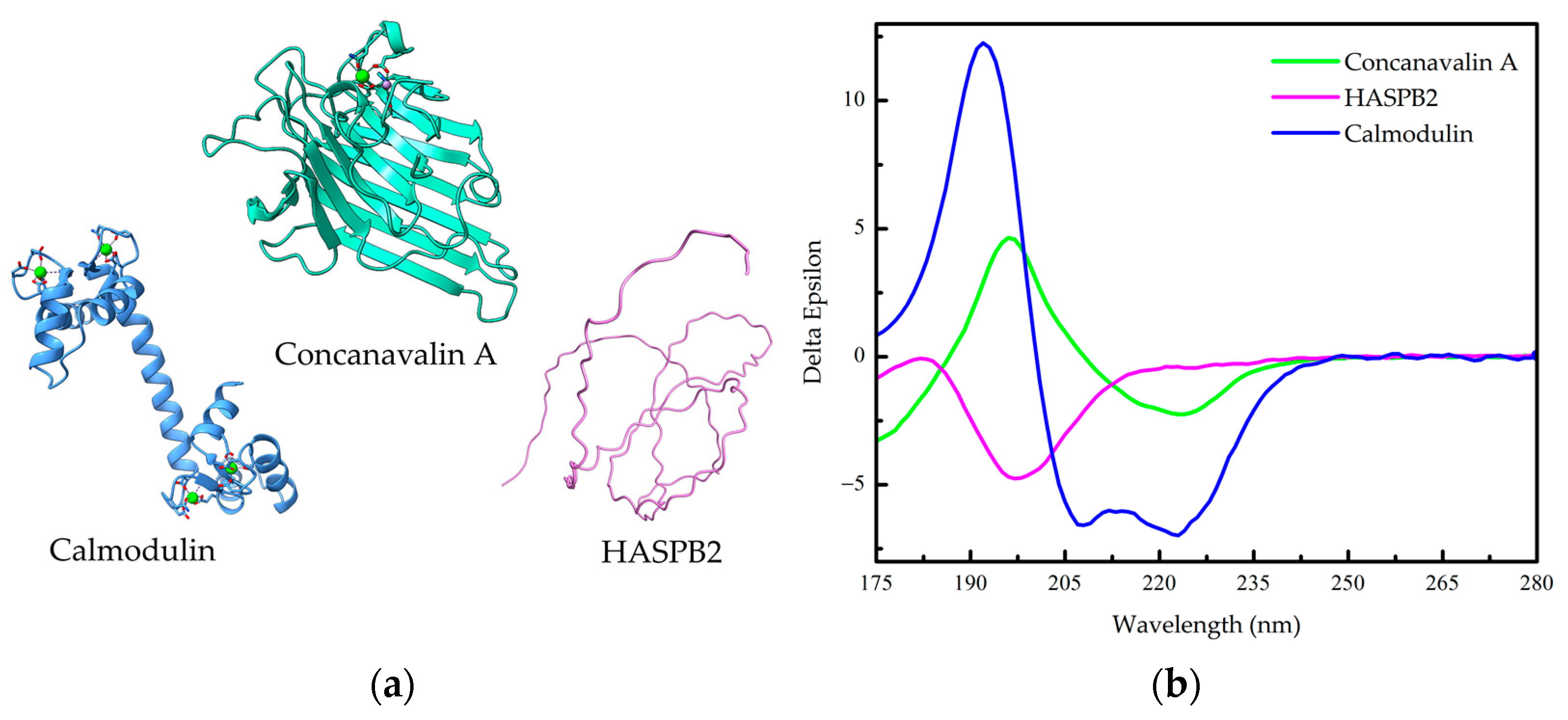
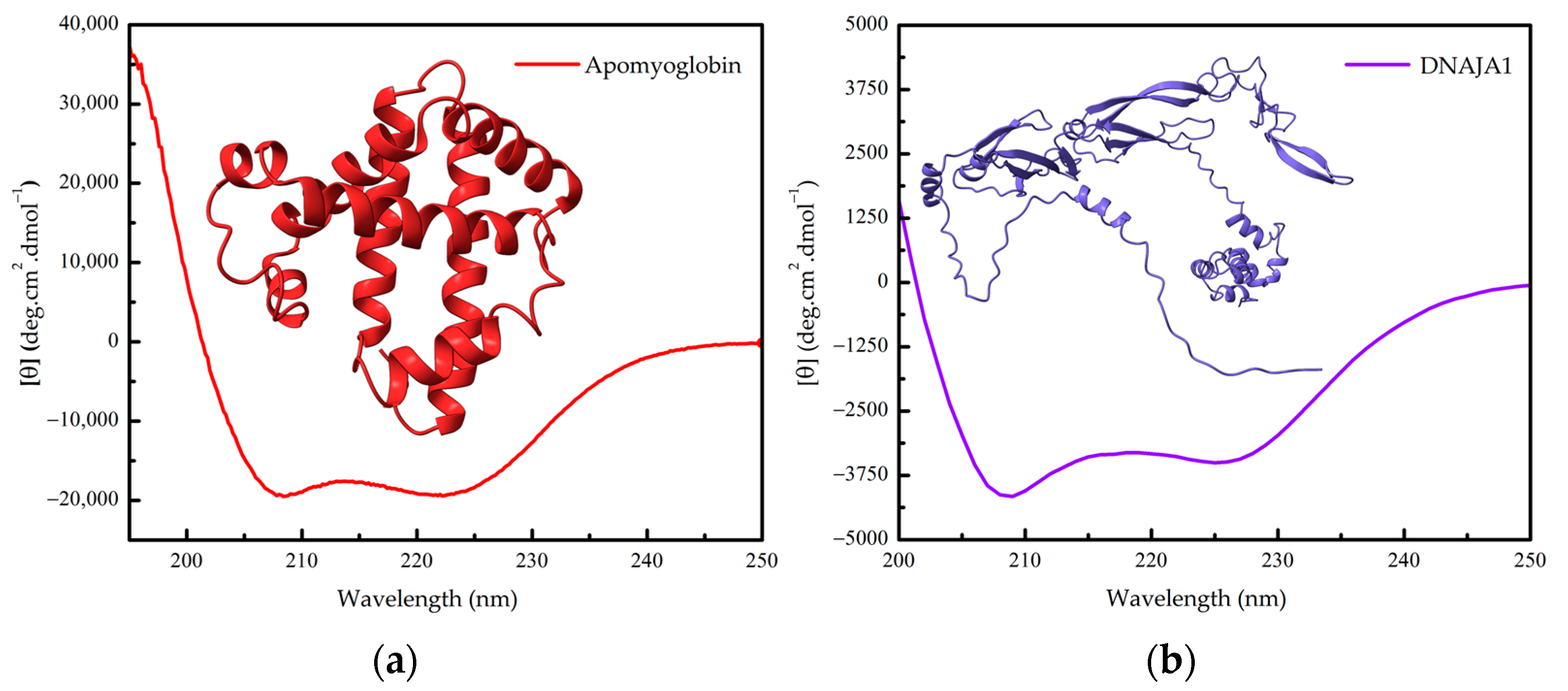
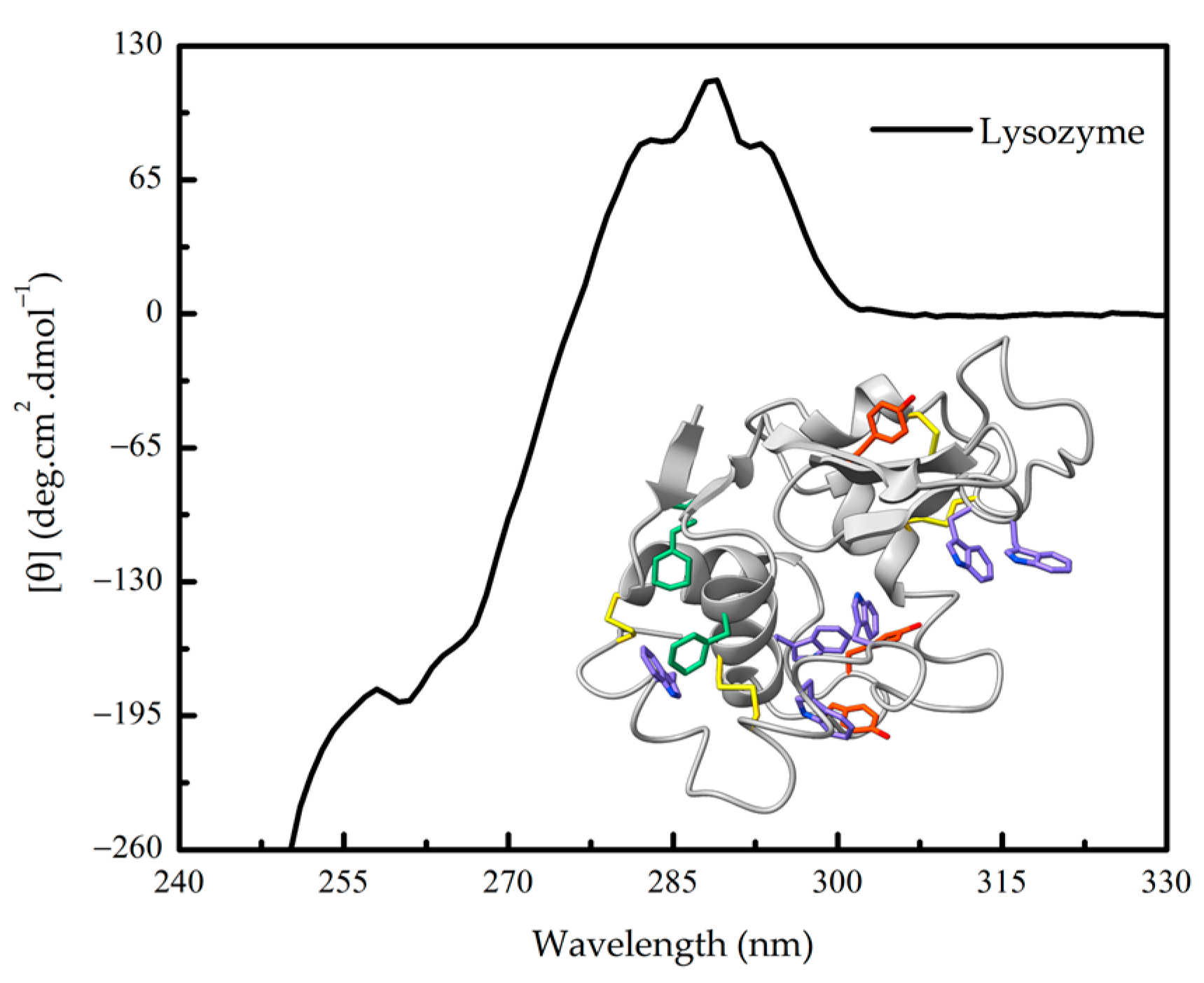
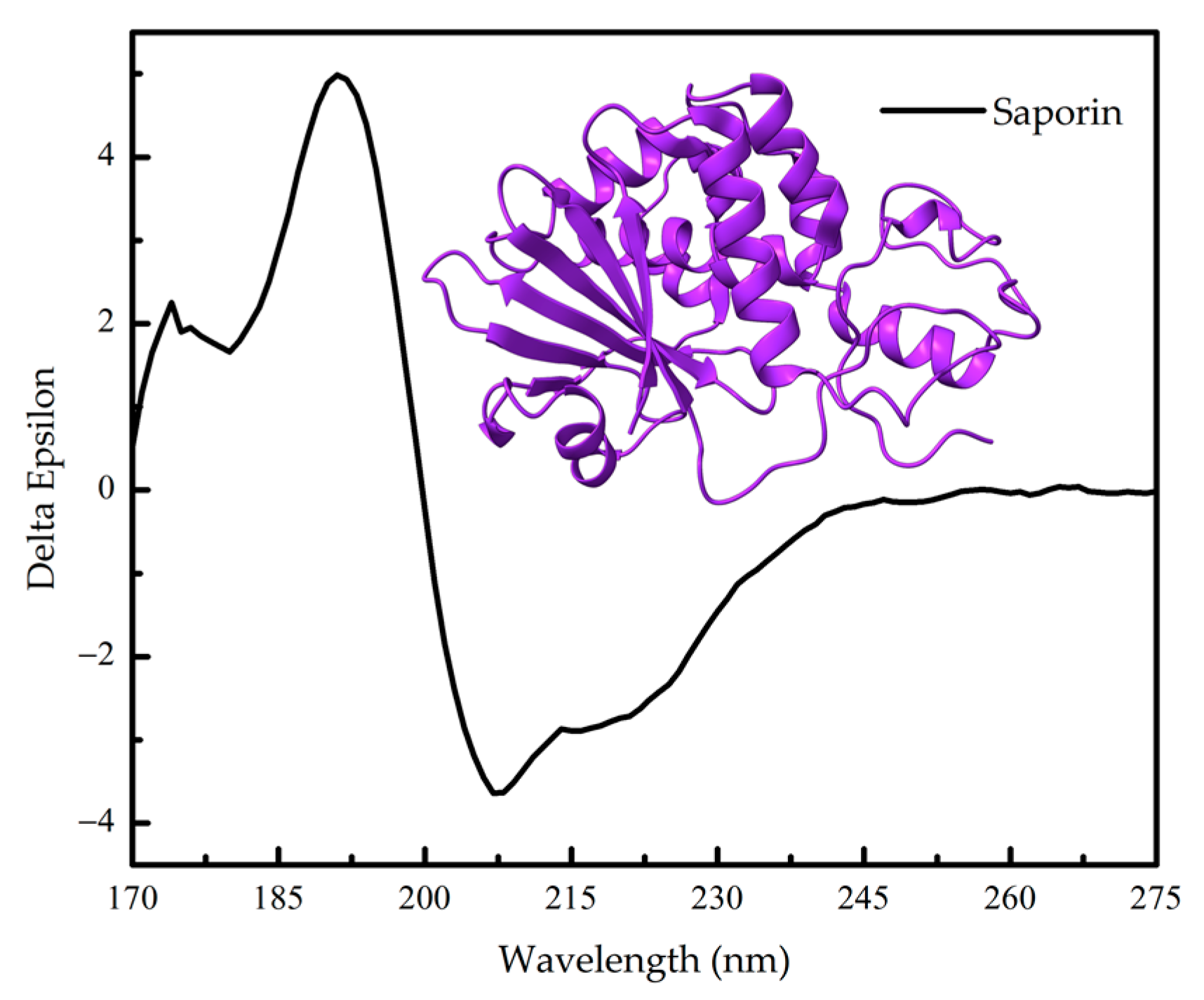
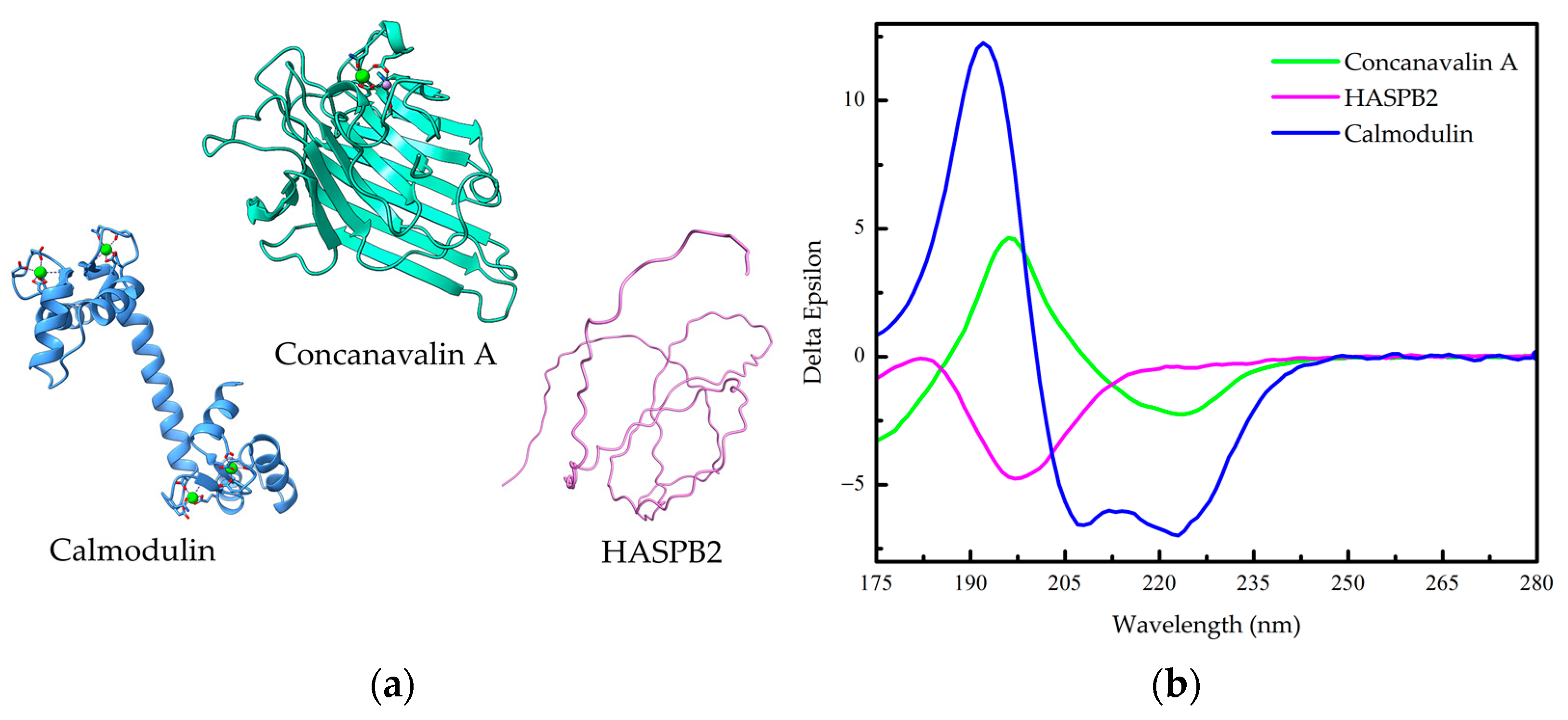
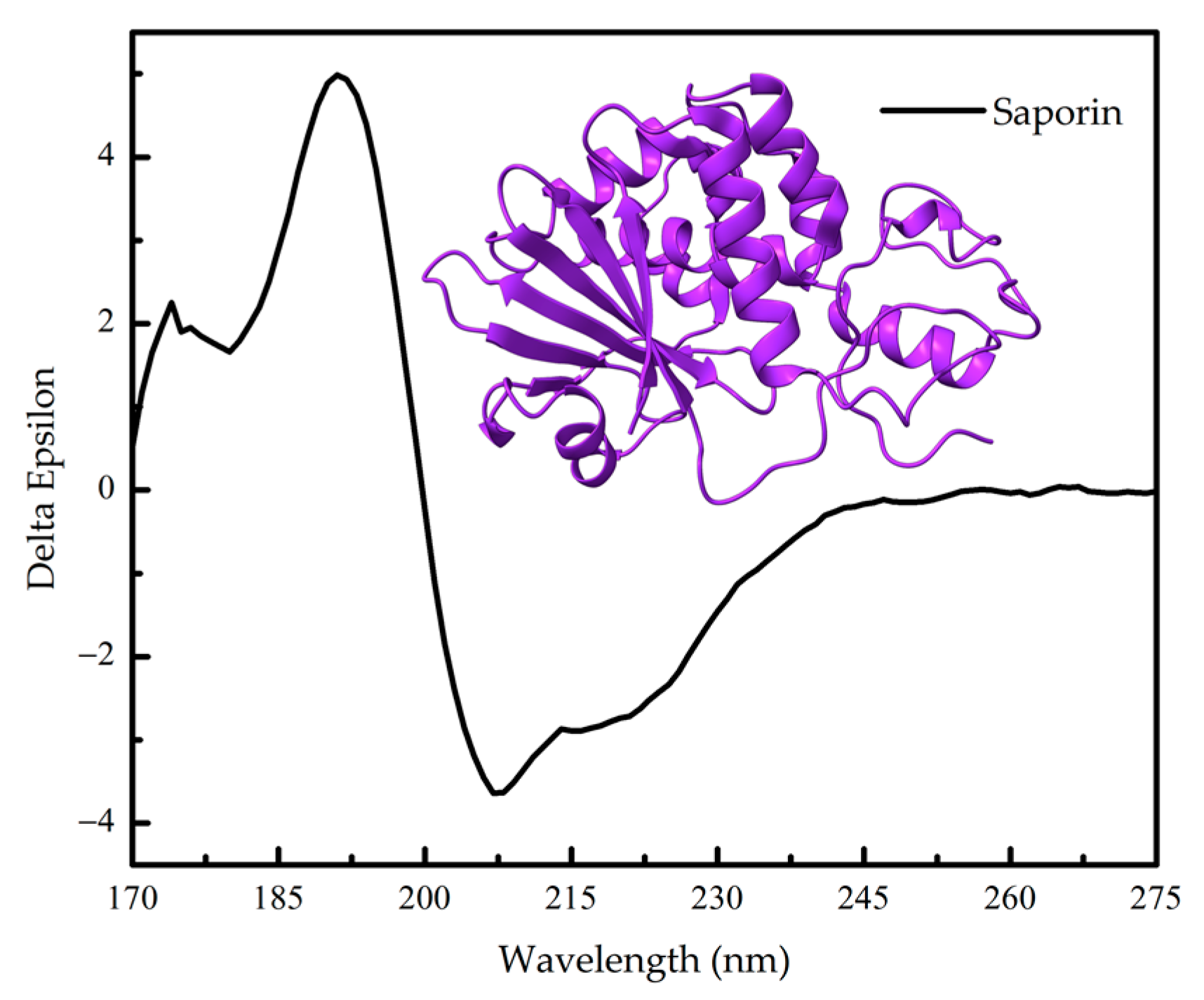
1.2. Protein Conformation
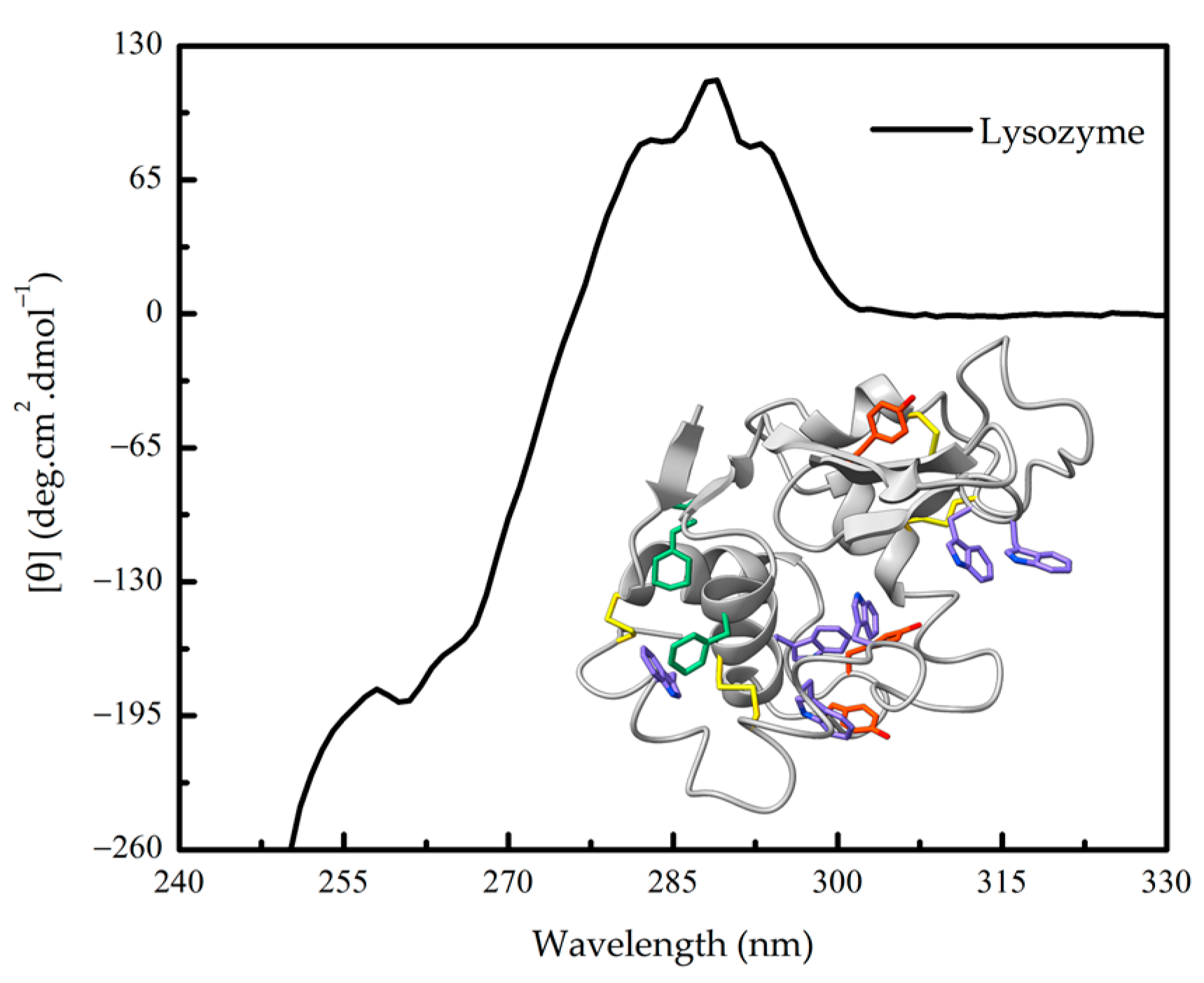
1.3. Further Advantages of CD Spectroscopy in Protein Research
1.4. Predicting Protein Secondary Structure: Computational Analysis of CD Spectra
2. Fine-Tuning Experimental Parameters for Accurate Results
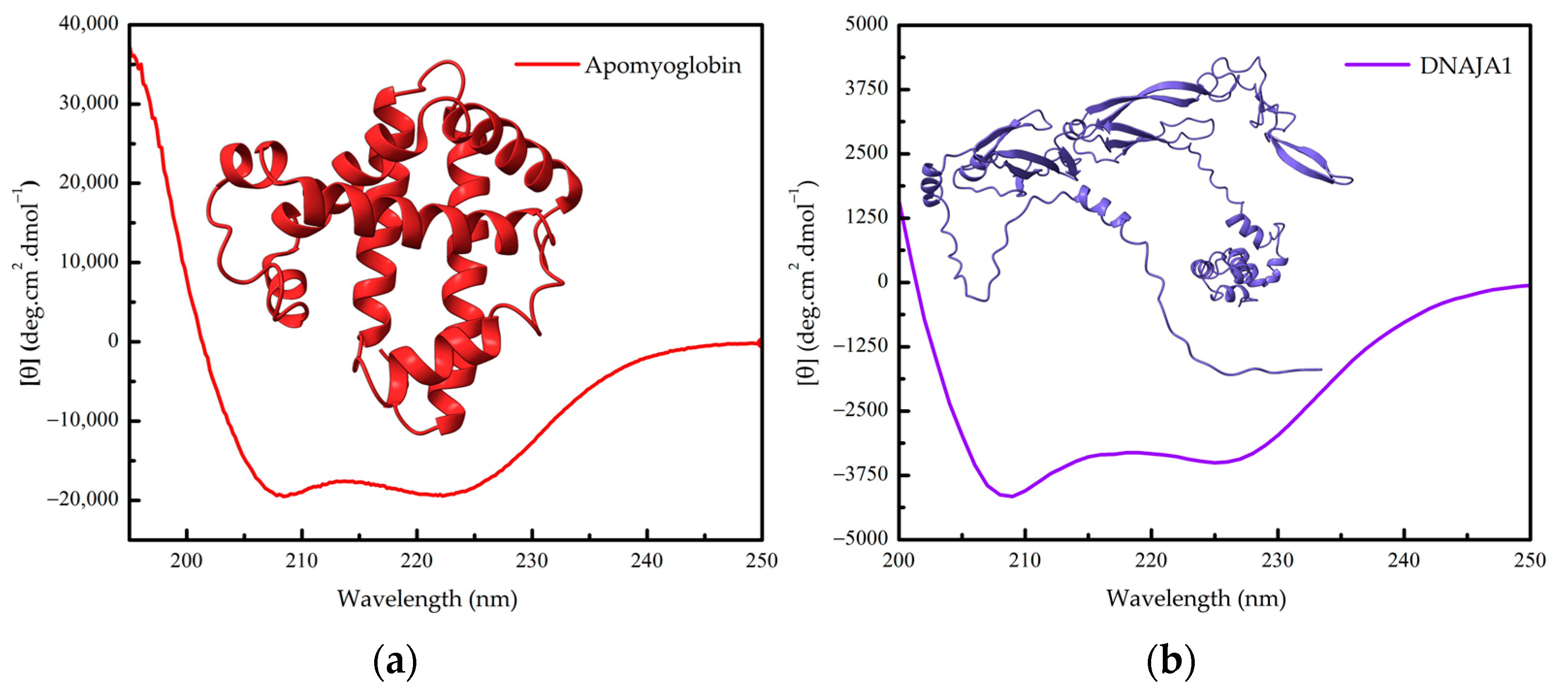
3. Studying Protein Unfolding and Aggregation Dynamics under Denaturing Conditions
4. Synchrotron Radiation Circular Dichroism (SRCD) Spectroscopy
5. Solid-State (SSCD), Vibrational (VCD) and High-Throughput (HTC) Circular Dichroism Spectroscopy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, W.C. Circular dichroism and its empirical application to biopolymers. Methods Biochem. Anal. 1985, 31, 61–163. [Google Scholar] [CrossRef]
- Kuwajima, K. Circular Dichroism. Methods Mol. Biol. 1995, 40, 115–135. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, A.; Sreerama, N.; Woody, R.W. Theoretical Approach to Electronic Optical Activity. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2000; pp. 55–96. [Google Scholar]
- Woody, R.W. Circular dichroism. Methods Enzymol. 1995, 246, 34–71. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W. Theory of circular dichroism of proteins. In Circular Dichroism and the Conformational Analysis of Biomolecules; Fasman, G.D., Ed.; Plenum Press: New York, NY, USA, 1996; pp. 25–67. [Google Scholar]
- Woody, R.W.; Koslowski, A. Recent developments in the electronic spectroscopy of amides and α-helical polypeptides. Biophys. Chem. 2002, 101–102, 535–551. [Google Scholar] [CrossRef]
- Whitmore, L.; Woollett, B.; Miles, A.J.; Klose, D.P.; Janes, R.W.; Wallace, B.A. PCDDB: The protein circular dichroism data bank, a repository for circular dichroism spectral and metadata. Nucleic Acids Res. 2010, 39, D480–D486. [Google Scholar] [CrossRef] [PubMed]
- Ramalli, S.G.; Miles, A.J.; Janes, R.W.; Wallace, B. The PCDDB (Protein Circular Dichroism Data Bank): A Bioinformatics Resource for Protein Characterisations and Methods Development. J. Mol. Biol. 2022, 434, 167441. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Miles, A.J.; Wien, F.; Wallace, B.A. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 2006, 22, 1955–1962. [Google Scholar] [CrossRef]
