
The Emergence of the Slc11 Clade MCbgut: A Parsimonious Hypothesis for the Dawn of Lactobacillales in the Gut of Early Vertebrates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
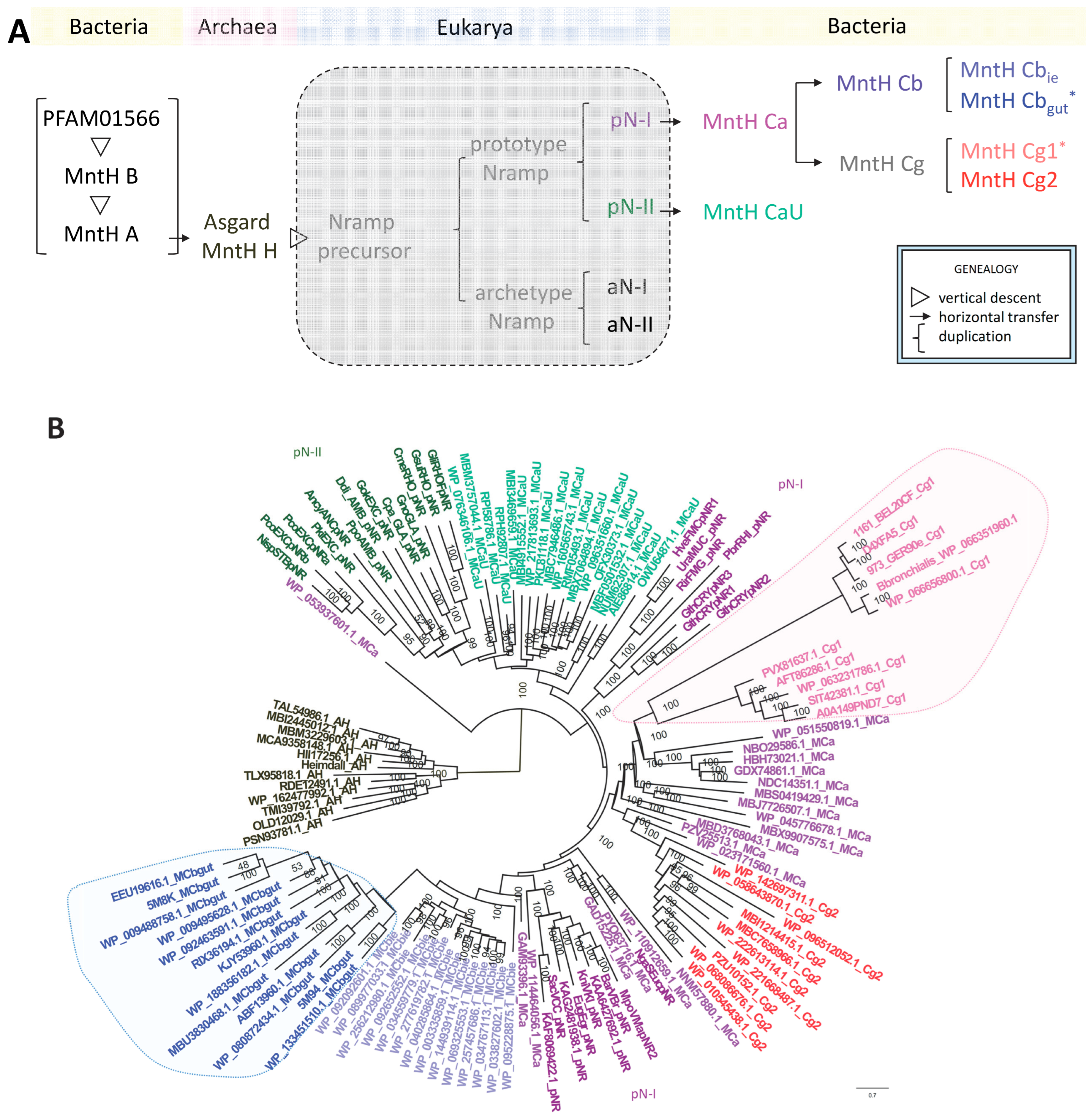
2. In Silico Results
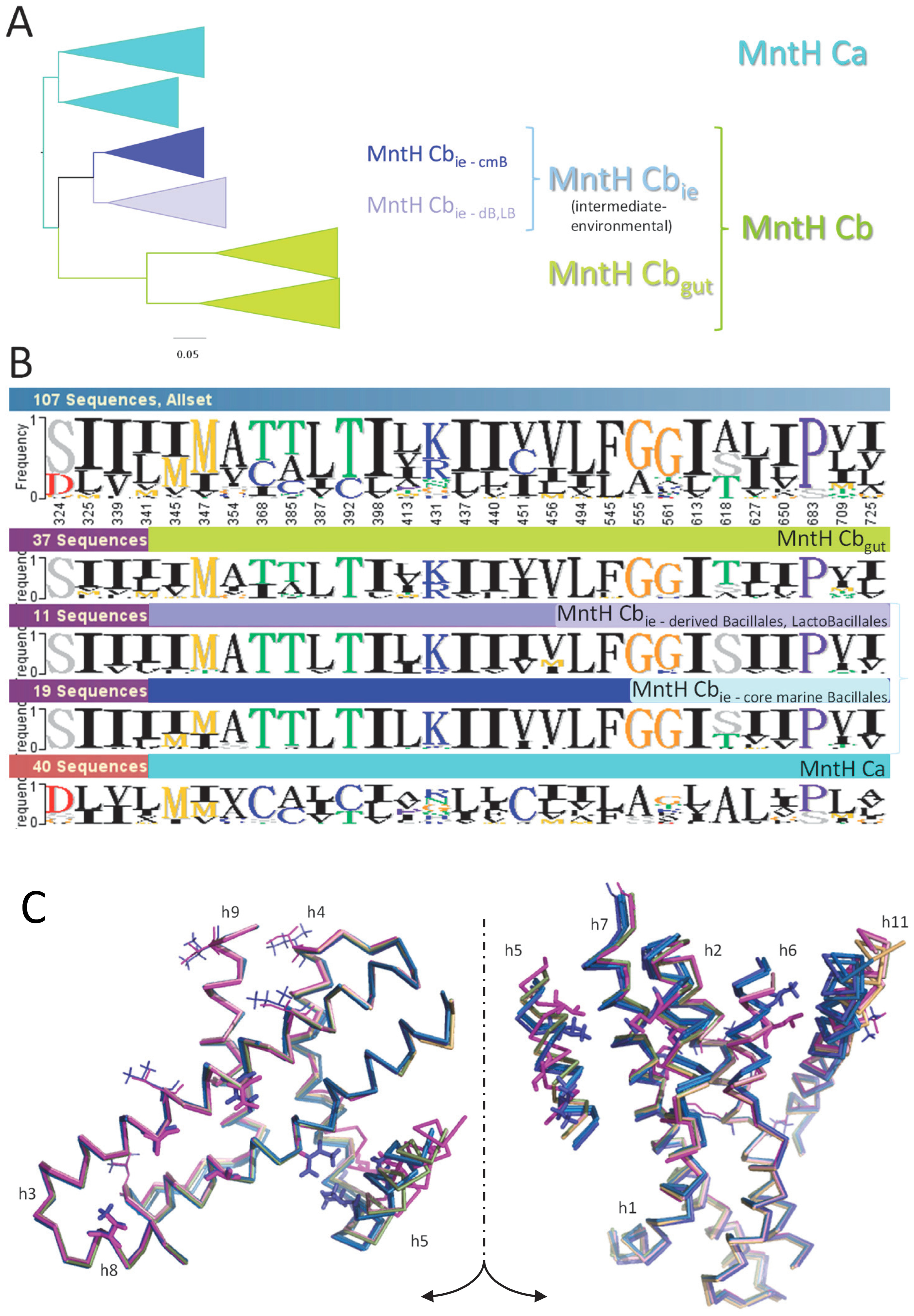
2.1. P. halotolerans A0A1I3CNB9 Identifies the Clade MCbie, Intermediate between MCa and MCbgut
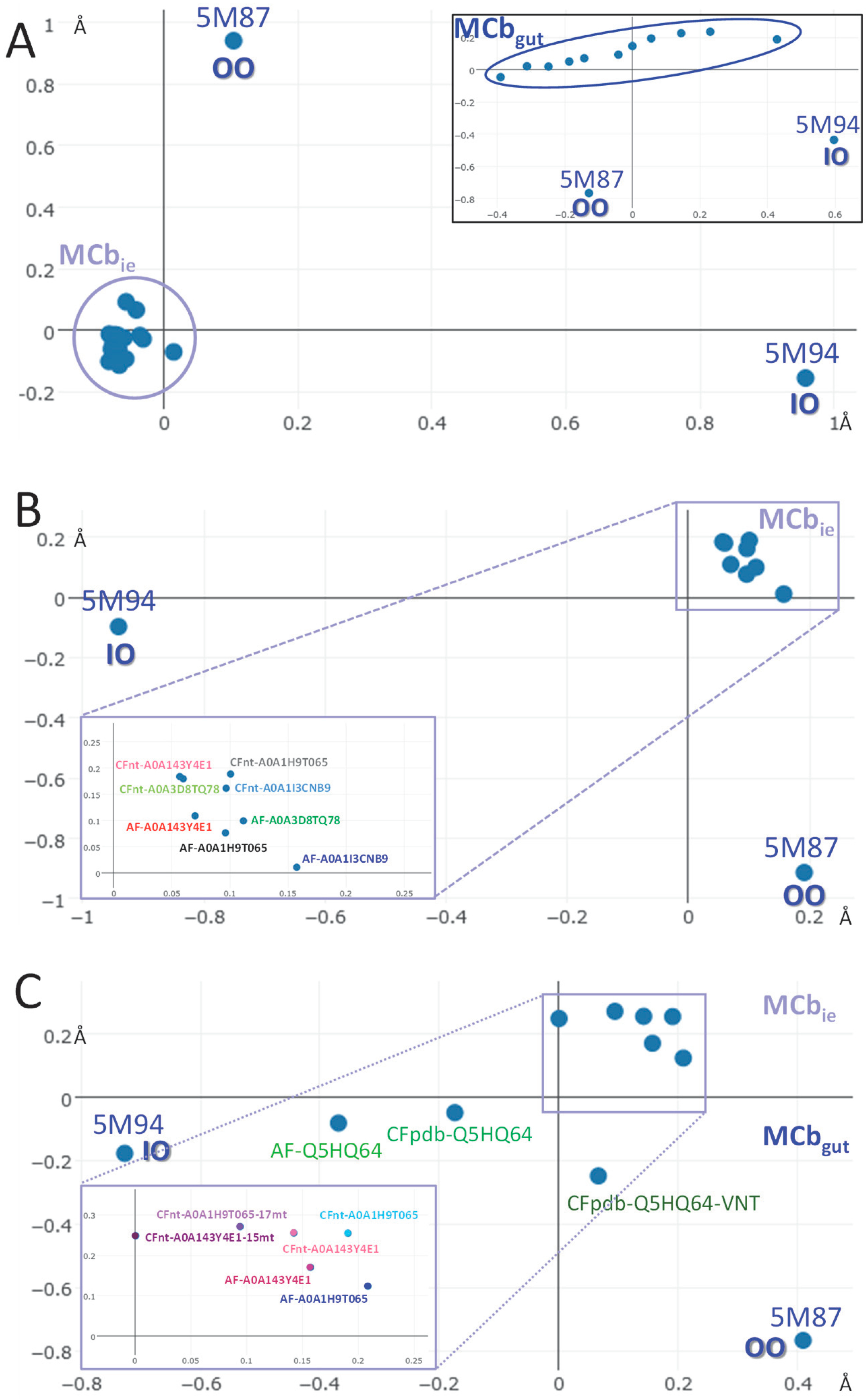
2.2. AF/CF 3D Modeling of Native MCbie and MCbgut Carriers Differs Significantly
2.3. Evolution of MCb Carrier Cycling between MCbie and MCbgut
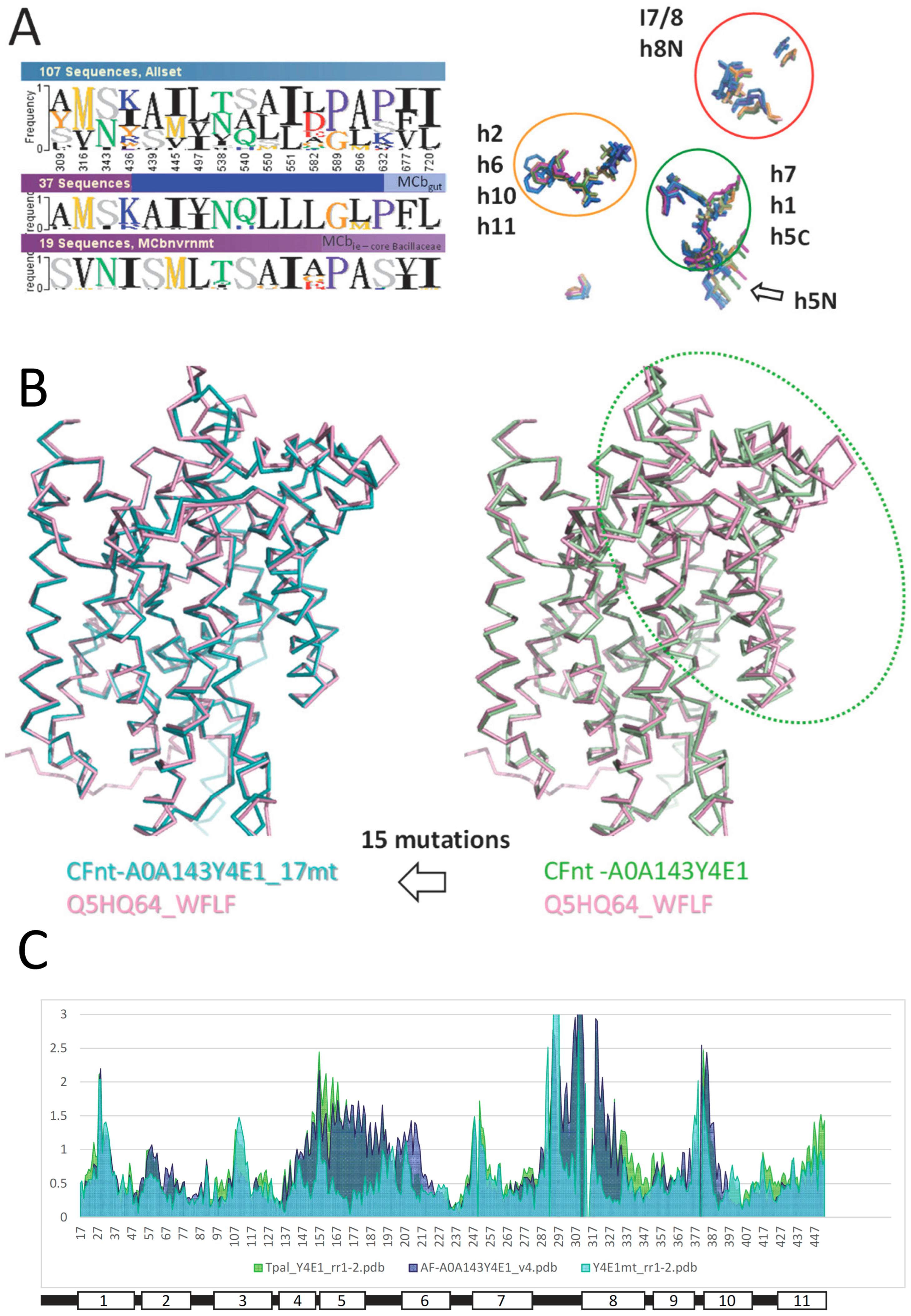
2.4. Mutagenesis of Slc11 Synapomorphy Supports Functional Divergence of MCbgut and MCbie
2.5. Conformation-Dependent Impact of Mutations Targeting MCb l 7/8 and l 8/9
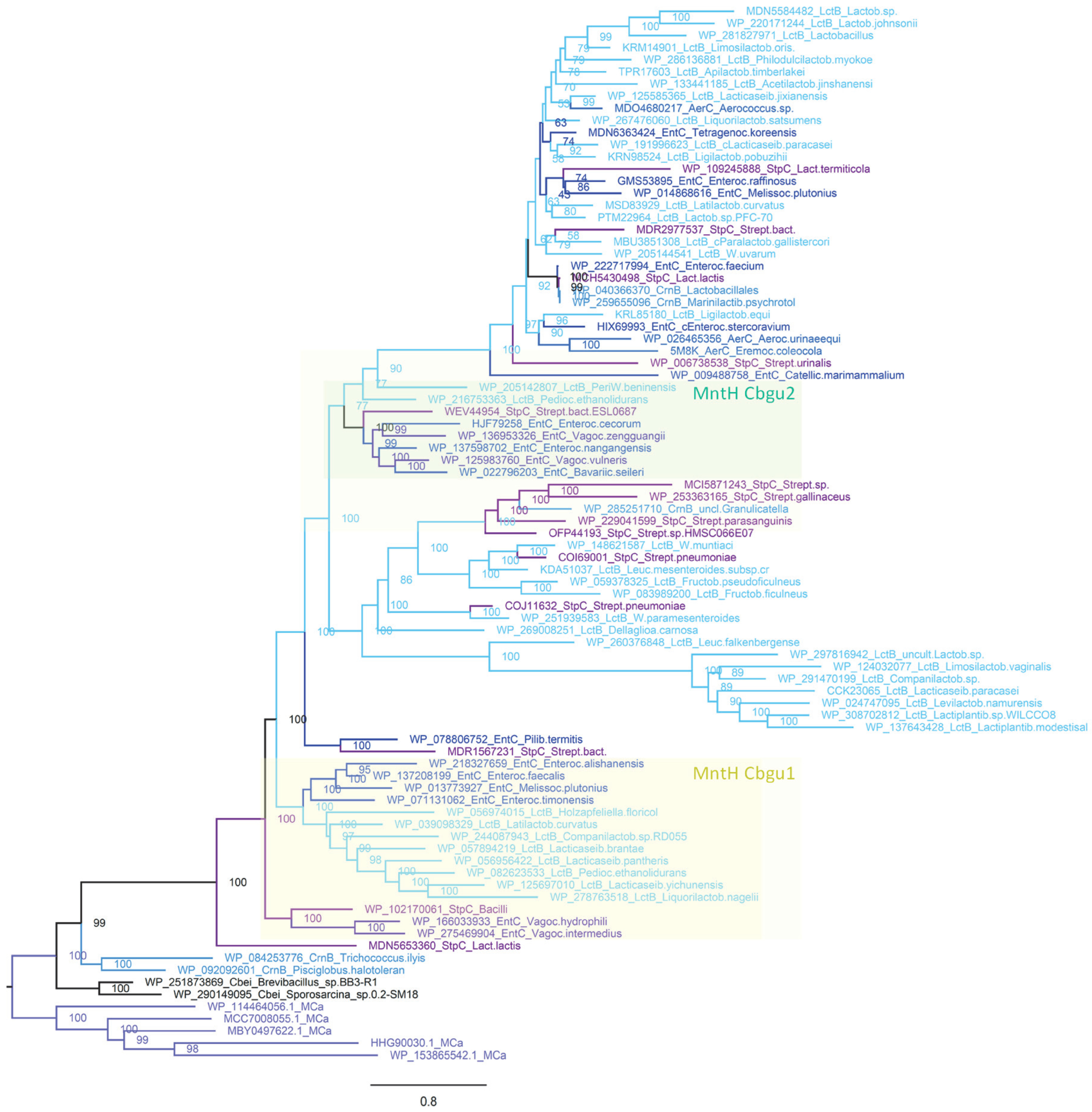
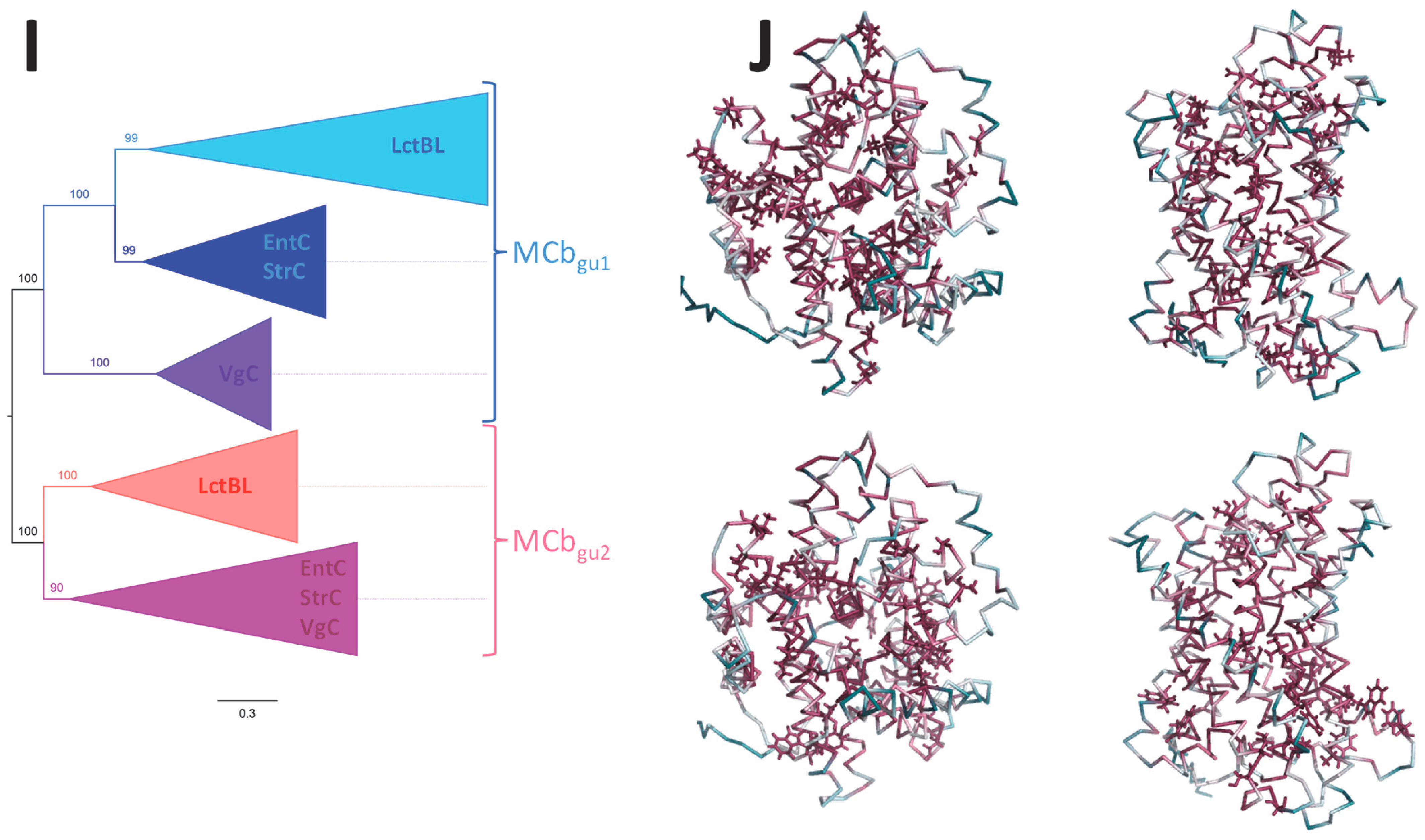
2.6. MCbgut Phylogeny Reveals a Pair of Distinct Carriers in the Common Ancestor of LB
3. Discussion
4. Materials and Methods
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Del Alamo, D.; Meiler, J.; McHaourab, H.S. Principles of Alternating Access in LeuT-fold Transporters: Commonalities and Divergences. J. Mol. Biol. 2022, 434, 167746. [Google Scholar] [CrossRef] [PubMed]
- Manatschal, C.; Dutzler, R. The Structural Basis for Metal Ion Transport in the SLC11/NRAMP Family. Chimia 2022, 76, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Gaudet, R. Structures and coordination chemistry of transporters involved in manganese and iron homeostasis. Biochem. Soc. Trans. 2023, 51, 897–923. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.F. Nramp: From sequence to structure and mechanism of divalent metal import. Curr. Top. Membr. 2012, 69, 249–293. [Google Scholar] [PubMed]
- Marinelli, F.; Faraldo-Gomez, J.D. Conformational free-energy landscapes of a Na+/Ca2+ exchanger explain its alternating-access mechanism and functional specificity. Proc. Natl. Acad. Sci. USA 2024, 121, e2318009121. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.F.M. Slc11 Synapomorphy: A Conserved 3D Framework Articulating Carrier Conformation Switch. Int. J. Mol. Sci. 2023, 24, 15076. [Google Scholar] [CrossRef]
- Cellier, M.F.M. Nramp: Deprive and conquer? Front. Cell Dev. Biol. 2022, 10, 988866. [Google Scholar] [CrossRef]
- Casanova, J.L.; MacMicking, J.D.; Nathan, C.F. Interferon-gamma and infectious diseases: Lessons and prospects. Science 2024, 384, eadl2016. [Google Scholar] [CrossRef]
