
Chromatic Bacteria v.2-A Himar1 Transposon-Based Delivery Vector to Extend the Host Range of a Toolbox to Fluorescently Tag Bacteria


Abstract
:1. Introduction
2. Results
2.1. Construction of pMRE-Himar Series
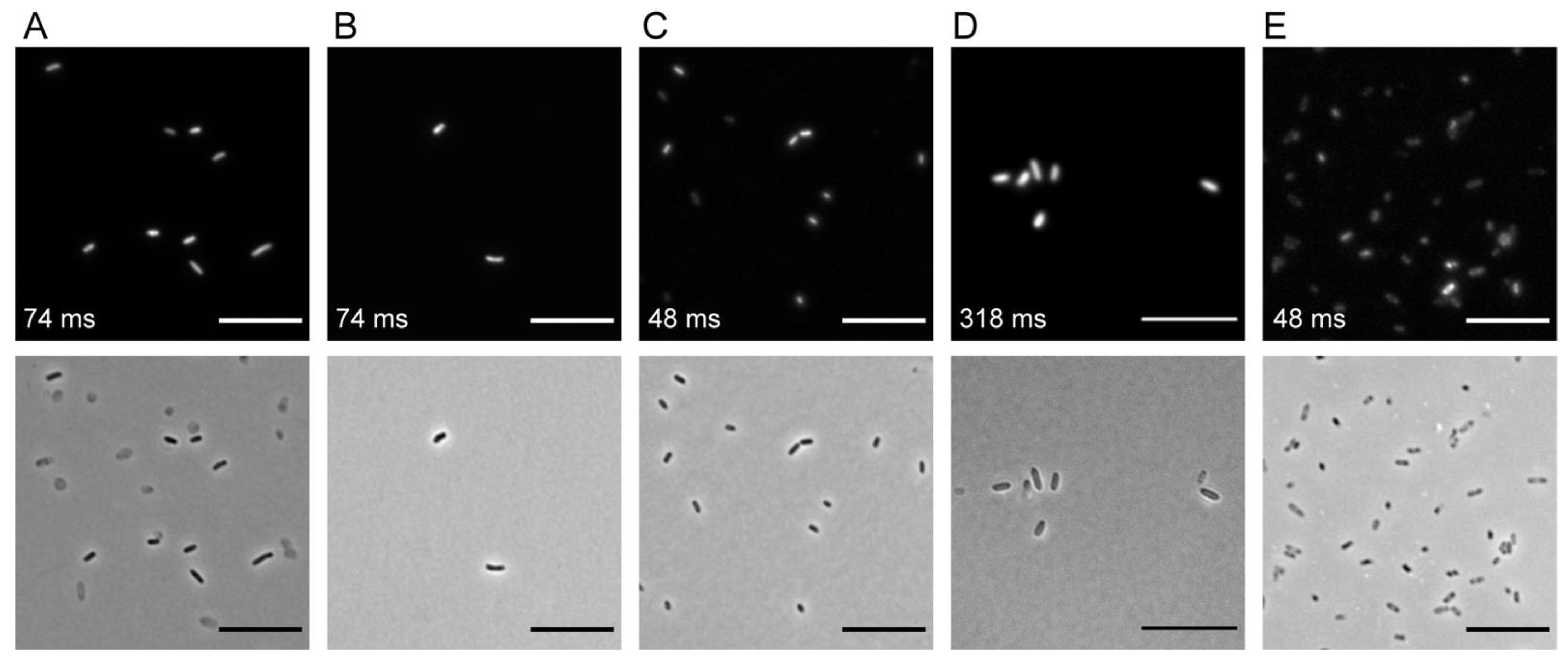
2.2. Transposon Delivery by Conjugation
3. Discussion
4. Materials and Methods
4.1. Strains and Media
4.2. Plasmid Construction
4.3. Transposon Delivery Using Conjugation
4.4. Screening for Fluorescent Colonies and Fluorescence Microscopy
4.5. Arbitrary PCR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlechter, R.O.; Jun, H.; Bernach, M.; Oso, S.; Boyd, E.; Muñoz-Lintz, D.A.; Dobson, R.C.J.; Remus, D.M.; Remus-Emsermann, M.N.P. Chromatic bacteria—A broad host-range plasmid and chromosomal insertion toolbox for fluorescent protein expression in bacteria. Front. Microbiol. 2018, 9, 3052. [Google Scholar] [CrossRef] [Green Version]
- Miebach, M.; Schlechter, R.; Clemens, J.; Jameson, P.E.; Remus-Emsermann, M.N.P. Litterbox—A gnotobiotic zeolite-clay system to investigate Arabidopsis-microbe interactions. Microorganisms 2020, 8, 464. [Google Scholar] [CrossRef] [Green Version]
- Bernach, M.; Soffe, R.; Remus-Emsermann, M.N.P.; Nock, V. Micropatterning of hybrid polydimethylsiloxane for replica leaves. Jpn. J. Appl. Phys. 2019, 58, SDDK01. [Google Scholar] [CrossRef]
- Remus-Emsermann, M.N.P.; Schlechter, R.O. Phyllosphere microbiology: At the interface between microbial individuals and the plant host. New Phytol. 2018, 218, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Nadell, C.D.; Drescher, K.; Foster, K.R. Spatial structure, cooperation and competition in biofilms. Nat. Rev. Microbiol. 2016, 14, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Tellier, M.; Bouuaert, C.C.; Chalmers, R. Mariner and the ITm superfamily of transposons. Microbiol. Spectr. 2015, 3, MDNA3–MDNA0033-2014. [Google Scholar] [CrossRef] [Green Version]
- Lampe, D.J. Bacterial genetic methods to explore the biology of mariner transposons. Genetica 2010, 138, 499–508. [Google Scholar] [CrossRef]
- Lampe, D.J.; Churchill, M.E.; Robertson, H.M. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996, 15, 5470–5479. [Google Scholar] [CrossRef]
- Lamberg, A.; Nieminen, S.; Qiao, M.; Savilahti, H. Efficient insertion mutagenesis strategy for bacterial genomes involving electroporation of in vitro-assembled DNA transposition complexes of bacteriophage Mu. Appl. Environ. Microbiol. 2002, 68, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Pajunen, M.I.; Pulliainen, A.T.; Finne, J.; Savilahti, H. Generation of transposon insertion mutant libraries for Gram-positive bacteria by electroporation of phage Mu DNA transposition complexes. Microbiology 2005, 151, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- McCully, L.M.; Bitzer, A.S.; Seaton, S.C.; Smith, L.M.; Silby, M.W. Interspecies social spreading: Interaction between two sessile soil bacteria leads to emergence of surface motility. mSphere 2019, 4, e00696-18. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.F.; Khubbar, M.K.; Saffarini, D.A.; McBride, M.J. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J. Bacteriol. 2005, 187, 6943–6952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.; Christiansen, N.; Høiby, N.; Twetman, S.; Givskov, M.; Tolker-Nielsen, T. A mariner transposon vector adapted for mutagenesis in oral streptococci. Microbiologyopen 2014, 3, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bouillaut, L.; Sonenshein, A.L.; Melville, S.B. Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J. Bacteriol. 2013, 195, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Rigby, K.; Lai, Y.; Nair, V.; Peschel, A.; Schittek, B.; Otto, M. Staphylococcus aureus mutant screen reveals interaction of the human antimicrobial peptide dermcidin with membrane phospholipids. Antimicrob. Agents Chemother. 2009, 53, 4200–4210. [Google Scholar] [CrossRef] [Green Version]
- Rubin, E.J.; Akerley, B.J.; Novik, V.N.; Lampe, D.J.; Husson, R.N.; Mekalanos, J.J. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. USA 1999, 96, 1645–1650. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in Gram negative bacteria. Biotechnology 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Thoma, S.; Schobert, M. An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol. Lett. 2009, 294, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]
- Innerebner, G.; Knief, C.; Vorholt, J.A. Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 2011, 77, 3202–3210. [Google Scholar] [CrossRef] [Green Version]
- Remus-Emsermann, M.N.P.; Kim, E.B.; Marco, M.L.; Tecon, R.; Leveau, J.H.J. Draft genome sequence of the phyllosphere model bacterium Pantoea agglomerans 299R. Genome Announc. 2013, 1, e00036-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, H.; Feil, W.S.; Chain, P.; Larimer, F.; DiBartolo, G.; Copeland, A.; Lykidis, A.; Trong, S.; Nolan, M.; Goltsman, E.; et al. Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. USA 2005, 102, 11064–11069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu Quoc, P.H.; Genevaux, P.; Pajunen, M.; Savilahti, H.; Georgopoulos, C.; Schrenzel, J.; Kelley, W.L. Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect. Immun. 2007, 75, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Laasik, E.; Ojarand, M.; Pajunen, M. Novel mutants of Erwinia carotovora subsp. carotovora defective in the production of plant cell wall degrading enzymes generated by Mu transpososome-mediated insertion mutagenesis. FEMS Microbiol. Lett. 2005, 243, 93–99. [Google Scholar] [PubMed]
- Goryshin, I.Y.; Jendrisak, J.; Hoffman, L.M.; Meis, R.; Reznikoff, W.S. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat. Biotechnol. 2000, 18, 97–100. [Google Scholar] [CrossRef]
