
Deciphering the Structural and Functional Paradigms of Clostridioides difficile Toxins TcdA and TcdB

 , , , , and
, , , , and
Abstract
1. Introduction
1.1. CTD Toxin
1.2. Regulation of Toxin Gene Loci (tcdA/tcdB)
1.3. Evolution of Hypervirulent Strains
1.4. Horizontal Gene Transfer and Recombination Events
2. Structural Biology of TcdA/B: Cryo-EM-Derived Conformational Dynamics, Receptor-Binding Domains, and Pore-Forming Motifs
2.1. Cryo-EM-Derived Conformational Dynamics of TcdA and TcdB
Structural Organization and Domain Architecture
2.2. pH-Induced Conformational Changes and Receptor Dissociation
2.3. Receptor-Binding Domains (RBDs) and Ligand Interactions
2.3.1. Ligand Specificity and Differential Receptor Utilization
2.3.2. Role of Receptor-Binding Variants in Hypervirulence
2.4. Pore-Forming Motifs and Membrane Insertion Mechanisms
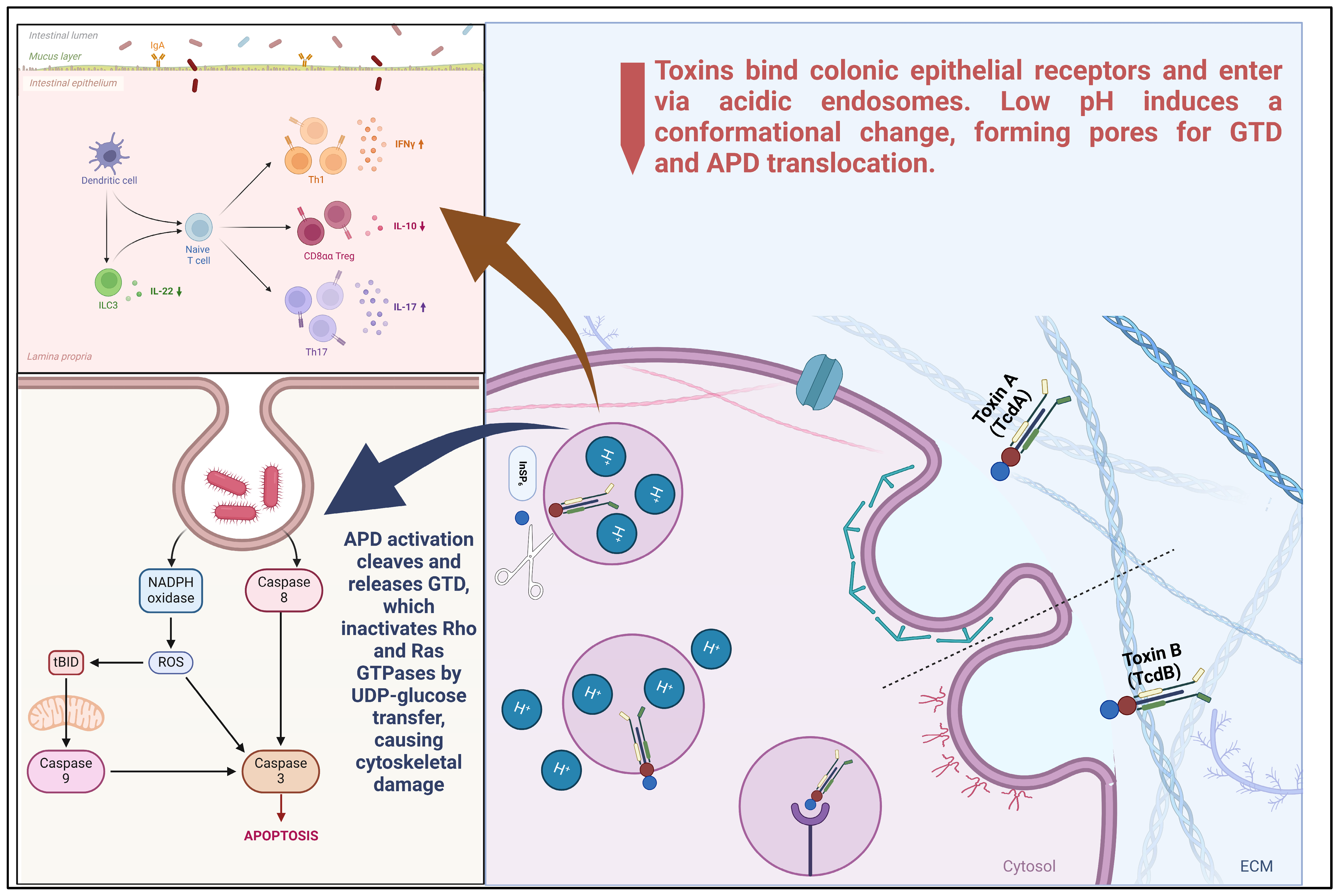
2.4.1. Receptor Binding and Endocytosis
2.4.2. Membrane Insertion and Pore Formation
3. Molecular Pathogenesis: Endocytic Trafficking, Cytosolic Delivery Mechanisms, and Rho GTPase Inactivation
3.1. Endocytic Trafficking of TcdA and TcdB
3.1.1. Receptor-Mediated Endocytosis and Receptor Specificity
3.1.2. Clathrin-Independent and PACSIN2-Dependent Endocytosis of TcdA
3.1.3. Endosomal Acidification and Toxin Translocation
3.2. Cytosolic Delivery and Autoprocessing Mechanisms
3.3. Rho GTPase Inactivation by TcdA and TcdB
4. Microbiota-Mediated Resistance to C. difficile Infection
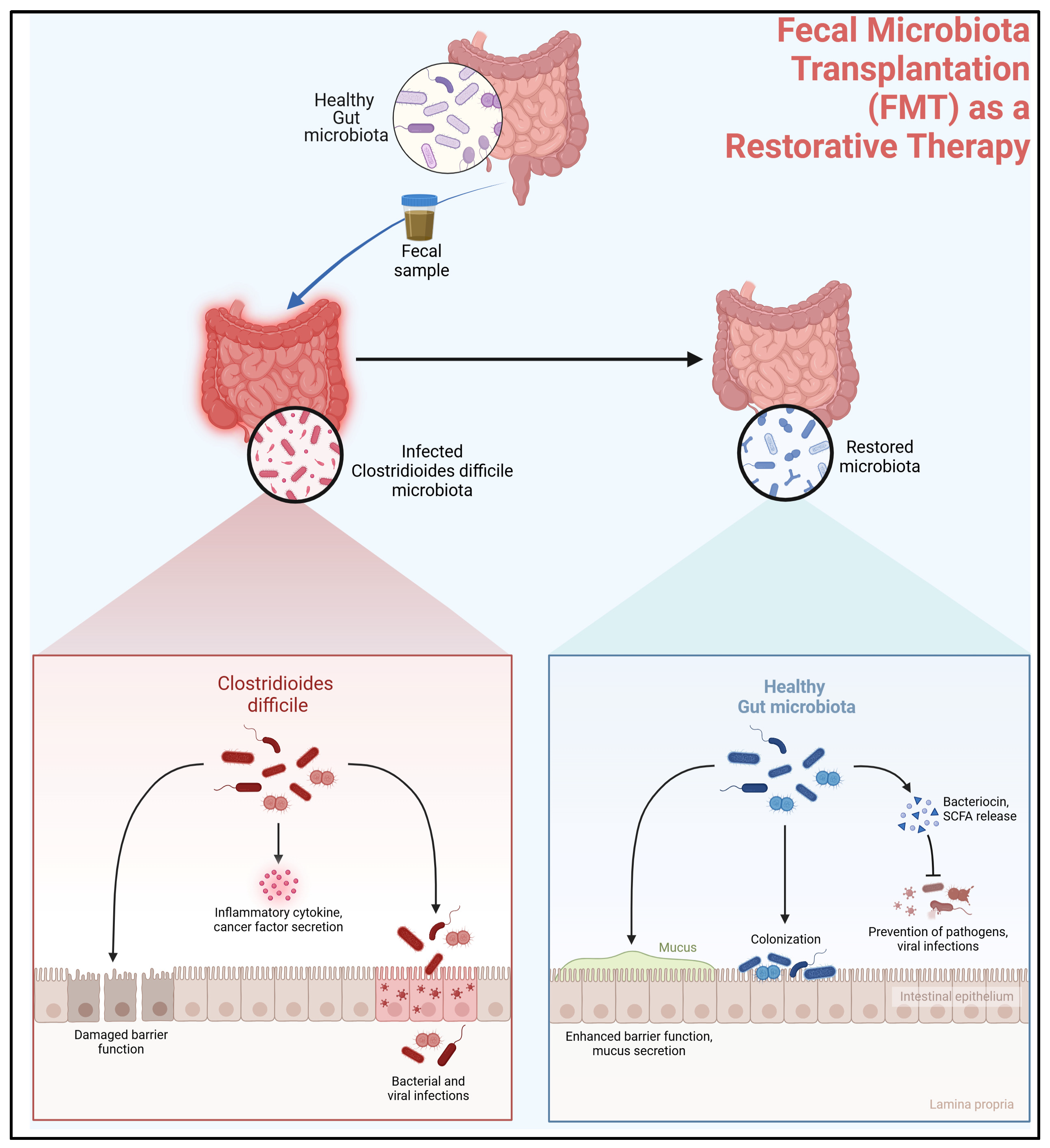
4.1. Role of Microbiota in Colonization Resistance
4.2. Fecal Microbiota Transplantation (FMT) as a Restorative Therapy
5. Future Directions: Integrative Models for Toxin–Host–Microbiome Crosstalk and Translational Challenges
5.1. Systems Biology Approaches to Toxin–Host–Microbiome Crosstalk
5.2. Translational Challenges in Therapeutic Development
5.3. Future Research Priorities
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APD | Adaptor Protein Domain |
| BAR | Bin/Amphiphysin/Rvs (domain) |
| CD | Clostridioides difficile |
| CDAD | Clostridioides difficile-Associated Diarrhea |
| CDI | Clostridioides difficile Infection |
| CDT | Clostridioides difficile Transferase (binary toxin) |
| CHC | Clathrin Heavy Chain |
| CHO | Chinese Hamster Ovary (cells) |
| CME | Clathrin-Mediated Endocytosis |
| CPD | Cysteine Protease Domain |
| CRD | Cysteine-Rich Domain |
| CROP | Combined Repetitive Oligopeptides (domain) |
| DCA | Deoxycholic Acid |
| DNA | Deoxyribonucleic Acid |
| DRBD | Double RNA-Binding Domain |
| EM | Electron Microscopy |
| FBD | Frizzled Binding Domain |
| FDA | Food and Drug Administration |
| FEME | Fast Endophilin-Mediated Endocytosis |
| FMT | Fecal Microbiota Transplantation |
| FZD | Frizzled (protein family) |
| GDP | Guanosine Diphosphate |
| GTD | Glucosyltransferase Domain |
| GTP | Guanosine Triphosphate |
| HGT | Horizontal Gene Transfer |
| HT | Human Toxin |
| IBD | Inflammatory Bowel Disease |
| II | Intrinsic Immunity |
| INDIA | Indian National Drug Authority (possible context) |
| IV | Intravenous |
| LCA | Lithocholic Acid |
| LDL | Low-Density Lipoprotein |
| LDLR | Low-Density Lipoprotein Receptor |
| MAPK | Mitogen-Activated Protein Kinase |
| MEF | Mouse Embryonic Fibroblast |
| NMDA | N-Methyl-D-Aspartate (receptor) |
| OFF | Inactive State |
| ON | Active State |
| PAK | p21-Activated Kinase |
| PCR | Polymerase Chain Reaction |
| PDGF | Platelet-Derived Growth Factor |
| RNA | Ribonucleic Acid |
| TEER | Trans-Epithelial Electrical Resistance |
| UDP | Uridine Diphosphate |
References
- Balsells, E.; Shi, T.; Leese, C.; Lyell, I.; Burrows, J.; Wiuff, C.; Campbell, H.; Kyaw, M.H.; Nair, H. Global burden of Clostridium difficile infections: A systematic review and meta-analysis. J. Glob. Health 2018, 9, 010407. [Google Scholar] [CrossRef] [PubMed]
- Stoian, M.; Andone, A.; Boeriu, A.; Bândilă, S.R.; Dobru, D.; Laszlo, S.Ș.; Corău, D.; Arbănași, E.M.; Russu, E.; Stoian, A. COVID-19 and Clostridioides difficile Coinfection Analysis in the Intensive Care Unit. Antibiotics 2024, 13, 367. [Google Scholar] [CrossRef] [PubMed]
- Leffler, D.A.; Lamont, J.T. Clostridium difficile Infection. N. Engl. J. Med. 2015, 372, 1539–1548. [Google Scholar] [CrossRef]
- Manse, J.S.; Baldwin, M.R. Binding and entry of Clostridium difficile toxin B is mediated by multiple domains. FEBS Lett. 2015, 589, 3945–3951. [Google Scholar] [CrossRef] [PubMed]