- Miles, A.J.; Drew, E.D.; Wallace, B.A. DichroIDP: A method for analyses of intrinsically disordered proteins using circular dichroism spectroscopy. Commun. Biol. 2023, 6, 823. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Wallace, B.; Janes, R.W. Synchrotron radiation circular dichroism spectroscopy of proteins: Secondary structure, fold recognition and structural genomics. Curr. Opin. Chem. Biol. 2001, 5, 567–571. [Google Scholar] [CrossRef]
- Ramos, C.H.I.; Kay, M.S.; Baldwin, R.L. Putative Interhelix Ion Pairs Involved in the Stability of Myoglobin. Biochemistry 1999, 38, 9783–9790. [Google Scholar] [CrossRef] [PubMed]
- A Ribeiro, E.; Ramos, C.H. Origin of the anomalous circular dichroism spectra of many apomyoglobin mutants. Anal. Biochem. 2004, 329, 300–306. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, J.R.; Linhares, L.A.; Aragão, A.Z.; Arruda, M.A.; Ramos, C.H. The stability and function of human cochaperone Hsp40/DNAJA1 are affected by zinc removal and partially restored by copper. Biochimie 2023, 213, 123–129. [Google Scholar] [CrossRef]
- Ramos, C.H.I. A spectroscopic-based laboratory experiment for protein conformational studies. Biochem. Mol. Biol. Educ. 2004, 32, 31–34. [Google Scholar] [CrossRef]
- Silva, J.L.; Oliveira, A.C.; Vieira, T.C.R.G.; de Oliveira, G.A.P.; Suarez, M.C.; Foguel, D. High-Pressure Chemical Biology and Biotechnology. Chem. Rev. 2014, 114, 7239–7267. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.A.; Gava, L.M.; Pinheiro, G.M.S.; Ramos, C.H.; Borges, J.C. From Conformation to Interaction: Techniques to Explore the Hsp70/ Hsp90 Network. Curr. Protein Pept. Sci. 2015, 16, 735–753. [Google Scholar] [CrossRef]
- Borges, J.C.; Ramos, C.H. Analysis of molecular targets of Mycobacterium tuberculosis by analytical ultracentrifugation. Curr. Med. Chem. 2011, 18, 1276–1285. [Google Scholar] [CrossRef]
- Guo, H.; An, S.; Ward, R.; Yang, Y.; Liu, Y.; Guo, X.-X.; Hao, Q.; Xu, T.-R. Methods used to study the oligomeric structure of G-protein-coupled receptors. Biosci. Rep. 2017, 37, BSR20160547. [Google Scholar] [CrossRef]
- Borges, J.C.; Seraphim, T.V.; Dores-Silva, P.R.; Barbosa, L.R.S. A review of multi-domain and flexible molecular chaperones studies by small-angle X-ray scattering. Biophys. Rev. 2016, 8, 107–120. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Hilario, E.; da Silva, S.L.F.; Ramos, C.H.I.; Bertolini, M.C. Effects of cardiomyopathic mutations on the biochemical and biophysical properties of the human α-tropomyosin. JBIC J. Biol. Inorg. Chem. 2004, 271, 4132–4140. [Google Scholar] [CrossRef]
- Rosli, N.E.; Ali, M.S.M.; Kamarudin, N.H.A.; Masomian, M.; Latip, W.; Saadon, S.; Rahman, R.N.Z.R.A. Structure Prediction and Characterization of Thermostable Aldehyde Dehydrogenase from Newly Isolated Anoxybacillus geothermalis Strain D9. Microorganisms 2022, 10, 1444. [Google Scholar] [CrossRef]
