Abstract
Deregulated transcription is a well-known characteristic of cancer cells, with differentially expressed genes being a common feature of several cancers. Often, deregulated transcription is a consequence of alterations in transcription factors (TFs), which play a crucial role in gene expression and can act as tumour suppressors or proto-oncogenes. In eukaryotic organisms, transcription is carried out by three distinct RNA polymerase complexes: Pol I, Pol II, and Pol III. Pol II, specifically, is responsible for transcribing messenger RNA (mRNA), the protein coding part of the genome, as well as long non-coding RNAs (lncRNAs). While there is considerable research on the impact of specific deregulated transcription factors in cancer development, there is a lack of studies focusing on defects within the RNA polymerase complexes and their subunits. This review aims to shed light in particular on the Pol II complex and highlight the deregulation of its subunits that have a significant impact on tumour development, prognosis, and survival. By providing a comprehensive overview of our current understanding of Pol II subunits in cancer, this review emphasizes the importance of further research in this area. It suggests that exploring these subunits’ deregulations could lead to the identification of valuable biomarkers and potential therapeutic targets, making it a topic of collective interest.
1. Introduction
Eukaryotic transcription is carried out by three different RNA polymerase machineries: RNA Polymerase I (Pol I), RNA Polymerase II (Pol II), and RNA Polymerase III (Pol III). Pol I is entitled to transcribe ribosomal RNAs (28S rRNA, 18S rRNA, and 5.8S rRNA); Pol II is devoted to the transcription of the coding part of the genome, messenger RNA (mRNA), long non-coding RNAs (lncRNAs), and small nuclear RNAs (snRNAs); Pol III is responsible for the transcription of transfer RNAs (tRNAs), 5S rRNA, and small nuclear RNAs (snRNAs). RNA Polymerases I, II, and III are complexes of 14, 12, and 17 subunits, respectively [1,2]. Five of the subunits, named DNA-directed RNA Polymerase subunits, RPB5-6-8-10-12 are shared among all three polymerase complexes. These subunits are encoded by the genes POLR2E-F-H-K-L, respectively.
- In Pol I, RPB5, RPB6, RPB8, RPB10, and RPB12 together with subunits RPA190, RPA135, RPAC40, RPAC19, and RPA12.2 form the core of the enzyme. Four other subunits, RPA43-RPA14, and the RPA49-RPA34.5 complete the complex [3]. The subunits specific to RNA Pol I are encoded by the genes POLR1A-H.
- In the Pol II complex (built up of 12 subunits RPB1–12), RPB1, RPB2, RPB3, and RPB11 share sequence homology with other Pol I and III subunits. RPB4, RPB7, RPB9, and the disordered c-terminal domain (CTD) of RPB1 subunit are specific to Pol II [4]. The genes encoding for Pol II 12 subunits are POLR2A-K.
- RNA Pol III is comprised of a core of 10 subunits and a peripheral heterodimeric stalk. The TFIIF-like RPC4/5 and the TFIIE-like RPC3/6/7 subcomplexes are Pol III specific. The subunits specific to Pol III are encoded by POLR3A-K genes [5].
RNA polymerase complexes work in concert with transcription activators and repressors. Sequence-specific DNA binding transcription factors (TFs) bind regulatory elements and interact via protein–protein interaction with the polymerases, in order to regulate the assembly of the transcription machinery and ensure the smooth course of transcription [6,7,8,9,10]. The activity of TFs is also controlled by coactivators and corepressors, which are multiprotein complexes with multiple enzymatic functions. Coactivators and corepressors’ actions can be summarised in two categories: (i) bridging sequence-specific DNA binding TFs with the general transcription machinery, like, for example, the case of the Mediator complex [11]; (ii) altering chromatin accessibility, as, for example, through nucleosome remodelling by the ATP-dependent complex hSWI/SNF, or post-translational modifications of histone tails like acetylation by histone acetyl transferases (HATs) associated with transcription activation, deacetylation by histone deacetylases (HDACs) associated with repression, or methylation associated with both activation or repression, depending on the site and the complex responsible [12,13,14,15].
In 2000, a seminal work by Hanahan and Weinberg defined the six hallmarks of cancer as alterations in the physiology of the cell that represent novel capabilities acquired by cancer cells during tumorigenesis: self-sufficiency in growth signals, insensitivity to antigrowth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis [16]. In the subsequent years, genome instability, among others, has also emerged as new hallmark of cancer, together with deregulated transcription (Figure 1) [17,18]. Because of their role as major regulators of gene expression, many transcription factors, activators and repressors, are deregulated (altered gene expression) or mutated in cancers and are well-established oncogenes/tumour suppressor genes [19]. Among them, it is worthwhile to mention the retinoblastoma (RB) tumour suppressor gene and its binding partner E2F1 (and other E2F genes). Rb binds E2F in its phosphorylated form (pRb) and represses E2F transcriptional activity. Indeed, Rb is phosphorylated by cell cycle-dependent kinases (CDKs), and in this way, its action and its activity on E2F is controlled through the cell cycle, tightly regulated to control cell proliferation. Cancers commonly bear a disruption of this pRB-E2F regulation [20,21]; TP53, commonly referred to as the guardian of the genome, is a tumour suppressor gene involved in response to stress signals including replication stress, metabolic stress, and DNA damage. It promotes the transcription of genes involved in the cell stress response and it is a central node in cell cycle control and arrest, cell senescence, and apoptosis, found to be mutated/deleted across a wide variety of cancer types [22]. Oncogenes of the MYC family (C-L-N- Myc), which regulate about 10% of the expressed genes and also some Pol I and Pol III transcripts, are usually upregulated in a wide variety of tumours [23,24,25,26,27,28,29,30]. Beyond deregulations of TFs, there is also evidence of transcription deregulations of RNA Pol I and Pol III complexes in cancers [19,31,32], while specific Pol II deregulations in relation to cancer remain elusive. Moreover, although the transcription regulation mediated by TFs occurs via their interaction with the subunits composing the RNA polymerase complexes [6,7,8,9], studies on the subunits of the RNA Polymerase II machinery itself remain elusive. The purpose of this review is to describe which are the mutations/deregulations affecting Pol II subunits linked to cancer development identified by research so far.
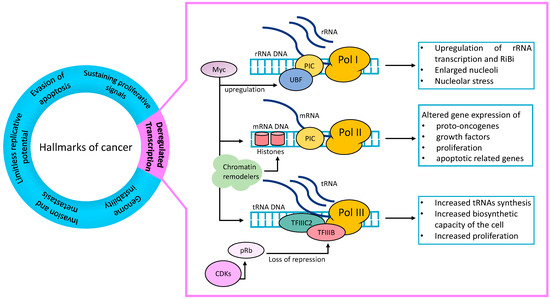
Figure 1.
Deregulated transcription is a hallmark of cancer: Transcription factors (TFs), which can be both oncogenes and tumour suppressor genes, and transcription complexes are deregulated in cancer. Pol I rRNA transcription and Ribosomal Biosynthesis (RiBi) can be upregulated in cancer cells. The TF and oncogene Myc upregulates UBF, a component of Pol I transcription machinery, thus increasing Pol I transcription. TFs and chromatin remodellers deregulate the Pol II genes transcription and result in altered gene expression. Cancer cells’ increased demand of cellular components to sustain their high proliferation need is satisfied by increased Pol III transcription and increased tRNAs synthesis. TFIIIC2 and TFIIIB upregulations also increase transcription rates. The tumour suppressor gene Rb, when phosphorylated by CDKs (pRb), is no longer able to exercise its repressive function on TFIIIB and results in TFIIIB derepression and increased Pol III transcription. UBF = Upstream Binding Factor; PIC = preinitiation complex; CDKs = cyclin-dependent kinases; Rb = retinoblastoma tumour suppressor gene; pRb = phospho-Rb.
2. Pol I
Hyperactivation of ribosomal DNA (rDNA) transcription and enlarged nucleoli (nucleolar hypertrophy) are common features of aggressive tumours, together with deregulation of ribosome biosynthesis (RiBi) and overexpression of rRNA [33]. These observations led to suggestions that Pol I could be directly targeted in cancer therapy. Indeed, in recent years, Pol I inhibitors have been developed and tested in a wide range of tumours. Additionally, many oncogenes and oncoproteins, such as AKT, PI3K, and Ras, play important roles in ribosome biogenesis, making the activation of the ribosomal surveillance pathway and consequent p53 activation a useful therapeutic means for a wide range of tumours [31,32,34]. Furthermore, well-known transcription factors which are tumour suppressor genes or oncogenes, for example, RB and MYC, regulate the transcription of Upstream Binding Factor (UBF), an essential component of the Pol I pre-initiation complex, resulting in repression or upregulation of Pol I transcription, respectively (Figure 1) [32,35,36]. Myc stimulates the expression of UBF, upregulating ribosome biosynthesis, and thus sensitizing cells to nucleolar stress. Therefore, it has been proposed that Pol I transcription in MYC-driven tumour cells could be a promising target, as inhibition of rDNA transcription could disrupt the ability of Myc to suppress p53 accumulation in tumour cells more than in normal cells [36]. Among the Pol I inhibitors, CX-5461 prevents the formation of Pol I pre-initiation complex and inhibits transcription initiation by impeding Pol I release from the promoter, and is very specific to Pol I and does not affect Pol II or III [36,37]. This small-molecule inhibitor has shown promising results in killing Eμ-MYC lymphoma cells by activating p53-dependent apoptosis [34]. Interestingly, CX-5461 has also been successful in the treatment of chemoresistant ovarian cancers, showing promising results in taxane resistant cells, which are instead sensitive to CX-5461 [38,39]. Another inhibitor, CX-3543, binds quadruplex DNA and inhibits Pol I transcription by inhibiting nucleolin-G-quadruplex complex formation, which leads to the translocation of nucleolin into the nucleoplasm and the triggering of apoptosis [35]. BMH-21 is a potent compound capable of inducing p53 activation selectively in different cancer cell lines but not in normal cells, which works by binding DNA GC-rich regions. Being rDNA highly GC-rich, it has been shown that BMH-21 functions by disassembling the Pol I complex at rDNA promoter regions to inhibit rDNA transcription [40]. Recently, it was also found that Pol I activity is upregulated in prostate cancers. CX-5461 blocked proliferation and suppressed prostate cancer tumorigenesis in a p53-dependent manner [41]. Similarly, BMH-21 decreased prostate cancer cell growth in vitro and in vivo in different cell lines, also in a p53-independent manner [41].