- Cellier, M.F.; Bergevin, I.; Boyer, E.; Richer, E. Polyphyletic origins of bacterial Nramp transporters. Trends. Genet. 2001, 17, 365–370. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Bosma, E.F.; Rau, M.H.; van Gijtenbeek, L.A.; Siedler, S. Regulation and distinct physiological roles of manganese in bacteria. FEMS Microbiol. Rev. 2021, 45, fuab028. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, S.T.; Rau, M.H.; Sanchez, B.J.; Jensen, K.; Zeidan, A.A. From genotype to phenotype: Computational approaches for inferring microbial traits relevant to the food industry. FEMS Microbiol. Rev. 2023, 47, fuab028, fuad030. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.; Slesarev, A.; Wolf, Y.; Sorokin, A.; Mirkin, B.; Koonin, E.; Pavlov, A.; Pavlova, N.; Karamychev, V.; Polouchine, N.; et al. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 15611–15616. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Evolutionary genomics of lactic acid bacteria. J. Bacteriol. 2007, 189, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Huynh, U.; Qiao, M.; King, J.; Trinh, B.; Valdez, J.; Haq, M.; Zastrow, M.L. Differential Effects of Transition Metals on Growth and Metal Uptake for Two Distinct Lactobacillus Species. Microbiol. Spectr. 2022, 10, e0100621. [Google Scholar] [CrossRef] [PubMed]
- Elli, M.; Zink, R.; Rytz, A.; Reniero, R.; Morelli, L. Iron requirement of Lactobacillus spp. in completely chemically defined growth media. J. Appl. Microbiol. 2000, 88, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, X.; Cheng, C.C.; Bujdos, D.; Tollenaar, S.; Simpson, D.J.; Tasseva, G.; Perez-Munoz, M.E.; Frese, S.; Ganzle, M.G.; et al. phylogenomic analysis of Limosilactobacillus reuteri reveals ancient and stable evolutionary relationships with rodents and birds and zoonotic transmission to humans. BMC Biol. 2023, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y. Microbiota in the stomach and application of probiotics to gastroduodenal diseases. World J. Gastroenterol. 2022, 28, 6702–6715. [Google Scholar] [CrossRef]
- Lebreton, F.; Manson, A.L.; Saavedra, J.T.; Straub, T.J.; Earl, A.M.; Gilmore, M.S. Tracing the Enterococci from Paleozoic Origins to the Hospital. Cell 2017, 169, 849–861.e13. [Google Scholar] [CrossRef]
- Weller, D.; Andrus, A.; Wiedmann, M.; den Bakker, H.C. Listeria booriae sp. nov. and Listeria newyorkensis sp. nov., from food processing environments in the USA. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 1, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Maayer, P.; Aliyu, H.; Cowan, D.A. Reorganising the order Bacillales through phylogenomics. Syst. Appl. Microbiol. 2019, 42, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.D.; Wallbanks, S.; Lane, D.J.; Shah, J.; Nietupski, R.; Smida, J.; Dorsch, M.; Stackebrandt, E. Phylogenetic analysis of the genus Listeria based on reverse transcriptase sequencing of 16S rRNA. Int. J. Syst. Bacteriol. 1991, 41, 240–246. [Google Scholar] [CrossRef]
- Tian, R.; Imanian, B. VBCG: 20 validated bacterial core genes for phylogenomic analysis with high fidelity and resolution. Microbiome 2023, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef]
