1. Introduction
Glucagon-like peptide1 receptor (GLP-1R) is of particular interest due to its role in the treatment of type 2 diabetes mellitus (T2D) and appetite regulation [
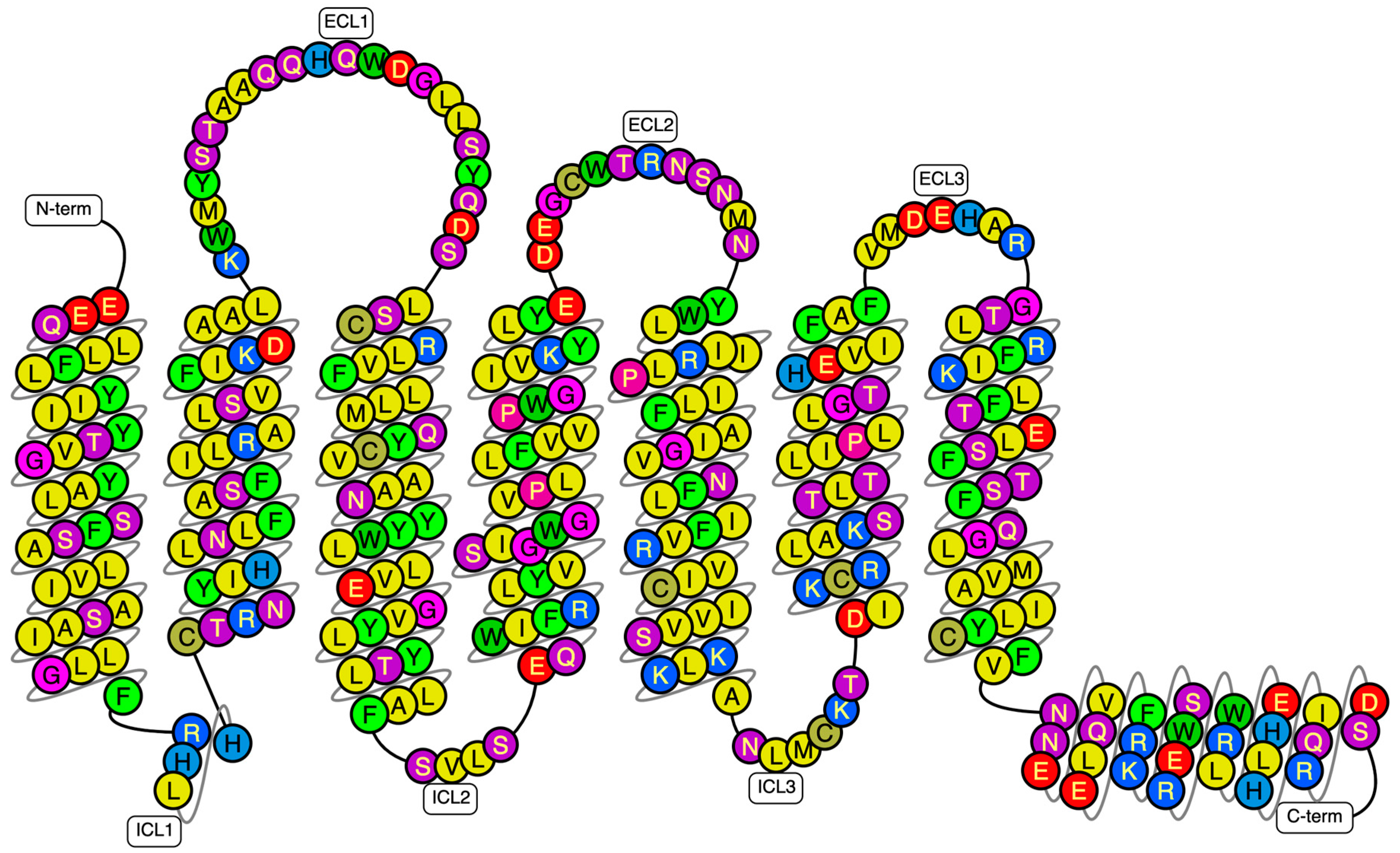
1]. This receptor belongs to the small family (or class) B of G-protein-coupled receptors (GPCRs), which consist structurally of seven transmembrane (TM) domains of 310–420 residues, interconnected by three intracellular (IL) and extracellular (EL) loops, and an extracellular N-terminal domain (NTD) of 120–160 residues (
Figure 1) [
2,
3].
Glucagon-like peptide 1 (GLP-1) is the endogenous ligand of GLP-1R. In response to food intake, it is produced from the gastrointestinal tract and has vital roles in regulating insulin secretion, appetite control, and carbohydrate metabolism [
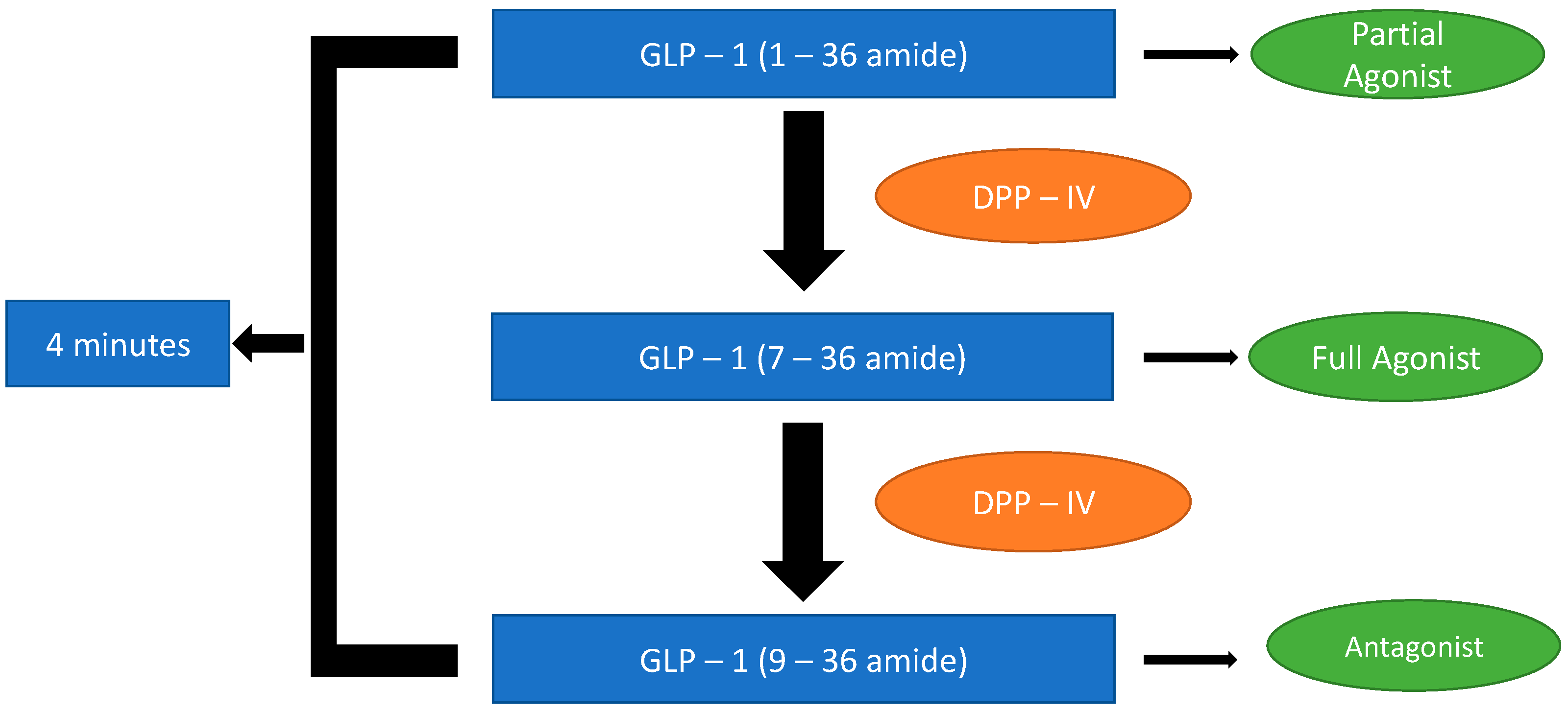
6]. As a result of the distinctive processing of its precursor glucagon, GLP-1 occurs in two active forms: GLP-1 (7–37 amide) and GLP-1 (7–36 amide). GLP-1 (7–36 amide) is the primary circulatory form, which exerts insulinotropic and glucoregulatory functions. However, within 1 to 4 min of secretion, both active forms are degraded by dipeptidyl peptidase 4 (DPP-IV) through cleavage of NH2-terminal amino acids to their respective principal metabolites; 9–37 amide and 9–36 amide (
Figure 2), of which each remains in circulation for about 30 min [
7]. Contrary to the antecedent belief that GLP-1 (9–36 amide) is pharmacologically inactive as a result of its weak or no insulinotropic activity, recent findings have demonstrated that GLP-1 (9–36 amide) possess distinctive extra-pancreatic insulin-like actions in the heart, liver and vasculature, which are autonomously mediated irrespective of the GLP-1R [
8,
9,
10,
11]. GLP-1 (7–36 amide) is a 30-amino-acid peptide hormone released from intestinal L-cells following supplement ingestion [
12]. The peptide GLP-1 has numerous functions, including potentiation of the glucose-actuated release of insulin from pancreatic beta cells, heightening insulin articulations, obstruction of beta-cell apoptosis, progression of beta-cell neogenesis, diminishing glucagon emission, conceding gastric discharging, supporting satiety, and intensifying peripheral glucose disposal. It is thus also a physiological regulator of appetite and food intake [
13].
Fasting blood concentration of GLP-1 (9–36 amide) in humans usually ranges from 5 to 15 pmol/L, and there is usually a two- to four-fold increase after ingesting food. There is an increase in blood GLP-1 concentration 15 min after food ingestion, and peak concentration is reached after 60 min. The blood GLP-1 concentration decreases gradually in the second hour until the next time food is ingested [