Deregulations specifically affecting some of the subunits of Pol I complex are also reported to have a role in cancer. POLR1A is overexpressed in tumoral and metastatic cell lines of colorectal cancer [42]. Similarly, POLR1B is usually upregulated in a broad range of human cancers including non-small cells lung cancer (NSCLC), where knockdown of POLR1B resulted in reduced cell proliferation [43]. Furthermore, in ER-positive breast cancer, it can be considered a biomarker for resistance to fulvestrant [44]. The POLR1C subunit is differentially expressed in breast cancer patients compared to normal tissue and either amplified or overexpressed in gastric cancer [45]. In colorectal cancer, a region in 13q containing POLR1D is frequently amplified, resulting in POLR1D overexpression [46], which is associated with higher risk, poorer overall survival, and acquired resistance to bevacizumab, identifying POLR1D as a potential therapeutic target [47,48,49]. Finally, POLR1E expression correlates with increased tumour stage in bladder cancer [50].
3. RNA Polymerase III
Similar to Pol I transcription, Pol III transcription can also contribute to carcinogenesis. Indeed, it was found both in ovarian and breast cancer cells that Pol III transcription can lead to the aberrant production of its transcripts that contribute to cell growth or to the overproduction of tRNAs [51,52]. The accelerated proliferation of cancer cells necessitates an increase in the synthesis of cellular components to match their heightened rate of division. To achieve the necessary boost in biosynthetic capacity for rapid proliferation, the output of Pol III is enhanced [53]. Hyper activation of Pol III and components of Pol III transcription such as TFIIIB complex was observed in different cancer types and in mice with myeloma compared to tumour free mice [54], and TFIIIC2 was found to be overexpressed in ovarian carcinomas [52]. It was also found that Pol III activity is elevated in primary human fibroblasts carrying loss of function mutations in p53 [55], as well as in mice lacking Rb [56,57]. Indeed, many known mutations in RB lay in a region involved in binding TFIIIB, and in this way repress Pol III transcription. When this binding is compromised, usually by hyperphosphorylation of RB by CDK-E/D, TFIIIB is de-repressed and further activates Pol III. Hyperactivation of Pol III has also been associated with c-MYC upregulation (Figure 1) ([32] and citations therein). This supports the belief that limiting Pol III activity can impede robust cancer proliferation, as demonstrated by studies in which reduced Pol III activity in mouse xenograft models inhibited tumour formation. Pol III and Pol III-associated transcription factors could potentially serve as valuable biomarkers for cancer diagnosis [53,58]. More recently, it has been found that the subunit POLR3G is capable of binding the telomerase reverse transcriptase TERT [59], a central protein in stimulating the proliferative capabilities of many cancer cell types. In support and extension of this finding, it was shown that POLR3G inhibition resulted in prostate cancer cell-specific proliferation arrest and cell death [60] and that POLR3G overexpression correlated with poor prognosis of transitional cell carcinoma [61]. POLR3G was also identified in clinical data as one out of five genes in a risk signature predicting the prognosis of patients with hepatocellular carcinoma [62]. More recently, Lautré and colleagues showed that POLR3G is the only component of the Pol III transcription apparatus significantly overexpressed in triple-negative breast cancer (TNBC) but not in other types of breast cancer. Suppression of POLR3G in the MDA-MB231 TNBC cell line decreases colony formation and invasive growth in vitro. Importantly, POLR3G KO impairs tumour growth and metastasis of intraductal xenografts in mice [63]. In conclusion, POLR3G expression plays a significant role in regulating tumour formation and metastasis in TNBC, with its deletion leading to altered gene expression patterns and cell fate decisions. Further research will determine whether targeting POLR3G could emerge as a potential therapeutic strategy for TNBC. Other Pol III subunits have been linked to cancer, like mutations in POLR3B implicated in lung cancer, with the gene found to be differentially methylated, making POLR3B a susceptibility and risk gene in lung cancer [64,65,66,67]. Furthermore, POLR3K subunit high expression is correlated with poorer overall and disease free survival in ovarian cancer [68].
4. RNA Pol II
The Pol II active centre is formed by the subunits RPB1 and RPB2, which sit at two opposite sides of a positively charged “cleft”, the entry point of DNA for elongation, together with RPB3, -10, -11, and -12 [69,70]. RPB1 forms a mobile “clamp” on its side of the cleft, which can be in an open or closed state depending on whether the structure is devoid or not of DNA and RNA, respectively [1,69]. A small portion of RPB2 and the N-terminal of RPB6 contribute to the structure of the clamp [71]. From the base of the clamp protrudes the “linker”, connecting RPB1 with its C-terminal domain (CTD) [70], which is phosphorylated in order to coordinate transcription progression and co-transcriptional events. RPB2 side of the cleft constitutes the “lobe” and “protrusion domains”, blocking the end of the cleft [1,69].
Generally, Pol II is divided into functional domains defined as the clamp (comprehensive of RPB1 mainly and RPB2), the jaw lobe (RPB2, RPB1 and RPB9), the RPB5 jaw, and the stalk (RPB4, RPB7) [72]. The latter binds the polymerase core through the RPB7 N-terminal domain, called the “tip”. The stalk subunits could function in recruiting factors to the CTD and in mediating the interactions with transcription initiation factors, thus contributing to promoter specificity [70]. Additionally, RPB4 tunes the CTD phosphorylation levels [73] and guides nascent RNA co-transcriptional processing, by directly interacting with the nascent transcripts and recruiting co-transcriptional factors, thus being important in transcription termination [74,75,76].
For the purpose of this review, we will focus on the RNA Polymerase II complex and its subunits. Cancer cells rely on transcription to sustain their growth and survival. Therefore, blocking RNA synthesis via RNA Polymerase inhibitors like DRB or flavopiridol, or nucleotide analogues such as 3′-ethynylcytidine (Ecyd), is an effective strategy both in vitro and in vivo [77,78,79,80]. Definitely, it is possible to downregulate many oncogenes by reducing overall transcription levels. So far, little is known about Pol II subunits implications in human diseases with a particular focus on cancer. However, recent studies provide evidence of emerging roles of some Pol II subunits (Table 1).
5. POLR2A/RPB1
POLR2A subunit, being at the core of the catalytic Pol II active site, is considered an essential transcription gene and oncogene, being able to promote rapid cell growth and repress apoptosis in tumours [81,82]. In fact, usually POLR2A is highly expressed in tumour tissues compared to neighbouring normal tissues. For example, POLR2A overexpression has been found to induce proliferation in gastric cancer cells and inhibit apoptosis both in vitro and in vivo [82]. Furthermore, POLR2A expression level was found to be higher in patients derived xenograft models of ovarian cancer [83]. Interestingly, POLR2A high protein levels were found in AML cell lines compared to normal progenitor cells. Accordingly, mRNA expression levels were also higher in primary AML patients, resulted in reduced overall survival and increased tumour growth. Indeed, POLR2A KD reduced cell proliferation and tumour growth in vitro and in vivo [84]. Analysing POLR2A mRNA expression levels in TCGA dataset, it was also found that POLR2A upregulation occurs not only in AML patients, but also in cholangiocarcinoma and thymoma [84]. On the contrary, POLR2A was downregulated in adrenocortical carcinoma, testicular germ cell tumours and uterine corpus endometrial carcinoma. Additionally, polymorphisms in POLR2A were associated with poor outcomes in non-small cell lung cancer [85]. The POLR2A gene lays in a neighbouring region of TP53 on Chr 17 and is often co-deleted together with it as a “passenger gene”. In fact, in colorectal cancer POLR2A is commonly hemizygously deleted together with TP53, rendering cancer cells more sensitive to the transcription elongation inhibitor α-amanitin [86] and POLR2A depletion with siRNAs, suggesting the possibility of targeting POLR2A in p53−/+ cancer cells or cancers with a broader 17p loss [87,88,89,90]. Similarly, siRNA inhibition of POLR2A in hemizygous TP53 TNBCs where POLR2A is hemizygously co-deleted resulted in reduced tumour growth and enhanced tumour suppression [91].
6. POLR2B/RPB2
POLR2B is the second largest subunit of Pol II and, together with POLR2A, forms the catalytic site of the complex. There are not many studies regarding POLR2B deregulations in cancer. However, it has been shown that mutations in POLR2B are linked to sensitivity to JQ1, a bromodomain inhibitor targeting BET proteins BRD2-3-4 in breast cancer cell lines. JQ1 sensitivity was assessed in over 1000 cancer cell lines to identify possible markers of sensitivity, and POLR2B mutations were identified in breast cancer cell lines as predictors of JQ1 sensitivity [92,93]. Furthermore, POLR2B downregulation could play a role in cell resistance to 5-Azacytidine (AZA), a DNA demethylating agent widely used in cancer treatment and more specifically in treatment of patients with AML, with a moderately good response rate. Indeed, POLR2B mRNA expression level was found to be significantly decreased in AZA resistant cell lines compared to non-resistant ones [94]. More recent studies analysing TCGA datasets found upregulated POLR2B in hepatocellular carcinoma and high mRNA levels to be correlated with overall reduced patient survival rates and were associated with tumour staging. Consistently, high POLR2B protein expression levels were observed in the cancer tissue compared to normal liver tissue. Additionally, POLR2B expression was even higher in virus (HBV or HCV)-driven hepatocellular carcinoma. Therefore, POLR2B gene has a possible value for further development as a diagnostic biomarker in virus-related HCC [95].
