
Comparative Genomics and In Silico Evaluation of Genes Related to the Probiotic Potential of Bifidobacterium breve 1101A

 , ,
, ,  , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and DNA Extraction
2.2. Whole-Genome Sequencing of Bifidobacterium breve 1101A Comparative Genome Analysis
2.3. Taxonomy, Phylogenomics, and Evolutionary Analysis
2.4. Prediction of Mobile Elements, Insertion Sequences, Bacteriocins, and CRISPR-Cas Systems in Bifidobacterium breve 1101A
2.5. Prediction of Antibiotic Resistance Genes
2.6. Genomic Plasticity Analysis
2.7. Pangenome Analysis
2.8. Identification of Genes Related to Probiotic Features
3. Results
3.1. Whole-Genome Characterization of Bifidobacterium breve 1101A
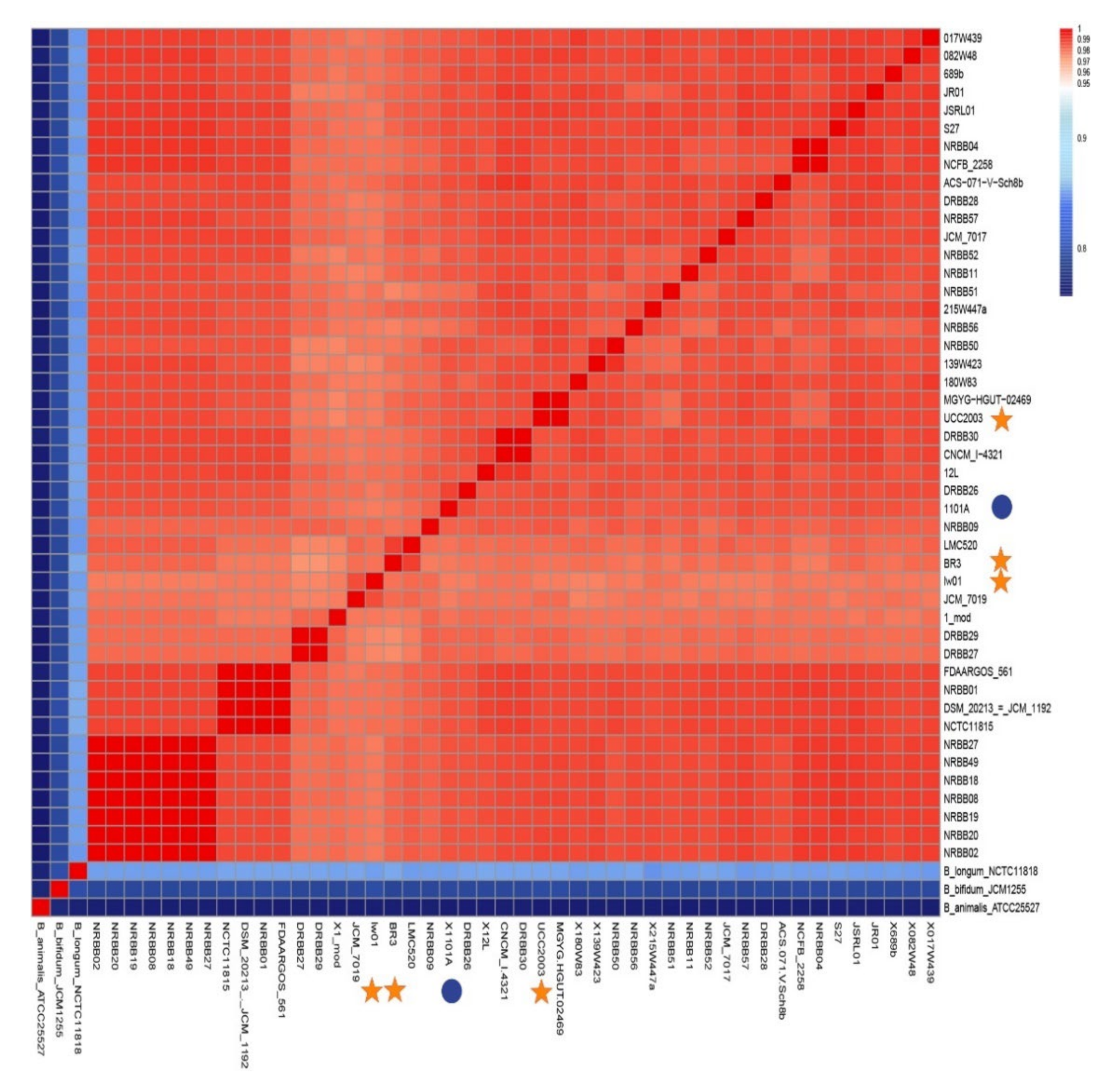
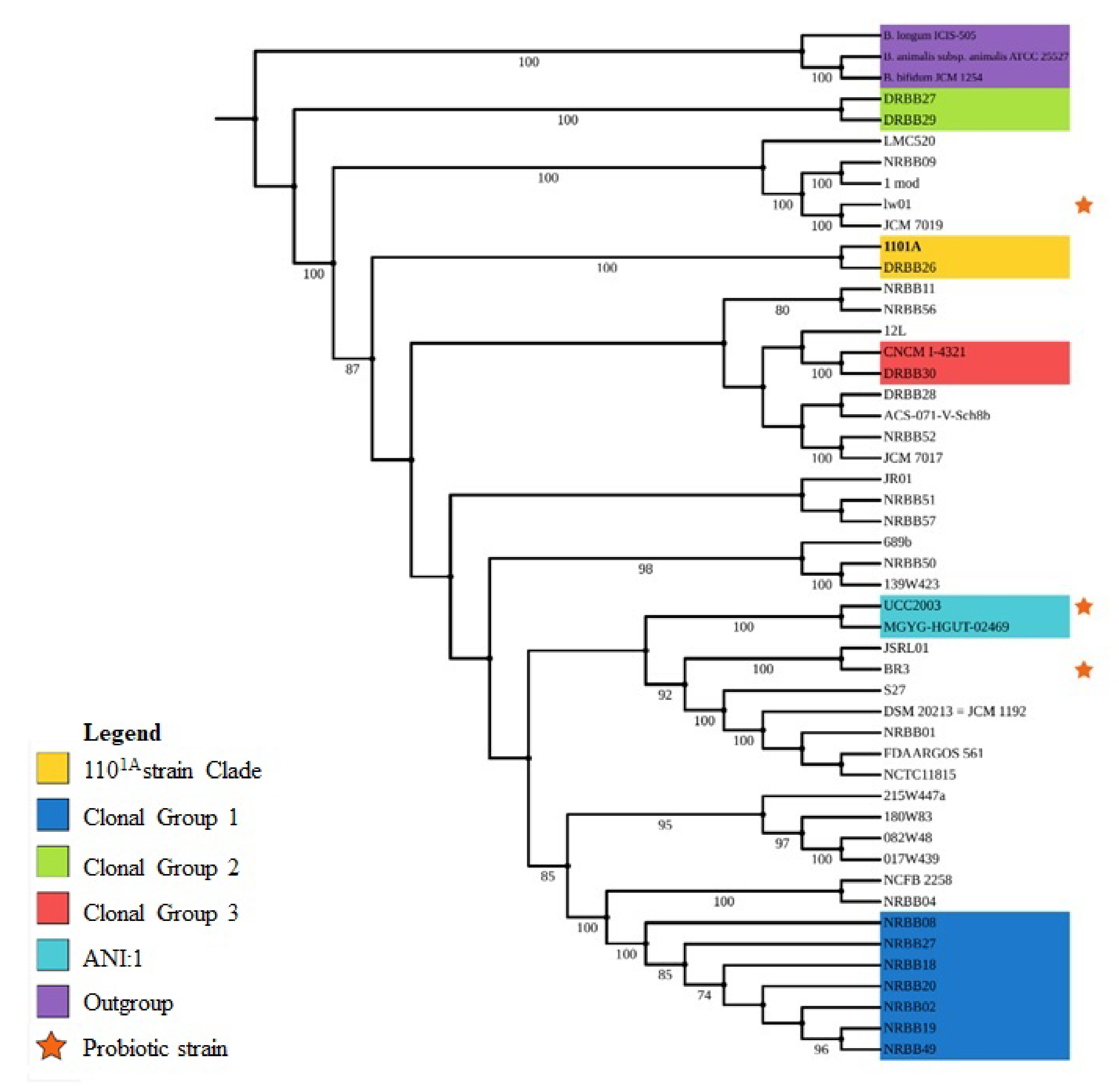
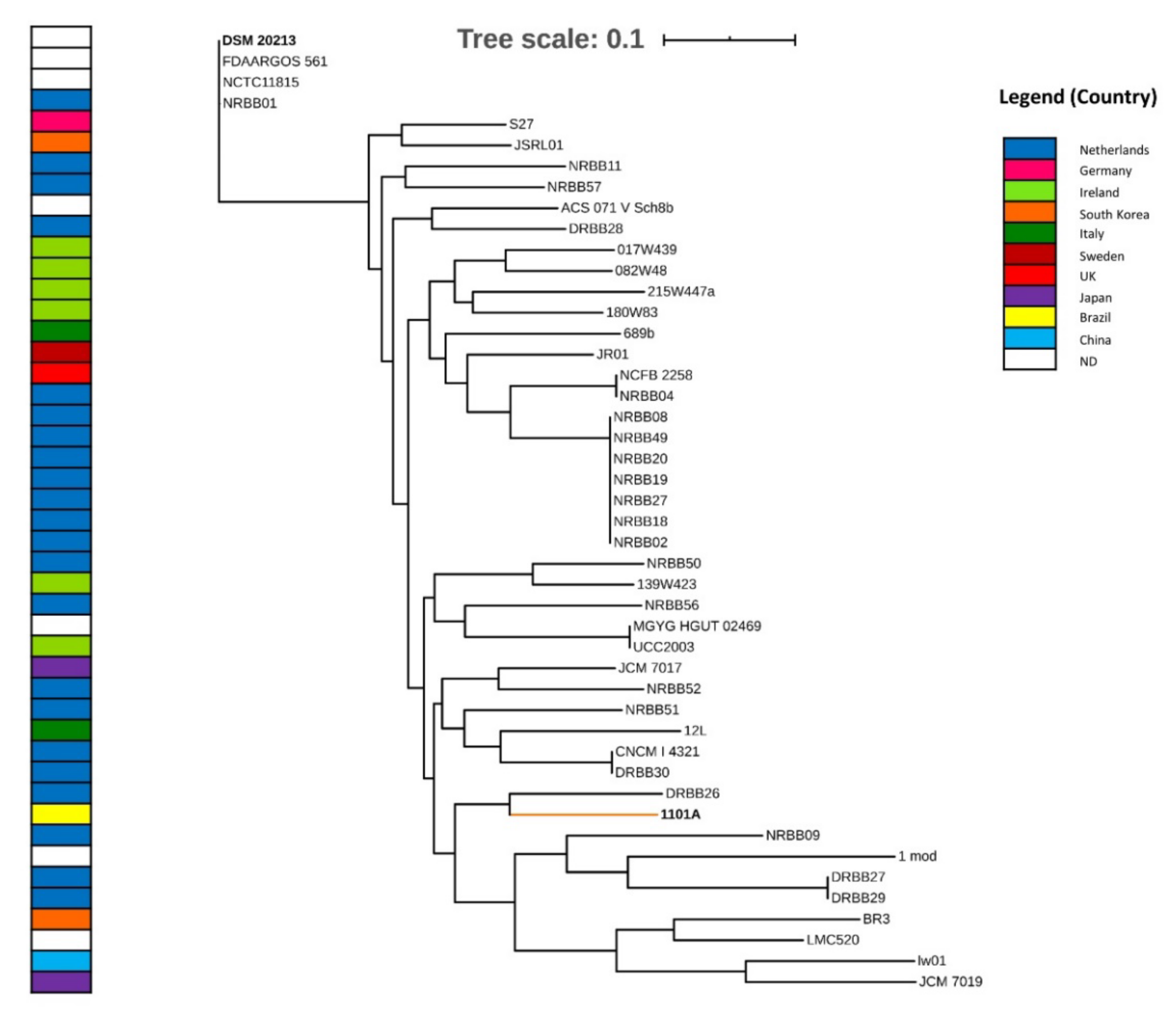
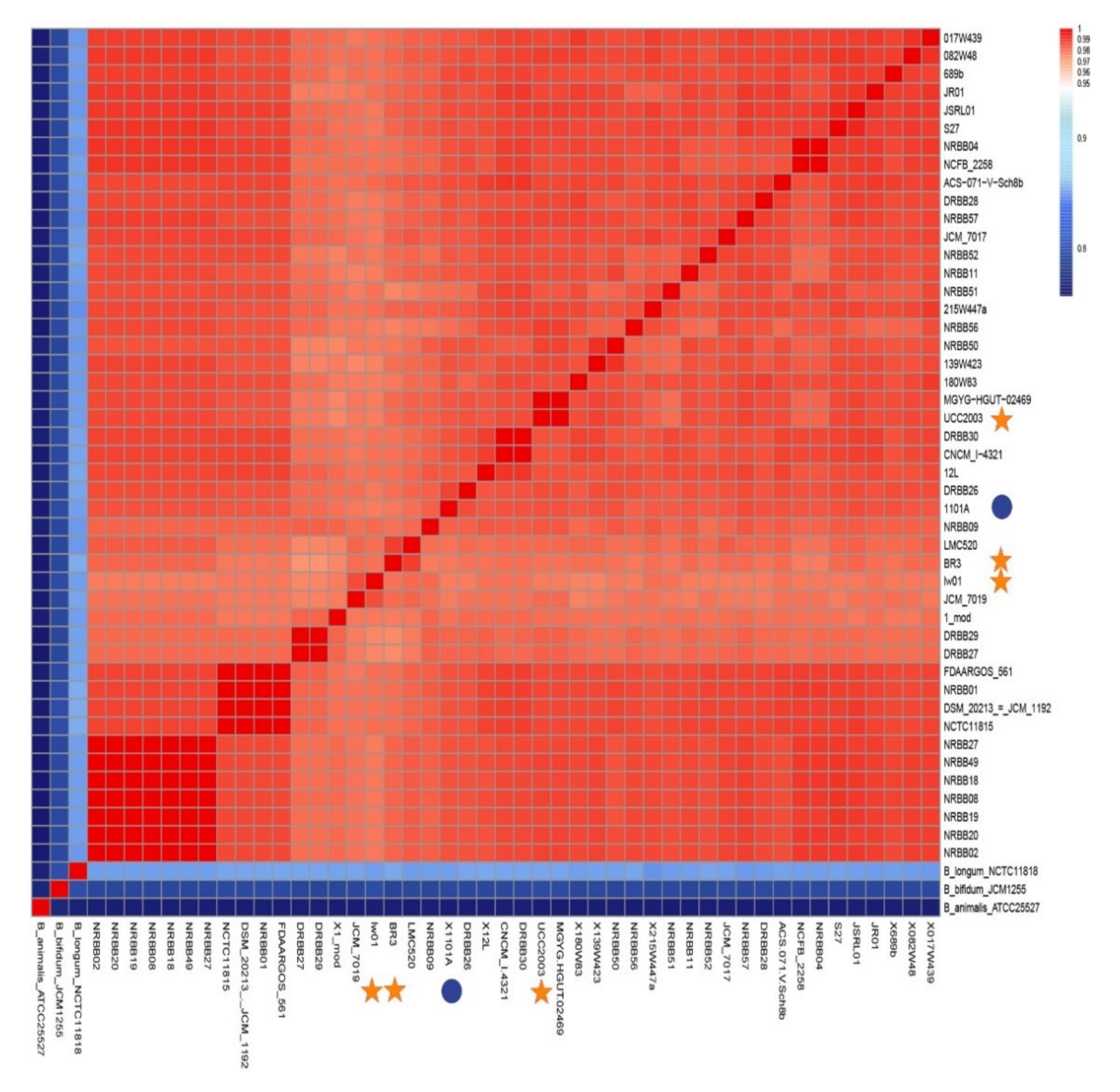
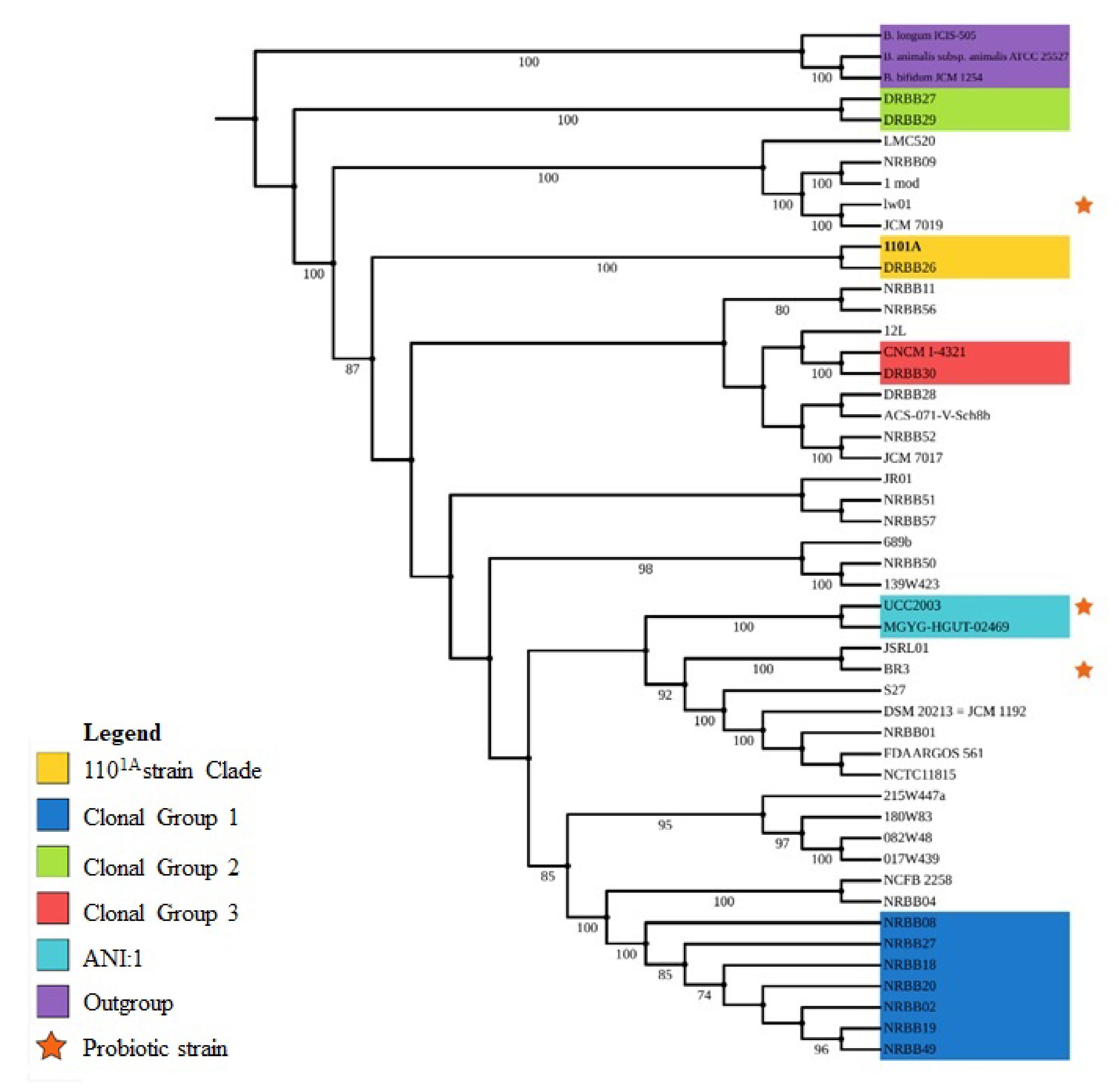
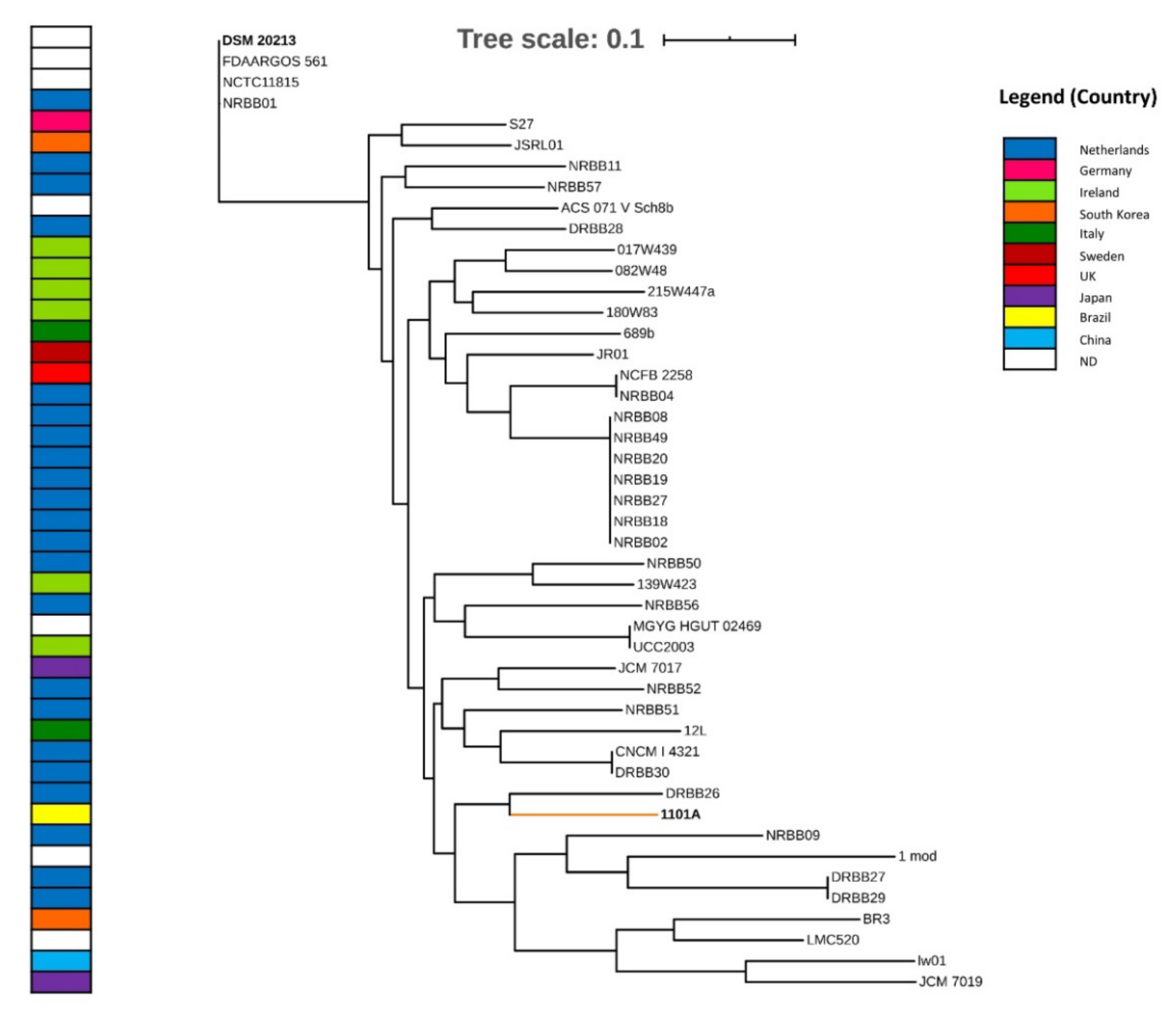
3.2. Taxonomy and Phylogenomics Analysis
3.3. Prediction of Mobile Elements, Bacteriocins, Insertion Sequences, CRISPR-Cas Systems, and Antibiotic Resistance Genes in Bifidobacterium breve 1101A
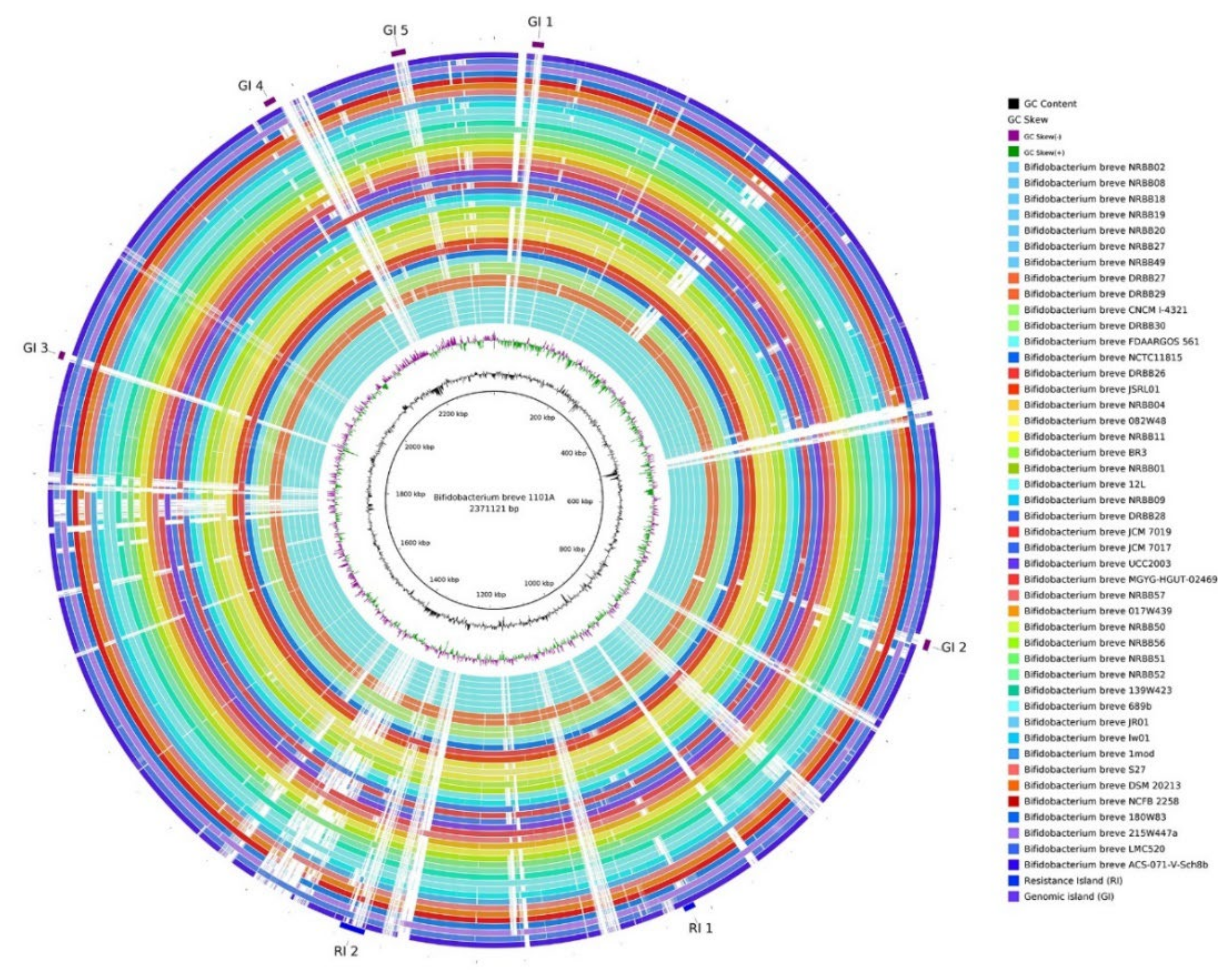
3.4. Genomic Plasticity Analysis
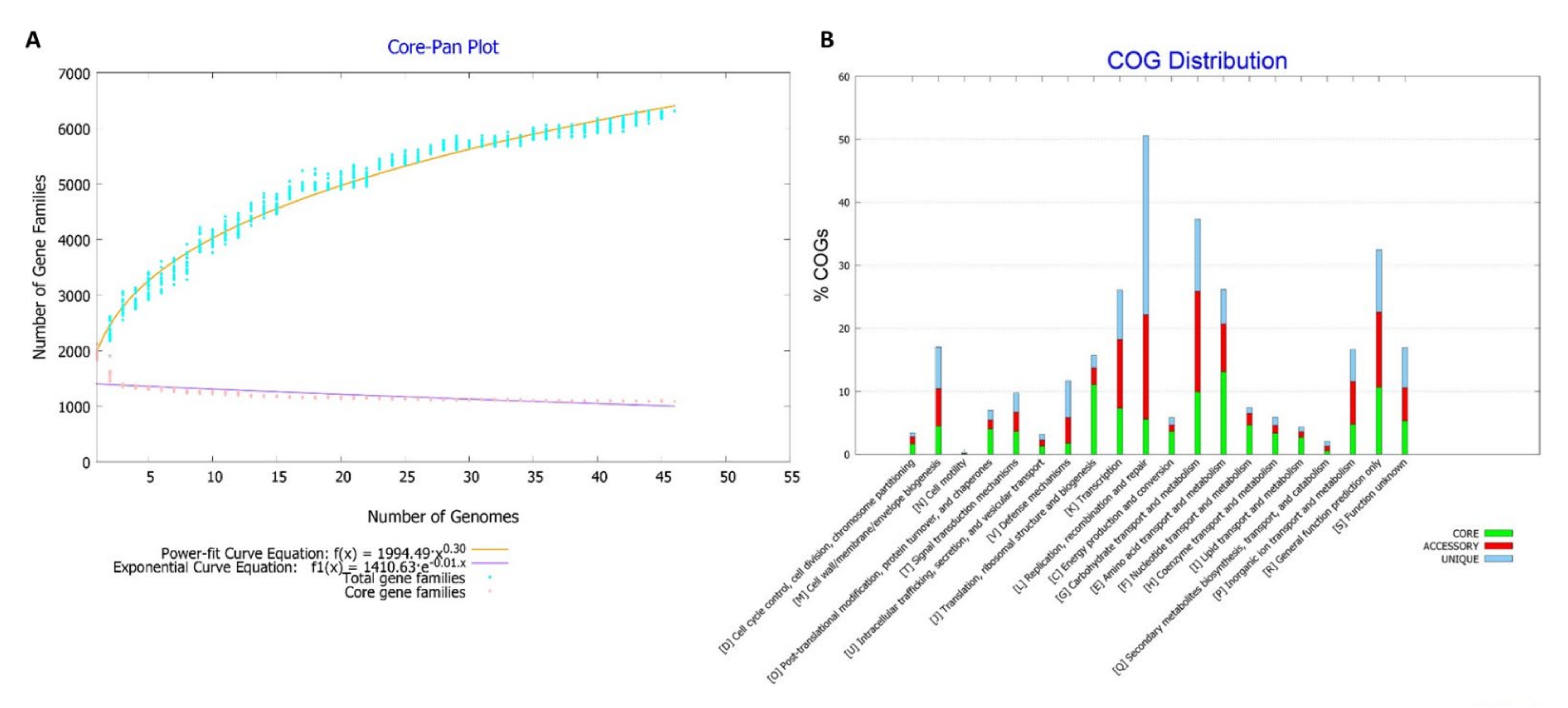
3.5. Pangenome Analysis
3.6. Identification of Genes Related to Probiotic Features
4. Discussion
4.1. Evolutionary Relationships from Phylogenomic Analysis
4.2. Assessing the Risk of Antibiotic Resistance Genes in Probiotic Bacteria
4.3. Genome Plasticity Analysis
4.4. Pangenome Analysis
4.5. Identification of Genes Related to Probiotic Features in B. breve 1101A
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klijn, A.; Mercenier, A.; Arigoni, F. Lessons from the genomes of bifidobacteria. FEMS Microbiol. Rev. 2005, 29, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Li, X.; O’Sullivan, D.J. Transcription Analysis of a Lantibiotic Gene Cluster from Bifidobacterium longum DJO10A. Appl. Environ. Microbiol. 2011, 77, 5879–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, A.; Van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F.; et al. Diversity of Bifidobacteria within the Infant Gut Microbiota. PLoS ONE 2012, 7, e36957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cionci, N.B.; Baffoni, L.; Gaggìa, F.; Di Gioia, D. Therapeutic Microbiology: The Role of Bifidobacterium breve as Food Supplement for the Prevention/Treatment of Paediatric Diseases. Nutrients 2018, 10, 1723. [Google Scholar] [CrossRef] [Green Version]
- FAO. Health and Nutritional Properties of Probiotics in Food Including Powder Milk with Live Lactic Acid Bacteria; Food and Agricultural Organization of the United Nations; World Health Organization: Rome, Italy, 2001. [Google Scholar]
- Foligne, B.; Daniel, C.; Pot, B. Probiotics from research to market: The possibilities, risks and challenges. Curr. Opin. Microbiol. 2013, 16, 284–292. [Google Scholar] [CrossRef]
- Ruiz, L.; Margolles, A.; Sánchez, B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front. Microbiol. 2013, 4, 396. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Mandal, S. Bifidobacteria—Insight into clinical outcomes and mechanisms of its probiotic action. Microbiol. Res. 2016, 192, 159–171. [Google Scholar] [CrossRef]
- Ohland, C.L.; Macnaughton, W.K. Probiotic bacteria and intestinal epithelial barrier function. Am. J. Physiol. Liver Physiol. 2010, 298, G807–G819. [Google Scholar] [CrossRef] [Green Version]
- Bermudez-Brito, M.; Plaza-Díaz, J.; Muñoz-Quezada, S.; Gómez-Llorente, C.; Gil, A. Probiotic Mechanisms of Action. Ann. Nutr. Metab. 2012, 61, 160–174. [Google Scholar] [CrossRef]