- van Dijk, M.C.; de Kruijff, R.M.; Hagedoorn, P.L. The Role of Iron in Staphylococcus aureus Infection and Human Disease: A Metal Tug of War at the Host-Microbe Interface. Front. Cell Dev. Biol. 2022, 10, 857237. [Google Scholar] [CrossRef]
- McLaughlin, H.P.; Hill, C.; Gahan, C.G. The impact of iron on Listeria monocytogenes; inside and outside the host. Curr. Opin. Biotechnol. 2011, 22, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Ehrnstorfer, I.A.; Manatschal, C.; Arnold, F.M.; Laederach, J.; Dutzler, R. Structural and mechanistic basis of proton-coupled metal ion transport in the SLC11/NRAMP family. Nat. Commun. 2017, 8, 14033. [Google Scholar] [CrossRef]
- Ehrnstorfer, I.A.; Geertsma, E.R.; Pardon, E.; Steyaert, J.; Dutzler, R. Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nat. Struct. Mol. Biol. 2014, 21, 990–996. [Google Scholar] [CrossRef]
- Schwartzman, J.A.; Lebreton, F.; Salamzade, R.; Shea, T.; Martin, M.J.; Schaufler, K.; Urhan, A.; Abeel, T.; Camargo, I.; Sgardioli, B.F.; et al. Global diversity of enterococci and description of 18 previously unknown species. Proc. Natl. Acad. Sci. USA 2024, 121, e2310852121. [Google Scholar] [CrossRef]
- Richer, E.; Courville, P.; Bergevin, I.; Cellier, M.F. Horizontal gene transfer of “prototype” Nramp in bacteria. J. Mol. Evol. 2003, 57, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Finke, N.; Simister, R.L.; O’Neil, A.H.; Nomosatryo, S.; Henny, C.; MacLean, L.C.; Canfield, D.E.; Konhauser, K.; Lalonde, S.V.; Fowle, D.A.; et al. Mesophilic microorganisms build terrestrial mats analogous to Precambrian microbial jungles. Nat. Commun. 2019, 10, 4323. [Google Scholar] [CrossRef]
- Lenton, T.M.; Daines, S.J. Matworld—The biogeochemical effects of early life on land. New Phytol. 2017, 215, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Ettoumi, B.; Guesmi, A.; Brusetti, L.; Borin, S.; Najjari, A.; Boudabous, A.; Cherif, A. Microdiversity of deep-sea Bacillales isolated from Tyrrhenian sea sediments as revealed by ARISA, 16S rRNA gene sequencing and BOX-PCR fingerprinting. Microbes Environ. 2013, 28, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Mandic-Mulec, I.; Stefanic, P.; van Elsas, J.D. Ecology of Bacillaceae. Microbiol. Spectr. 2015, 3, TBS-0017-2013. [Google Scholar] [CrossRef]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798, Erratum in Int. J. Syst. Evol. Microbiol. 2020, 70, 6531–6533. [Google Scholar]
- Shimeld, S.M.; Holland, P.W. Vertebrate innovations. Proc. Natl. Acad. Sci. USA 2000, 97, 4449–4452. [Google Scholar] [CrossRef] [PubMed]