7. POLR2C/RPB3
As for POLR2B, not much research highlights the role of POLR2C deregulations in general in cancer. However, a few studies conducted in hepatocellular carcinoma (HCC) cell lines and tissues found that POLR2C protein expression is increased by immunohistochemical staining in tissue samples of different stages compared to normal tissue, and higher expression (stronger staining) was progressively observed from low to high grade HCC. Furthermore, POLR2C overexpression in patients correlated with poorer overall and disease-free survival. HCC cell lines overexpressing POLR2C had also enhanced proliferation and migration capabilities, which translated into enhanced tumour growth in injected mice. POLR2C overexpression upregulated a series of mesenchymal markers, such as N-cadherin, and downregulated epithelial markers, such as E-cadherin, thus promoting epithelial to mesenchymal transition (EMT). Interestingly, POLR2C was found to be a regulator of E-cadherin expression through its interaction with Snail, a key regulator of E-cadherin transcription, via its N-terminal domain. Intriguingly, Pol II activity and the expression of the other subunits of the complex remained unchanged. Another mechanism through which POLR2C regulates HCC cell proliferation is due to its regulation of VOPP1 expression, which is upregulated in different cancers and promotes cell proliferation and migration while inhibiting apoptosis [96,97]. Other than in HCC, POLR2C expression alterations were also found in osteosarcoma tumour samples, where mRNA expression is significantly lower compared to normal osteoblasts [98], and in gastric cancer. In the latter, microarray analysis, confirmed from TCGA analysis of differentially expressed genes between gastric cancer patients showing cisplatin resistance and patients showing drug sensitivity, found POLR2C to be upregulated together with two other Pol II subunits, POLR2L and POLR2F, thus indicating that these subunit upregulations could be involved in mechanisms of cisplatin resistance in gastric cancer [99].
8. POLR2D/RPB4
Some research has been conducted on POLR2D functions in tumour development. The first evidence comes from colorectal cancer, where it was found that POLR2D mRNA expression levels significantly correlated with increased expression of the CA 19-9 tumour marker in patients, indicating that this gene could be used to evaluate disease state [100]. POLR2D was also found to be upregulated in one sample of single cell RNA-Seq analysis performed on malignant ascites cells from ovarian cancer patients [101]. The upregulation of POLR2D is also important in prostate cancer, where, analysing patients on the TCGA database, it was shown that high POLR2D expression associated with significantly lower disease-free survival compared to patients with low expression of POLR2D [102]. Consistent with these observations, the downregulation of POLR2D is considered to be a protective factor in highly grade serous ovarian cancer, being preferentially downregulated in tumour samples compared to normal ones and associated with better overall survival [103].
9. POLR2E/RPB5
So far, upregulation and overexpression have been identified as the main deregulations of Pol II subunits in a wide variety of tumour types. However, the alteration linking the POLR2E subunit and cancer is not related to a differential expression of the gene, but it is a well characterized polymorphism. It was firstly identified in 2011 by Jin and colleagues, who reported, in a prostate cancer genome wide association study, that the SNP rs3787016 in POLR2E is associated with increased risk of prostate cancer occurrence [104]. They proposed that this variant could interfere with correct splicing and therefore produce a dysfunctional POLR2E, thus impacting Pol II transcription. More recently, these results were further confirmed in a sample of prostate cancer in an Iranian population, and additionally, a new variant of POLR2E, rs1046040, was found to be a predisposition factor for cancer development [105]. Interestingly, other studies have shown how this polymorphism, located on the fourth intron of POLR2E gene, was also significantly associated with oesophageal cancer, breast cancer, papillary thyroid carcinoma, and liver cancer [106,107,108,109].
10. POLR2F/RPB6
POLR2F encodes the sixth largest subunit of Pol II complex, and it is also one of the shared subunits among the three polymerase complexes (Pol I, Pol II, Pol III). Early studies showed that POLR2F, together with two other genes, was significantly overexpressed in colorectal carcinoma tissues compared to normal tissues, and specifically its overexpression correlated with early disease occurrence and relapse [110]. Previous studies showed POLR2F to be upregulated in a colorectal cancer metastatic tumour cell line [111]. As previously mentioned for the POLR2C subunit, TCGA analysis of differentially expressed genes, comparing gastric cancer patients with resistance to cisplatin towards patients showing drug sensitivity, found POLR2F to be upregulated, indicating that these subunit upregulations could be involved in mechanisms of cisplatin resistance in gastric cancer [99]. Furthermore, an analysis of microarray datasets TNBC and non-TNBC identified 1075 differentially expressed genes, among which POLR2F was found to be upregulated significantly in TNBC and identified as a potential cancer-causing gene [112]. Lastly, a retrospective study on prostate cancer integrating transcriptome and clinical and pathological data from different databases identified POLR2F upregulation as being associated with poorer prognosis in patients and predictive of worse outcomes in androgen deprivation therapy treatment [113]. Not only does the upregulation/overexpression of POLR2F have a potential prognostic value in tumours, but there is evidence that downregulation of POLR2F could also be relevant. It was found that POLR2F expression was significantly lower in glioblastoma tumour tissues compared to normal tissues and that this impacted on overall survival [114]. However, in silico analysis to identify genes important for tumour progression and survival identified POLR2F as a survival gene in glioblastoma, and specifically in tumours with IDH1 mutations, POLR2F appears to be significantly overexpressed [115]. Moreover, it was also found that lower levels of the protein are associated with increased risk in colorectal cancer, and POLR2F lower expression was found to be associated with a poorer prognosis in cervical cancer in patients positive for HPV18 [116,117].
11. POLR2G/RPB7
Findings on POLR2G subunits are controversial. Li et al. conducted an analysis matching clinical data with mRNA expression in patients with hepatocellular carcinoma, leading to the identification of high-risk factors and protective factors. POLR2G, whose expression was upregulated in tumour tissues compared to adjacent normal tissues, was identified as a protective factor in the subset of patients studied, associated therefore with better prognosis and overall survival [118]. However, a more recent study combining single cell RNA-seq and TCGA RNA-seq data identified POLR2G (together with another subunit of Pol II, POLR2L) as a gene expressed at higher levels in HCC cancer stem cells, which are important for HCC heterogeneity and resistance to treatment. Importantly, the increased expression of mRNA was also associated with a progressive increase in tumour grade (from G1 to G4) and nodal metastasis status, which are associated with tumour progression and aggressiveness [119].
12. POLR2H/RPB8
POLR2H is another subunit common to all three RNA polymerase complexes. As for most of the other subunits, its upregulation/overexpression is particularly relevant in different tumours. The first evidence shows that high mRNA expression of POLR2H is found in HPV+ head and neck carcinomas [120]. More recent research in rectal cancer, analysing both the TCGA and GEO databases, identified among a total of 18 prognostic associated genes, and a subset of 11 genes including the subunits POLR2H and POLR2J were associated with increased risk of poor overall survival in patients. Importantly, in the high risk group, POLR2H was overexpressed [48]. In agreement with this analysis, both POLR2H and POLR2J are overexpressed in rectal tumour organoids [121]. POLR2H high mRNA levels are also associated with patients’ overall survival in hepatocellular carcinoma [122]. Several groups have similarly shown how POLR2H is upregulated in prostate cancer and overexpressed in prostate cancer cells compared to the normal tissue, indicating the potential role of POLR2H as a biomarker for prostate cancer prognosis and diagnosis, as well as a potential drug target [123,124,125]. Furthermore, POLR2H mRNA levels are significantly higher in lung cancer tissue samples compared to normal samples [126], and its high expression significantly correlates with patients’ survival in lung squamous cell carcinoma [127,128]. Interestingly, more recent data showed that POLR2H is upregulated in a subset of breast cancer patients resistant to radiotherapy, potentially implicating its role in the acquisition of radio-resistance [129,130].
13. POLR2I/RPB9
POLR2I is a subunit specific of Pol II, found to be either amplified or upregulated in a subset of ovarian cancer and head and neck cancer cells. It was speculated that POLR2I could contribute to mechanisms of cancer cells’ resistance to DNA-damaging chemotherapeutics like cisplatin, 5-fluorouracil, and PARP inhibitor olaparib and also radiation therapy [131]. Furthermore, in a meta-analysis study of microarray datasets aimed at identifying potential prognostic candidates in colorectal cancer cell lines with different levels of aggressiveness, POLR2I was found as a candidate gene downregulated in less aggressive cell lines [132]. At the same time, down-regulation of POLR2I expression was observed in hepatocellular carcinomas, where it correlated with mutations in the mRNA of other genes (FKH and FOXP3) which are not present in the genomic DNA, suggesting that POLR2I down-regulation could impair Pol II transcription fidelity and hence contribute to producing aberrant transcripts [133,134].
14. POLR2L/RPB10
POLR2L is one the Pol II subunits shared with the other polymerases, and as for most of the other Pol II subunits, its upregulation has been identified in different cancers. As previously mentioned regarding the POLR2C subunit, after screening for thousands of genes and 300 miRNAs associated with cisplatin resistance in gastric cancer, six hub genes were identified and confirmed in TCGA databases. Among these genes, POLR2L, together with POLR2C, POLR2F, and POLR2K, were significantly upregulated and associated with cisplatin resistance [99]. Furthermore, POLR2L was among the seven candidate genes found to be upregulated to significantly promote cell proliferation compared to control cells, identified via CRISPR/Cas9 activation library screening in two hepatocellular carcinoma cell lines. Zhang and colleagues showed that cell survival is increased by the gain of function of POLR2L in both cell lines. Additionally, the upregulation of these genes promoted liver tumour growth and its colonization potential with the formation of metastasis to the lung. Indeed, these cell lines showed increased invasion capability compared to the control [135].