- Filić, V.; Mijanović, L.; Putar, D.; Talajić, A.; Ćetković, H.; Weber, I. Regulation of the Actin Cytoskeleton via Rho GTPase Signalling in Dictyostelium and Mammalian Cells: A Parallel Slalom. Cells 2021, 10, 1592. [Google Scholar] [CrossRef]
- Pruitt, R.N.; Chagot, B.; Cover, M.; Chazin, W.J.; Spiller, B.; Lacy, D.B. Structure-Function Analysis of Inositol Hexakisphosphate-induced Autoprocessing in Clostridium difficile Toxin A. J. Biol. Chem. 2009, 284, 21934–21940. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhan, X.; Luo, J.; Li, D.; Zhou, R.; Zhang, J.; Pan, Z.; Zhang, Y.; Jia, T.; Zhang, X.; et al. Structural dynamics of the CROPs domain control stability and toxicity of Paeniclostridium sordellii lethal toxin. Nat. Commun. 2023, 14, 8426. [Google Scholar] [CrossRef]
- Kordus, S.L.; Thomas, A.K.; Lacy, D.B. Clostridioides difficile toxins: Mechanisms of action and antitoxin therapeutics. Nat. Rev. Microbiol. 2021, 20, 285–298. [Google Scholar] [CrossRef]
- Gerding, D.N.; Johnson, S.; Rupnik, M.; Aktories, K. Clostridium difficile binary toxin CDT. Gut Microbes 2013, 5, 15–27. [Google Scholar] [CrossRef]
- Alam, Z.; Madan, R. Clostridioides difficile Toxins: Host Cell Interactions and Their Role in Disease Pathogenesis. Toxins 2024, 16, 241. [Google Scholar] [CrossRef]
- Carter, G.P.; Douce, G.R.; Govind, R.; Howarth, P.M.; Mackin, K.E.; Spencer, J.; Buckley, A.M.; Antunes, A.; Kotsanas, D.; Jenkin, G.A.; et al. The Anti-Sigma Factor TcdC Modulates Hypervirulence in an Epidemic BI/NAP1/027 Clinical Isolate of Clostridium difficile. PLoS Pathog. 2011, 7, e1002317. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Govind, R. Regulation of Clostridioides difficile toxin production. Curr. Opin. Microbiol. 2021, 65, 95–100. [Google Scholar] [CrossRef]
- Ransom, E.M.; Kaus, G.M.; Tran, P.M.; Ellermeier, C.D.; Weiss, D.S. Multiple factors contribute to bimodal toxin gene expression in Clostridioides (Clostridium) difficile. Mol. Microbiol. 2018, 110, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Perkins, M.L.; Norstad, M.; Garcia, H.G. A bistable autoregulatory module in the developing embryo commits cells to binary expression fates. Curr. Biol. 2023, 33, 2851–2864.e11. [Google Scholar] [CrossRef]
- Martin-Verstraete, I.; Peltier, J.; Dupuy, B. The Regulatory Networks That Control Clostridium difficile Toxin Synthesis. Toxins 2016, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Anjuwon-Foster, B.R.; Tamayo, R. A genetic switch controls the production of flagella and toxins in Clostridium difficile. PLoS Genet. 2017, 13, e1006701. [Google Scholar] [CrossRef]
- Norris, M.A.W.; Plaskon, D.M.; Tamayo, R. Phase Variation of Flagella and Toxins in Clostridioides difficile is Mediated by Selective Rho-dependent Termination. J. Mol. Biol. 2024, 436, 168456. [Google Scholar] [CrossRef]
- Edwards, A.N.; McBride, S.M. Initiation of sporulation in Clostridium difficile: A twist on the classic model. FEMS Microbiol. Lett. 2014, 358, 110–118. [Google Scholar] [CrossRef]
- Buddle, J.E.; Fagan, R.P. Pathogenicity and virulence of Clostridioides difficile. Virulence 2023, 14, 2150452. [Google Scholar] [CrossRef]
- Hasan, K.; Alaribe, O.; Govind, R. Regulatory networks: Linking toxin production and sporulation in Clostridioides difficile. Anaerobe 2024, 91, 102920. [Google Scholar] [CrossRef]
- Lin, W.N.; Tay, M.Z.; Lu, R.; Liu, Y.; Chen, C.-H.; Cheow, L.F. The Role of Single-Cell Technology in the Study and Control of Infectious Diseases. Cells 2020, 9, 1440. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.J.; Tremblay, B.J.-M.; Zeng, J.; Wei, X.; Hodgins, H.; Worley, J.; Bry, L.; Dong, M.; Doxey, A.C. Phylogenomics of 8,839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin A and B. PLoS Pathog. 2020, 16, e1009181. [Google Scholar] [CrossRef]
- Shen, E.; Zhu, K.; Li, D.; Pan, Z.; Luo, Y.; Bian, Q.; He, L.; Song, X.; Zhen, Y.; Jin, D.; et al. Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B. Commun. Biol. 2020, 3, 347. [Google Scholar] [CrossRef] [PubMed]
- Aptekorz, M.; Szczegielniak, A.; Wiechuła, B.; Harmanus, C.; Kuijper, E.; Martirosian, G. Occurrence of Clostridium difficile ribotype 027 in hospitals of Silesia, Poland. Anaerobe 2017, 45, 106–113. [Google Scholar] [CrossRef]