- Jesus, C.N.; Evans, R.; Forth, J.; Estarellas, C.; Gervasio, F.L.; Battaglia, G. Amphiphilic Histidine-Based Oligopeptides Exhibit pH-Reversible Fibril Formation. ACS Macro Lett. 2021, 10, 984–989. [Google Scholar] [CrossRef]
- Regis, W.C.; Fattori, J.; Santoro, M.M.; Jamin, M.; Ramos, C.H. On the difference in stability between horse and sperm whale myoglobins. Arch. Biochem. Biophys. 2005, 436, 168–177. [Google Scholar] [CrossRef]
- Opassi, G.; Nordström, H.; Lundin, A.; Napolitano, V.; Magari, F.; Dzus, T.; Klebe, G.; Danielson, U.H. Establishing Trypanosoma cruzi farnesyl pyrophosphate synthase as a viable target for biosensor driven fragment-based lead discovery. Protein Sci. 2020, 29, 977–989. [Google Scholar] [CrossRef]
- dos Santos, R.V.; Grillo, G.; Fonseca, H.; Stanisic, D.; Tasic, L. Hesperetin as an inhibitor of the snake venom serine protease from Bothrops jararaca. Toxicon 2021, 198, 64–72. [Google Scholar] [CrossRef]
- Ruzza, P.; Honisch, C.; Hussain, R.; Siligardi, G. Free Radical Generation in Far-UV Synchrotron Radiation Circular Dichroism Assays—Protein and Buffer Composition Contribution. Int. J. Mol. Sci. 2021, 22, 11325. [Google Scholar] [CrossRef]
- Spöttel, J.; Brockelt, J.; Falke, S.; Rohn, S. Characterization of Conjugates between α-Lactalbumin and Benzyl Isothiocyanate—Effects on Molecular Structure and Proteolytic Stability. Molecules 2021, 26, 6247. [Google Scholar] [CrossRef]
- Shiratori, T.; Goto, S.; Sakaguchi, T.; Kasai, T.; Otsuka, Y.; Higashi, K.; Makino, K.; Takahashi, H.; Komatsu, K. Singular value decomposition analysis of the secondary structure features contributing to the circular dichroism spectra of model proteins. Biochem. Biophys. Rep. 2021, 28, 101153. [Google Scholar] [CrossRef]
- Rajkovic, A.; Kanchugal, S.; Abdurakhmanov, E.; Howard, R.; Wärmländer, S.; Erwin, J.; Saldaña, H.A.B.; Gräslund, A.; Danielson, H.; Flores, S.C. Amino acid substitutions in human growth hormone affect secondary structure and receptor binding. PLoS ONE 2023, 18, e0282741. [Google Scholar] [CrossRef]
- Ramos, C.H.I.; Weisbuch, S.; Jamin, M. Diffusive Motions Control the Folding and Unfolding Kinetics of the Apomyoglobin pH 4 Molten Globule Intermediate. Biochemistry 2007, 46, 4379–4389. [Google Scholar] [CrossRef]