15. POLR2J/RPB11
There are different gene copies of POLR2J1–4 on the human Chr 7, and at least 11 alternative mRNAs encoding different POLR2J isoforms. The first evidence of a role of POLR2J in cancer came from expression studies in lung tumours, where 50 cases were analysed and nine genes, including POLR2J, were found to show a two-fold increase in expression compared with normal bronchial epithelial cells. POLR2J showed higher expression in nine of the tumour samples, with two matched samples showing more than double expression levels in the tumours [136]. TCGA characterized colorectal cancer genome using 224 tumour samples with their corresponding normal tissues, to identify new biomarkers and potential therapeutic targets, found that POLR2J expression increased progressively with the aggressiveness of the tumour, and similar results were also found in liver metastatic tumours [137]. As mentioned above, POLR2J was associated with higher risk and poorer survival in rectal cancers and was found to be overexpressed in rectal tumour organoids [48,122]. Furthermore, Li and colleagues observed that POLR2J is upregulated, both at the mRNA and protein levels, in glioblastoma cells compared to normal tissues and resulted in enhanced cell proliferation and metastatic capability [138]. It was already established that upregulation of POLR2J is correlated with poor prognosis in glioblastoma patients and that its downregulation is instead associated with better overall survival. It was shown that knockdown of POLR2J reduced cell proliferation and caused cell cycle arrest at G1/G0 phase in glioblastoma cells, and additionally, POLR2J knockdown was able to reduce the migrative and invasive abilities of cancer cells and to suppress epithelial to mesenchymal transition (EMT), indicative of invading cancer cells [139]. Consistently, the overexpression of POLR2J promoted the migrative and invasive abilities of cells, indicating that it could play a crucial role in EMT and increase metastatic potential in glioblastoma [138]. Finally, POLR2J has also been found associated with patients survival of grade 2 ovarian cancers [140], testicular germ cell tumour prognosis [141], and breast cancer susceptibility in a Belgian population [142].
16. POLR2K/RPB12
POLR2K is the last subunit shared among Pol I, II and III. Initial studies in liver cancer cell lines showed that siRNAs targeting POLR2K significantly reduced the viability of cells, indicating a potential role of the subunit in hepatocellular carcinoma [143]. Similar to other Pol II subunits, its increased expression is observed in different cancers. In HER2 breast cancers, POLR2K gain of expression by mRNA upregulation or gene amplification was observed [144]; more recent studies showed that low expression of POLR2K significantly prolonged patients survival and reduced cancer rates in breast cancer, while the high expression of POLR2K was associated with poor patients’ survival [145]. Recently, along with four other genes, higher POLR2K expression levels were found via machine learning approaches in a high-risk group of patients for breast cancer [146]. Using mRNA expression profiling, POLR2K was firstly identified in 2016 as a differentially expressed gene in prostate cancer tissues compared to normal tissues, and later, it was evident that POLR2K differential expression had a significant prognostic value for patients overall survival [147,148]. Finally, in bladder cancer patients, high levels of POLR2K were associated with poor progression free and overall survival. Furthermore, the knock down of the Pol II subunit reduced the viability of two bladder cancer cell lines, indicating that its decreased expression could slow down cell growth [149].
17. Conclusions
RNA Pol II transcription deregulation and transcription factors deregulations are well-established markers of cancer. In this review, we have collected evidence of Pol II subunits deregulations important for cancer development, progression, and aggressiveness. Notably, most of the alterations affecting the subunits found in cancer are up-regulations. However, in the same subset of patients, two subunits have rarely been found to be upregulated together, nor their upregulation shown to be concurrent with POLR2A subunit upregulation. This indicates that the phenotypes observed in patients and in cancer cells could be dependent on the single subunits upregulations and not strictly related to an overall increase in Pol II transcriptions. During transcription, Pol II subunits engage in protein–protein and protein–DNA/RNA interactions with TFs and template DNA and nascent mRNA. For instance, in the pre-initiation complex, POLR2A and POLR2B subunits directly contact the general TFIIB and TFIIF dimerization module [71,150,151,152]. The Mediator complex facilitates the crosstalk between TFs bound enhancers and promoters and Pol II, therefore positively regulating transcription through extensive interactions with general TFs and Pol II itself [7,10,153,154,155,156]. Once moving into elongation, the general TFs are released and Pol II establishes new interactions with elongation factors [8,9,157,158,159]. Not only does Pol II establish interactions with transcription factors and co-transcriptional activators, but also with components of co-transcriptional processes such as RNA splicing, capping, and 3′-end processing, which are regulated by CTD phosphorylation and CTD phosphatases [160,161,162,163,164,165,166]. Interestingly, studies in yeast have highlighted the involvement of RPB4 and RPB7 subunits (POLR2D and POLR2G) in recruiting CTD phosphatases Fcp1 and Ssu72, keeping the CTD phosphorylation levels under control [73]. Indeed, even if Pol II is always studied as a complex, it has recently become of interest how its subunits could play independent roles in biological processes and can operate outside of the complex [167,168]. We can speculate that the higher levels of Pol II subunits could interfere with transcription and co-transcriptional processes, being the subunits the interacting platform with general transcription factors and transcription activators/repressor. It is also conceivable that this is relevant not only for RNA Pol II transcription, but also for RNA Pol I and Pol III transcription. This because some subunits are shared between the three complexes, and because of RNA Pol II contributions to the transcription of the other RNA Polymerases [2,169]. We believe, therefore, that the study of single subunits should be further explored to determine their specific contribution to the transcription stages and the interchange of TFs, particularly relevant due to their possible valuable role as therapeutic targets or new biomarkers for high-risk patients in a wide variety of tumours.

Table 1.
Summarising RNA Pol II subunits deregulations in cancers.
Table 1.
Summarising RNA Pol II subunits deregulations in cancers.
| Gene | Deregulation | Associated Cancer | References |
|---|---|---|---|
| POLR2A | Upregulated/Overexpressed | Gastric cancer Ovarian cancer Acute Myeloid Leukaemia Cholangiocarcinoma Thymoma | [82,83,84] |
| Downregulated | Adrenocortical carcinoma Testicular germ cell carcinoma Endometrial carcinoma | [84] | |
| Polymorphisms | Non-small cell lung cancer | [85] | |
| Co-deletion with TP53 | Colorectal cancer Triple-negative breast cancer | [86,87,88,89,90,91] | |
| POLR2B | Mutated | Breast cancer | [92,93] |
| Upregulated/Overexpressed | Hepatocellular carcinoma | [95] | |
| Downregulated | Acute myeloid leukaemia | [94] | |
| POLR2C | Upregulated/Overexpressed | Hepatocellular carcinoma Gastric cancer | [96,97,99] |
| Downregulated | Osteosarcoma | [98] | |
| POLR2D | Upregulated/Overexpressed | Colorectal cancer Ovarian cancer Prostate cancer | [100,101,102] |
| POLR2E | Polymorphisms | Prostate cancer Oesophageal cancer Breast cancer Papillary thyroid carcinoma Liver cancer | [104,105,106,107,108,109] |
| POLR2F | Upregulated/Overexpressed | Colorectal cancer Gastric cancer Triple negative breast cancer Prostate cancer Glioblastoma | [99,110,111,112,113,114,115] |
| Downregulated | Cervical cancer (HPV18+) | [116,117] | |
| POLR2G | Upregulated/Overexpressed | Hepatocellular carcinoma | [118,119] |
| POLR2H | Upregulated/Overexpressed | Head and neck carcinomas Colorectal cancer Hepatocellular carcinoma Prostate cancer Lung cancer Breast cancer | [48,120,121,122,123,124,125,126,127,128,129,130] |
| POLR2I | Upregulated/Overexpressed | Ovarian cancer Head and neck cancer | [131,132] |
| Downregulated | Hepatocellular carcinoma | [133,134] | |
| POLR2L | Upregulated/Overexpressed | Gastric cancer Hepatocellular carcinoma | [99,135] |