- Fernandes, P.J.; Powell, J.A.C.; Archer, J.A.C. Construction of Rhodococcus random mutagenesis libraries using Tn5 transposition complexes. Microbiology 2001, 147, 2529–2536. [Google Scholar] [CrossRef] [Green Version]
- Vidal, J.E.; Chen, J.; Li, J.; McClane, B.A. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS ONE 2009, 4, e6232. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, W.W.; Jiang, W.; Wanner, B.L. Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6K gamma origin plasmids at different copy numbers. Gene 1994, 138, 1–7. [Google Scholar] [CrossRef]
- Wang, P.; Robert, L.; Pelletier, J.; Dang, W.L.; Taddei, F.; Wright, A.; Jun, S. Robust growth of Escherichia coli. Curr. Biol. 2010, 20, 1099–1103. [Google Scholar] [CrossRef] [Green Version]
- Diard, M.; Garcia, V.; Maier, L.; Remus-Emsermann, M.N.P.; Regoes, R.R.; Ackermann, M.; Hardt, W.-D. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 2013, 494, 353–356. [Google Scholar] [CrossRef]
- Ledermann, R.; Bartsch, I.; Remus-Emsermann, M.N.; Vorholt, J.A.; Fischer, H.-M. Stable fluorescent and enzymatic tagging of Bradyrhizobium diazoefficiens to analyze host-plant infection and colonization. Mol. Plant. Microbe Interact. 2015, 28, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlechter, R.O.; Kear, E.J.; Remus, D.M.; Remus-Emsermann, M.N.P. Fluorescent protein expression as a proxy for bacterial fitness in a high-throughput assay. Appl. Environ. Microbiol. 2021, 87, e0098221. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Schlechter, R.; Remus-Emsermann, M. Delivering “chromatic bacteria” fluorescent protein tags to proteobacteria using conjugation. Bio-Protocol 2019, 9, e3199. [Google Scholar] [CrossRef] [PubMed]
- Remus-Emsermann, M.N.P.; Gisler, P.; Drissner, D. MiniTn7-transposon delivery vectors for inducible or constitutive fluorescent protein expression in Enterobacteriaceae. FEMS Microbiol. Lett. 2016, 363, fnw178. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Noe, J.C.; Paik, S.; Kitten, T. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 2005, 63, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name | Fluorescent Protein Gene | Selectable Marker(s) |
|---|---|---|
| pMRE-Himar-131 | mTurquoise2 | CmR |
| pMRE-Himar-133 | sYFP2 | CmR |
| pMRE-Himar-134 | mOrange2 | CmR |
| pMRE-Himar-135 | mScarlet-I | CmR |
| pMRE-Himar-136 | mCardinal | CmR |
| pMRE-Himar-137 | mClover3 | CmR |
| pMRE-Himar-140 | mTagBFP2 | CmR, GmR |
| pMRE-Himar-141 | mTurquoise2 | CmR, GmR |
| pMRE-Himar-142 | sGFP2 | CmR, GmR |
| pMRE-Himar-143 | sYFP2 | CmR, GmR |
| pMRE-Himar-144 | mOrange2 | CmR, GmR |
| pMRE-Himar-145 | mScarlet-I | CmR, GmR |
| pMRE-Himar-146 | mCardinal | CmR, GmR |
| pMRE-Himar-147 | mClover3 | CmR, GmR |
| pMRE-Himar-151 | mTurquoise2 | CmR, KmR |
| pMRE-Himar-153 | sYFP2 | CmR, KmR |