- Janezic, S.; Dingle, K.; Alvin, J.; Accetto, T.; Didelot, X.; Crook, D.W.; Lacy, D.B.; Rupnik, M. Comparative genomics of Clostridioides difficile toxinotypes identifies module-based toxin gene evolution. Microb. Genom. 2020, 6, e000449. [Google Scholar] [CrossRef]
- Pan, Z.; Zhang, Y.; Luo, J.; Li, D.; Zhou, Y.; He, L.; Yang, Q.; Dong, M.; Tao, L. Functional analyses of epidemic Clostridioides difficile toxin B variants reveal their divergence in utilizing receptors and inducing pathology. PLoS Pathog. 2021, 17, e1009197. [Google Scholar] [CrossRef]
- Knight, D.R.; Elliott, B.; Chang, B.J.; Perkins, T.T.; Riley, T.V. Diversity and Evolution in the Genome of Clostridium difficile. Clin. Microbiol. Rev. 2015, 28, 721–741. [Google Scholar] [CrossRef]
- Brouwer, M.S.; Roberts, A.P.; Hussain, H.; Williams, R.J.; Allan, E.; Mullany, P. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat. Commun. 2013, 4, 2601. [Google Scholar] [CrossRef]
- Brouwer, M.S.M.; Warburton, P.J.; Roberts, A.P.; Mullany, P.; Allan, E. Genetic Organisation, Mobility and Predicted Functions of Genes on Integrated, Mobile Genetic Elements in Sequenced Strains of Clostridium difficile. PLoS ONE 2011, 6, e23014. [Google Scholar] [CrossRef]
- Newcomer, E.P.; Fishbein, S.R.S.; Zhang, K.; Hink, T.; Reske, K.A.; Cass, C.; Iqbal, Z.H.; Struttmann, E.L.; Burnham, C.-A.D.; Dubberke, E.R.; et al. Genomic surveillance of Clostridioides difficile transmission and virulence in a healthcare setting. mBio 2024, 15, e0330023. [Google Scholar] [CrossRef]
- Monot, M.; Eckert, C.; Lemire, A.; Hamiot, A.; Dubois, T.; Tessier, C.; Dumoulard, B.; Hamel, B.; Petit, A.; Lalande, V.; et al. Clostridium difficile: New Insights into the Evolution of the Pathogenicity Locus. Sci. Rep. 2015, 5, 15023. [Google Scholar] [CrossRef] [PubMed]
- Mehner-Breitfeld, D.; Rathmann, C.; Riedel, T.; Just, I.; Gerhard, R.; Overmann, J.; Brüser, T. Evidence for an Adaptation of a Phage-Derived Holin/Endolysin System to Toxin Transport in Clostridioides difficile. Front. Microbiol. 2018, 9, 2446. [Google Scholar] [CrossRef]
- Murata, K.; Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 324–334. [Google Scholar] [CrossRef]
- Aminzadeh, A.; Larsen, C.E.; Boesen, T.; Jørgensen, R. High-resolution structure of native toxin A from Clostridioides difficile. Embo Rep. 2021, 23, e53597. [Google Scholar] [CrossRef]
- Jiang, M.; Shin, J.; Simeon, R.; Chang, J.-Y.; Meng, R.; Wang, Y.; Shinde, O.; Li, P.; Chen, Z.; Zhang, J. Structural dynamics of receptor recognition and pH-induced dissociation of full-length Clostridioides difficile Toxin B. PLoS Biol. 2022, 20, e3001589. [Google Scholar] [CrossRef]
- Childress, K.O.; Cencer, C.S.; Tyska, M.J.; Lacy, D.B. Nectin-3 and shed forms of CSPG4 can serve as epithelial cell receptors for Clostridioides difficile TcdB. mBio 2023, 14, e0185723. [Google Scholar] [CrossRef] [PubMed]
- Bland, S.J.; Larabee, J.L.; Shadid, T.M.; Lang, M.L.; Ballard, J.D. Deletion of a 19-Amino-Acid Region in Clostridioides difficile TcdB2 Results in Spontaneous Autoprocessing and Reduced Cell Binding and Provides a Nontoxic Immunogen for Vaccination. Infect. Immun. 2019, 87, e00210-19. [Google Scholar] [CrossRef] [PubMed]
- Lanis, J.M.; Hightower, L.D.; Shen, A.; Ballard, J.D. TcdB from hypervirulent Clostridium difficile exhibits increased efficiency of autoprocessing. Mol. Microbiol. 2012, 84, 66–76. [Google Scholar] [CrossRef]
- Zhang, Y.; Hamza, T.; Gao, S.; Feng, H. Masking autoprocessing of Clostridium difficile toxin A by the C-terminus combined repetitive oligo peptides. Biochem. Biophys. Res. Commun. 2015, 459, 259–263. [Google Scholar] [CrossRef]
- Pruitt, R.N.; Chambers, M.G.; Ng, K.K.-S.; Ohi, M.D.; Lacy, D.B. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc. Natl. Acad. Sci. USA 2010, 107, 13467–13472. [Google Scholar] [CrossRef]
- Larabee, J.L.; Hauck, G.D.; Ballard, J.D. Cell-penetrating peptides derived from Clostridium difficile TcdB2 and a related large clostridial toxin. J. Biol. Chem. 2018, 293, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Cho, K.-H.; Song, H.; Yi, H.; Kim, H.S. Deletion Mutations Conferring Substrate Spectrum Extension in the Class A β-Lactamase. Antimicrob. Agents Chemother. 2014, 58, 6265–6269. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, Q.; Luo, J.; Zhang, Y.; Li, X.; He, L.; Ma, C.; Tao, L. Identification of a hemorrhagic determinant in Clostridioides difficile TcdA and Paeniclostridium sordellii TcsH. Microbiol. Spectr. 2024, 12, e0035424. [Google Scholar] [CrossRef]