- Azad, M.A.K.; Sarker, M.; Li, T.; Yin, J. Probiotic Species in the Modulation of Gut Microbiota: An Overview. BioMed Res. Int. 2018, 2018, 9478630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jesus, L.C.L.; Drumond, M.M.; Aburjaile, F.F.; de Sousa, T.J.; Coelho-Rocha, N.D.; Profeta, R.; Brenig, B.; Mancha-Agresti, P.; Azevedo, V. Probiogenomics of Lactobacillus delbrueckii subsp. lactis CIDCA 133: In Silico, In Vitro, and In Vivo Approaches. Microorganisms 2021, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Abriouel, H.; Montoro, B.P.; de la Fuente Ordoñez, J.J.; Lerma, M.L.; Knapp, C.W.; Benomar, N. New insights into the role of plasmids from probiotic Lactobacillus pentosus MP-10 in Aloreña table olive brine fermentation. Sci. Rep. 2019, 9, 10938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennedsen, M.; Stuer-Lauridsen, B.; Danielsen, M.; Johansen, E. Screening for Antimicrobial Resistance Genes and Virulence Factors via Genome Sequencing. Appl. Environ. Microbiol. 2011, 77, 2785–2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo-Cantabrana, C.; Crawley, A.; Sanchez, B.; Barrangou, R. Characterization and Exploitation of CRISPR Loci in Bifidobacterium longum. Front. Microbiol. 2017, 8, 1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alayande, K.A.; Aiyegoro, O.A.; Nengwekhulu, T.M.; Katata-Seru, L.; Ateba, C.N. Integrated genome-based probiotic relevance and safety evaluation of Lactobacillus reuteri PNW1. PLoS ONE 2020, 15, e0235873. [Google Scholar] [CrossRef]
- Pereira, J.Q.; Ritter, A.C.; Cibulski, S.; Brandelli, A. Functional genome annotation depicts probiotic properties of Bacillus velezensis FTC01. Gene 2019, 713, 143971. [Google Scholar] [CrossRef]
- Kapse, N.; Engineer, A.; Gowdaman, V.; Wagh, S.; Dhakephalkar, P. Functional annotation of the genome unravels probiotic potential of Bacillus coagulans HS243. Genomics 2019, 111, 921–929. [Google Scholar] [CrossRef]
- Debmalya Barh, D.; Soares, S.; Tiwari, S.; Azevedo, V. Pan-Genomics: Applications, Challenges, and Future Prospects; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 978-0-12-817076-2. [Google Scholar]
- Garrigues, C.; Johansen, E.; Crittenden, R. Pangenomics—An avenue to improved industrial starter cultures and probiotics. Curr. Opin. Biotechnol. 2013, 24, 187–191. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, J.; Zhang, D.; Liu, H.; Wang, S.; Wang, Y.; Ji, H. Complete Genome Sequencing and Comparative Genome Characterization of Lactobacillus johnsonii ZLJ010, a Potential Probiotic With Health-Promoting Properties. Front. Genet. 2019, 10, 812. [Google Scholar] [CrossRef] [Green Version]
- Fontana, A.; Falasconi, I.; Molinari, P.; Treu, L.; Basile, A.; Vezzi, A.; Campanaro, S.; Morelli, L. Genomic Comparison of Lactobacillus helveticusstrains Highlights Probiotic Potential. Front. Microbiol. 2019, 10, 1380. [Google Scholar] [CrossRef] [Green Version]
- Souza, T.; Silva, A.; Drews, J.; Gomes, D.; Vinderola, C.; Nicoli, J. In vitro evaluation of Bifidobacterium strains of human origin for potential use in probiotic functional foods. Benef. Microbes 2013, 4, 179–186. [Google Scholar] [CrossRef]
- De, S.; Kaur, G.; Roy, A.; Dogra, G.; Kaushik, R.; Yadav, P.; Singh, R.; Datta, T.K.; Goswami, S.L. A Simple Method for the Efficient Isolation of Genomic DNA from Lactobacilli Isolated from Traditional Indian Fermented Milk (dahi). Indian J. Microbiol. 2010, 50, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Galardini, M.; Biondi, E.G.; Bazzicalupo, M.; Mengoni, A. CONTIGuator: A bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol. Med. 2011, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Piro, V.C.; Faoro, H.; Weiss, V.A.; Steffens, M.B.; Pedrosa, F.O.; Souza, E.M.; Raittz, R.T. FGAP: An automated gap closing tool. BMC Res. Notes 2014, 7, 371. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.; Lund, O.; Voldby Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heel, A.J.; De Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2019, 48, D561–D569. [Google Scholar] [CrossRef]