- Grillner, S. Evolution of the vertebrate motor system—From forebrain to spinal cord. Curr. Opin. Neurobiol. 2021, 71, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ren, Y.; Uesaka, M.; Beavan, A.J.S.; Muffato, M.; Shen, J.; Li, Y.; Sato, I.; Wan, W.; Clark, J.W.; et al. Hagfish genome elucidates vertebrate whole-genome duplication events and their evolutionary consequences. Nat. Ecol. Evol. 2024, 8, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Marletaz, F.; Timoshevskaya, N.; Timoshevskiy, V.A.; Parey, E.; Simakov, O.; Gavriouchkina, D.; Suzuki, M.; Kubokawa, K.; Brenner, S.; Smith, J.J.; et al. The hagfish genome and the evolution of vertebrates. Nature 2024, 627, 811–820. [Google Scholar] [CrossRef]
- Sassa, M.; Takagi, T.; Kinjo, A.; Yoshioka, Y.; Zayasu, Y.; Shinzato, C.; Kanda, S.; Murakami-Sugihara, N.; Shirai, K.; Inoue, K. Divalent metal transporter-related protein restricts animals to marine habitats. Commun. Biol. 2021, 4, 463. [Google Scholar] [CrossRef]
- Richer, E.; Campion, C.G.; Dabbas, B.; White, J.H.; Cellier, M.F. Transcription factors Sp1 and C/EBP regulate NRAMP1 gene expression. FEBS J. 2008, 275, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Cellier, M.F.M. Developmental Control of NRAMP1 (SLC11A1) Expression in Professional Phagocytes. Biology 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Raberg, L. Human and pathogen genotype-by-genotype interactions in the light of coevolution theory. PLoS Genet. 2023, 19, e1010685. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Shawki, A.; Anthony, S.R.; Nose, Y.; Engevik, M.A.; Niespodzany, E.J.; Barrientos, T.; Ohrvik, H.; Worrell, R.T.; Thiele, D.J.; Mackenzie, B. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver. Physiol. 2015, 309, G635–G647. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Z.; Guo, X.; Guo, R.; Zhu, L.; Qiu, X.; Yu, X.; Chai, J.; Gu, C.; Feng, Z. Changes in nutrient consumption patterns of Lactobacillus fermentum mediated by sodium lactate. J. Sci. Food Agric. 2023, 103, 1775–1783. [Google Scholar] [CrossRef]
- de Hoon, M.J.; Eichenberger, P.; Vitkup, D. Hierarchical evolution of the bacterial sporulation network. Curr. Biol. 2010, 20, R735–R745. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sereti, I.; Verburgh, M.L.; Gifford, J.; Lo, A.; Boyd, A.; Verheij, E.; Verhoeven, A.; Wit, F.; Schim van der Loeff, M.F.; Giera, M.; et al. Impaired gut microbiota-mediated short-chain fatty acid production precedes morbidity and mortality in people with HIV. Cell Rep. 2023, 42, 113336. [Google Scholar] [CrossRef]
- Nakayama, S.; Sekiguchi, T.; Ogasawara, M. Molecular and evolutionary aspects of the protochordate digestive system. Cell Tissue Res. 2019, 377, 309–320. [Google Scholar] [CrossRef]