- Menard, L.M.; Wood, N.B.; Vigoreaux, J.O. Secondary Structure of the Novel Myosin Binding Domain WYR and Implications within Myosin Structure. Biology 2021, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Stifter, S.A.; Matthews, A.Y.; Mangan, N.E.; Fung, K.Y.; Drew, A.; Tate, M.D.; da Costa, T.P.S.; Hampsey, D.; Mayall, J.; Hansbro, P.M.; et al. Defining the distinct, intrinsic properties of the novel type I interferon, IFNε. J. Biol. Chem. 2018, 293, 3168–3179. [Google Scholar] [CrossRef] [PubMed]
- Ishak, S.N.H.; Kamarudin, N.H.A.; Ali, M.S.M.; Leow, A.T.C.; Rahman, R.N.Z.R.A. Ion-Pair Interaction and Hydrogen Bonds as Main Features of Protein Thermostability in Mutated T1 Recombinant Lipase Originating from Geobacillus zalihae. Molecules 2020, 25, 3430. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, V.V.; Yampolskaya, D.S.; Kleymenov, S.Y.; Chebotareva, N.A.; Matyushenko, A.M.; Levitsky, D.I. Effect of Neurodegenerative Mutations in the NEFL Gene on Thermal Denaturation of the Neurofilament Light Chain Protein. Biochemistry 2023, 88, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, C.A.; Baase, W.A.; Johnson, W.C. Conformation and circular dichroism of DNA. Biopolymers 1979, 18, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Marqusee, S.; Baldwin, R.L. Helix stabilization by Glu-...Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. USA 1987, 84, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Morrisett, J.D.; David, J.S.K.; Pownall, H.J.; Gotto, A.M. Interaction of an apolipoprotein (apoLP-alanine) with phosphatidylcholine. Biochemistry 1973, 12, 1290–1299. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Yang, J.T.; Chau, K.H. Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry 1974, 13, 3350–3359. [Google Scholar] [CrossRef]
- Greenfield, N.J.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.J.; Ramalli, S.G.; Wallace, B.A. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2021, 31, 37–46. [Google Scholar] [CrossRef]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins: Struct. Funct. Bioinform. 2011, 80, 374–381. [Google Scholar] [CrossRef]
- Nagy, G.; Igaev, M.; Jones, N.C.; Hoffmann, S.V.; Grubmüller, H. SESCA: Predicting Circular Dichroism Spectra from Protein Molecular Structures. J. Chem. Theory Comput. 2019, 15, 5087–5102. [Google Scholar] [CrossRef]
- Drew, E.D.; Janes, R.W. PDBMD2CD: Providing predicted protein circular dichroism spectra from multiple molecular dynamics-generated protein structures. Nucleic Acids Res. 2020, 48, W17–W24. [Google Scholar] [CrossRef]
- Brookes, E.; Rocco, M. A database of calculated solution parameters for the AlphaFold predicted protein structures. Sci. Rep. 2022, 12, 7349. [Google Scholar] [CrossRef]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef]