- Soares, S.C.; Geyik, H.; Ramos, R.T.; de Sá, P.H.; Barbosa, E.G.; Baumbach, J.; Figueiredo, H.C.; Miyoshi, A.; Tauch, A.; Silva, A.; et al. GIPSy: Genomic island prediction software. J. Biotechnol. 2015, 232, 2–11. [Google Scholar] [CrossRef]
- Esaiassen, E.; Hjerde, E.; Cavanagh, J.P.; Simonsen, G.S.; Klingenberg, C. Bifidobacterium bacteremia: Clinical Characteristics and a Genomic Approach To Assess Pathogenicity. J. Clin. Microbiol. 2017, 55, 2234–2248. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Araujo, F.A.; Barh, D.; Silva, A.; Guimarães, L.; Ramos, R.T.J. GO FEAT: A rapid web-based functional annotation tool for genomic and transcriptomic data. Sci. Rep. 2018, 8, 1794. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.; Ondov, B.; Koren, S.; Phillippy, A. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; O’Sullivan, D.J. Genomic Insights into Bifidobacteria. Microbiol. Mol. Biol. Rev. 2010, 74, 378–416. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, S.; Goh, Y.J.; Klaenhammer, T.R. Genomics of Probiotic Bacteria. In Prebiotics and Probiotics Science and Technology; Charalampopoulos, D., Rastall, R.A., Eds.; Springer: New York, NY, USA, 2009; pp. 681–723. ISBN 978-0-387-79057-2. [Google Scholar]
- Bottacini, F.; Morrissey, R.; Esteban-Torres, M.; James, K.; Van Breen, J.; Dikareva, E.; Egan, M.; Lambert, J.; Van Limpt, K.; Knol, J.; et al. Comparative genomics and genotype-phenotype associations in Bifidobacterium breve. Sci. Rep. 2018, 8, 10633. [Google Scholar] [CrossRef] [Green Version]
- Bottacini, F.; O’Connell Motherway, M.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A.; et al. Comparative Genomics of the Bifidobacterium Brevetaxon. BMC Genomics 2014, 15, 170. [Google Scholar] [CrossRef] [Green Version]
- Ventura, M.; Canchaya, C.; Del Casale, A.; Dellaglio, F.; Neviani, E.; Fitzgerald, G.F.; Van Sinderen, D. Analysis of bifidobacterial evolution using a multilocus approach. Int. J. Syst. Evol. Microbiol. 2006, 56, 2783–2792. [Google Scholar] [CrossRef]
- Fanning, S.; Hall, L.J.; Van Sinderen, D. Bifidobacterium breve UCC2003 surface exopolysaccharide production is a beneficial trait mediating commensal-host interaction through immune modulation and pathogen protection. Gut Microbes 2012, 3, 420–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, Y.; Wang, Y.; Xu, L.; Guo, Y.; Wang, Y.; Wang, L.; Guo, C. Oral Administration of Bifidobacterium breve Promotes Antitumor Efficacy via Dendritic Cells-Derived Interleukin 12. OncoImmunology 2021, 10, 1868122. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Li, Q.; Tian, K.; Xu, L.; Liu, G.; Guo, C. Exopolysaccharide, Isolated From a Novel Strain Bifidobacterium breve Lw01 Possess an Anticancer Effect on Head and Neck Cancer – Genetic and Biochemical Evidences. Front. Microbiol. 2019, 10, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-J.; Yoon, J.-K.; Kwon, S.-K.; Chung, M.-J.; Seo, J.-G.; Kim, J.F. Complete genome sequence of the probiotic bacterium Bifidobacterium breve KCTC 12201BP isolated from a healthy infant. J. Biotechnol. 2015, 214, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Chung, M.-J.; Seo, J.-G. In Vitro Evaluation of Antimicrobial Activity of Lactic Acid Bacteria against Clostridium difficile. Toxicol. Res. 2013, 29, 99–106. [Google Scholar] [CrossRef]