- Castro, L.F.; Goncalves, O.; Mazan, S.; Tay, B.H.; Venkatesh, B.; Wilson, J.M. Recurrent gene loss correlates with the evolution of stomach phenotypes in gnathostome history. Proc. Biol. Sci. 2014, 281, 20132669. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- Suzuki-Hashido, N.; Tsuchida, S.; Hayakawa, T.; Sakamoto, M.; Azumano, A.; Seino, S.; Matsuda, I.; Ohkuma, M.; Ushida, K. Lactobacillus nasalidis sp. nov., isolated from the forestomach of a captive proboscis monkey (Nasalis larvatus). Int. J. Syst. Evol. Microbiol. 2021, 71, 004787. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; He, Y.; Xie, K.; Feng, L.; Gao, S.; Cai, L. Review of microbiota gut brain axis and innate immunity in inflammatory and infective diseases. Front. Cell. Infect. Microbiol. 2023, 13, 1282431. [Google Scholar] [CrossRef] [PubMed]
- Duar, R.M.; Lin, X.B.; Zheng, J.; Martino, M.E.; Grenier, T.; Perez-Munoz, M.E.; Leulier, F.; Ganzle, M.; Walter, J. Lifestyles in transition: Evolution and natural history of the genus Lactobacillus. FEMS Microbiol. Rev. 2017, 41 (Suppl. S1), S27–S48. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Calles-Enriquez, M.; Nes, I.; Martin, M.C.; Fernandez, M.; Ladero, V.; Alvarez, M.A. Tyramine biosynthesis is transcriptionally induced at low pH and improves the fitness of Enterococcus faecalis in acidic environments. Appl. Microbiol. Biotechnol. 2015, 99, 3547–3558. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.I.; Matos, D.; San Romao, M.V.; Crespo, M.T. Dual role for the tyrosine decarboxylation pathway in Enterococcus faecium E17: Response to an acid challenge and generation of a proton motive force. Appl. Environ. Microbiol. 2009, 75, 345–352. [Google Scholar] [CrossRef]
- Goulty, M.; Botton-Amiot, G.; Rosato, E.; Sprecher, S.G.; Feuda, R. The monoaminergic system is a bilaterian innovation. Nat. Commun. 2023, 14, 3284. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Decaestecker, E.; Van de Moortel, B.; Mukherjee, S.; Gurung, A.; Stoks, R.; De Meester, L. Hierarchical eco-evo dynamics mediated by the gut microbiome. Trends Ecol. Evol. 2024, 39, 165–174. [Google Scholar] [CrossRef]
- Tannock, G.W.; Wilson, C.M.; Loach, D.; Cook, G.M.; Eason, J.; O’Toole, P.W.; Holtrop, G.; Lawley, B. Resource partitioning in relation to cohabitation of Lactobacillus species in the mouse forestomach. ISME J. 2012, 6, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Shankregowda, A.M.; Siriyappagouder, P.; Kuizenga, M.; Bal, T.M.P.; Abdelhafiz, Y.; Eizaguirre, C.; Fernandes, J.M.O.; Kiron, V.; Raeymaekers, J.A.M. Host habitat rather than evolutionary history explains gut microbiome diversity in sympatric stickleback species. Front. Microbiol. 2023, 14, 1232358. [Google Scholar] [CrossRef] [PubMed]
- Baldo, L.; Pretus, J.L.; Riera, J.L.; Musilova, Z.; Bitja Nyom, A.R.; Salzburger, W. Convergence of gut microbiotas in the adaptive radiations of African cichlid fishes. ISME J. 2017, 11, 1975–1987. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Khoshbouei, H. Dopamine and norepinephrine are embracing their immune side and so should we. Curr. Opin. Neurobiol. 2022, 77, 102626. [Google Scholar] [CrossRef] [PubMed]