| POLR2J | Upregulated/Overexpressed | Lung cancer Colorectal cancer Glioblastoma Ovarian cancer Testicular germ cell carcinoma Breast cancer | [48,136,137,138,139,140,141,142] |
| POLR2K | Upregulated/Overexpressed | Hepatocellular carcinoma Breast cancer Prostate cancer Bladder cancer | [143,144,145,146,147,148,149] |
Author Contributions
Writing—original draft preparation, M.M.S.; writing—review and editing, M.M.S. and M.S.; supervision, M.S.; funding acquisition, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by BBSRC grant number BB/S016155/1 and Cancer Research UK grant number C17422/A25154.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cramer, P.; Armache, K.-J.; Baumli, S.; Benkert, S.; Brueckner, F.; Buchen, C.; Damsma, G.; Dengl, S.; Geiger, S.; Jasiak, A.; et al. Structure of Eukaryotic RNA Polymerases. Annu. Rev. Biophys. 2008, 37, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.; Ito, K.; Chu, C.-S.; Roeder, R.G. Gene-Specific Control of tRNA Expression by RNA Polymerase II. Mol. Cell 2020, 78, 765–778.e7. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Cramer, P. Conservation between the RNA Polymerase I, II, and III Transcription Initiation Machineries. Mol. Cell 2012, 45, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Chiang, C.-M. The General Transcription Machinery and General Cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef]
- Ramsay, E.P.; Abascal-Palacios, G.; Daiß, J.L.; King, H.; Gouge, J.; Pilsl, M.; Beuron, F.; Morris, E.; Gunkel, P.; Engel, C.; et al. Structure of human RNA polymerase III. Nat. Commun. 2020, 11, 6409. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rucobo, F.W.; Kohler, R.; van de Waterbeemd, M.; Heck, A.J.; Hemann, M.; Herzog, F.; Stark, H.; Cramer, P. Molecular Basis of Transcription-Coupled Pre-mRNA Capping. Mol. Cell 2015, 58, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Plaschka, C.; Larivière, L.; Wenzeck, L.; Seizl, M.; Hemann, M.; Tegunov, D.; Petrotchenko, E.V.; Borchers, C.H.; Baumeister, W.; Herzog, F.; et al. Architecture of the RNA polymerase II–Mediator core initiation complex. Nature 2015, 518, 376–380. [Google Scholar] [CrossRef]
- Ehara, H.; Yokoyama, T.; Shigematsu, H.; Yokoyama, S.; Shirouzu, M.; Sekine, S.-I. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 2017, 357, 921–924. [Google Scholar] [CrossRef]
- Bernecky, C.; Plitzko, J.M.; Cramer, P. Structure of a transcribing RNA polymerase II–DSIF complex reveals a multidentate DNA–RNA clamp. Nat. Struct. Mol. Biol. 2017, 24, 809–815. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2017, 19, 262–274. [Google Scholar] [CrossRef]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Näär, A.M.; Lemon, B.D.; Tjian, R. Transcriptional Coactivator Complexes. Annu. Rev. Biochem. 2001, 70, 475–501. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L. Chromatin Remodeling: Nucleosomes Bulging at the Seams. Curr. Biol. 2002, 12, R245–R247. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Heinrich, R. Biological Control through Regulated Transcriptional Coactivators. Cell 2004, 119, 157–167. [Google Scholar] [CrossRef]
- Rosenfeld, M.G.; Lunyak, V.V.; Glass, C.K. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006, 20, 1405–1428. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bywater, M.J.; Pearson, R.B.; McArthur, G.A.; Hannan, R.D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer 2013, 13, 299–314. [Google Scholar] [CrossRef]
- Johnson, D.G. The paradox of E2F1: Oncogene and tumor suppressor gene. Mol. Carcinog. 2000, 27, 151–157. [Google Scholar] [CrossRef]
- Manickavinayaham, S.; Velez-Cruz, R.; Biswas, A.K.; Chen, J.; Guo, R.; Johnson, D.G. The E2F1 transcription factor and RB tumor suppressor moonlight as DNA repair factors. Cell Cycle 2020, 19, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Borrero, L.J.H.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, L.; Perna, F.; Nimer, S.D. Beyond transcription factors: How oncogenic signalling reshapes the epigenetic landscape. Nat. Rev. Cancer 2016, 16, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.; Gorgoulis, V. Involvement of E2F transcription factor family in cancer. Eur. J. Cancer 2005, 41, 2403–2414. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nature 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef]
- Cole, M.D.; Cowling, V.H. Transcription-independent functions of MYC: Regulation of translation and DNA replication. Nat. Rev. Mol. Cell Biol. 2008, 9, 810–815. [Google Scholar] [CrossRef]
- Dang, C.V.; Resar, L.M.; Emison, E.; Kim, S.; Li, Q.; Prescott, J.E.; Wonsey, D.; Zeller, K. Function of the c-Myc Oncogenic Transcription Factor. Exp. Cell Res. 1999, 253, 63–77. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed]
- White, R.J. RNA polymerase III transcription and cancer. Oncogene 2004, 23, 3208–3216. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Treré, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA Polymerase I Transcription Machinery: An Emerging Target for the Treatment of Cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef]
- Poortinga, G.; Quinn, L.M.; Hannan, R.D. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene 2014, 34, 403–412. [Google Scholar] [CrossRef]
- Mars, J.-C.; Tremblay, M.G.; Valere, M.; Sibai, D.S.; Sabourin-Felix, M.; Lessard, F.; Moss, T. The chemotherapeutic agent CX-5461 irreversibly blocks RNA polymerase I initiation and promoter release to cause nucleolar disruption, DNA damage and cell inviability. NAR Cancer 2020, 2, zcaa032. [Google Scholar] [CrossRef]
- Cornelison, R.; Dobbin, Z.C.; Katre, A.A.; Jeong, D.H.; Zhang, Y.; Chen, D.; Petrova, Y.; Llaneza, D.C.; Steg, A.D.; Parsons, L.; et al. Targeting RNA-Polymerase I in Both Chemosensitive and Chemoresistant Populations in Epithelial Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6529–6540. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef]
- Ferreira, R.; Schneekloth, J.S.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef]
- Low, J.; Sirajuddin, P.; Bs, M.M.; Bs, S.A.; Rege, A.; Guner, G.; Liu, H.; Yang, Z.; De Marzo, A.M.; Bieberich, C.; et al. Effective targeting of RNA polymerase I in treatment-resistant prostate cancer. Prostate 2019, 79, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, V.; Schneider, D.A.; Ortiz-Gonzalez, M.; Soriano-Lerma, A.; Linde-Rodriguez, A.; Perez-Carrasco, V.; Gutierrez-Fernandez, J.; Cuadros, M.; Morales, J.C.; González, C.; et al. Targeting ribosomal G-quadruplexes with naphthalene-diimides as RNA polymerase I inhibitors for colorectal cancer treatment. Cell Chem. Biol. 2021, 28, 1590–1601.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, H.; Zhao, J.; Ma, X.; Qi, W. POLR1B is upregulated and promotes cell proliferation in non-small cell lung cancer. Oncol. Lett. 2019, 19, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Qi, L.; Kong, X.; Wang, Z.; Fang, Y.; Wang, J. Identification of the Significant Genes Regulated by Estrogen Receptor in Estrogen Receptor-Positive Breast Cancer and Their Expression Pattern Changes When Tamoxifen or Fulvestrant Resistance Occurs. Front. Genet. 2020, 11, 538734. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.A.; Araujo, J.; Cardenas, N.K.; Morante, Z.; Doimi, F.; Vidaurre, T.; Balko, J.M.; Gomez, H.L. A prognostic signature based on three-genes expression in triple-negative breast tumours with residual disease. npj Genom. Med. 2016, 1, 15015. [Google Scholar] [CrossRef]
- Sheffer, M.; Bacolod, M.D.; Zuk, O.; Giardina, S.F.; Pincas, H.; Barany, F.; Paty, P.B.; Gerald, W.L.; Notterman, D.A.; Domany, E. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 7131–7136. [Google Scholar] [CrossRef]
- Wang, M.; Niu, W.; Hu, R.; Wang, Y.; Liu, Y.; Liu, L.; Zhong, J.; Zhang, C.; You, H.; Zhang, J.; et al. POLR1D promotes colorectal cancer progression and predicts poor prognosis of patients. Mol. Carcinog. 2018, 58, 735–748. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Q.; Liu, H.; Guo, Q.; Qiu, P.; Chen, J.; Lin, J. Metabolic reprogramming-associated genes predict overall survival for rectal cancer. J. Cell. Mol. Med. 2020, 24, 5842–5849. [Google Scholar] [CrossRef]
- Tian, Y.; Sun, F.; Zhong, Y.; Huang, W.; Wang, G.; Liu, C.; Xiao, Y.; Wu, J.; Mu, L. Expression and Clinical Significance of POLR1D in Colorectal Cancer. Oncology 2019, 98, 138–145. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Nekoohesh, L.; Motevaseli, E. Bladder Cancer Biomarkers: Review and Update. Asian Pac. J. Cancer Prev. 2014, 15, 2395–2403. [Google Scholar] [CrossRef]
- Pavon-Eternod, M.; Gomes, S.; Geslain, R.; Dai, Q.; Rosner, M.R.; Pan, T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009, 37, 7268–7280. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.G.; Sourvinos, G.; Allison, S.J.; Tosh, K.; Scott, P.H.; Spandidos, D.A.; White, R.J. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 12619–12624. [Google Scholar] [CrossRef] [PubMed]