- Lee, J.-H.; Karamychev, V.; Kozyavkin, S.; Mills, D.; Pavlov, A.; Pavlova, N.; Polouchine, N.; Richardson, P.; Shakhova, V.; Slesarev, A.; et al. Comparative genomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genom. 2008, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the Evolutionary History of an Ancient Phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [Green Version]
- LoCascio, R.G.; Desai, P.; Sela, D.A.; Weimer, B.; Mills, D.A. Broad Conservation of Milk Utilization Genes in Bifidobacterium longum subsp. Infantis as Revealed by Comparative Genomic Hybridization. Appl. Environ. Microbiol. 2010, 76, 7373–7381. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Hu, T.; Qu, X.; Zhang, L.; Ding, Z.; Dong, A. Plasmids from Food Lactic Acid Bacteria: Diversity, Similarity, and New Developments. Int. J. Mol. Sci. 2015, 16, 13172–13202. [Google Scholar] [CrossRef] [Green Version]
- Iwata, M.; Morishita, T. The presence of plasmids in Bifidobacterium breve. Lett. Appl. Microbiol. 1989, 9, 165–168. [Google Scholar] [CrossRef]
- Sgorbati, B.; Scardovi, V.; Leblanc, D.J. Plasmids in the Genus Bifidobacterium. Microbiology 1982, 128, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Riordan, K.; Fitzgerald, G.F. Molecular characterisation of a 5.75-kb cryptic plasmid from Bifidobacterium breve NCFB 2258 and determination of mode of replication. FEMS Microbiol. Lett. 1999, 174, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Mahony, J.; Lugli, G.A.; van Sinderen, D.; Ventura, M. Impact of gut-associated bifidobacteria and their phages on health: Two sides of the same coin? Appl. Microbiol. Biotechnol. 2018, 102, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.-M.; Touchon, M.; Rocha, E.P.C. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef] [Green Version]
- Mancino, W.; Lugli, G.A.; van Sinderen, D.; Ventura, M.; Turroni, F. Mobilome and Resistome Reconstruction from Genomes Belonging to Members of the Bifidobacterium Genus. Microorganisms 2019, 7, 638. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Ren, L.; Song, Z.; Wang, C.; Sun, B. Purification and characteristics of bifidocin A, a novel bacteriocin produced by Bifidobacterium animals BB04 from centenarians’ intestine. Food Control 2015, 50, 889–895. [Google Scholar] [CrossRef]
- Walsh, C.J.; Guinane, C.; Hill, C.; Ross, R.P.; O’Toole, P.W.; Cotter, P.D. In silico identification of bacteriocin gene clusters in the gastrointestinal tract, based on the Human Microbiome Project’s reference genome database. BMC Microbiol. 2015, 15, 183. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Gänzle, M.G.; Lin, X.B.; Ruan, L.; Sun, M. Diversity and dynamics of bacteriocins from human microbiome. Environ. Microbiol. 2015, 17, 2133–2143. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; Ruiz-Ojeda, F.J.; Gil-Campos, M.; Gil, A. Mechanisms of Action of Probiotics. Adv. Nutr. Int. Rev. J. 2019, 10, S49–S66. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, S.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, W.; Ujiroghene, O.J.; Yang, Y.; Lu, J.; Zhang, S.; Pang, X.; Lv, J. Occurrence and Diversity of CRISPR Loci in Lactobacillus casei Group. Front. Microbiol. 2020, 11, 624. [Google Scholar] [CrossRef]
- Briner, A.E.; Lugli, G.A.; Milani, C.; Duranti, S.; Turroni, F.; Gueimonde, M.; Margolles, A.; Van Sinderen, U.; Ventura, M.; Barrangou, R. Occurrence and Diversity of CRISPR-Cas Systems in the Genus Bifidobacterium. PLoS ONE 2015, 10, e0133661. [Google Scholar] [CrossRef] [Green Version]
