
Pyrvinium Pamoate and BCL-XL Inhibitors Act Synergistically to Kill Patient-Derived Colorectal Adenoma Organoids

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics and Consent
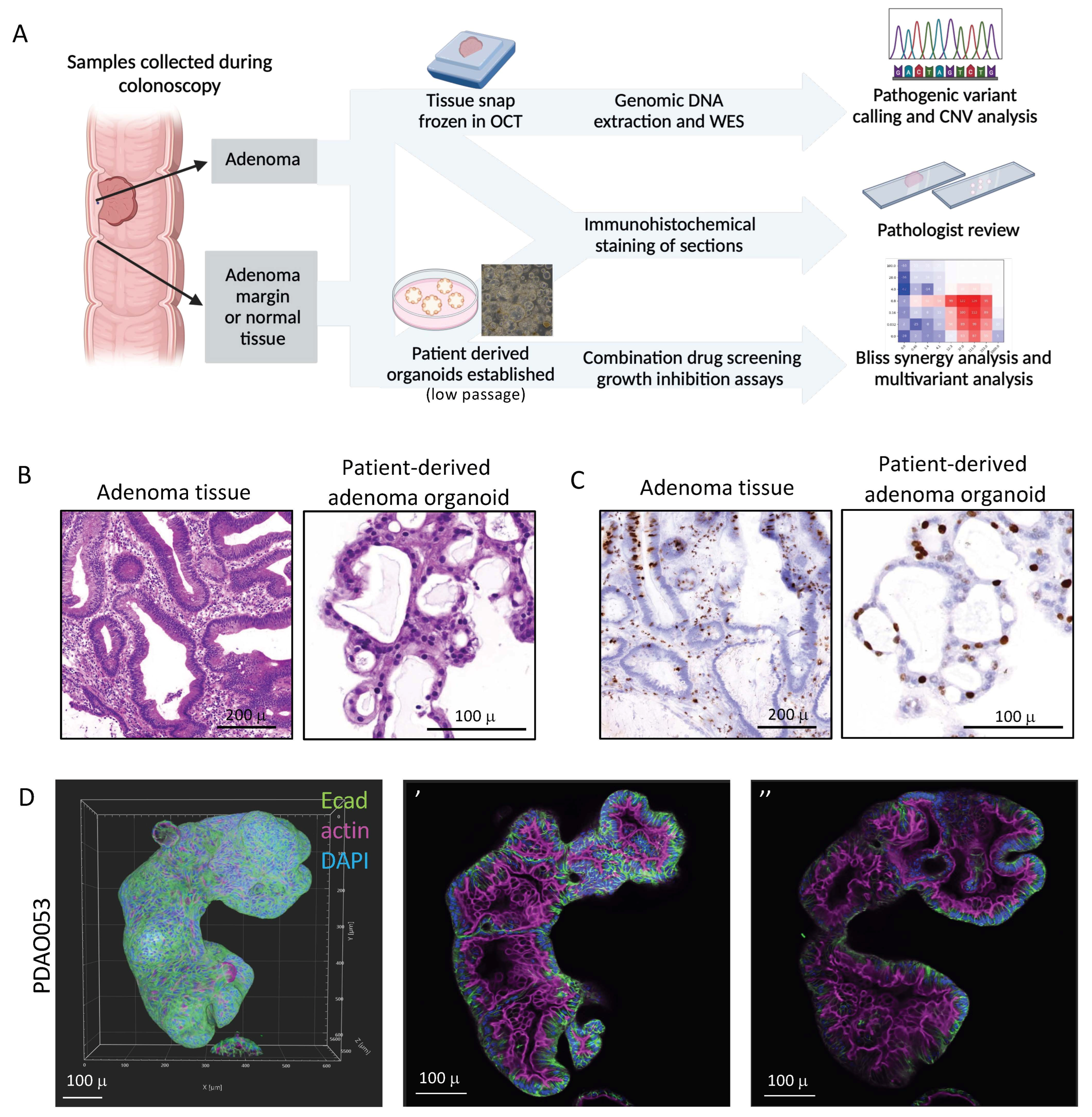
2.2. Establishing Colorectal Adenoma Organoids
2.3. Histology and Immunohistochemistry
2.4. Immunofluorescence and Confocal Microscopy
2.5. Whole Exome Sequencing (WES) and Genetic Alteration Analysis
2.6. Adenoma Organoid Drug Sensitivity and Analysis
2.7. Statistical Analysis
3. Results
3.1. Patient-Derived Adenoma Organoids Display Similar Histological Features to Their Primary Tissue
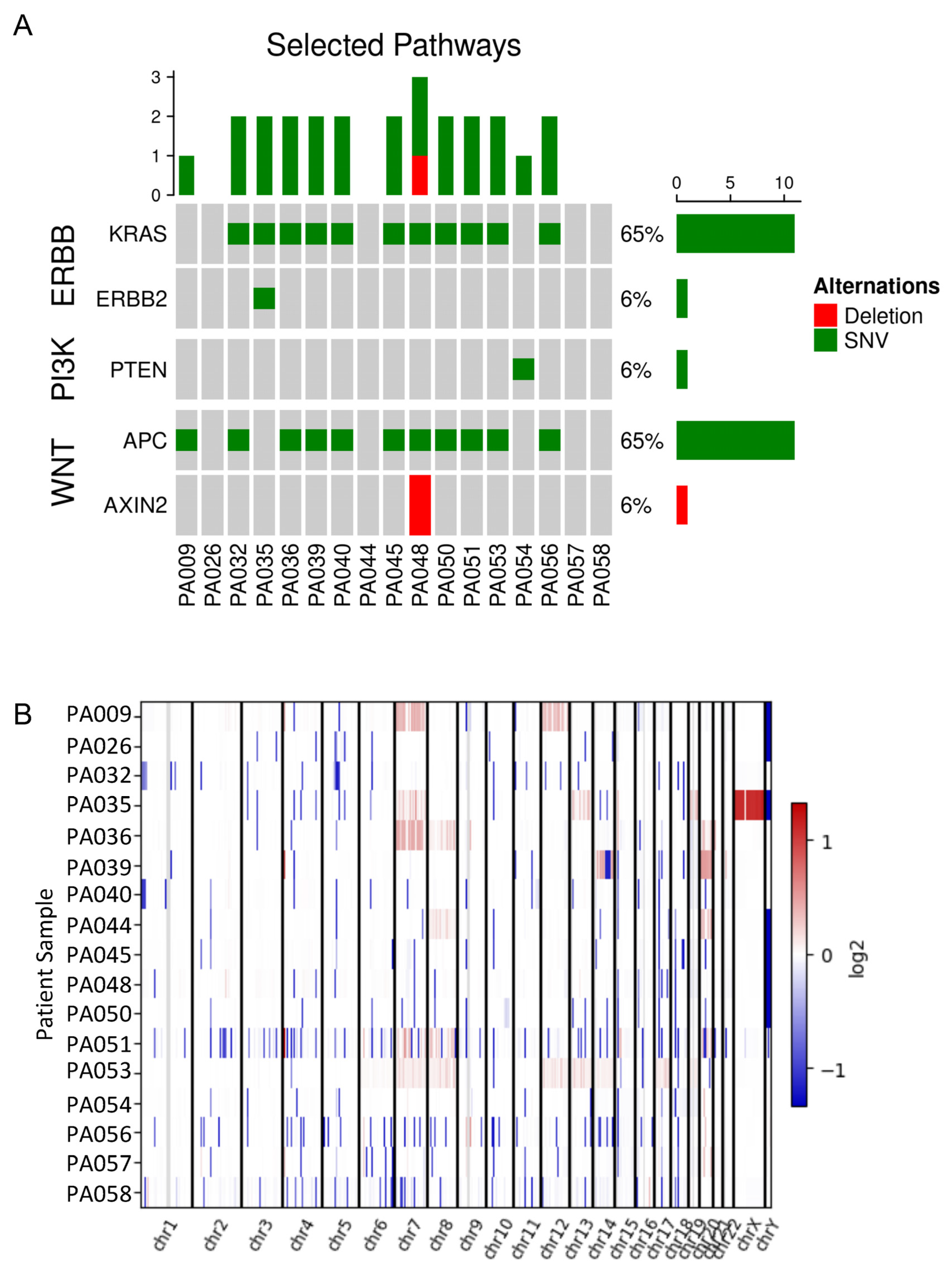
3.2. Genomic Analysis of Colorectal Adenomas
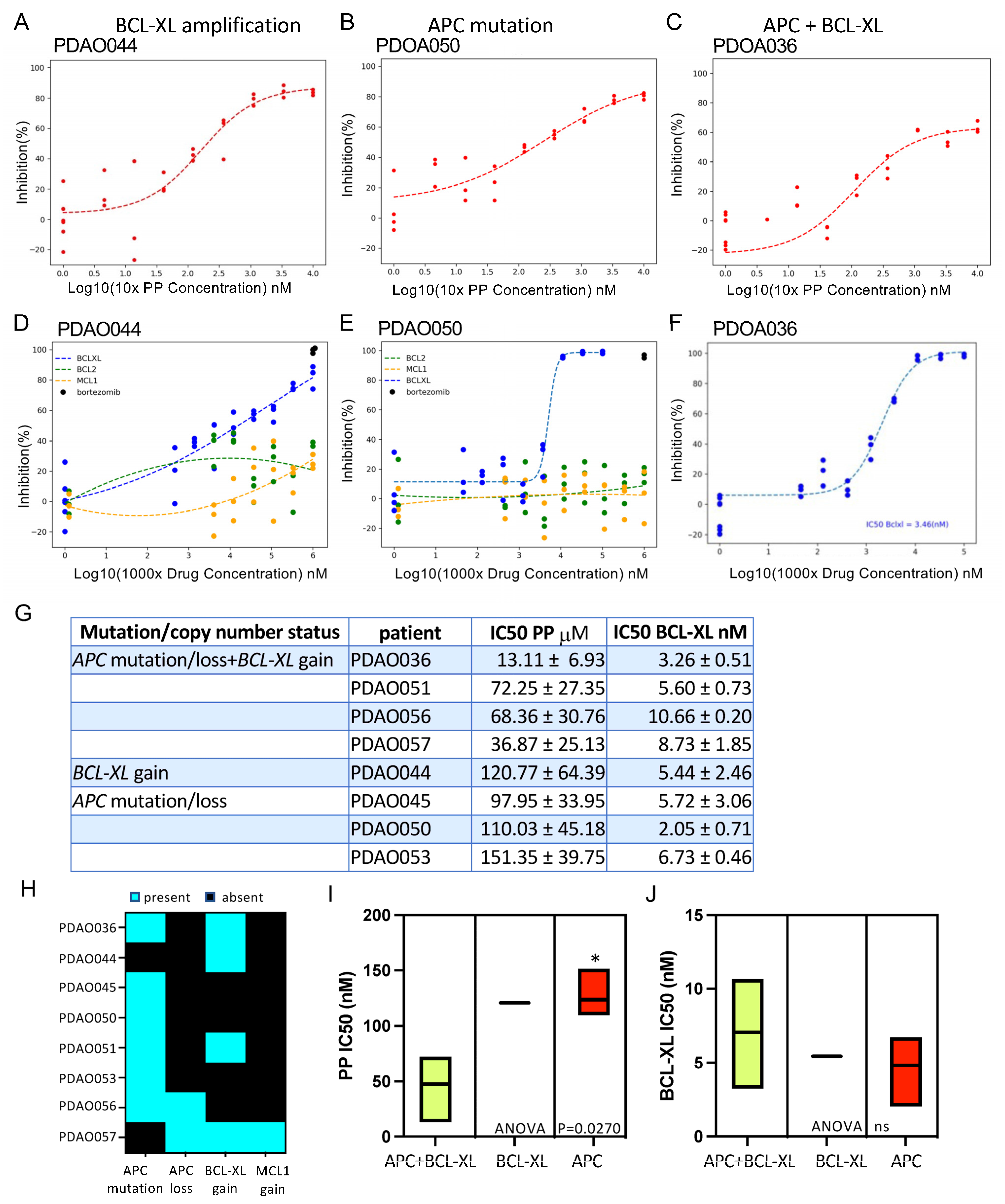
3.3. Patient-Derived Adenoma Organoid Sensitivity to Wnt and BCL-2 Family Inhibitors