- Larabee, J.L.; Bland, S.J.; Hunt, J.J.; Ballard, J.D. Intrinsic Toxin-Derived Peptides Destabilize and Inactivate Clostridium difficile TcdB. mBio 2017, 8, e00503-17. [Google Scholar] [CrossRef]
- Zhang, K.; Zhou, Q.; Gu, H.; Yang, M.; Lin, X.; Wang, M.; Zhai, H.; Zhang, F.; Luo, Y.; Chen, L.; et al. TcdB From Hypervirulent Clostridioides difficile Induces Neuronal Loss and Neurotransmitter Alterations in the Intrinsic Enteric Nervous System. J. Infect. Dis. 2024. [Google Scholar] [CrossRef]
- Tao, L.; Zhang, J.; Meraner, P.; Tovaglieri, A.; Wu, X.; Gerhard, R.; Zhang, X.; Stallcup, W.B.; Miao, J.; He, X.; et al. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 2016, 538, 350–355. [Google Scholar] [CrossRef]
- Gupta, P.; Zhang, Z.; Sugiman-Marangos, S.N.; Tam, J.; Raman, S.; Julien, J.-P.; Kroh, H.K.; Lacy, D.B.; Murgolo, N.; Bekkari, K.; et al. Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor-binding determinants. J. Biol. Chem. 2017, 292, 17290–17301. [Google Scholar] [CrossRef]
- Gray, V.E.; Hause, R.J.; Fowler, D.M. Analysis of Large-Scale Mutagenesis Data To Assess the Impact of Single Amino Acid Substitutions. Genetics 2017, 207, 53–61. [Google Scholar] [CrossRef]
- Doyle, D.A.; DeAngelis, P.L.; Ballard, J.D. CSPG4-dependent cytotoxicity for C. difficile TcdB is influenced by extracellular calcium and chondroitin sulfate. mSphere 2024, 9, e0009424. [Google Scholar] [CrossRef]
- Mentula, S.; Laakso, S.; Lyytikäinen, O.; Kirveskari, J. Differentiating virulent 027 and non-027 Clostridium difficile strains by molecular methods. Expert Rev. Mol. Diagn. 2015, 15, 1225–1229. [Google Scholar] [CrossRef]
- Gerding, D.N.; Kelly, C.P.; Rahav, G.; Lee, C.; Dubberke, E.R.; Kumar, P.N.; Yacyshyn, B.; Kao, D.; Eves, K.; Ellison, M.C.; et al. Bezlotoxumab for Prevention of Recurrent Clostridium difficile Infection in Patients at Increased Risk for Recurrence. Clin. Infect. Dis. 2018, 67, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Krueger, E.; Brown, A.C. Inhibition of bacterial toxin recognition of membrane components as an anti-virulence strategy. J. Biol. Eng. 2019, 13, 4. [Google Scholar] [CrossRef]
- Luo, J.; Yang, Q.; Zhang, X.; Zhang, Y.; Wan, L.; Zhan, X.; Zhou, Y.; He, L.; Li, D.; Jin, D.; et al. TFPI is a colonic crypt receptor for TcdB from hypervirulent clade 2 C. difficile. Cell 2022, 185, 980–994.e15. [Google Scholar] [CrossRef] [PubMed]
- Pourliotopoulou, E.; Karampatakis, T.; Kachrimanidou, M. Exploring the Toxin-Mediated Mechanisms in Clostridioides difficile Infection. Microorganisms 2024, 12, 1004. [Google Scholar] [CrossRef]
- Kinsolving, J.; Bous, J.; Kozielewicz, P.; Košenina, S.; Shekhani, R.; Grätz, L.; Masuyer, G.; Wang, Y.; Stenmark, P.; Dong, M.; et al. Structural and functional insight into the interaction of Clostridioides difficile toxin B and FZD7. Cell Rep. 2024, 43, 113727. [Google Scholar] [CrossRef]
- Kim, J.-S.; Choi, D.-K.; Shin, J.-Y.; Shin, S.-M.; Park, S.-W.; Cho, H.-S.; Kim, Y.-S. Endosomal acidic pH-induced conformational changes of a cytosol-penetrating antibody mediate endosomal escape. J. Control. Release 2016, 235, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Park, M.; Tam, J.; Auger, A.; Beilhartz, G.L.; Lacy, D.B.; Melnyk, R.A. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc. Natl. Acad. Sci. USA 2014, 111, 3721–3726. [Google Scholar] [CrossRef]
- Chandrasekaran, R.; Lacy, D.B. The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 2017, 41, 723–750. [Google Scholar] [CrossRef]
- Chen, P.; Zeng, J.; Liu, Z.; Thaker, H.; Wang, S.; Tian, S.; Zhang, J.; Tao, L.; Gutierrez, C.B.; Xing, L.; et al. Structural basis for CSPG4 as a receptor for TcdB and a therapeutic target in Clostridioides difficile infection. Nat. Commun. 2021, 12, 3748. [Google Scholar] [CrossRef]
- Chandrasekaran, R.; Kenworthy, A.K.; Lacy, D.B. Clostridium difficile Toxin A Undergoes Clathrin-Independent, PACSIN2-Dependent Endocytosis. PLoS Pathog. 2016, 12, e1006070. [Google Scholar] [CrossRef]
- Schöttelndreier, D.; Langejürgen, A.; Lindner, R.; Genth, H. Low Density Lipoprotein Receptor-Related Protein-1 (LRP1) Is Involved in the Uptake of Clostridioides difficile Toxin A and Serves as an Internalizing Receptor. Front. Cell. Infect. Microbiol. 2020, 10, 565465. [Google Scholar] [CrossRef]