- Gueimonde, M.; Sánchez, B.; de los Reyes-Gavilán, C.G.; Margolles, A. Antibiotic Resistance in Probiotic Bacteria. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Lokesh, D.; Parkesh, R.; Kammara, R. Bifidobacterium adolescentis is intrinsically resistant to antitubercular drugs. Sci. Rep. 2018, 8, 11897. [Google Scholar] [CrossRef] [PubMed]
- Serafini, F.; Bottacini, F.; Viappiani, A.; Baruffini, E.; Turroni, F.; Foroni, E.; Lodi, T.; van Sinderen, D.; Ventura, M. Insights into Physiological and Genetic Mupirocin Susceptibility in Bifidobacteria. Appl. Environ. Microbiol. 2011, 77, 3141–3146. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yu, Z.; Michel, F.C.; Wittum, T.; Morrison, M. Development and Application of Real-Time PCR Assays for Quantification of erm Genes Conferring Resistance to Macrolides-Lincosamides-Streptogramin B in Livestock Manure and Manure Management Systems. Appl. Environ. Microbiol. 2007, 73, 4407–4416. [Google Scholar] [CrossRef] [Green Version]
- Martínez, N.; Luque, R.; Milani, C.; Ventura, M.; Bañuelos, O.; Margolles, A. A Gene Homologous to rRNA Methylase Genes Confers Erythromycin and Clindamycin Resistance in Bifidobacterium breve. Appl. Environ. Microbiol. 2018, 84, e02888-17. [Google Scholar] [CrossRef] [Green Version]
- van Hoek, A.H.; Mayrhofer, S.; Domig, K.J.; Aarts, H.J. Resistance determinant erm(X) is borne by transposon Tn5432 in Bifidobacterium thermophilum and Bifidobacterium animalis subsp. lactis. Int. J. Antimicrob. Agents 2008, 31, 544–548. [Google Scholar] [CrossRef]
- Bottacini, F.; Morrissey, R.; Roberts, R.J.; James, K.; van Breen, J.; Egan, M.; Lambert, J.; van Limpt, K.; Knol, J.; Motherway, M.O.; et al. Comparative Genome and Methylome Analysis Reveals Restriction/Modification System Diversity in the Gut Commensal Bifidobacterium breve. Nucleic Acids Res. 2018, 46, 1860–1877. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, O.; Lau Baggesen, D.; Bolton, D.; Butaye, P.; Cook, P.; Davies, R.; Sofos, J. Scientific Opinion on the maintenance of the list of QPS biological agents intentionally added to food and feed. EFSA J. 2012, 10, 3020. [Google Scholar] [CrossRef]
- Liu, R.; Yang, B.; Stanton, C.; Paul Ross, R.; Zhao, J.; Zhang, H.; Chen, W. Comparative genomics and gene-trait matching analysis of Bifidobacterium breve from Chinese children. Food Biosci. 2020, 36, 100631. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, Z.; Wu, J.; Yu, J. A Brief Review of Software Tools for Pangenomics. Genom. Proteom. Bioinform. 2015, 13, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Klassen, J.L.; Currie, C.R. Gene fragmentation in bacterial draft genomes: Extent, consequences and mitigation. BMC Genom. 2012, 13, 14. [Google Scholar] [CrossRef] [Green Version]
- Rouli, L.; Merhej, V.; Fournier, P.-E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Naveed, M.; Chaudhry, Z.; Ali, Z.; Amjad, M.; Zulfiqar, F.; Numan, A. Annotation and curation of hypothetical proteins: Prioritizing targets for experimental study. Adv. Life Sci. 2018, 5, 73–87. [Google Scholar]
- Vonk, R.; Reckman, G.; Harmsen, H.; Priebe, M. Probiotics and Lactose Intolerance. In Probiotics; Rigobelo, E., Ed.; InTech: London, UK, 2012; ISBN 978-953-51-0776-7. [Google Scholar]
- De Vrese, M.; Stegelmann, A.; Richter, B.; Fenselau, S.; Laue, C.; Schrezenmeir, J. Probiotics-compensation for lactase insufficiency. Am. J. Clin. Nutr. 2001, 73 (Suppl. S2), 421s–429s. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.R.; Youn, S.Y.; Ji, G.E.; Park, M.S. Production of α- and β-galactosidases from Bifidobacterium longum subsp. longum RD47. J. Microbiol. Biotechnol. 2014, 24, 675–682. [Google Scholar] [CrossRef]