7]. It is clear from these varied activities that GLP-1 plays a central role in controlling postprandial glucose levels and, in that capacity, drugs that stimulate the GLP-1 receptor, such as dipeptidyl peptidase 4 inhibitors or GLP-1 analogues, have been manufactured for use in the treatment of type 2 diabetes mellitus (T2D) [
14,
15]. The excessive secretion of GLP-1 has been hypothesised to be responsible for postprandial reactive hypoglycaemia, while diminished secretion might lead to obesity [
12]. T2D treatment needs the positive allosteric modulation of GLP-1R to inhibit glucagon secretion, thus stimulating insulin secretion in a glucose-dependent routine [
16]. A structural study carried out by Song et al. [
16] showed similarity in the transmembrane domain (TMD) architecture of GLP-1R and Glucagon receptor (GCGR), which was also consistent with the overlap in their primary sequences (45% similar in their TMDs). The human GLP-1R TMD was crystallised with two negative allosteric modulators, NNC0640 and PF-06372222, respectively, at 3.0 and 2.7 Å resolution. The crystallised structures of GLP-1R and GCGR showed a common binding pocket for the negative allosteric modulators, which is located outside helices V–VII, close to the receptor’s intracellular domain [
16]. A molecular-modelling and mutagenesis study has shown that agonist positive allosteric modulators also target the same region but in a clear-cut sub-pocket at the interface between helices V and VI, which may aid the formation of an intracellular binding site that enhances G-protein coupling [
16]. The secretin receptors have immense potential in drug discovery due to their importance in fundamental homeostatic functions. To date, three of these hormones are used clinically: glucagon, parathyroid hormone and calcitonin, for the treatment of hypoglycaemia, osteoporosis, and hypercalcaemia, respectively [
17]. This study aims to determine the allosteric binding site and molecular mechanism of allosteric binding to GLP1-R, using allosteric modulators identified through a literature survey. We also perform in silico evaluation of the ADME/Tox properties of the allosteric modulators.
4. Discussion
Recent work has shown that T2D treatment needs the positive allosteric modulation of GLP-1R to inhibit glucagon secretion, thus stimulating insulin secretion [
16]. It has also been suggested that the novel agonist human monoclonal antibody IRAB-A binds allosterically to the insulin receptor and thereby activates and enhances the signalling of insulin [
27]. We demonstrate an application of these techniques in identifying the interactions occurring at the allosteric binding site of the GLP-1R using known allosteric modulators of GLP-1R. We also carried out in silico ADME/Tox evaluations of the compounds used in this study. Molecular docking is a virtual screening method used to discover new ligands for GPCRs, it should rank active molecules high and produce poses which will inform chemists which compounds to purchase for further screening [
28].