- Decarie-Spain, L.; Hayes, A.M.R.; Lauer, L.T.; Kanoski, S.E. The gut-brain axis and cognitive control: A role for the vagus nerve. Semin. Cell Dev. Biol. 2024, 156, 201–209. [Google Scholar] [CrossRef]
- Kasarello, K.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Communication of gut microbiota and brain via immune and neuroendocrine signaling. Front. Microbiol. 2023, 14, 1118529. [Google Scholar] [CrossRef]
- Liu, Y.; Sanderson, D.; Mian, M.F.; McVey Neufeld, K.A.; Forsythe, P. Loss of vagal integrity disrupts immune components of the microbiota-gut-brain axis and inhibits the effect of Lactobacillus rhamnosus on behavior and the corticosterone stress response. Neuropharmacology 2021, 195, 108682. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.P.; Mileto, S.J.; Lyras, D. Impact of enteric bacterial infections at and beyond the epithelial barrier. Nat. Rev. Microbiol. 2023, 21, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Gierynska, M.; Szulc-Dabrowska, L.; Struzik, J.; Mielcarska, M.B.; Gregorczyk-Zboroch, K.P. Integrity of the Intestinal Barrier: The Involvement of Epithelial Cells and Microbiota-A Mutual Relationship. Animals 2022, 12, 145. [Google Scholar] [CrossRef]
- Brescia, C.; Audia, S.; Pugliano, A.; Scaglione, F.; Iuliano, R.; Trapasso, F.; Perrotti, N.; Chiarella, E.; Amato, R. Metabolic drives affecting Th17/Treg gene expression changes and differentiation: Impact on immune-microenvironment regulation. APMIS 2024. [Google Scholar] [CrossRef]
- de Candia, P.; Procaccini, C.; Russo, C.; Lepore, M.T.; Matarese, G. Regulatory T cells as metabolic sensors. Immunity 2022, 55, 1981–1992. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Heredia, M.; Samsom, J.; Huehn, J. Impact of gut microenvironment on epigenetic signatures of intestinal T helper cell subsets. Immunol. Lett. 2022, 246, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Cassmann, T.J.; Bhagwate, A.V.; Hitosugi, T.; Ip, W.K.E. Lactic acid induces transcriptional repression of macrophage inflammatory response via histone acetylation. Cell Rep. 2024, 43, 113746. [Google Scholar] [CrossRef]
- Jeffrey, M.P.; Saleem, L.; MacPherson, C.W.; Tompkins, T.A.; Clarke, S.T.; Green-Johnson, J.M. A Lacticaseibacillus rhamnosus secretome induces immunoregulatory transcriptional, functional and immunometabolic signatures in human THP-1 monocytes. Sci. Rep. 2024, 14, 8379. [Google Scholar] [CrossRef]
- Jordan, C.K.I.; Brown, R.L.; Larkinson, M.L.Y.; Sequeira, R.P.; Edwards, A.M.; Clarke, T.B. Symbiotic Firmicutes establish mutualism with the host via innate tolerance and resistance to control systemic immunity. Cell Host Microbe 2023, 31, 1433–1449.e9. [Google Scholar] [CrossRef] [PubMed]