- Tucker, K.D.; Wilkins, T.D. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 1991, 59, 73–78. [Google Scholar] [CrossRef]
- Maier, W.; Bednorz, M.; Meister, S.; Roebroek, A.; Weggen, S.; Schmitt, U.; Pietrzik, C.U. LRP1 is critical for the surface distribution and internalization of the NR2B NMDA receptor subtype. Mol. Neurodegener. 2013, 8, 25. [Google Scholar] [CrossRef]
- Tao, L.; Tian, S.; Zhang, J.; Liu, Z.; Robinson-McCarthy, L.; Miyashita, S.-I.; Breault, D.T.; Gerhard, R.; Oottamasathien, S.; Whelan, S.P.J.; et al. Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat. Microbiol. 2019, 4, 1760–1769. [Google Scholar] [CrossRef]
- Henkel, D.; Tatge, H.; Schöttelndreier, D.; Tao, L.; Dong, M.; Gerhard, R. Receptor Binding Domains of TcdB from Clostridioides difficile for Chondroitin Sulfate Proteoglycan-4 and Frizzled Proteins Are Functionally Independent and Additive. Toxins 2020, 12, 736. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.J.; van Dissel, J.T.; Wilcox, M.H. Clostridium difficile: Changing epidemiology and new treatment options. Curr. Opin. Infect. Dis. 2007, 20, 376–383. [Google Scholar] [CrossRef]
- Orrell, K.E.; Mansfield, M.J.; Doxey, A.C.; Melnyk, R.A. The C. difficile toxin B membrane translocation machinery is an evolutionarily conserved protein delivery apparatus. Nat. Commun. 2020, 11, 432. [Google Scholar] [CrossRef]
- Marquardt, I.; Jakob, J.; Scheibel, J.; Hofmann, J.D.; Klawonn, F.; Neumann-Schaal, M.; Gerhard, R.; Bruder, D.; Jänsch, L. Clostridioides difficile Toxin CDT Induces Cytotoxic Responses in Human Mucosal-Associated Invariant T (MAIT) Cells. Front. Microbiol. 2021, 12, 752549. [Google Scholar] [CrossRef]
- Chen, P.; Jin, R. Receptor binding mechanisms of Clostridioides difficile toxin B and implications for therapeutics development. FEBS J. 2021, 290, 962–969. [Google Scholar] [CrossRef]
- Schorch, B.; Song, S.; van Diemen, F.R.; Bock, H.H.; May, P.; Herz, J.; Brummelkamp, T.R.; Papatheodorou, P.; Aktories, K. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. USA 2014, 111, 6431–6436. [Google Scholar] [CrossRef]
- Pinheiro, D.H.; Vélez, A.M.; Fishilevich, E.; Wang, H.; Carneiro, N.P.; Valencia-Jiménez, A.; Valicente, F.H.; Narva, K.E.; Siegfried, B.D. Clathrin-dependent endocytosis is associated with RNAi response in the western corn rootworm, Diabrotica virgifera virgifera LeConte. PLoS ONE 2018, 13, e0201849. [Google Scholar] [CrossRef] [PubMed]
- Paparella, A.S.; Aboulache, B.L.; Harijan, R.K.; Potts, K.S.; Tyler, P.C.; Schramm, V.L. Inhibition of Clostridium difficile TcdA and TcdB toxins with transition state analogues. Nat. Commun. 2021, 12, 6285. [Google Scholar] [CrossRef] [PubMed]
- Castro-Córdova, P.; Otto-Medina, M.; Montes-Bravo, N.; Brito-Silva, C.; Lacy, D.B.; Paredes-Sabja, D. Redistribution of the Novel Clostridioides difficile Spore Adherence Receptor E-Cadherin by TcdA and TcdB Increases Spore Binding to Adherens Junctions. Infect. Immun. 2023, 91, e00476-22. [Google Scholar] [CrossRef]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cell. Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-Independent Pathways of Endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef]
- Wint, H.; Li, J.; Abe, T.; Yamada, H.; Higaki, T.; Nasu, Y.; Watanabe, M.; Takei, K.; Takeda, T. Pacsin 2-dependent N-cadherin internalization regulates the migration behaviour of malignant cancer cells. J. Cell Sci. 2023, 136, jcs260827. [Google Scholar] [CrossRef]
- Papatheodorou, P.; Kindig, S.; Badilla-Lobo, A.; Fischer, S.; Durgun, E.; Thuraisingam, T.; Witte, A.; Song, S.; Aktories, K.; Chaves-Olarte, E.; et al. The Compound U18666A Inhibits the Intoxication of Cells by Clostridioides difficile Toxins TcdA and TcdB. Front. Microbiol. 2021, 12, 784856. [Google Scholar] [CrossRef]
- Heber, S.; Barthold, L.; Baier, J.; Papatheodorou, P.; Fois, G.; Frick, M.; Barth, H.; Fischer, S. Inhibition of Clostridioides difficile Toxins TcdA and TcdB by Ambroxol. Front. Pharmacol. 2022, 12, 809595. [Google Scholar] [CrossRef]
- Ruhe, F.; Olling, A.; Abromeit, R.; Rataj, D.; Grieschat, M.; Zeug, A.; Gerhard, R.; Alekov, A. Overexpression of the Endosomal Anion/Proton Exchanger ClC-5 Increases Cell Susceptibility toward Clostridium difficile Toxins TcdA and TcdB. Front. Cell. Infect. Microbiol. 2017, 7, 67. [Google Scholar] [CrossRef]