Molecular docking has been used extensively in GPCR drug discovery to identify compounds (hit and lead generation) which target different receptors in the GPCR family [
29]. Jenkins et al., utilised Flare to study 2-mercaptoacetamide (2MA) a structural analogue of urea, their findings showed that 2-MA is a competitive inhibitor and flare is a robust software for performing docking simulations [
30]. Egorov et al., in their in silico docking study performed using flare showed that the synthesised compounds would play a significant role in the treatment of ailments such as breast cancer, neurodegenerative diseases etc [
31]. Carlsson et al., applied molecular docking to perform a screening of over six million commercially available compounds against the active like conformations of A
2AAR. Their findings showed that nine of the 20 predicted agonists were confirmed to be A
2AAR ligands [
32]. Docking based programs can generate 3D conformations of binding structures which is very useful for function and drug-based analysis [
33]. Hou et al., used techniques such as homology, molecular dynamics, and molecular docking to access prediction accuracy of ligand-binding poses and screening power of docking-based virtual screening. Their findings showed that the crystal structures outperformed the homology models before any refinement through molecular dynamics. However, the optimised homology models show a similar performance to the crystal structures following a docking assessment [
34]. Shoichet et al., applied ligand docking to screen a large library of compounds to identify compounds with joint activity against on-targets and selectivity versus anti-targets using selected GPCRs (dopamine D
2, serotonin 5-HT
2A, histamine H
1, κ-opioid and μ-opioid receptors) [
35]. Their findings showed a hit range of 40% to 65% for the on-targets with very reliable calculated binding affinities [