- Nagahata, Y.; Masuda, K.; Nishimura, Y.; Ikawa, T.; Kawaoka, S.; Kitawaki, T.; Nannya, Y.; Ogawa, S.; Suga, H.; Satou, Y.; et al. Tracing the evolutionary history of blood cells to the unicellular ancestor of animals. Blood 2022, 140, 2611–2625. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Opazo, J.C.; Hoffmann, F.G. Gene duplication, genome duplication, and the functional diversification of vertebrate globins. Mol. Phylogenet. Evol. 2013, 66, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Mussini, G.; Dunn, F.S. Decline and fall of the Ediacarans: Late-Neoproterozoic extinctions and the rise of the modern biosphere. Biol. Rev. Camb. Philos. Soc. 2024, 99, 110–130. [Google Scholar] [CrossRef]
- Evans, S.D.; Tu, C.; Rizzo, A.; Surprenant, R.L.; Boan, P.C.; McCandless, H.; Marshall, N.; Xiao, S.; Droser, M.L. Environmental drivers of the first major animal extinction across the Ediacaran White Sea-Nama transition. Proc. Natl. Acad. Sci. USA 2022, 119, e2207475119. [Google Scholar] [CrossRef]
- Antell, G.T.; Saupe, E.E. Bottom-up controls, ecological revolutions and diversification in the oceans through time. Curr. Biol. 2021, 31, R1237–R1251. [Google Scholar] [CrossRef]
- Mangano, M.G.; Buatois, L.A. The rise and early evolution of animals: Where do we stand from a trace-fossil perspective? Interface Focus 2020, 10, 20190103. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.; Liu, A.G.; Bowyer, F.; Wilby, P.R.; Dunn, F.S.; Kenchington, C.G.; Cuthill, J.F.H.; Mitchell, E.G.; Penny, A. Integrated records of environmental change and evolution challenge the Cambrian Explosion. Nat. Ecol. Evol. 2019, 3, 528–538, Erratum in Nat. Ecol. Evol. 2019, 3, 858. [Google Scholar]
- Darroch, S.A.F.; Smith, E.F.; Laflamme, M.; Erwin, D.H. Ediacaran Extinction and Cambrian Explosion. Trends Ecol. Evol. 2018, 33, 653–663. [Google Scholar] [CrossRef]
- Boem, F.; Greslehner, G.P.; Konsman, J.P.; Chiu, L. Minding the gut: Extending embodied cognition and perception to the gut complex. Front. Neurosci. 2023, 17, 1172783. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P. Adaptability and evolution. Interface Focus 2017, 7, 20160126. [Google Scholar] [CrossRef]
- Tetlock, A.; Yost, C.K.; Stavrinides, J.; Manzon, R.G. Changes in the gut microbiome of the sea lamprey during metamorphosis. Appl. Environ. Microbiol. 2012, 78, 7638–7644. [Google Scholar] [CrossRef]
- Mathai, P.P.; Byappanahalli, M.N.; Johnson, N.S.; Sadowsky, M.J. Gut Microbiota Associated with Different Sea Lamprey (Petromyzon marinus) Life Stages. Front. Microbiol. 2021, 12, 706683, Erratum in Front. Microbiol. 2021, 12, 807068. [Google Scholar] [CrossRef]
- Salzer, J.L.; Zalc, B. Myelination. Curr. Biol. 2016, 26, R971–R975. [Google Scholar] [CrossRef] [PubMed]
- Wyles, J.S.; Kunkel, J.G.; Wilson, A.C. Birds, behavior, and anatomical evolution. Proc. Natl. Acad. Sci. USA 1983, 80, 4394–4397. [Google Scholar] [CrossRef]