- Peritore-Galve, F.C.; Shupe, J.A.; Cave, R.J.; Childress, K.O.; Washington, M.K.; Kuehne, S.A.; Lacy, D.B. Glucosyltransferase-dependent and independent effects of Clostridioides difficile toxins during infection. PLoS Pathog. 2022, 18, e1010323. [Google Scholar] [CrossRef]
- Di Bella, S.; Ascenzi, P.; Siarakas, S.; Petrosillo, N.; Di Masi, A. Clostridium difficile Toxins A and B: Insights into Pathogenic Properties and Extraintestinal Effects. Toxins 2016, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, C.; Wang, H.; Wang, J. The Role of Rho GTPases in Toxicity of Clostridium difficile Toxins. Toxins 2015, 7, 5254–5267. [Google Scholar] [CrossRef]
- Paparella, A.S.; Cahill, S.M.; Aboulache, B.L.; Schramm, V.L. Clostridioides difficile TcdB Toxin Glucosylates Rho GTPase by an SNi Mechanism and Ion Pair Transition State. ACS Chem. Biol. 2022, 17, 2507–2518. [Google Scholar] [CrossRef]
- Pruitt, R.N.; Chumbler, N.M.; Rutherford, S.A.; Farrow, M.A.; Friedman, D.B.; Spiller, B.; Lacy, D.B. Structural Determinants of Clostridium difficile Toxin A Glucosyltransferase Activity. J. Biol. Chem. 2012, 287, 8013–8020. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, F.; Gerhard, R.; Hönig, R.; Giehl, K.; Pich, A. TcdB of Clostridioides difficile Mediates RAS-Dependent Necrosis in Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 4258. [Google Scholar] [CrossRef]
- Quesada-Gómez, C.; López-Ureña, D.; Chumbler, N.; Kroh, H.K.; Castro-Peña, C.; Rodríguez, C.; Orozco-Aguilar, J.; González-Camacho, S.; Rucavado, A.; Guzmán-Verri, C.; et al. Analysis of TcdB Proteins within the Hypervirulent Clade 2 Reveals an Impact of RhoA Glucosylation on Clostridium difficile Proinflammatory Activities. Infect. Immun. 2016, 84, 856–865. [Google Scholar] [CrossRef]
- Petersen, L.; Stroh, S.; Schöttelndreier, D.; Grassl, G.A.; Rottner, K.; Brakebusch, C.; Fahrer, J.; Genth, H. The Essential Role of Rac1 Glucosylation in Clostridioides difficile Toxin B-Induced Arrest of G1-S Transition. Front. Microbiol. 2022, 13, 846215. [Google Scholar] [CrossRef]
- Chen, B.; Liu, Z.; Perry, K.; Jin, R. Structure of the glucosyltransferase domain of TcdA in complex with RhoA provides insights into substrate recognition. Sci. Rep. 2022, 12, 9028. [Google Scholar] [CrossRef]
- Barthold, L.; Heber, S.; Schmidt, C.Q.; Gradl, M.; Weidinger, G.; Barth, H.; Fischer, S. Human α-Defensin-6 Neutralizes Clostridioides difficile Toxins TcdA and TcdB by Direct Binding. Int. J. Mol. Sci. 2022, 23, 4509. [Google Scholar] [CrossRef]
- Seekatz, A.M.; Young, V.B. Clostridium difficile and the microbiota. J. Clin. Investig. 2014, 124, 4182–4189. [Google Scholar] [CrossRef]
- Kachrimanidou, M.; Tsintarakis, E. Insights into the Role of Human Gut Microbiota in Clostridioides difficile Infection. Microorganisms 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, N.A.; Schubert, A.M.; Flynn, K.J.; Leslie, J.L.; Sinani, H.; Bergin, I.L.; Young, V.B.; Schloss, P.D. The Gut Bacterial Community Potentiates Clostridioides difficile Infection Severity. mBio 2022, 13, e0118322. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.J.; Subramanian, A.; Lopansri, B.; Goodman, B.; Jones, P.B.; Ferraro, J.; Stenehjem, E.; Brown, S.M. Antibiotic Exposure and Risk for Hospital-Associated Clostridioides difficile Infection. Antimicrob. Agents Chemother. 2020, 64, e02169-19. [Google Scholar] [CrossRef]
- Schubert, A.M.; Sinani, H.; Schloss, P.D. Antibiotic-Induced Alterations of the Murine Gut Microbiota and Subsequent Effects on Colonization Resistance against Clostridium difficile. mBio 2015, 6, e00974-15. [Google Scholar] [CrossRef]
- Mills, J.P.; Rao, K.; Young, V.B. Probiotics for prevention of Clostridium difficile infection. Curr. Opin. Gastroenterol. 2018, 34, 3–10. [Google Scholar] [CrossRef]
- McMillan, A.S.; Theriot, C.M. Bile acids impact the microbiota, host, and C. difficile dynamics providing insight into mechanisms of efficacy of FMTs and microbiota-focused therapeutics. Gut Microbes 2024, 16, 2393766. [Google Scholar] [CrossRef] [PubMed]
- Weingarden, A.R.; Chen, C.; Bobr, A.; Yao, D.; Lu, Y.; Nelson, V.M.; Sadowsky, M.J.; Khoruts, A. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Liver Physiol. 2014, 306, G310–G319. [Google Scholar] [CrossRef]
- Kisthardt, S.C.; Thanissery, R.; Pike, C.M.; Foley, M.H.; Theriot, C.M. The microbial-derived bile acid lithocholate and its epimers inhibit Clostridioides difficile growth and pathogenicity while sparing members of the gut microbiota. J. Bacteriol. 2023, 205, e0018023. [Google Scholar] [CrossRef]