35]. Docking into a crystal structure produces an accurate ligand binding pose prediction without any refinement [
36]. The widescale application of molecular docking in drug development makes it a preferred method for this study; this has been paired with binding affinity predictions to determine the feasibility of the complexes.
The conformational transitions observed from the inactive to the active structures were similar to the findings of Liang et al. [
20] who reported that the movement of TM 6 in the inactive state upon signalling created a binding pocket. The docking of the ligands to the inactive structure (PDB ID: 5VEW) is corroborated by Song et al. [
16]; they reported that positive allosteric modulators (PAM) of the GLP-1R bind outside helices 5–7 near the intracellular part of the receptor, but in a distinct sub-pocket between helices 5 and 6.
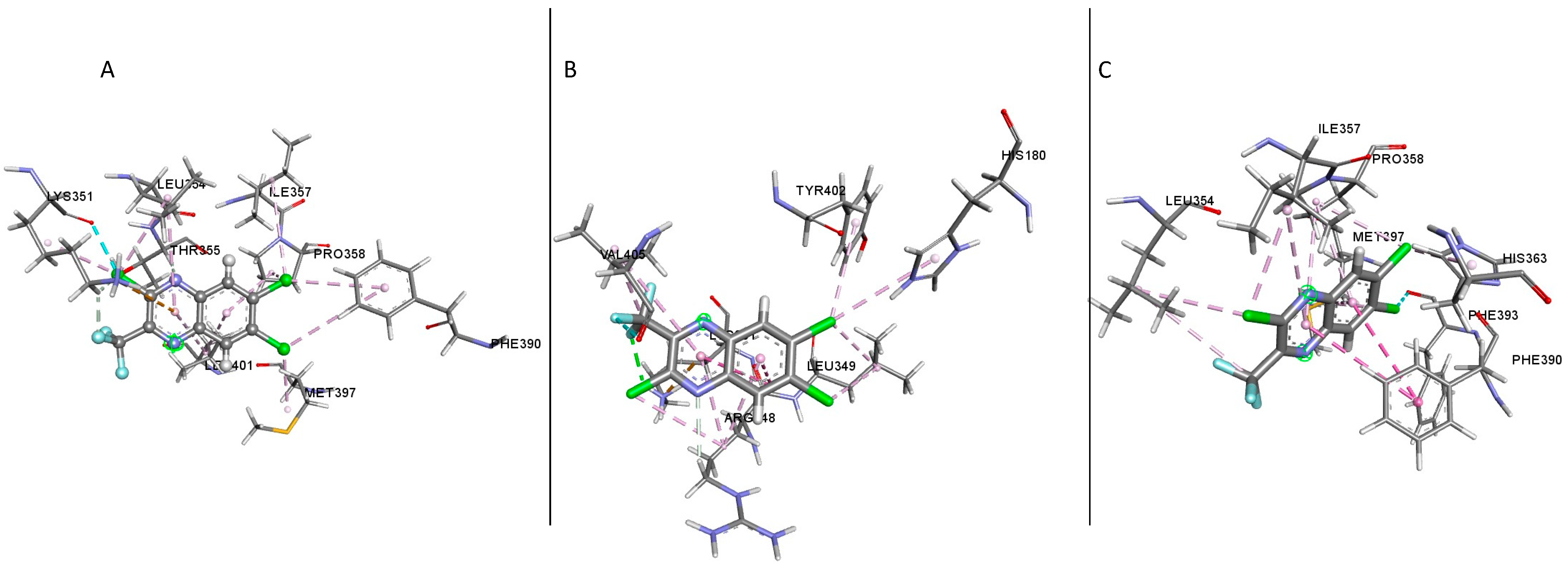
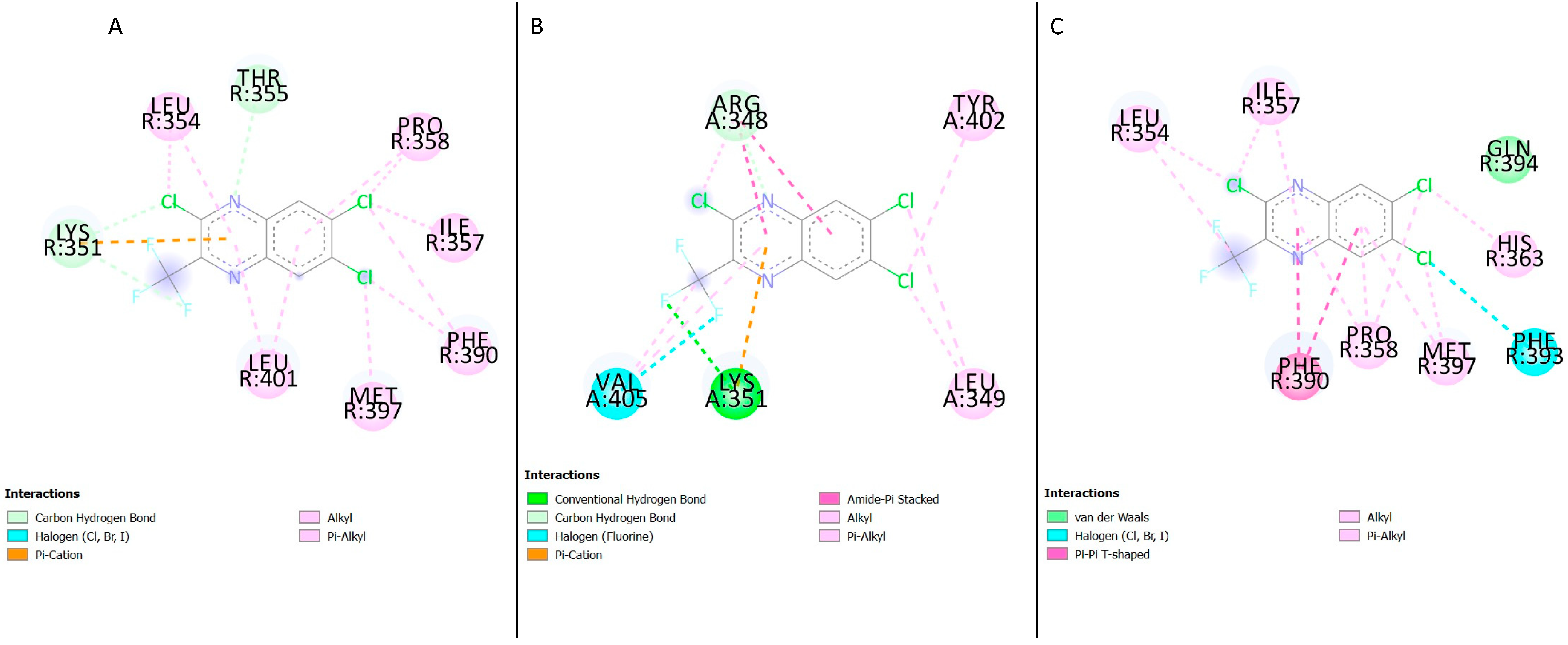
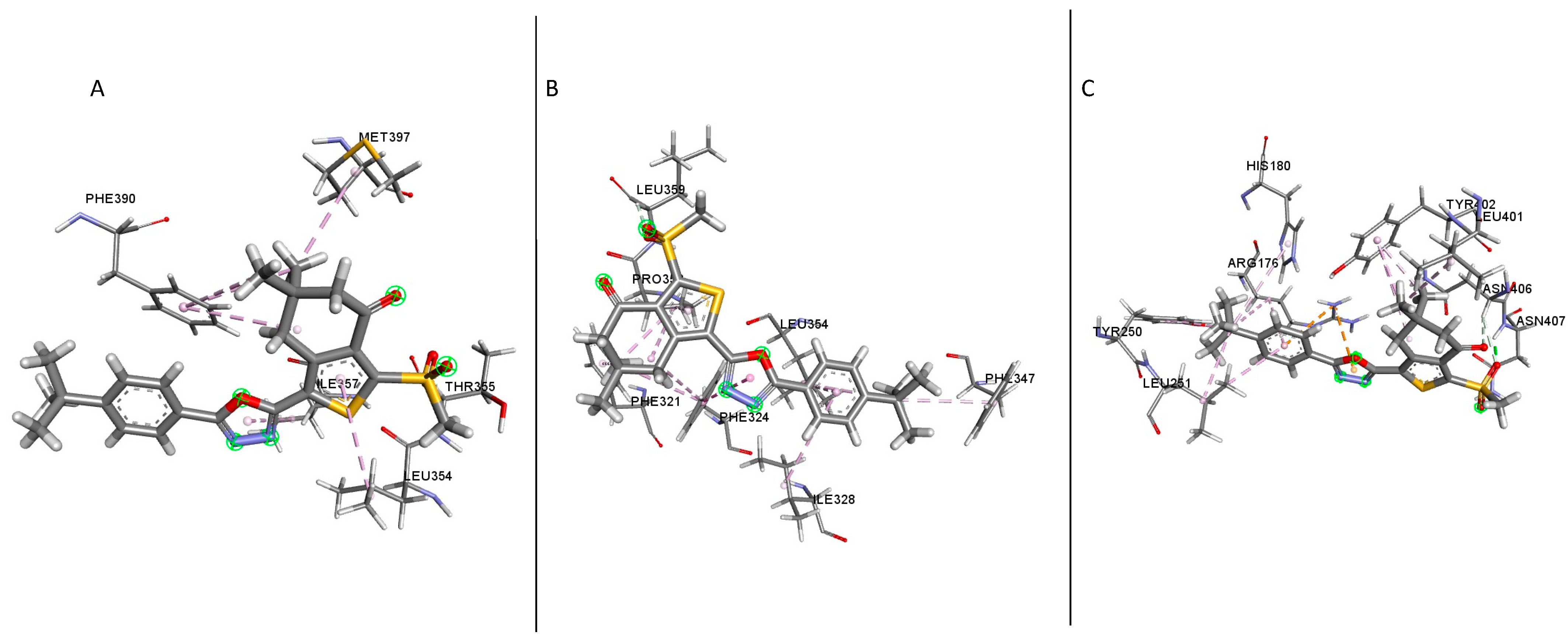
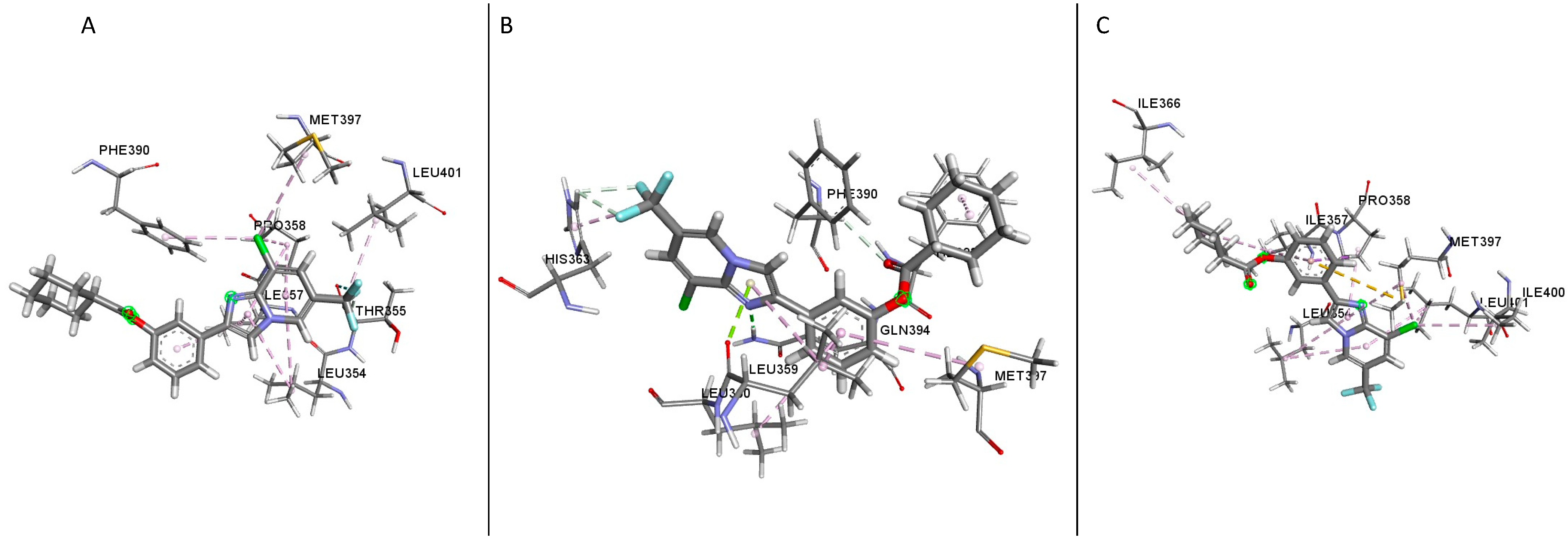
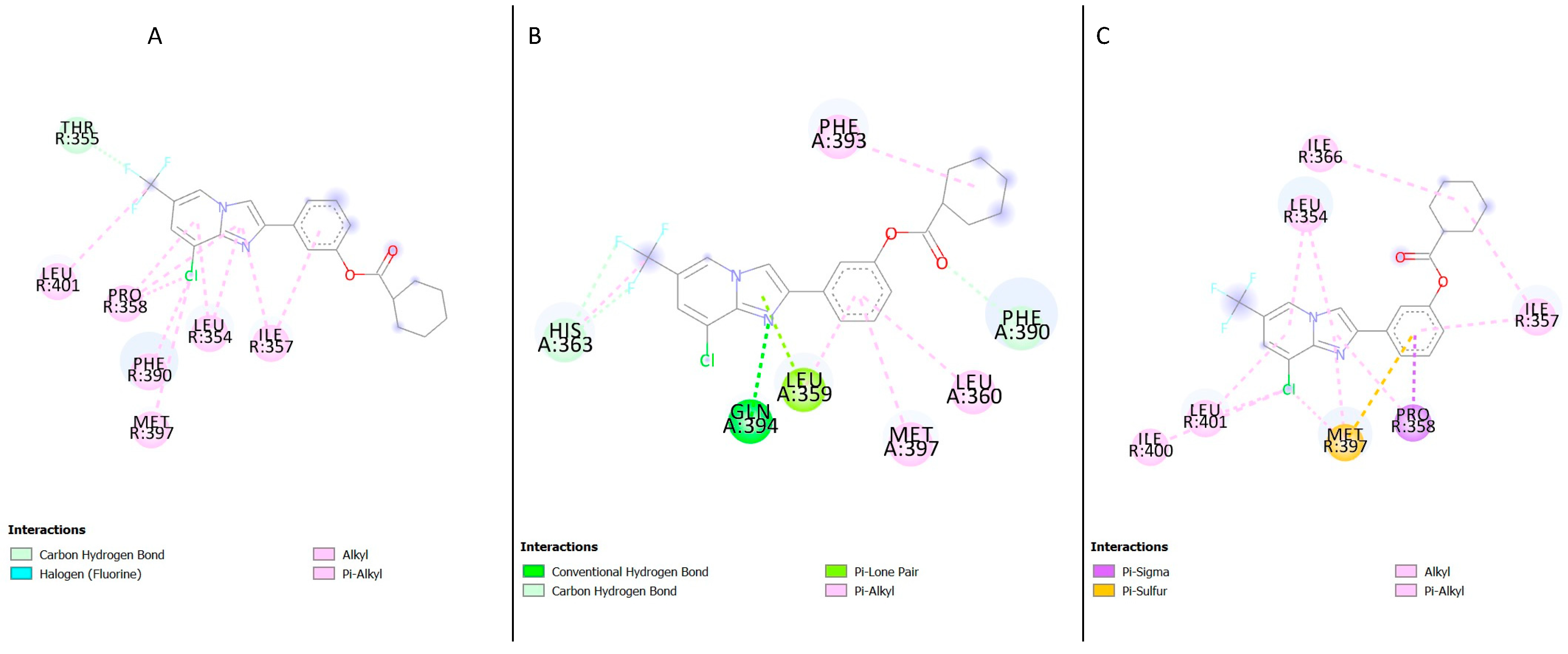
The docking result of compound
1 showed several residues present in the binding sites of the different GLP-1R crystal structures; in the structure 5VAI, the following residues were identified (
Table 1); Leu354, Lys351, Leu401, Thr355, Met397, Pro355, Phe390, Ile357 and Pro358 (
Figure 5A). Most of these residues were hydrophobic amino acids except for Thr355 and Lys351; both are polar and positively charged, respectively. In the inactive structure 5VEW, the following residues were present in the binding site (
Table 1), Arg348, Tyr402, Val405, Lys351, Leu349 and His180 (
Figure 5B,
Table 1). All the residues present in the binding site were polar. Upon examination of the structure 6B3J, the following residues interacted in the binding site (
Table 1); Leu354, Ile357, Gln394, His363, Phe393, Met397, Pro358 and Phe390 (
Figure 5C,