- Edelhoch, H. Spectroscopic Determination of Tryptophan and Tyrosine in Proteins. Biochemistry 1967, 6, 1948–1954. [Google Scholar] [CrossRef]
- Schmid, F. Optical spectroscopy to characterize protein conformation and conformational changes. In Protein Structure: A Practical Approach; Creighton, T.E., Ed.; Oxford University Press: New York, NY, USA, 1997; pp. 261–297. [Google Scholar]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Corrêa, D.H.A.; Ramos, C.H.I. The use of circular dichroism spectroscopy to study protein folding, form and function. Afr. J. Biochem. Res. 2009, 3, 164–173. [Google Scholar]
- Buck, M. Trifluoroethanol and colleagues: Cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998, 31, 297–355. [Google Scholar] [CrossRef]
- Mares-Guia, T.R.; Maigret, B.; Martins, N.F.; Maia, A.L.T.; Vilela, L.; Ramos, C.H.I.; Neto, L.J.; Juliano, M.A.; dos Mares-Guia, M.L.; Santoro, M.M. Molecular dynamics and circular dichroism studies of human and rat C-peptides. J. Mol. Graph. Model. 2006, 25, 532–542. [Google Scholar] [CrossRef]
- Timasheff, S.N. The Control of Protein Stability and Association by Weak Interactions with Water: How Do Solvents Affect These Processes? Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 67–97. [Google Scholar] [CrossRef]
- Ramos, C.H.I. Mapping Subdomains in the C-terminal Region of Troponin I Involved in Its Binding to Troponin C and to Thin Filament. J. Biol. Chem. 1999, 274, 18189–18195. [Google Scholar] [CrossRef]
- Ramos, C.H.; Ferreira, S.T. Protein Folding, Misfolding and Aggregation: Evolving Concepts and Conformational Diseases. Protein Pept. Lett. 2005, 12, 213–222. [Google Scholar] [CrossRef]
- Ramos, C.H.; Lima, M.V.; Silva, S.L.; Borin, P.F.; Régis, W.C.; Santoro, M.M. Stability and folding studies of the N-domain of troponin C. Evidence for the formation of an intermediate. Arch. Biochem. Biophys. 2004, 427, 135–142. [Google Scholar] [CrossRef]
- Benjwal, S.; Verma, S.; Röhm, K.-H.; Gursky, O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006, 15, 635–639. [Google Scholar] [CrossRef]
- Miles, A.J.; Wallace, B.A. Synchrotron radiation circular dichroism spectroscopy of proteins and applications in structural and functional genomics. Chem. Soc. Rev. 2005, 35, 39–51. [Google Scholar] [CrossRef]
- Wallace, B.; Janes, R.W. Synchrotron radiation circular dichroism (SRCD) spectroscopy: An enhanced method for examining protein conformations and protein interactions. Biochem. Soc. Trans. 2010, 38, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, P.S.; Araujo, A.P.U.; Lopes, J.L.S. Going deep into protein secondary structure with synchrotron radiation circular dichroism spectroscopy. Biophys. Rev. 2017, 9, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Andersson, E.; Jones, N.C.; Hoffmann, S.V.; Jemth, P.; Kjaergaard, M. Coupled Binding and Helix Formation Monitored by Synchrotron-Radiation Circular Dichroism. Biophys. J. 2019, 117, 729–742. [Google Scholar] [CrossRef]