- Rea, M.C.; Sit, C.S.; Clayton, E.; O’Connor, P.M.; Whittal, R.M.; Zheng, J.; Vederas, J.C.; Ross, R.P.; Hill, C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc. Natl. Acad. Sci. USA 2010, 107, 9352–9357. [Google Scholar] [CrossRef]
- Ney, L.-M.; Wipplinger, M.; Grossmann, M.; Engert, N.; Wegner, V.D.; Mosig, A.S. Short chain fatty acids: Key regulators of the local and systemic immune response in inflammatory diseases and infections. Open Biol. 2023, 13, 230014. [Google Scholar] [CrossRef]
- Horrocks, V.; King, O.G.; Yip, A.Y.G.; Marques, I.M.; McDonald, J.A.K. Role of the gut microbiota in nutrient competition and protection against intestinal pathogen colonization. Microbiology 2023, 169, 001377. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Danhof, H.A.; Shrestha, R.; Chang-Graham, A.L.; Hyser, J.M.; Haag, A.M.; Mohammad, M.A.; Britton, R.A.; Versalovic, J.; Sorg, J.A.; et al. Reuterin disrupts Clostridioides difficile metabolism and pathogenicity through reactive oxygen species generation. Gut Microbes 2020, 12, 1795388. [Google Scholar] [CrossRef]
- Fletcher, J.R.; Pike, C.M.; Parsons, R.J.; Rivera, A.J.; Foley, M.H.; McLaren, M.R.; Montgomery, S.A.; Theriot, C.M. Clostridioides difficile exploits toxin-mediated inflammation to alter the host nutritional landscape and exclude competitors from the gut microbiota. Nat. Commun. 2021, 12, 462. [Google Scholar] [CrossRef]
- Deng, H.; Yang, S.; Zhang, Y.; Qian, K.; Zhang, Z.; Liu, Y.; Wang, Y.; Bai, Y.; Fan, H.; Zhao, X.; et al. Bacteroides fragilis Prevents Clostridium difficile Infection in a Mouse Model by Restoring Gut Barrier and Microbiome Regulation. Front. Microbiol. 2018, 9, 2976. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-S. Fecal Microbiota Transplantation Is Effective for the Treatment of Partially Treated Clostridioides difficile Infection. Gut Liver 2021, 15, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Yadegar, A.; Pakpour, S.; Ibrahim, F.F.; Nabavi-Rad, A.; Cook, L.; Walter, J.; Seekatz, A.M.; Wong, K.; Monaghan, T.M.; Kao, D. Beneficial effects of fecal microbiota transplantation in recurrent Clostridioides difficile infection. Cell Host Microbe 2023, 31, 695–711. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Y.; Lou, E.; Zhao, X.; Chen, X. The role of gut fungi in Clostridioides difficile infection. Biomed. J. 2023, 47, 100686. [Google Scholar] [CrossRef]
- Wortelboer, K.; Nieuwdorp, M.; Herrema, H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine 2019, 44, 716–729. [Google Scholar] [CrossRef]
- Song, J.H.; Kim, Y.S. Recurrent Clostridium difficile Infection: Risk Factors, Treatment, and Prevention. Gut Liver 2019, 13, 16–24. [Google Scholar] [CrossRef]
- Gupta, S.; Allen-Vercoe, E.; Petrof, E.O. Fecal microbiota transplantation: In perspective. Ther. Adv. Gastroenterol. 2016, 9, 229–239. [Google Scholar] [CrossRef]
- Ouyang, Z.-R.; Niu, X.-R.; Wang, W.-G.; Zhao, J.-H. The role of short-chain fatty acids in Clostridioides difficile infection: A review. Anaerobe 2022, 75, 102585. [Google Scholar] [CrossRef]
- Karimi, M.; Shirsalimi, N.; Hashempour, Z.; Omran, H.S.; Sedighi, E.; Beigi, F.; Mortezazadeh, M. Safety and efficacy of fecal microbiota transplantation (FMT) as a modern adjuvant therapy in various diseases and disorders: A comprehensive literature review. Front. Immunol. 2024, 15, 1439176. [Google Scholar] [CrossRef]
- Yang, Y.; An, Y.; Dong, Y.; Chu, Q.; Wei, J.; Wang, B.; Cao, H. Fecal microbiota transplantation: No longer cinderella in tumour immunotherapy. EBioMedicine 2024, 100, 104967. [Google Scholar] [CrossRef]
- Mullish, B.H.; Allegretti, J.R. The contribution of bile acid metabolism to the pathogenesis of Clostridioides difficile infection. Ther. Adv. Gastroenterol. 2021, 14, 17562848211017725. [Google Scholar] [CrossRef]
- Porcari, S.; Benech, N.; Valles-Colomer, M.; Segata, N.; Gasbarrini, A.; Cammarota, G.; Sokol, H.; Ianiro, G. Key determinants of success in fecal microbiota transplantation: From microbiome to clinic. Cell Host Microbe 2023, 31, 712–733. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.S.; Nicholson, M.R.; Khoruts, A.; Kahn, S.A. Fecal Microbiota Transplantation Across the Lifespan: Balancing Efficacy, Safety, and Innovation. Am. J. Gastroenterol. 2022, 118, 435–439. [Google Scholar] [CrossRef]