Table 1). His363 and Gln394 were polar, while the remaining residues were hydrophobic. An analysis of the ligand’s second and third top poses and the protein structures showed the residues Thr355 and Leu401 were present in the first three poses of the 5VAI-Compound
1 complex (
Table 1,
Tables S2 and S3). The other protein structures used in the study did not have any similar interacting residues across the three poses analysed.
The presence of polar residues in the binding sites of the three structures examined suggests that these polar residues play a vital role in ligand binding [
20]. The presence of these polar residues in the crystal structures of the receptor indicates the presence of a significant polar binding network around the peptide bindings site [
20]. Bueno et al. [
18] reported the capability of 2,6,7-trichloro-3-(trifluoromethyl)quinoxaline to potentiate GLP-1(9–36)-NH2-mediated cAMP accumulation in GLP-1R-expressing cells. The study results showed that compound
1 potentiated the activity of GLP-1(9–36)-NH2 on the wild-type receptor but failed to exert the same effect in cells expressing the mutant GLP-1R, which lacks the cysteine-347 residue [
18]. A comparative molecular dynamics simulation showed that the cysteine-3476.36bF (C3476.36bF) mutant maximises van der Waals interactions with all the three negative allosteric modulators (NAMs) PF-06372222, NNC0640 and MK-0893 through the stabilisation of the aliphatic side chain of Lysine-3516.40b (K3516.40b) in an optimal conformation for hydrophobic interactions with NAMs [
16,
21]. An analysis of the findings from this study showed the residue Lys351(K351) forming hydrogen bonds and pi-cation interactions (
Figure 5A,B and
Figure 7B). The mutation S352→A terminates the inhibition of GLP-1R by NAMs while the T355→A eliminates the inhibition by NAMs, NNC0640 and PF-06372222 but doesn’t inhibit the activity of MK-0893 [
16]. The findings from previous research stated that positions 352–355 play a crucial role in binding an allosteric inhibitor to GLP-1R [
16].