- Makumire, S.; Zininga, T.; Vahokoski, J.; Kursula, I.; Shonhai, A. Biophysical analysis of Plasmodium falciparum Hsp70-Hsp90 organising protein (PfHop) reveals a monomer that is characterised by folded segments connected by flexible linkers. PLoS ONE 2020, 15, e0226657. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.V.; Jacinto, J.P.; Guerra, J.P.L.; Vieira, B.J.C.; Waerenborgh, J.C.; Jones, N.C.; Hoffmann, S.V.; Pereira, A.S.; Tavares, P. Structural features and stability of apo- and holo-forms of a simple iron–sulfur protein. Eur. Biophys. J. 2021, 50, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Théron, L.; Bonifacie, A.; Delabre, J.; Sayd, T.; Aubry, L.; Gatellier, P.; Ravel, C.; Chambon, C.; Astruc, T.; Rouel, J.; et al. Investigation by Synchrotron Radiation Circular Dichroism of the Secondary Structure Evolution of Pepsin under Oxidative Environment. Foods 2021, 10, 998. [Google Scholar] [CrossRef]
- Buckley, A.; Warren, J.; Hussain, R.; Smith, R. Synchrotron radiation circular dichroism spectroscopy reveals that gold and silver nanoparticles modify the secondary structure of a lung surfactant protein B analogue. Nanoscale 2023, 15, 4591–4603. [Google Scholar] [CrossRef] [PubMed]
- Wien, F.; Kubiak, K.; Turbant, F.; Mosca, K.; Węgrzyn, G.; Arluison, V. Synchrotron Radiation Circular Dichroism, a New Tool to Probe Interactions between Nucleic Acids Involved in the Control of ColE1-Type Plasmid Replication. Appl. Sci. 2022, 12, 2639. [Google Scholar] [CrossRef]
- Cappannini, A.; Mosca, K.; Mukherjee, S.; Moafinejad, S.N.; Sinden, R.R.; Arluison, V.; Bujnicki, J.; Wien, F. NACDDB: Nucleic Acid Circular Dichroism Database. Nucleic Acids Res. 2022, 51, D226–D231. [Google Scholar] [CrossRef]
- Harada, T.; Moriyama, H. Solid-State Circular Dichroism Spectroscopy. In Encyclopedia of Polymer Science and Technology, 3rd ed.; Mark, H.F., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–29. [Google Scholar]
- Castiglioni, E.; Biscarini, P.; Abbate, S. Experimental aspects of solid state circular dichroism. Chirality 2009, 21, E28–E36. [Google Scholar] [CrossRef]
- Sanchez-Rexach, E.; Smith, P.T.; Gomez-Lopez, A.; Fernandez, M.; Cortajarena, A.L.; Sardon, H.; Nelson, A. 3D-Printed Bioplastics with Shape-Memory Behavior Based on Native Bovine Serum Albumin. ACS Appl. Mater. Interfaces 2021, 13, 19193–19199. [Google Scholar] [CrossRef]