- Petrof, E.O.; Khoruts, A. From Stool Transplants to Next-Generation Microbiota Therapeutics. Gastroenterology 2014, 146, 1573–1582. [Google Scholar] [CrossRef]
- Alam, Z.; Markantonis, J.E.; Fallon, J.T. Host Immune Responses to Clostridioides difficile Infection and Potential Novel Therapeutic Approaches. Trop. Med. Infect. Dis. 2023, 8, 506. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. Modeling host-pathogen interactions: H. sapiens as a host and C. difficile as a pathogen. J. Mol. Recognit. 2012, 25, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Phelps, E.; Ganapini, V.; Khan, N.; Ouyang, F.; Xu, H.; Khanna, S.; Tariq, R.; Friedman-Moraco, R.J.; Woodworth, M.H.; et al. Fecal microbiota transplantation for the treatment of recurrent and severe Clostridium difficile infection in solid organ transplant recipients: A multicenter experience. Am. J. Transplant. 2018, 19, 501–511. [Google Scholar] [CrossRef]
- Jafari, N.V.; Kuehne, S.A.; Minton, N.P.; Allan, E.; Bajaj-Elliott, M. Clostridium difficile-mediated effects on human intestinal epithelia: Modelling host-pathogen interactions in a vertical diffusion chamber. Anaerobe 2016, 37, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Nibbering, B.; Gerding, D.N.; Kuijper, E.J.; Zwittink, R.D.; Smits, W.K. Host Immune Responses to Clostridioides difficile: Toxins and Beyond. Front. Microbiol. 2021, 12, 804949. [Google Scholar] [CrossRef]
- Raeisi, H.; Azimirad, M.; Nabavi-Rad, A.; Aghdaei, H.A.; Yadegar, A.; Zali, M.R. Application of recombinant antibodies for treatment of Clostridioides difficile infection: Current status and future perspective. Front. Immunol. 2022, 13, 972930. [Google Scholar] [CrossRef]
- Di Bella, S.; Sanson, G.; Monticelli, J.; Zerbato, V.; Principe, L.; Giuffrè, M.; Pipitone, G.; Luzzati, R. Clostridioides difficile infection: History, epidemiology, risk factors, prevention, clinical manifestations, treatment, and future options. Clin. Microbiol. Rev. 2024, 37, e0013523. [Google Scholar] [CrossRef]
- Pruitt, R.N.; Lacy, D.B. Toward a structural understanding of Clostridium difficile toxins A and B. Front. Cell. Infect. Microbiol. 2012, 2, 28. [Google Scholar] [CrossRef]
- Fujimoto, K.; Uematsu, S. Phage therapy for Clostridioides difficile infection. Front. Immunol. 2022, 13, 1057892. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Therapy | Targeted Toxin Domain (TcdA/TcdB) | Mechanism | Advantages | Clinical Relevance | Limitation | Reference |
|---|---|---|---|---|---|---|
| Recombinant sdAbs | CROPs domain (TcdB) | Blocks CSPG4/PVR receptor binding | Neutralizes hypervirulent RT027/RT078 strains | Phase II | Standardization needed, Limited efficacy against TcdA−B+ strains | [123] |
| Bezlotoxumab | Enzymatic Domain (TcdB) | Masking CSPG4 binding | ↓ recurrence | Approved | High cost, only IV route | [124] |
| Glucosyltransferase Inhibitors | DXD motif (TcdA/TcdB) | Blocks Rho/Ras GTPase inactivation | Oral bioavailability | Preclinical | Off-target effects on host GTPases | [125] |
| Bacteriophage therapy | C. difficle cell lysis | Selectively targets C. difficles cells | Highly specific, ↓ bacterial burden without disrupting microbiota | Preclinical | Limited data, potential for phage resistance | [126] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qutub, M.; Tatode, A.; Hussain, U.M.; Premchandani, T.; Taksande, J.; Umekar, M.; Thakre, D. Deciphering the Structural and Functional Paradigms of Clostridioides difficile Toxins TcdA and TcdB. Bacteria 2025, 4, 21. https://doi.org/10.3390/bacteria4020021
Qutub M, Tatode A, Hussain UM, Premchandani T, Taksande J, Umekar M, Thakre D. Deciphering the Structural and Functional Paradigms of Clostridioides difficile Toxins TcdA and TcdB. Bacteria. 2025; 4(2):21. https://doi.org/10.3390/bacteria4020021
Chicago/Turabian StyleQutub, Mohammad, Amol Tatode, Ujban Md Hussain, Tanvi Premchandani, Jayshree Taksande, Milind Umekar, and Deepak Thakre. 2025. "Deciphering the Structural and Functional Paradigms of Clostridioides difficile Toxins TcdA and TcdB" Bacteria 4, no. 2: 21. https://doi.org/10.3390/bacteria4020021
APA StyleQutub, M., Tatode, A., Hussain, U. M., Premchandani, T., Taksande, J., Umekar, M., & Thakre, D. (2025). Deciphering the Structural and Functional Paradigms of Clostridioides difficile Toxins TcdA and TcdB. Bacteria, 4(2), 21. https://doi.org/10.3390/bacteria4020021