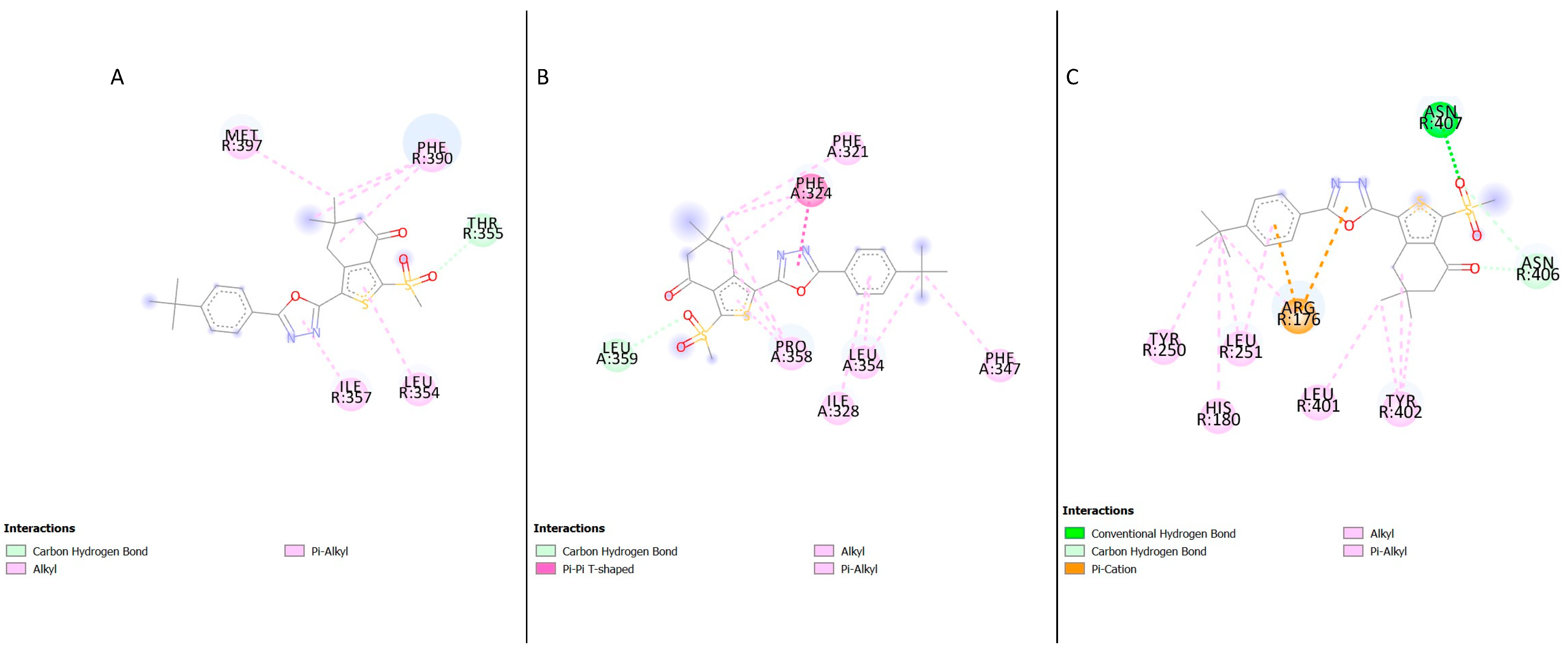
The examination of the docking results of compound
2 and the GLP-1R structures demonstrated several residues present in the active site of the crystal structures. In the active structure 5VAI, the following hydrophobic residues were present in the active site; Met397, Ile357, Leu354, Phe390. Thr355 was identified as the polar residues (
Table 2,
Figure 7A). All the residues interacting with the ligand in the binding site of the inactive crystal structure 5VEW were hydrophobic (
Table 2,
Figure 7B). Three of the residues, namely: Tyr402, His180 and Arg176 identified in the binding site of 6B3J, were polar, whilst the other residues were hydrophobic (
Table 2,
Figure 7C). The amino acid residue Phe390 interacted with compound
2, and the structure 5VAI in the top three poses was analysed (
Table 2,
Tables S5 and S6). The analysis of the top three poses of the compound
2-5VEW complex showed the amino acid residues Phe324, Leu354 and Pro 358 present in all the poses analysed (
Table 2,
Tables S5 and S6).
The presence of polar residues in the binding site of the GLP-1R crystal structures suggests critical hydrogen bond interactions which maintain receptor integrity and apo state [
20]. A previous study has demonstrated the capacity of compound
2 to potentiate GLP-1(9–36)-NH2-mediated cAMP accumulation in GLP-1R-expressing cells. The study showed that compound
2 potentiated the activity of GLP-1(9–36)-NH2 on the wild-type receptor but failed to exert the same effect in cells expressing the mutant GLP-1R, which lacks the Cysteine-347 residue [
18].
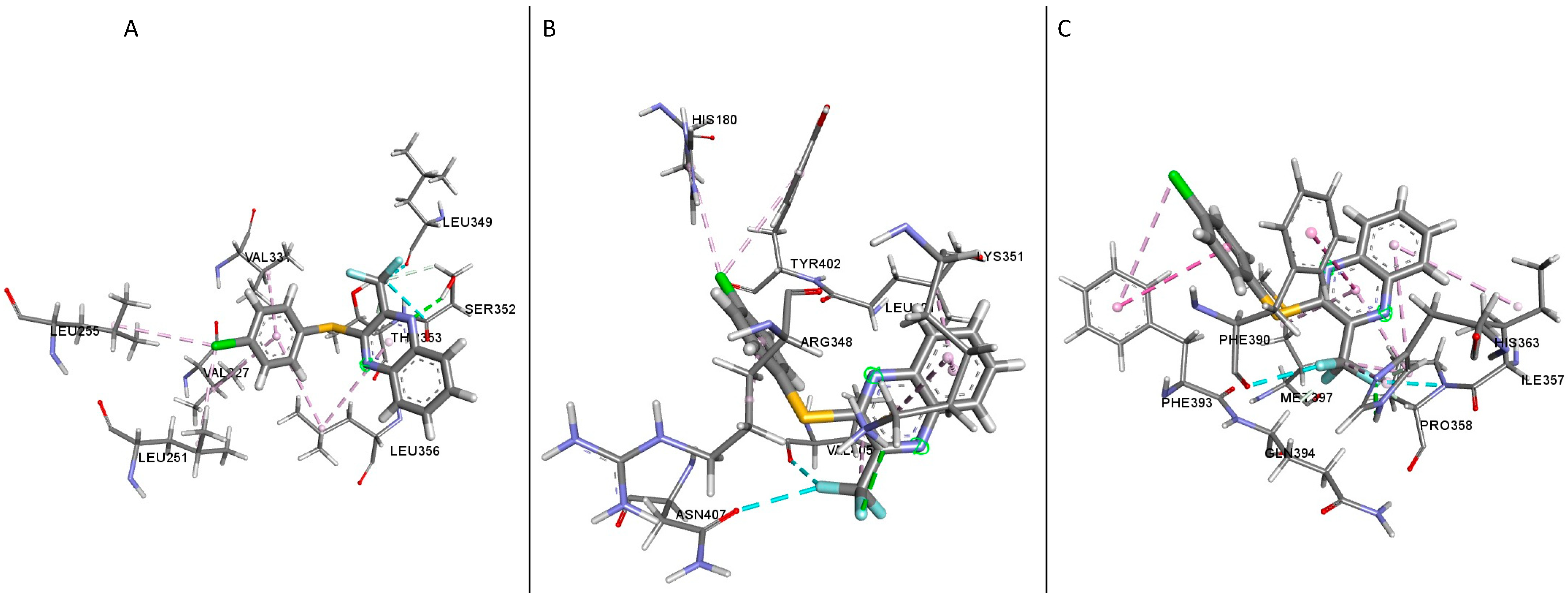
In the active structure (PDB ID: 5VAI [
19]-Compound
3 complex), eight residues were identified in the active site, most of which were hydrophobic residues. The active site consists of hydrophobic residues Val327, Val331, Leu251, Leu255, Leu356, and Leu349. Thr353 and Ser352 are the residues responsible for the polar region of the binding site (
Table 3,
Figure 9A). Upon examination of the inactive structure (PDB ID: 5VEW [
16]), there was an uneven distribution of polar (Tyr402, Asn407, His180, Arg348 and Lys351) and hydrophobic (Leu401 and Val405) amino acid residues (
Table 3,
Figure 9B). The active site of the other structure (PDB ID: 6B3J [
20]) examined had the majority of hydrophobic residues present (Ile357, Phe393, Ile356, Pro358, Phe390, Met397) while the remaining residues (His363, Gln394) were polar (
Table 3,
Figure 9C). The presence of polar residues in the active sites of the structures examined implies a critical hydrogen bonding between the ligand and the residues [
20].