| pMRE-Himar-155 | mScarlet-I | CmR, KmR |
| pMRE-Himar-157 | mClover3 | CmR, KmR |
| pMRE-Himar-171 | mTurquoise2 | CmR, EmR |
| pMRE-Himar-174 | mOrange2 | CmR, EmR |
| pMRE-Himar-175 | mScarlet-I | CmR, EmR |
| pMRE-Himar-176 | mCardinal | CmR, EmR |
| pMRE-Himar-177 | mClover3 | CmR, EmR |
| pMRE-Himar-191 | mTurquoise2 | CmR, KmR, EmR |
| pMRE-Himar-192 | sGFP2 | CmR, KmR, EmR |
| pMRE-Himar-193 | sYFP2 | CmR, KmR, EmR |
| pMRE-Himar-194 | mOrange2 | CmR, KmR, EmR |
| pMRE-Himar-195 | mScarlet-I | CmR, KmR, EmR |
| pMRE-Himar-196 | mCardinal | CmR, KmR, EmR |
| pMRE-Himar-197 | mClover3 | CmR, KmR, EmR |
| Strain | Features, Notes | Growth Medium | Growth Temperature | Transconjugants (Yes/No) | Source |
|---|---|---|---|---|---|
| Escherichia coli S17-1 | Cloning host for R6K replicon plasmids; :RP4-2 pro thi hsdR+ Tpr Smr Tc::Mu-Kan::Tn7/λpir | LB | 37 °C | n.a. | [17] |
| Escherichia coli ST18 | Conjugation donor; Genotype: S17-1 λpirΔhemA | LB, 5-ala | 37 °C | n.a. | [18] |
| Aeromicrobium sp. Leaf245 | Transposon recipient (Actinobacteria) | NB | 30 °C | no | [19] |
| Agreia sp. Leaf335 | Transposon recipient (Actinobacteria) | NB | 30 °C | no | [19] |
| Arthrobacter sp. Leaf145 | Transposon recipient (Actinobacteria) | NB | 30 °C | no | [19] |
| Microbacterium sp. Leaf320 | Transposon recipient (Actinobacteria) | R2A | 30 °C | no | [19] |
| Microbacterium sp. Leaf347 | Transposon recipient (Actinobacteria) | R2A | 30 °C | no | [19] |
| Plantibacter sp. Leaf1 | Transposon recipient (Actinobacteria) | NB | 30 °C | no | [19] |
| Rathayibacter sp. Leaf296 | Transposon recipient (Actinobacteria) | NA | 30 °C | no | [19] |
| Rhodococcus sp. Leaf225 | Transposon recipient (Actinobacteria) | NA | 30 °C | no | [19] |
| Williamsia sp. Leaf354 | Transposon recipient (Actinobacteria) | NB | 30 °C | no | [19] |
| Acidovorax sp. Leaf84 | Transposon recipient (Proteobacteria) | R2A | 30 °C | yes | [19] |
| Sphingomonas melonis FR1 | Transposon recipient (Proteobacteria) | NB | 30 °C | yes | [20] |
| Pantoea eucalypti 299R | Transposon recipient (Proteobacteria) | LB | 30 °C | yes | [21] |
| Pseudomonas syringae B728a | Transposon recipient (Proteobacteria) | LB | 30 °C | no | [22] |
| Sphingomonas sp. Leaf17 | Transposon recipient (Proteobacteria) | NB | 30 °C | no | [19] |
| Sphingomonas sp. Leaf34 | Transposon recipient (Proteobacteria) | R2A | 30 °C | no | [19] |
| Sphingomonas sp. Leaf357 | Transposon recipient (Proteobacteria) | R2A | 30 °C | yes | [19] |
| Pedobacter sp. Leaf194 | Transposon recipient (Bacteroidetes) | R2A | 30 °C | no | [19] |
| Strain Name | Transposon Insertion Flanking Region Sequence | Region Hit |
|---|---|---|
| Sphingomonas sp. Leaf357:: MRE-Himar-145/1 | AGGGGCTCGCAGTCGATTTACCGGTTCGCATGATCGTAACCGCACAGGGGAAGGAAACATGGGCTCTCTTCCGCCAGCGCGGTGGGATGTACCCTGAG | Beta-hexosaminidase CDS |