- Gjidoda, A.; Henry, R.W. RNA polymerase III repression by the retinoblastoma tumor suppressor protein. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1829, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Cabarcas, S.; Schramm, L. RNA polymerase III transcription in cancer: The BRF2 connection. Mol. Cancer 2011, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Crighton, D.; Boyle, J.M.; Varley, J.M.; White, R.J. RNA polymerase III transcription can be derepressed by oncogenes or mutations that compromise p53 function in tumours and Li-Fraumeni syndrome. Oncogene 2002, 21, 2961–2970. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Trouchet, D.; Martint, K.; Jackson, S.P.; Kouzaridest, T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 1996, 382, 88–90. [Google Scholar] [CrossRef]
- Scott, P.H.; Cairns, C.A.; Sutcliffe, J.E.; Alzuherri, H.M.; McLees, A.; Winter, A.G.; White, R.J.; Wang, S.; Gustafson, E.; Pang, L.; et al. Regulation of RNA Polymerase III Transcription during Cell Cycle Entry. J. Biol. Chem. 2001, 276, 1005–1014. [Google Scholar] [CrossRef]
- Liang, X.; Xie, R.; Su, J.; Ye, B.; Wei, S.; Liang, Z.; Bai, R.; Chen, Z.; Li, Z.; Gao, X. Inhibition of RNA polymerase III transcription by Triptolide attenuates colorectal tumorigenesis. J. Exp. Clin. Cancer Res. 2019, 38, 217. [Google Scholar] [CrossRef]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akıncılar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.P.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef]
- Petrie, J.L.; Swan, C.; Ingram, R.M.; Frame, F.M.; Collins, A.T.; Dumay-Odelot, H.; Teichmann, M.; Maitland, N.J.; White, R.J. Effects on prostate cancer cells of targeting RNA polymerase III. Nucleic Acids Res. 2019, 47, 3937–3956. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, W.; Li, J.; Zhu, H.; Yu, J. Identification of Survival-Associated Alternative Splicing Signatures in Lung Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 587343. [Google Scholar] [CrossRef]
- Dai, X.; Jiang, W.; Ma, L.; Sun, J.; Yan, X.; Qian, J.; Wang, Y.; Shi, Y.; Ni, S.; Yao, N. A metabolism-related gene signature for predicting the prognosis and therapeutic responses in patients with hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Lautré, W.; Richard, E.; Feugeas, J.-P.; Dumay-Odelot, H.; Teichmann, M. The POLR3G Subunit of Human RNA Polymerase III Regulates Tumorigenesis and Metastasis in Triple-Negative Breast Cancer. Cancers 2022, 14, 5732. [Google Scholar] [CrossRef] [PubMed]
- Musolf, A.M.; Moiz, B.A.; Sun, H.; Pikielny, C.W.; Bossé, Y.; Mandal, D.; de Andrade, M.; Gaba, C.; Yang, P.; Li, Y.; et al. Whole Exome Sequencing of Highly Aggregated Lung Cancer Families Reveals Linked Loci for Increased Cancer Risk on Chromosomes 12q, 7p, and 4q. Cancer Epidemiol. Biomark. Prev. 2020, 29, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.-M.; Wang, Z.-Y.; Zhang, X. Identification of four differentially methylated genes as prognostic signatures for stage I lung adenocarcinoma. Cancer Cell Int. 2018, 18, 60. [Google Scholar] [CrossRef]
- Yunlei, Z.; Zhe, C.; Yan, L.; Pengcheng, W.; Yanbo, Z.; Le, S.; Qianjin, L. INMAP, a novel truncated version of POLR3B, represses AP-1 and p53 transcriptional activity. Mol. Cell. Biochem. 2012, 374, 81–89. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Thomas, C.F.; Dong, J.; Schulte, S.C.; Khadka, P.; Sun, Z.; Kosari, F.; Jen, J.; Molina, J.R.; Vasmatzis, G.; et al. Pathways Impacted by Genomic Alterations in Pulmonary Carcinoid Tumors. Clin. Cancer Res. 2018, 24, 1691–1704. [Google Scholar] [CrossRef]
- An, Y.; Duan, H. The Comprehensive Analysis of Interferon-Related Prognostic Signature with regard to Immune Features in Ovarian Cancer. Dis. Markers 2022, 2022, 7900785. [Google Scholar] [CrossRef]
- Hahn, S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 2004, 11, 394–403. [Google Scholar] [CrossRef]
- Cramer, P. RNA polymerase II structure: From core to functional complexes. Curr. Opin. Genet. Dev. 2004, 14, 218–226. [Google Scholar] [CrossRef]
- Woychik, N.A.; Hampsey, M. The RNA Polymerase II Machinery. Cell 2002, 108, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Kostek, S.A.; Grob, P.; De Carlo, S.; Lipscomb, J.S.; Garczarek, F.; Nogales, E. Molecular Architecture and Conformational Flexibility of Human RNA Polymerase II. Structure 2006, 14, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Allepuz-Fuster, P.; Martínez-Fernández, V.; Garrido-Godino, A.I.; Alonso-Aguado, S.; Hanes, S.D.; Navarro, F.; Calvo, O. Rpb4/7 facilitates RNA polymerase II CTD dephosphorylation. Nucleic Acids Res. 2014, 42, 13674–13688. [Google Scholar] [CrossRef]
- Garrido-Godino, A.; García-López, M.; García-Martínez, J.; Pelechano, V.; Medina, D.; Pérez-Ortín, J.; Navarro, F. Rpb1 foot mutations demonstrate a major role of Rpb4 in mRNA stability during stress situations in yeast. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 731–743. [Google Scholar] [CrossRef]
- Richard, S.; Gross, L.; Fischer, J.; Bendalak, K.; Ziv, T.; Urim, S.; Choder, M. Numerous Post-translational Modifications of RNA Polymerase II Subunit Rpb4/7 Link Transcription to Post-transcriptional Mechanisms. Cell Rep. 2021, 34, 108578. [Google Scholar] [CrossRef] [PubMed]
- Allepuz-Fuster, P.; O’brien, M.J.; González-Polo, N.; Pereira, B.; Dhoondia, Z.; Ansari, A.; Calvo, O. RNA polymerase II plays an active role in the formation of gene loops through the Rpb4 subunit. Nucleic Acids Res. 2019, 47, 8975–8987. [Google Scholar] [CrossRef]
- Fuda, N.J.; Ardehali, M.B.; Lis, J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 2009, 461, 186–192. [Google Scholar] [CrossRef]
- Turinetto, V.; Porcedda, P.; Orlando, L.; De Marchi, M.; Amoroso, A.; Giachino, C. The cyclin-dependent kinase inhibitor 5, 6-dichloro-1-beta-D-ribofuranosylbenzimidazole induces nongenotoxic, DNA replication-independent apoptosis of normal and leukemic cells, regardless of their p53 status. BMC Cancer 2009, 9, 281. [Google Scholar] [CrossRef]
- Fukushima, H.; Abe, T.; Sakamoto, K.; Tsujimoto, H.; Mizuarai, S.; Oie, S. 3′-Ethynylcytidine, an RNA polymerase inhibitor, combined with cisplatin exhibits a potent synergistic growth-inhibitory effect via Vaults dysfunction. BMC Cancer 2014, 14, 562. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2018, 10, 118–136. [Google Scholar] [CrossRef]
- Serra, O.; Galán, M.; Ginesta, M.; Calvo, M.; Sala, N.; Salazar, R. Comparison and applicability of molecular classifications for gastric cancer. Cancer Treat. Rev. 2019, 77, 29–34. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, J.; Li, F.; Ma, X.; Wu, F.; Miao, J.; Li, Q.; Wang, X.; Sun, R.; Yang, Y.; et al. POLR2A Promotes the Proliferation of Gastric Cancer Cells by Advancing the Overall Cell Cycle Progression. Front. Genet. 2021, 12, 688575. [Google Scholar] [CrossRef]
- Li, L.Y.; Kim, H.J.; Park, S.A.; Lee, S.H.; Kim, L.K.; Lee, J.Y.; Kim, S.; Kim, Y.T.; Kim, S.W.; Nam, E.J. Genetic Profiles Associated with Chemoresistance in Patient-Derived Xenograft Models of Ovarian Cancer. Cancer Res. Treat. 2019, 51, 1117–1127. [Google Scholar] [CrossRef]
- Yu, Q.; Xu, Y.; Zhuang, H.; Wu, Z.; Zhang, L.; Li, J.; Yang, L.; Wu, B.; Wang, P.; Zhang, X.; et al. Aberrant activation of RPB1 is critical for cell overgrowth in acute myeloid leukemia. Exp. Cell Res. 2019, 384, 111653. [Google Scholar] [CrossRef]
- Yoo, S.S.; Hong, M.J.; Lee, J.H.; Choi, J.E.; Lee, S.Y.; Lee, J.; Cha, S.I.; Kim, C.H.; Seok, Y.; Lee, E.; et al. Association between polymorphisms in microRNA target sites and survival in early-stage non-small cell lung cancer. Thorac. Cancer 2017, 8, 682–686. [Google Scholar] [CrossRef]
- Liu, X.; Farnung, L.; Wigge, C.; Cramer, P. Cryo-EM structure of a mammalian RNA polymerase II elongation complex inhibited by α-amanitin. J. Biol. Chem. 2018, 293, 7189–7194. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Han, C.; Wan, G.; Huang, X.; Ivan, C.; Jiang, D.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Rao, P.H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701. [Google Scholar] [CrossRef]
- Bradner, J.E. An essential passenger with p53. Nature 2015, 520, 626–627. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, Y.; Kulke, M.; Hechler, T.; Van der Jeught, K.; Dong, T.; He, B.; Miller, K.D.; Radovich, M.; Schneider, B.P.; et al. Targeted immunotherapy for HER2-low breast cancer with 17p loss. Sci. Transl. Med. 2021, 13, eabc6894. [Google Scholar] [CrossRef] [PubMed]