- Zhang, Z.; Schäffer, A.A.; Miller, W.; Madden, T.L.; Lipman, D.J.; Koonin, E.V.; Altschul, S.F. Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res. 1998, 26, 3986–3990. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Soding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Shih, A.C.; Lee, D.T.; Peng, C.L.; Wu, Y.W. Phylo-mLogo: An interactive and hierarchical multiple-logo visualization tool for alignment of many sequences. BMC Bioinform. 2007, 8, 63. [Google Scholar] [CrossRef]
- Van de Peer, Y.; De Wachter, R. TREECON for Windows: A software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput. Appl. Biosci. 1994, 10, 569–570. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Le, S.Q.; Lartillot, N.; Gascuel, O. Phylogenetic mixture models for proteins. Philos. Trans. R Soc. Lond. B Biol. Sci. 2008, 363, 3965–3976. [Google Scholar] [CrossRef]
- Brandt, B.W.; Feenstra, K.A.; Heringa, J. Multi-Harmony: Detecting functional specificity from sequence alignment. Nucleic Acids Res. 2010, 38, W35–W40. [Google Scholar] [CrossRef]
- Yariv, B.; Yariv, E.; Kessel, A.; Masrati, G.; Chorin, A.B.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Using evolutionary data to make sense of macromolecules with a “face-lifted” ConSurf. Protein Sci. 2023, 32, e4582. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar]
- Drew, E.D.; Janes, R.W. 2StrucCompare: A webserver for visualizing small but noteworthy differences between protein tertiary structures through interrogation of the secondary structure content. Nucleic Acids Res. 2019, 47, W477–W481. [Google Scholar] [CrossRef]
- Holm, L.; Laiho, A.; Törönen, P.; Salgado, M. DALI shines a light on remote homologs: One hundred discoveries. Protein Sci. 2023, 32, e4519. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Natarajan, P.; Ye, Y.; Hrabe, T.; Godzik, A. POSA: A user-driven, interactive multiple protein structure alignment server. Nucleic Acids Res. 2014, 42, W240–W245. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cellier, M.F.M. The Emergence of the Slc11 Clade MCbgut: A Parsimonious Hypothesis for the Dawn of Lactobacillales in the Gut of Early Vertebrates. Bacteria 2024, 3, 223-255. https://doi.org/10.3390/bacteria3030016
Cellier MFM. The Emergence of the Slc11 Clade MCbgut: A Parsimonious Hypothesis for the Dawn of Lactobacillales in the Gut of Early Vertebrates. Bacteria. 2024; 3(3):223-255. https://doi.org/10.3390/bacteria3030016
Chicago/Turabian StyleCellier, Mathieu F. M. 2024. "The Emergence of the Slc11 Clade MCbgut: A Parsimonious Hypothesis for the Dawn of Lactobacillales in the Gut of Early Vertebrates" Bacteria 3, no. 3: 223-255. https://doi.org/10.3390/bacteria3030016
APA StyleCellier, M. F. M. (2024). The Emergence of the Slc11 Clade MCbgut: A Parsimonious Hypothesis for the Dawn of Lactobacillales in the Gut of Early Vertebrates. Bacteria, 3(3), 223-255. https://doi.org/10.3390/bacteria3030016