| Sphingomonas sp. Leaf357:: MRE-Himar-145/2 | TGTTATAACCCGGGGCCCAGAAGCGCGCGAGGTAGTCTTTGAATGGATACATGGGCAGATATGCGATAACGCCGTCGAGCTTCCGGTTGGCGACGTCTCAGTCCGCGTCCATGACGACCCCGAGCGTGT | Hypothetical protein |
| Sphingomonas sp. Leaf357:: MRE-Himar-145/4 | CTCGGCGCGCAGGCCAATCTGTGGGCCGAATATATCGTGACGCCCACCGAATCCCAACATGCGCTGTTCCCGCGCGTCGACGCGCTGGCCGAGATCGCCTG | Hypothetical protein |
| Acidovorax sp. Leaf84:: MRE-Himar-145/2 | AGTCAACATCGAAAAGCTCGGAGACTATGTGAATCGCTATGGCGTCAATAGCTTTTTCGACGCATCCGATGATGCCCATC | Intergenic region |
| Acidovorax sp. Leaf84:: MRE-Himar-145/5 | ATCTGATCTTCAGACAGTCTGTCGGTAGCTCCCTCGCGCCTTGCAGAGCAGATGATGTGTTCCCCTTGAAAACGCCCTTGACATCATGCACCTCGACG | Hypothetical protein |
| Acidovorax sp. Leaf84:: MRE-Himar-145/6 | TAATCGGTGGATGGTAAATAGATAGGAAATTTATCACTGTGTTTCATAACAGGTTG | Intergenic region |
| Name | Sequence (5′ to 3′) 1 | Tm (°C) |
|---|---|---|
| pHimarEm1+XmaI_FWD | cccgggCAATTCGAGGGGTATCGCTCT | 67 |
| pHimarEm1_XbaI_RVS2 | tctagaGCACGAGGAAATTGCGCAAAAA | 67 |
| pMRE-HimarEm1+XbaI_overhang_FW2 | gcaatttcctcgtgctctagaATATAAACTGCCAGGAATTGGG | 62 |
| pntpII_1_REV | GCCATGTAAGCCCACTGCAAGCTAC | 73 |
| pntpII_1_FWD | GTAGCTTGCAGTGGGCTTACATGGC | 73 |
| pMRE-HimarEm1+XmaI_overhang_RV | gatacccctcgaattgcccgggCTGGCGGCCGCAAGCTCC | 74 |
| pMRE-HimarEm1_EmR+XbaI_overhang_RV | tttcatccttcgtagtctagaCATAAACTGCCAGGAATTGGGGAT | 67 |
| pHimarEm1_EmR+XbaI_RV | tctagaCTACGAAGGATGAAATTTTTCAGGG | 63 |
| ARB-RB-PCR1 | CTGGGGTAATGACTCTCTAGC | 59 |
| ARB-RB-PCR2 | CTGAGTAGGACAAATCCGCCG | 62 |
| PCR2 AP-PCR | GGCCACGCGTCGACTAGTCA | 66 |
| Arb1 | GGCCACGCGTCGACTAGTCANNNNNNNNNNGCTCG | n.a. |
| Arb2 | GGCCACGCGTCGACTAGTCANNNNNNNNNNGACTC | n.a. |
| Arb3 | GGCCACGCGTCGACTAGTCANNNNNNNNNNGATAC | n.a. |
| Sequencing primer | CTGGTTCCGCGCACATTTC | 61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stocks, C.; Schlechter, R.O.; Remus-Emsermann, M.N.P. Chromatic Bacteria v.2-A Himar1 Transposon-Based Delivery Vector to Extend the Host Range of a Toolbox to Fluorescently Tag Bacteria. Bacteria 2022, 1, 56-65. https://doi.org/10.3390/bacteria1010006
Stocks C, Schlechter RO, Remus-Emsermann MNP. Chromatic Bacteria v.2-A Himar1 Transposon-Based Delivery Vector to Extend the Host Range of a Toolbox to Fluorescently Tag Bacteria. Bacteria. 2022; 1(1):56-65. https://doi.org/10.3390/bacteria1010006
Chicago/Turabian StyleStocks, Christian, Rudolf O. Schlechter, and Mitja N. P. Remus-Emsermann. 2022. "Chromatic Bacteria v.2-A Himar1 Transposon-Based Delivery Vector to Extend the Host Range of a Toolbox to Fluorescently Tag Bacteria" Bacteria 1, no. 1: 56-65. https://doi.org/10.3390/bacteria1010006
APA StyleStocks, C., Schlechter, R. O., & Remus-Emsermann, M. N. P. (2022). Chromatic Bacteria v.2-A Himar1 Transposon-Based Delivery Vector to Extend the Host Range of a Toolbox to Fluorescently Tag Bacteria. Bacteria, 1(1), 56-65. https://doi.org/10.3390/bacteria1010006