- Van Der Jeught, K.; Xu, H.-C.; Li, Y.-J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y.; Li, Y.; Wang, H.; Stewart, S.; Van der Jeught, K.; Agarwal, P.; Zhang, Y.; Liu, S.; Zhao, G.; et al. Precise targeting of POLR2A as a therapeutic strategy for human triple negative breast cancer. Nat. Nanotechnol. 2019, 14, 388–397. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, S.; Umezu, T.; Ohtsuki, K.; Kobayashi, C.; Ohyashiki, K.; Ohyashiki, J.H. Constitutive activation of the ATM/BRCA1 pathway prevents DNA damage-induced apoptosis in 5-azacytidine-resistant cell lines. Biochem. Pharmacol. 2014, 89, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Zeb, A.; Cheng, L.F. Exploring the molecular mechanism of hepatitis virus inducing hepatocellular carcinoma by microarray data and immune infiltrates analysis. Front. Immunol. 2022, 13, 1032819. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.-P.; Jiang, B.-G.; Zhang, F.-B.; Wang, A.-D.; Ji, Y.-M.; Xu, Y.-F.; Li, J.-C.; Zhou, W.-P.; Han, H.-X. Rpb3 promotes hepatocellular carcinoma through its N-terminus. Oncotarget 2014, 5, 9256–9268. [Google Scholar] [CrossRef]
- Hu, P.; Wang, B.; Chen, T.; Xu, Y.; Zheng, G.; Zhu, Y.; Du, X. RNA polymerase II subunit 3 regulates vesicular, overexpressed in cancer, prosurvival protein 1 expression to promote hepatocellular carcinoma. J. Int. Med. Res. 2021, 49, 0300060521990512. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, N.; Zheng, Y.; Tong, Z.; Yang, T.; Kang, X.; He, Y.; Dong, L. Identification of Key Genes and Pathways in Osteosarcoma by Bioinformatics Analysis. Comput. Math. Methods Med. 2022, 2022, 7549894. [Google Scholar] [CrossRef]
- Zhou, D.; Li, X.; Zhao, H.; Sun, B.; Liu, A.; Han, X.; Cui, Z.; Yuan, L. Combining multi-dimensional data to identify a key signature (gene and miRNA) of cisplatin-resistant gastric cancer. J. Cell. Biochem. 2018, 119, 6997–7008. [Google Scholar] [CrossRef]
- Abba, M.; Laufs, S.; Aghajany, M.; Korn, B.; Benner, A.; Allgayer, H. Look who’s talking: Deregulated signaling in colorectal cancer. Cancer Genom. Proteom. 2012, 9, 15–25. [Google Scholar]
- Kim, S.; Han, Y.; Kim, S.I.; Lee, J.; Jo, H.; Wang, W.; Cho, U.; Park, W.; Rando, T.A.; Dhanasekaran, D.N.; et al. Computational modeling of malignant ascites reveals CCL5–SDC4 interaction in the immune microenvironment of ovarian cancer. Mol. Carcinog. 2021, 60, 297–312. [Google Scholar] [CrossRef]
- Yamada, Y.; Nishikawa, R.; Kato, M.; Okato, A.; Arai, T.; Kojima, S.; Yamazaki, K.; Naya, Y.; Ichikawa, T.; Seki, N. Regulation of HMGB3 by antitumor miR-205-5p inhibits cancer cell aggressiveness and is involved in prostate cancer pathogenesis. J. Hum. Genet. 2017, 63, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Li, Q.; Zhou, P.; Huang, J.; Zhuang, H.; Wu, H.; Chen, B. Analysis of Omics Data Reveals Nucleotide Excision Repair-Related Genes Signature in Highly-Grade Serous Ovarian Cancer to Predict Prognosis. Front. Cell Dev. Biol. 2022, 10, 874588. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Sun, J.; Isaacs, S.D.; Wiley, K.E.; Kim, S.-T.; Chu, L.W.; Zhang, Z.; Zhao, H.; Zheng, S.L.; Isaacs, W.B.; et al. Human polymorphisms at long non-coding RNAs (lncRNAs) and association with prostate cancer risk. Carcinogenesis 2011, 32, 1655–1659. [Google Scholar] [CrossRef] [PubMed]
- Sattarifard, H.; Hashemi, M.; Hassanzarei, S.; Basiri, A.; Narouie, B.; Ghavami, S. Long non-coding RNA POLR2E gene polymorphisms increased the risk of prostate cancer in a sample of the Iranian population. Nucleosides Nucleotides Nucleic Acids 2018, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chen, Y.; Yuan, Q.; Hua, Q.; Zhang, X.; Wang, M.; Tong, N.; Zhang, W.; Chen, J.; Zhang, Z. The HOTAIR, PRNCR1 and POLR2E polymorphisms are associated with cancer risk: A meta-analysis. Oncotarget 2017, 8, 43271–43283. [Google Scholar] [CrossRef][Green Version]
- Chen, B.; Jiao, Y.; Yaolong, F.; Li, T.; Liu, Y.; Wang, M.; Xiuli, G.; Feng, X. The POLR2E rs3787016 polymorphism is strongly associated with the risk of female breast and cervical cancer. Pathol.-Res. Pr. 2019, 215, 1061–1065. [Google Scholar] [CrossRef]
- Chen, B.; Wang, S.; Ma, G.; Han, J.; Zhang, J.; Gu, X.; Feng, X. The association of POLR2E rs3787016 polymorphism and cancer risk: A Chinese case–control study and meta-analysis. Biosci. Rep. 2018, 38, BSR20180853. [Google Scholar] [CrossRef]
- Chen, B.; Li, J.; Yi, C.; Jiao, Y.; Gu, X.; Feng, X. Long non-coding RNA POLR2E rs3787016 is associated with the risk of papillary thyroid carcinoma in Chinese population. Pathol.-Res. Pr. 2018, 214, 1040–1044. [Google Scholar] [CrossRef]
- Antonacopoulou, A.G.; Grivas, P.D.; Skarlas, L.; Kalofonou, M.; Scopa, C.D.; Kalofonos, H. POLR2F, ATP6V0A1 and PRNP expression in colorectal cancer: New molecules with prognostic significance? Anticancer Res. 2008, 28, 1221–1227. [Google Scholar]
- Orian-Rousseau, V.; Mink, S.; Mengwasser, J.; HogenEsch, H.; Guo, F.; Thies, W.-G.; Hofmann, M.; Herrlich, P.; Ponta, H. Genes upregulated in a metastasizing human colon carcinoma cell line. Int. J. Cancer 2004, 113, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Naorem, L.D.; Muthaiyan, M.; Venkatesan, A. Integrated network analysis and machine learning approach for the identification of key genes of triple-negative breast cancer. J. Cell. Biochem. 2018, 120, 6154–6167. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lv, Z.; Xia, H.; Guo, X.; Wang, J.; Wang, J.; Liu, M. Biochemical recurrence related metabolic novel signature associates with immunity and ADT treatment responses in prostate cancer. Cancer Med. 2022, 12, 862–878. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, R.; Zhang, L.; Meng, X.; Sun, W. Primary glioblastoma transcriptome data analysis for screening survival-related genes. J. Cell. Biochem. 2019, 121, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Masica, D.L.; Karchin, R. Correlation of Somatic Mutation and Expression Identifies Genes Important in Human Glioblastoma Progression and Survival. Cancer Res. 2011, 71, 4550–4561. [Google Scholar] [CrossRef]
- Sample, K.M. DNA repair gene expression is associated with differential prognosis between HPV16 and HPV18 positive cervical cancer patients following radiation therapy. Sci. Rep. 2020, 10, 2774. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, J.; Jiang, F.; Wang, L.; Xiao, Q.; Han, F.; Chen, J.; Yuan, S.; Wei, J.; Larsson, S.; et al. Identification of novel protein biomarkers and drug targets for colorectal cancer by integrating human plasma proteome with genome. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Li, N.; Zhao, L.; Guo, C.; Liu, C.; Liu, Y. Identification of a novel DNA repair-related prognostic signature predicting survival of patients with hepatocellular carcinoma. Cancer Manag. Res. 2019, 11, 7473–7484. [Google Scholar] [CrossRef]
- Yao, F.; Zhan, Y.; Li, C.; Lu, Y.; Chen, J.; Deng, J.; Wu, Z.; Li, Q.; Song, Y.; Chen, B.; et al. Single-Cell RNA Sequencing Reveals the Role of Phosphorylation-Related Genes in Hepatocellular Carcinoma Stem Cells. Front. Cell Dev. Biol. 2022, 9, 734287. [Google Scholar] [CrossRef]
- Slebos, R.J.; Yi, Y.; Ely, K.; Carter, J.; Evjen, A.; Zhang, X.; Shyr, Y.; Murphy, B.M.; Cmelak, A.J.; Burkey, B.B.; et al. Gene Expression Differences Associated with Human Papillomavirus Status in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2006, 12, 701–709. [Google Scholar] [CrossRef]
- Costales-Carrera, A.; Fernández-Barral, A.; Bustamante-Madrid, P.; Domínguez, O.; Guerra-Pastrián, L.; Cantero, R.; Del Peso, L.; Burgos, A.; Barbáchano, A.; Muñoz, A. Comparative Study of Organoids from Patient-Derived Normal and Tumor Colon and Rectal Tissue. Cancers 2020, 12, 2302. [Google Scholar] [CrossRef]
- Ke, R.-S.; Zhang, K.; Lv, L.-Z.; Dong, Y.-P.; Pan, F.; Yang, F.; Cai, Q.-C.; Jiang, Y. Prognostic value and oncogene function of heterogeneous nuclear ribonucleoprotein A1 overexpression in HBV-related hepatocellular carcinoma. Int. J. Biol. Macromol. 2019, 129, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Liang, Z.; Gao, Z.; Pan, Z.; Han, S.; Liu, X.; Zhao, C.; Yang, W.; Pan, Z.; Feng, W. Identification of the key genes and pathways in prostate cancer. Oncol. Lett. 2018, 16, 6663–6669. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, H. Identification of prognosis biomarkers of prostatic cancer in a cohort of 498 patients from TCGA. Curr. Probl. Cancer 2019, 43, 100503. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-H.; Lee, C.-Y.; Lee, T.-Y.; Huang, H.-D.; Hsu, J.B.-K.; Chang, T.-H. Biomarker Identification through Multiomics Data Analysis of Prostate Cancer Prognostication Using a Deep Learning Model and Similarity Network Fusion. Cancers 2021, 13, 2528. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Wang, Z.-W.; Ni, N.; Li, L.; Kong, X.-Y. Similarity and difference of pathogenesis among lung cancer subtypes suggested by expression profile data. Pathol.-Res. Pr. 2021, 220, 153365. [Google Scholar] [CrossRef]