- Pfeiler, E.A.; Klaenhammer, T.R. Role of Transporter Proteins in Bile Tolerance of Lactobacillus acidophilus. Appl. Environ. Microbiol. 2009, 75, 6013–6016. [Google Scholar] [CrossRef] [Green Version]
- Motherway, M.O.; Zomer, A.; Leahy, S.C.; Reunanen, J.; Bottacini, F.; Claesson, M.J.; O’Brien, F.; Flynn, K.; Casey, P.G.; Munoz, J.A.M.; et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc. Natl. Acad. Sci. USA 2011, 108, 11217–11222. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.-F.; Zheng, H.-Q.; Sun, S.-R.; Pang, X.-H.; Liang, Y.; Shang, J.-C.; Zhu, Z.-T.; Meng, X.-C. Role of luxS in Stress Tolerance and Adhesion Ability in Lactobacillus plantarum KLDS1.0391. BioMed Res. Int. 2018, 2018, 4506829. [Google Scholar] [CrossRef] [Green Version]
- Chou, L.-S.; Weimer, B. Isolation and Characterization of Acid- and Bile-Tolerant Isolates from Strains of Lactobacillus acidophilus. J. Dairy Sci. 1999, 82, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Hamon, E.; Horvatovich, P.; Izquierdo, E.; Bringel, F.; Marchioni, E.; Aoudé-Werner, D.; Ennahar, S. Comparative proteomic analysis of Lactobacillus plantarum for the identification of key proteins in bile tolerance. BMC Microbiol. 2011, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Kullen, M.J.; Klaenhammer, T.R. Identification of the pH-inducible, proton-translocating F1F0-ATPase (atpBEFHAGDC) operon of Lactobacillus acidophilus by differential display: Gene structure, cloning and characterization. Mol. Microbiol. 2002, 33, 1152–1161. [Google Scholar] [CrossRef]
- Matsumoto, M.; Ohishi, H.; Benno, Y. H+-ATPase activity in Bifidobacterium with special reference to acid tolerance. Int. J. Food Microbiol. 2004, 93, 109–113. [Google Scholar] [CrossRef]
- Pfeiler, E.A.; Azcarate-Peril, M.A.; Klaenhammer, T.R. Characterization of a Novel Bile-Inducible Operon Encoding a Two-Component Regulatory System in Lactobacillus acidophilus. J. Bacteriol. 2007, 189, 4624–4634. [Google Scholar] [CrossRef] [Green Version]
- Zomer, A.; Fernandez, M.; Kearney, B.; Fitzgerald, G.F.; Ventura, M.; Van Sinderen, D. An interactive regulatory network controls stress response in Bifidobacterium breve UCC2003. J. Bacteriol. 2009, 191, 7039–7049. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, K.; Versalovic, J.; Roos, S.; Britton, R.A. Genomic and Genetic Characterization of the Bile Stress Response of Probiotic Lactobacillus reuteri ATCC 55730. Appl. Environ. Microbiol. 2008, 74, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Chagnaud, P.; Tannock, G.W.; Loach, D.M.; Bello, F.D.; Jenkinson, H.F.; Hammes, W.P.; Hertel, C. A High-Molecular-Mass Surface Protein (Lsp) and Methionine Sulfoxide Reductase B (MsrB) Contribute to the Ecological Performance of Lactobacillus reuteri in the Murine Gut. Appl. Environ. Microbiol. 2005, 71, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Bron, P.A.; Grangette, C.; Mercenier, A.; de Vos, W.M.; Kleerebezem, M. Identification of Lactobacillus plantarum Genes That Are Induced in the Gastrointestinal Tract of Mice. J. Bacteriol. 2004, 186, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Cappa, F.; Cattivelli, D.; Cocconcelli, P.S. The uvrA gene is involved in oxidative and acid stress responses in Lactobacillus helveticus CNBL1156. Res. Microbiol. 2005, 156, 1039–1047. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | GC% | Size (Mb) | Prokka Annotation | Source | Host | Country | Accession Number | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CDS | tRNA | rRNA | tmRNA | CRISPR | |||||||
| 1101A | 58.76 | 2.37 | 1986 | 55 | 9 | 1 | - | Fecal | Children | Brazil | Present study |