An earlier study carried out by Gong et al. [
37], reported that compounds which possess a 3-(8-chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridine2-yl)phenyl acetate moiety, including compound
3 (3-(8-chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridine-2-yl) phenylcyclo propane carboxylate), are selective GLP-1R agonists, and have potential as anti-diabetic treatment agents.
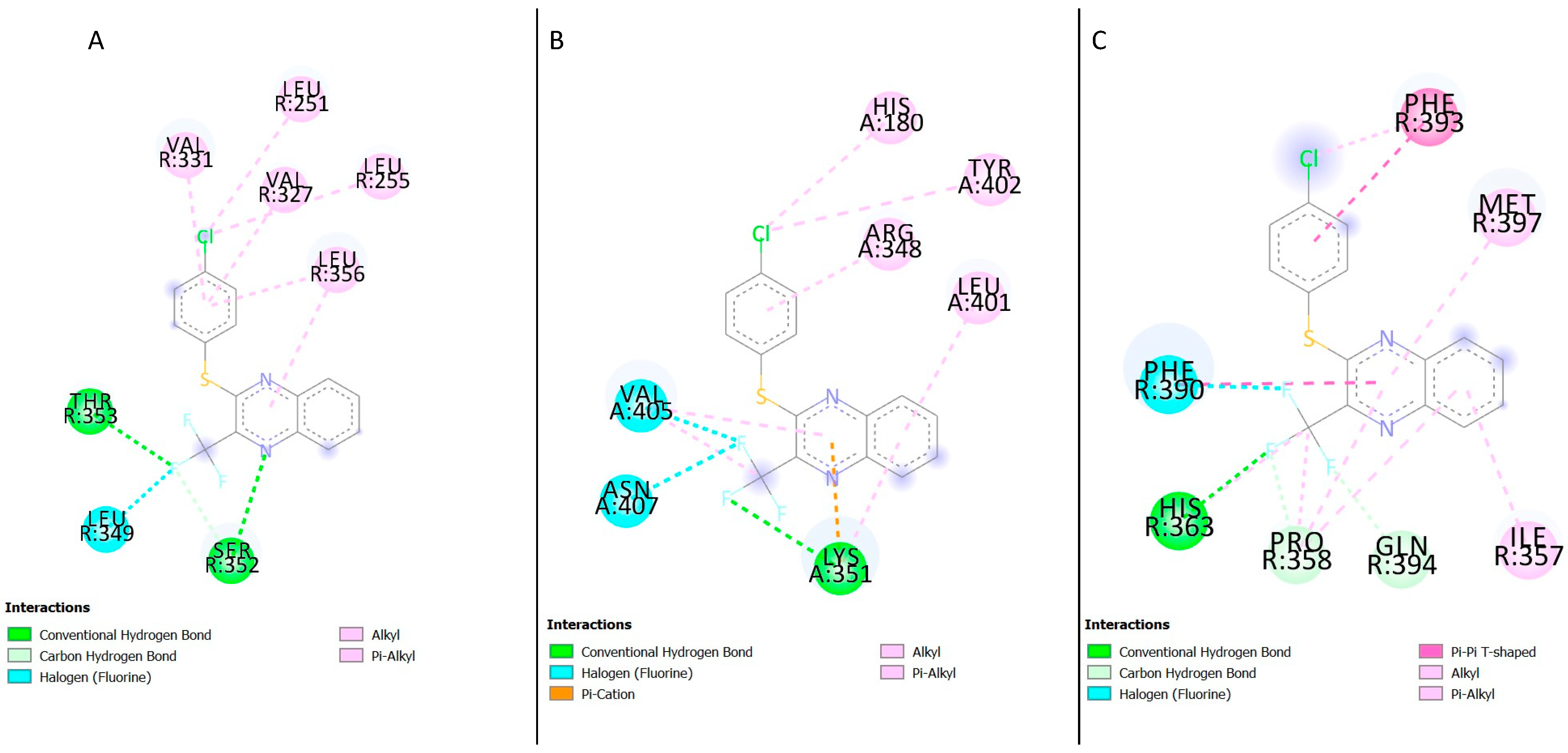
In the active structure (PDB ID: 5VAI [
19]), seven residues were found in the compound
4 binding site; the majority of the residues were hydrophobic except Thr355 (
Figure 11A). The docking result of the inactive structure (PDB ID: 5VEW [
16]) showed the majority of the residues found in the active site being hydrophobic (
Table 4,
Figure 11B), His363 and Gln394 were the only polar residues identified in the binding site. Examination of the docking results with the crystal structure 6B3J (PDB ID: 6B3J [
20]) showed that the residues interacting with the ligand were hydrophobic (
Table 4,
Figure 11C).
In a study carried out by Gong et al. [
37], with screened heterocyclic small molecules, they discovered a novel skeleton of 3-(8-chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridine2-yl)phenyl acetate derivative (compound
4), which on further characterisation proved to be selective GLP-1R agonists which potentially had a therapeutic effect on diabetes. In a later study, performed by Bueno et al. [
18], their findings inferred that compound
4 failed to activate GLP-1R through a covalent mechanism as it remained unaltered after 2 h and thus was considered non-reactive. An analysis of the top three binding poses of the all the protein–ligand complexes showed that the residues interacting with the ligands were slightly different albeit docking at TM6 for most of the poses (
Table 1,
Table 2,
Table 3 and
Table 4 and
Tables S3–S10). This however would suggest a flexibility of the ligand within the binding pocket of the receptor.