- Kuroda, R.; Harada, T.; Shindo, Y. A solid-state dedicated circular dichroism spectrophotometer: Development and application. Rev. Sci. Instrum. 2001, 72, 3802–3810. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C.C.F. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys. 1998, 31, 1–39. [Google Scholar] [CrossRef]
- Hu, H.Y.; Li, Q.; Cheng, H.C.; Du, H.N. β-sheet structure formation of proteins in solid state as revealed by circular dichroism spectroscopy. Biopolym. Orig. Res. Biomol. 2001, 62, 15–21. [Google Scholar] [CrossRef]
- Hu, H.-Y.; Jiang, L.-L.; Hong, J.-Y. Study of Protein Amyloid-Like Aggregates by Solid-State Circular Dichroism Spectroscopy. Curr. Protein Pept. Sci. 2016, 18, 100–103. [Google Scholar] [CrossRef]
- Brown, C.P.; Hughes, M.D.G.; Mahmoudi, N.; Brockwell, D.J.; Coletta, P.L.; Peyman, S.A.; Evans, S.D.; Dougan, L. Structural and mechanical properties of folded protein hydrogels with embedded microbubbles. Biomater. Sci. 2023, 11, 2726–2737. [Google Scholar] [CrossRef]
- Hughes, M.D.G.; Cussons, S.; Mahmoudi, N.; Brockwell, D.J.; Dougan, L. Single molecule protein stabilisation translates to macromolecular mechanics of a protein network. Soft Matter 2020, 16, 6389–6399. [Google Scholar] [CrossRef]
- Adams, Z.C.; Olson, E.J.; Lopez-Silva, T.L.; Lian, Z.; Kim, A.Y.; Holcomb, M.; Zimmermann, J.; Adhikary, R.; Dawson, P.E. Direct observation of peptide hydrogel self-assembly. Chem. Sci. 2022, 13, 10020–10028. [Google Scholar] [CrossRef]
- Baumruk, V.; Keiderling, T.A. Vibrational circular dichroism of proteins in water solution. J. Am. Chem. Soc. 1993, 115, 6939–6942. [Google Scholar] [CrossRef]
- Yang, G.; Xu, Y. Vibrational Circular Dichroism Spectroscopy of Chiral Molecules. In Electronic and Magnetic Properties of Chiral Molecules and Supramolecular Architectures; Topics in Current Chemistry; Naaman, R., Beratan, D., Waldeck, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 298, pp. 189–236. [Google Scholar] [CrossRef]
- Li, Z.; Hirst, J.D. Quantitative first principles calculations of protein circular dichroism in the near-ultraviolet. Chem. Sci. 2017, 8, 4318–4333. [Google Scholar] [CrossRef]
- Kurouski, D. Advances of Vibrational Circular Dichroism (VCD) in bioanalytical chemistry. A review. Anal. Chim. Acta 2017, 990, 54–66. [Google Scholar] [CrossRef]
- Xu, C.; Ren, Z.; Zhou, H.; Zhou, J.; Ho, C.P.; Wang, N.; Lee, C. Expanding chiral metamaterials for retrieving fingerprints via vibrational circular dichroism. Light Sci. Appl. 2023, 12, 154. [Google Scholar] [CrossRef]