| JCM 1192 | 58.9 | 2.27 | 1930 | 54 | 6 | 1 | - | Fecal | Children | Japan | NZ_AP012324 |
| NCTC11815 | 58.9 | 2.28 | 1927 | 54 | 9 | 1 | - | Intestine | Children | UK | NZ_LR134348 |
| 215W447a | 59.3 | 2.59 | 2258 | 72 | 9 | 1 | - | Gut * | Children | Ireland | NZ_CP021558 |
| NRBB57 | 59.4 | 2.51 | 2162 | 72 | 9 | 1 | - | Gut * | Children | Netherlands | NZ_CP021389 |
| DRBB30 | 58.9 | 2.47 | 2139 | 56 | 9 | 1 | - | Gut * | Children | Netherlands | NZ_CP023199 |
| CNCM I-4321 | 59 | 2.46 | 2142 | 56 | 6 | 1 | - | Gut * | Children | Netherlands | NZ_CP021559 |
| DRBB28 | 59 | 2.46 | 2140 | 54 | 9 | 1 | - | Gut * | Children | Netherlands | NZ_CP021553 |
| DRBB29 | 58.9 | 2.44 | 2132 | 56 | 6 | 1 | - | Gut * | Children | Netherlands | NZ_CP023198 |
| DRBB27 | 58.9 | 2.44 | 2135 | 56 | 6 | 1 | - | Gut * | Children | Netherlands | NZ_CP021552 |
| NRBB56 | 58.9 | 2.43 | 2030 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP021394 |
| UCC2003 | 58.7 | 2.42 | 2026 | 55 | 6 | 1 | 2 | Fecal | Children (breastfed) ** | Ireland | NC_020517 |
| MGYG-HGUT-02469 | 58.7 | 2.42 | 2026 | 55 | 6 | 1 | 2 | Gut | Human | - | NZ_LR699003 |
| BR3 | 59.1 | 2.43 | 2098 | 55 | 9 | 1 | 1 | Fecal | Human ** | Korea | NZ_CP010413 |
| 139W423 | 58.6 | 2.41 | 2056 | 54 | 9 | 1 | 1 | Gut * | Children | Ireland | NZ_CP021556 |
| NRBB50 | 58.8 | 2.41 | 2057 | 55 | 9 | 1 | 1 | Gut * | Children | Netherlands | NZ_CP021391 |
| LMC520 | 59 | 2.4 | 2050 | 56 | 9 | 1 | - | Environment | - | - | NZ_CP019596 |
| NRBB51 | 59 | 2.4 | 2001 | 54 | 9 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP021392 |
| DRBB26 | 58.5 | 2.4 | 2021 | 54 | 9 | 1 | - | Gut * | Children | Netherlands | NZ_CP021390 |
| NRBB52 | 58.9 | 2.38 | 2012 | 53 | 9 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP021393 |
| NRBB11 | 58.7 | 2.38 | 1951 | 54 | 9 | 1 | 1 | Gut * | Children | Netherlands | NZ_CP021388 |
| 1 mod | 58.8 | 2.36 | 1975 | 53 | 6 | 1 | - | - | - | - | NZ_LR655209 |
| JCM 7019 | 58.6 | 2.36 | 2017 | 57 | 6 | 1 | 1 | Fecal | Adult | Japan | NZ_CP006713 |
| 689b | 58.7 | 2.33 | 1929 | 53 | 6 | 1 | - | Fecal | Children | Italy | NZ_CP006715 |
| ACS-071-V-Sch8b | 58.7 | 2.33 | 1929 | 54 | 9 | 1 | 3 | Vagina | Human | USA | NC_017218 |
| NRBB04 | 58.7 | 2.32 | 1932 | 53 | 9 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP021386 |
| NCFB 2258 | 58.7 | 2.32 | 1920 | 53 | 6 | 1 | 2 | Fecal | Children | UK | NZ_CP006714 |
| lw01 | 58.8 | 2.31 | 1953 | 54 | 6 | 1 | 1 | Fecal | Children ** | China | NZ_CP034192 |
| JR01 | 58.9 | 2.3 | 1959 | 54 | 9 | 1 | - | Stool | Human | Sweden | NZ_CP040931 |
| 017W439 | 58.7 | 2.3 | 1955 | 54 | 6 | 1 | - | Gut * | Children | Ireland | NZ_CP021554 |
| S27 | 58.7 | 2.29 | 1887 | 55 | 9 | 1 | 2 | Fecal | Children (breastfed) | Germany | NZ_CP006716 |
| NRBB20 | 58.6 | 2.29 | 1917 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP023195 |
| NRBB02 | 58.6 | 2.29 | 1914 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP021385 |
| NRBB27 | 58.6 | 2.29 | 1916 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP023196 |
| NRBB49 | 58.6 | 2.29 | 1917 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP023197 |
| NRBB08 | 58.6 | 2.29 | 1919 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP023192 |
| NRBB19 | 58.6 | 2.29 | 1921 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP023194 |
| NRBB18 | 58.6 | 2.29 | 1919 | 55 | 6 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP023193 |
| JCM 7017 | 58.7 | 2.29 | 1883 | 54 | 6 | 1 | 2 | Fecal | Children | Japan | NZ_CP006712 |
| 082W48 | 58.8 | 2.29 | 1919 | 53 | 9 | 1 | - | Gut * | Children | Ireland | NZ_CP021555 |