- Jia, B.; Gong, T.; Sun, B.; Zhang, Z.; Zhong, D.; Wang, C. Identification of a DNA damage repair gene-related signature for lung squamous cell carcinoma prognosis. Thorac. Cancer 2022, 13, 1143–1152. [Google Scholar] [CrossRef]
- Guttapadu, R.; Katte, T.; Sayeeram, D.; Bhatia, S.; Abraham, A.R.; Rajeev, K.; Amara, A.R.R.; Siri, S.; Bommana, K.; Rasalkar, A.A.; et al. Identification of novel biomarkers for lung squamous cell carcinoma. 3 Biotech 2023, 13, 72. [Google Scholar] [CrossRef]
- Jia, Z.; Ai, X.; Sun, F.; Zang, T.; Guan, Y.; Gao, F. Identification of New Hub Genes Associated with Bladder Carcinoma via Bioinformatics Analysis. Tumori J. 2015, 101, 117–122. [Google Scholar] [CrossRef]
- Miao, W.; Bade, D.; Wang, Y. Targeted Proteomic Analysis Revealed Kinome Reprogramming during Acquisition of Radioresistance in Breast Cancer Cells. J. Proteome Res. 2021, 20, 2830–2838. [Google Scholar] [CrossRef]
- Gaponova, A.V.; Deneka, A.Y.; Beck, T.N.; Liu, H.; Andrianov, G.; Nikonova, A.S.; Nicolas, E.; Einarson, M.B.; Golemis, E.A.; Serebriiskii, I.G. Identification of evolutionarily conserved DNA damage response genes that alter sensitivity to cisplatin. Oncotarget 2016, 8, 19156–19171. [Google Scholar] [CrossRef] [PubMed]
- Long, N.P.; Lee, W.J.; Huy, N.T.; Lee, S.J.; Park, J.H.; Kwon, S.W. Novel Biomarker Candidates for Colorectal Cancer Metastasis: A Meta-analysis of In Vitro Studies. Cancer Inform. 2016, 15s4, CIN.S40301-17. [Google Scholar] [CrossRef]
- Walmacq, C.; Kireeva, M.L.; Irvin, J.; Nedialkov, Y.; Lubkowska, L.; Malagon, F.; Strathern, J.N.; Kashlev, M. Rpb9 Subunit Controls Transcription Fidelity by Delaying NTP Sequestration in RNA Polymerase II. J. Biol. Chem. 2009, 284, 19601–19612. [Google Scholar] [CrossRef]
- Ren, J.; Liu, Y.; Wang, S.; Wang, Y.; Li, W.; Chen, S.; Cui, D.; Yang, S.; Li, M.-Y.; Feng, B.; et al. The FKH domain in FOXP3 mRNA frequently contains mutations in hepatocellular carcinoma that influence the subcellular localization and functions of FOXP3. J. Biol. Chem. 2020, 295, 5484–5495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ren, Z.; Zheng, H.; Lin, M.; Chen, G.; Luo, O.J.; Zhu, G. CRISPR activation screening in a mouse model for drivers of hepatocellular carcinoma growth and metastasis. iScience 2023, 26, 106099. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Lockwood, W.W.; Buys, T.P.H.; Chari, R.; Coe, B.P.; Lam, S.; Lam, W.L. Integrative genomic and gene expression analysis of chromosome 7 identified novel oncogene loci in non-small cell lung cancer. Genome 2008, 51, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Agarwal, E.; Leiphrakpam, P.; Brattain, M.G.; Chowdhury, S. Establishment and Validation of an Orthotopic Metastatic Mouse Model of Colorectal Cancer. ISRN Hepatol. 2013, 2013, 206875. [Google Scholar] [CrossRef]
- Li, Z. POLR2J is a potential biomarker for abnormal tumor progression, vorinostat sensitization, immune infiltration, and prognosis of glioblastoma multiform. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef]
- Zhao, J.; Song, X.; Xu, T.; Yang, Q.; Liu, J.; Jiang, B.; Wu, J. Identification of Potential Prognostic Competing Triplets in High-Grade Serous Ovarian Cancer. Front. Genet. 2021, 11, 607722. [Google Scholar] [CrossRef]
- Yao, L.; Cong, R.; Ji, C.; Zhou, X.; Luan, J.; Meng, X.; Song, N. RNA-Binding Proteins Play an Important Role in the Prognosis of Patients With Testicular Germ Cell Tumor. Front. Genet. 2021, 12, 610291. [Google Scholar] [CrossRef] [PubMed]
- Farahmand, S.; Goliaei, S.; Ansari-Pour, N.; Razaghi-Moghadam, Z. GTA: A game theoretic approach to identifying cancer subnetwork markers. Mol. Biosyst. 2015, 12, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Park, E.S.; Lee, J.-S.; Lee, Y.-H.; Ishikawa, T.; Kim, Y.J.; Thorgeirsson, S.S. Identification of Potential Driver Genes in Human Liver Carcinoma by Genomewide Screening. Cancer Res. 2009, 69, 4059–4066. [Google Scholar] [CrossRef]
- Natrajan, R.; Weigelt, B.; Mackay, A.; Geyer, F.C.; Grigoriadis, A.; Tan, D.S.P.; Jones, C.; Lord, C.J.; Vatcheva, R.; Rodriguez-Pinilla, S.M.; et al. An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancers. Breast Cancer Res. Treat. 2009, 121, 575–589. [Google Scholar] [CrossRef]
- Teng, X.; Yang, T.; Yuan, B.; Yang, Y.; Liu, J.; Wang, X.; Wang, Y.; Ma, T.; Yin, X.; Yu, H.; et al. Prognostic analysis of patients with breast cancer based on tumor mutational burden and DNA damage repair genes. Front. Oncol. 2023, 13, 1177133. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, J.; Dong, C.; Lim, D.; Feng, Z. Development of a risk model to predict prognosis in breast cancer based on cGAS-STING-related genes. Front. Genet. 2023, 14, 1121018. [Google Scholar] [CrossRef]
- Kelly, R.S.; Sinnott, J.A.; Rider, J.R.; Ebot, E.M.; Gerke, T.; Bowden, M.; Pettersson, A.; Loda, M.; Sesso, H.D.; Kantoff, P.W.; et al. The role of tumor metabolism as a driver of prostate cancer progression and lethal disease: Results from a nested case-control study. Cancer Metab. 2016, 4, 22. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, Z.; Wang, Y.; Xue, S. Identification of crucial genes and pathways associated with prostate cancer in multiple databases. J. Int. Med. Res. 2021, 49, 03000605211016624. [Google Scholar] [CrossRef]
- Yang, L.; Wang, K.; Guo, W.; Chen, X.; Guo, Q.; Wei, L.; Zhou, Y. Gene Expression and Regulatory Webwork of POLR2K in Bladder Carcinogenesis by Integrated Bioinformatics Approaches. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Kornberg, R.D. Eukaryotic transcriptional control. Trends Biochem. Sci. 1999, 24, M46–M49. [Google Scholar] [CrossRef]
- Liu, X.; Bushnell, D.A.; Kornberg, R.D. RNA polymerase II transcription: Structure and mechanism. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2012, 1829, 2–8. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef]
- Gupta, K.; Sari-Ak, D.; Haffke, M.; Trowitzsch, S.; Berger, I. Zooming in on Transcription Preinitiation. J. Mol. Biol. 2016, 428, 2581–2591. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef]
- Xu, Y.; Bernecky, C.; Lee, C.-T.; Maier, K.C.; Schwalb, B.; Tegunov, D.; Plitzko, J.M.; Urlaub, H.; Cramer, P. Architecture of the RNA polymerase II-Paf1C-TFIIS transcription elongation complex. Nat. Commun. 2017, 8, 15741. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Urlaub, H.; Cramer, P. Structure of paused transcription complex Pol II–DSIF–NELF. Nature 2018, 560, 601–606. [Google Scholar] [CrossRef]
- Compe, E.; Genes, C.M.; Braun, C.; Coin, F.; Egly, J.-M. TFIIE orchestrates the recruitment of the TFIIH kinase module at promoter before release during transcription. Nat. Commun. 2019, 10, 2084. [Google Scholar] [CrossRef]
- Ghosh, A.; Shuman, S.; Lima, C.D. Structural Insights to How Mammalian Capping Enzyme Reads the CTD Code. Mol. Cell 2011, 43, 299–310. [Google Scholar] [CrossRef]
- Eick, D.; Geyer, M. The RNA Polymerase II Carboxy-Terminal Domain (CTD) Code. Chem. Rev. 2013, 113, 8456–8490. [Google Scholar] [CrossRef] [PubMed]
- Heidemann, M.; Hintermair, C.; Voß, K.; Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2013, 1829, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Harlen, K.M.; Churchman, L.S. The code and beyond: Transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 2017, 18, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Robles, C.M.G.; Coin, F. Conducting the CTD orchestra. Nat. Chem. Biol. 2018, 15, 97–98. [Google Scholar] [CrossRef]
- Maita, H.; Nakagawa, S. What is the switch for coupling transcription and splicing? RNA Polymerase II C-terminal domain phosphorylation, phase separation and beyond. Wiley Interdiscip. Rev. RNA 2019, 11, e1574. [Google Scholar] [CrossRef]
- Garg, G.; Dienemann, C.; Farnung, L.; Schwarz, J.; Linden, A.; Urlaub, H.; Cramer, P. Structural insights into human co-transcriptional capping. Mol. Cell 2023, 83, 2464–2477.e5. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Zhu, J.; Bao, L.; Wang, H.; Jiang, Y.; Tian, K.; Wang, R.; Zheng, H.; Duan, W.; et al. Targeted protein degradation reveals RNA Pol II heterogeneity and functional diversity. Mol. Cell 2022, 82, 3943–3959.e11. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Bao, L.; Zhu, J.; Duan, W.; Zheng, H.; Wang, H.; Jiang, Y.; Liu, W.; Zhang, M.; et al. RNA Pol II preferentially regulates ribosomal protein expression by trapping disassociated subunits. Mol. Cell 2023, 83, 1280–1297.e11. [Google Scholar] [CrossRef]
- Abraham, K.J.; Khosraviani, N.; Chan, J.N.Y.; Gorthi, A.; Samman, A.; Zhao, D.Y.; Wang, M.; Bokros, M.; Vidya, E.; Ostrowski, L.A.; et al. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 2020, 585, 298–302. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).