- Kessler, J.; Andrushchenko, V.; Kapitán, J.; Bouř, P. Insight into vibrational circular dichroism of proteins by density functional modeling. Phys. Chem. Chem. Phys. 2018, 20, 4926–4935. [Google Scholar] [CrossRef]
- Litwińczuk, A.; Ryu, S.R.; Nafie, L.A.; Lee, J.W.; Kim, H.I.; Jung, Y.M.; Czarnik-Matusewicz, B. The transition from the native to the acid-state characterized by multi-spectroscopy approach: Study for the holo-form of bovine α-lactalbumin. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2014, 1844, 593–606. [Google Scholar] [CrossRef]
- Rudd, T.R.; Nichols, R.J.; Yates, E.A. Selective Detection of Protein Secondary Structural Changes in Solution Protein−Polysaccharide Complexes Using Vibrational Circular Dichroism (VCD). J. Am. Chem. Soc. 2008, 130, 2138–2139. [Google Scholar] [CrossRef]
- Kurouski, D.; Lu, X.; Popova, L.; Wan, W.; Shanmugasundaram, M.; Stubbs, G.; Dukor, R.K.; Lednev, I.K.; Nafie, L.A. Is Supramolecular Filament Chirality the Underlying Cause of Major Morphology Differences in Amyloid Fibrils? J. Am. Chem. Soc. 2014, 136, 2302–2312. [Google Scholar] [CrossRef]
- Pazderková, M.; Pazderka, T.; Shanmugasundaram, M.; Dukor, R.K.; Lednev, I.K.; Nafie, L.A. Origin of enhanced VCD in amyloid fibril spectra: Effect of deuteriation and pH. Chirality 2017, 29, 469–475. [Google Scholar] [CrossRef]
- Keiderling, T.A. Structure of Condensed Phase Peptides: Insights from Vibrational Circular Dichroism and Raman Optical Activity Techniques. Chem. Rev. 2020, 120, 3381–3419. [Google Scholar] [CrossRef]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linhares, L.A.; Ramos, C.H.I. Unlocking Insights into Folding, Structure, and Function of Proteins through Circular Dichroism Spectroscopy—A Short Review. Appl. Biosci. 2023, 2, 639-655. https://doi.org/10.3390/applbiosci2040040
Linhares LA, Ramos CHI. Unlocking Insights into Folding, Structure, and Function of Proteins through Circular Dichroism Spectroscopy—A Short Review. Applied Biosciences. 2023; 2(4):639-655. https://doi.org/10.3390/applbiosci2040040
Chicago/Turabian StyleLinhares, Leonardo A., and Carlos H. I. Ramos. 2023. "Unlocking Insights into Folding, Structure, and Function of Proteins through Circular Dichroism Spectroscopy—A Short Review" Applied Biosciences 2, no. 4: 639-655. https://doi.org/10.3390/applbiosci2040040
APA StyleLinhares, L. A., & Ramos, C. H. I. (2023). Unlocking Insights into Folding, Structure, and Function of Proteins through Circular Dichroism Spectroscopy—A Short Review. Applied Biosciences, 2(4), 639-655. https://doi.org/10.3390/applbiosci2040040