The binding affinity (ΔG
noelec) values for the 5VAI-Compound
1, 5VEW-Compound
1, 6B3J-Compound
1, 5VAI-Compound
2, 5VEW-Compound
2, 6B3J-Compound
2, 5VAI-Compound
3, 5VEW-Compound
3, 6B3J-Compound
3, 5VAI-Compound
4, 5VEW-Compound
4, 6B3J-Compound
4 was revealed to be −6.8 Kcal/mol, −7.2 Kcal/mol, −7.2 Kcal/mol, −8.5 Kcal/mol, −8.1 Kcal/mol, −8.8 Kcal/mol, −7.5 Kcal/mol, −7.9 Kcal/mol, −7.9 Kcal/mol, −8.5 Kcal/mol, −8.6 Kcal/mol, −9.2 Kcal/mol respectively (
Table 5). The results showed that the dockings were feasible energetically; this was demonstrated by the negative Gibbs free energy (ΔG) values (
Table 5). The findings showed that compound
4-protein structure (PDB IDs: 5VAI [
19], 6B3J [
20], 5VEW [
16]) complexes had the highest predicted binding affinities. This suggests that compound
4 would be bound tightly to the allosteric binding site of the receptor. The findings on
Table 5, also show that compound
2 and compound
4 would be bound strongly to the allosteric binding site of the GLP-1R.
The in silico prediction of ADME/Tox properties of any new drug candidate is essential in drug development, thus allowing scientists to investigate its properties to optimise the drug candidates to acceptable ADME/Tox standards [
38]. The findings in
Table 6 showed that the allosteric modulators complied with Lipinski’s rule of five except compound
4. According to the ADMET Predictor software, compound
4 violated the rule because of a high logP value of over 5.6. This translates to compound
4 not being a likely drug candidate according to the traditional method of evaluating drug-likeness; Lipinski’s rule of 5. The molecular descriptors HBA and HBD were found to comply with the cut off limits of Ro5 (
Table 6). The ligands evaluated in this study complied with the Veber drug-likeness filter (rotatable bonds ≤ 10, TPSA ≤ 140) [
39]. Based on the rule of three for fragment-based drug discovery (molecular weight < 300, ClogP < 3, the number of hydrogen bond donors and acceptors < 3 and the number of rotatable bonds < 3), the ligands violated all the rules [
40]. Lipophilicity is often expressed as the distribution coefficient in water/octanol (logD); this parameter influences some processes like plasma protein binding, oral absorption and VD [
41]. Nevertheless, higher logD values translate to higher vulnerability to P450 metabolism leading to higher clearance [
41]. The predicted logD values presented in
Table 5 showed compound
4 with the highest logD value while compound
1 had the least. However, all the ligands also possessed high logD values of over 3.5 which leads to low aqueous solubility, this makes these ligands potentially promiscuous as high lipophilicity often leads to low metabolic clearance and toxicity. The predicted human jejunal permeability (P
eff) values (
Table 6) show that the allosteric modulators had values of over 1.5 cm/s × 10
4; this indicates that the allosteric modulators would be absorbed entirely. This finding is corroborated by previous research, which reports that drug candidates with a P
eff value of >1.5 would be wholly absorbed irrespective of transport mechanism(s) being utilised [
42].
The volume of distribution (VD) is essential in ADME studies [
43]. It relates to the amount of a drug in the body to the measured concentration in a suitable biological fluid [
43]. The findings for the VD parameter (
Table 6) showed compound
2 having the lowest value (0.86 L/kg) while compound
1 had the highest. Hassan et al. reported that VD values of <5.5 L/kg guaranteed decreased deep tissue penetration [
44]. Nonetheless, compounds that enter tissues and bind extensively will show VD above the total body water (i.e., any value greater than 1 L/kg) [
45]. The allosteric modulators would remain in the bloodstream for an extended period, thus exerting their effects over a longer period due to their predicted high plasma protein binding (% unbound) of over 90% (
Table 6). The ADME/Tox software predicted high BBB penetration for all the allosteric modulators investigated. This implies that the allosteric modulators investigated may potentially treat ailments affecting the brain (
Table 6). The predicted TPSA values (
Table 6) shows compounds
1,
3 and
4 with TPSA values of less than 60 Å
2, while the compound
2 TPSA value was above 60 Å
2 but less than 140 Å
2. In 2009, Fernandes and Gattass [
46] reported that molecules with PSA values greater than 140 Å
2 are believed to have low cell membrane penetrating capacity, while those with PSA ≤ 60 Å
2 are easily absorbed.
All the allosteric modulators investigated were not substrates of P-glycoprotein (P-gp); compound
3 and compound
4 were identified as inhibitors of P-gp (
Table 7). Even though most compounds do not inhibit P-gp, there is a possibility that the allosteric modulators can be transported out of the cell by it [
47]. The allosteric modulators were not inhibitors of organic anion transporting polypeptide 1B1 (OATP1B1), but the prediction showed that the allosteric modulators were inhibitors of organic cation transporter 2 (OCT2) (
Table 7). All the ligands inhibited bile salt export pump (BSEP) except compound
1, while the rest were substrates of breast cancer resistance protein (BCRP) except the allosteric modulator compound
3. Cytochrome P450 (CYP450) are the enzymes that catalyse the oxidation of organic substances [
44]. The evaluation of the allosteric modulators on the various isoforms of hepatic CYP450 was also predicted; the findings are shown in
Table 8.
Table 8 demonstrates that all the allosteric modulators were substrates of CYP3A4, albeit compound
3 was an isoform inhibitor. Allosteric modulators showed inhibitory tendencies and substrate specificities for the various isoforms investigated (
Table 8). The ADMET predictor software inferred that the clearance pathway for the allosteric modulators studied is via metabolism. Drug toxicity has resulted in the failure of drug candidates in clinical trials, hence, the use of in silico models to predict the potential toxicity of new drug candidates [
43].
The predicted toxicity parameters were shown in
Table 9; all allosteric modulators evaluated reported negative for AMES toxicity. The hERG filter parameter returned negative, implying that the allosteric modulators do not have an affinity for the hERG potassium channel in humans. All the allosteric modulators studied could cause potential reproductive/developmental toxicity except compound
1 (
Table 9). The allosteric modulators were all skin sensitisers except compound
3 (
Table 9). The liver function parameters (
Table 9) showed some of the allosteric modulators causing elevations in levels of liver enzymes studied. Compound
1 and compound
2 elevated the levels of the liver functions studied (
Table 9), resulting in them being classed as hepatotoxic by the prediction software.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}