| FDAARGOS 561 | 58.9 | 2.28 | 1932 | 54 | 9 | 1 | - | Clinical Isolate | Human | - | NZ_CP033841 |
| JSRL01 | 58.6 | 2.27 | 1860 | 54 | 9 | 1 | 2 | Fecal | Baby | South Korea | NZ_CP045646 |
| 180W83 | 58.8 | 2.27 | 1922 | 54 | 9 | 1 | 1 | Gut * | Children | Ireland | NZ_CP021557 |
| NRBB01 | 58.9 | 2.27 | 1937 | 54 | 6 | 1 | - | Gut * | Children | Netherlands | NZ_CP021384 |
| NRBB09 | 58.7 | 2.27 | 1916 | 54 | 9 | 1 | 2 | Gut * | Children | Netherlands | NZ_CP021387 |
| 12L | 58.9 | 2.24 | 1845 | 53 | 6 | 1 | - | Human Milk | Human | Italy | NZ_CP006711 |
| Start | End | Orientation | Description | Completeness | Identity |
|---|---|---|---|---|---|
| 1112602 | 1113381 | Forward | Fe-S cluster assembly ATPase SufC | 100 | 100 |
| 1113550 | 1114824 | Forward | Cysteine desulfurase * | 100 | 100 |
| 1114836 | 1115390 | Forward | SUF system NifU family Fe-S cluster assembly protein | 100 | 100 |
| 1115398 | 1115982 | Forward | Metal-sulfur cluster assembly factor * | 100 | 100 |
| 1116102 | 1117346 | Reverse | Glucose-1-phosphate adenylyltransferase | 100 | 99.76 |
| 1117550 | 1118428 | Reverse | RNA methyltransferase | 100 | 100 |
| 1118671 | 1119468 | Forward | RNA methyltransferase * | 100 | 100 |
| 1119579 | 1119917 | Forward | Histidine triad domain protein | 100 | 100 |
| 1119936 | 1121120 | Forward | PhoH family protein | 100 | 99.75 |
| CRISPR ID | Start | End | Spacer | Repeat Sequences | Evidence Level |
|---|---|---|---|---|---|
| 1101A_1 | 140420 | 140756 | 6 | TACTGGTGGTTTTGCCCCGCTGAGG | 2 |
| 1101A_2 | 1104813 | 1104898 | 1 | GCTTAGTGCAATAAATTCTCGAAAT | 1 |
| 1101A_3 | 2095179 | 2095325 | 2 | AATCTCCTAAAATCCTGTCACTAAG | 1 |
| Database | ileS | rpoB | erm(X) |
|---|---|---|---|
| ARG-ANNOT | - | - | 99.18 (99.88) |
| CARD | 88.16 (99.22) | 88.56 (99.86) | 99.18 (99.88) |
| MEGARES | 88.16 (99.22) | - | 99.88 (100) |
| NCBI-AMRFinderPlus | - | - | 99.88 (100) |
| ResFinder | - | - | 99.18 (99.88) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdez-Baez, J.; da Costa, F.M.R.; Pinto Gomide, A.C.; Profeta, R.; da Silva, A.L.; Sousa, T.d.J.; Viana, M.V.C.; Bentes Kato, R.; Americo, M.F.; dos Santos Freitas, A.; et al. Comparative Genomics and In Silico Evaluation of Genes Related to the Probiotic Potential of Bifidobacterium breve 1101A. Bacteria 2022, 1, 161-182. https://doi.org/10.3390/bacteria1030013
Valdez-Baez J, da Costa FMR, Pinto Gomide AC, Profeta R, da Silva AL, Sousa TdJ, Viana MVC, Bentes Kato R, Americo MF, dos Santos Freitas A, et al. Comparative Genomics and In Silico Evaluation of Genes Related to the Probiotic Potential of Bifidobacterium breve 1101A. Bacteria. 2022; 1(3):161-182. https://doi.org/10.3390/bacteria1030013
Chicago/Turabian StyleValdez-Baez, Juan, Francielly Morais Rodrigues da Costa, Anne Cybelle Pinto Gomide, Rodrigo Profeta, Alessandra Lima da Silva, Thiago de Jesus Sousa, Marcus Vinícius Canário Viana, Rodrigo Bentes Kato, Monique Ferrary Americo, Andria dos Santos Freitas, and et al. 2022. "Comparative Genomics and In Silico Evaluation of Genes Related to the Probiotic Potential of Bifidobacterium breve 1101A" Bacteria 1, no. 3: 161-182. https://doi.org/10.3390/bacteria1030013
APA StyleValdez-Baez, J., da Costa, F. M. R., Pinto Gomide, A. C., Profeta, R., da Silva, A. L., Sousa, T. d. J., Viana, M. V. C., Bentes Kato, R., Americo, M. F., dos Santos Freitas, A., Carvalho, R. D. d. O., Brenig, B., Martins, F. S., Aburjaile, F., & Azevedo, V. (2022). Comparative Genomics and In Silico Evaluation of Genes Related to the Probiotic Potential of Bifidobacterium breve 1101A. Bacteria, 1(3), 161-182. https://doi.org/10.3390/bacteria1030013