3.4. Combination of PP and BCL-XL Induces Apoptosis in Adenoma Organoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Landscaping the cancer terrain. Science 1998, 280, 1036–1037. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.-K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Shaukat, A.; Kaltenbach, T.; Dominitz, J.A.; Robertson, D.J.; Anderson, J.C.; Cruise, M.; Burke, C.A.; Gupta, S.; Lieberman, D.; Syngal, S.; et al. Endoscopic Recognition and Management Strategies for Malignant Colorectal Polyps: Recommendations of the US Multi-Society Task Force on Colorectal Cancer. Gastrointest. Endosc. 2020, 92, 997–1015.e1. [Google Scholar] [CrossRef]
- Wang, L.; He, X.; Ugai, T.; Haruki, K.; Lo, C.-H.; Hang, D.; Akimoto, N.; Fujiyoshi, K.; Wang, M.; Fuchs, C.S.; et al. Risk Factors and Incidence of Colorectal Cancer According to Major Molecular Subtypes. JNCI Cancer Spectr. 2021, 5, pkaa089. [Google Scholar] [CrossRef]
- Delisle, M.; Helewa, R.M.; Ward, M.A.R.; Hochman, D.J.; Park, J.; McKay, A. The Association Between Wait Times for Colorectal Cancer Treatment and Health Care Costs: A Population-Based Analysis. Dis. Colon Rectum 2020, 63, 160–171. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Cottrell, S.; Bicknell, D.; Kaklamanis, L.; Bodmer, W. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet 1992, 340, 626–630. [Google Scholar] [CrossRef]
- Sugai, T.; Habano, W.; Takagi, R.; Yamano, H.; Eizuka, M.; Arakawa, N.; Takahashi, Y.; Yamamoto, E.; Kawasaki, K.; Yanai, S.; et al. Analysis of molecular alterations in laterally spreading tumors of the colorectum. J. Gastroenterol. 2017, 52, 715–723. [Google Scholar] [CrossRef]
- Sugimoto, T.; Ohta, M.; Ikenoue, T.; Yamada, A.; Tada, M.; Fujishiro, M.; Ogura, K.; Yamaji, Y.; Okamoto, M.; Kanai, F.; et al. Macroscopic morphologic subtypes of laterally spreading colorectal tumors showing distinct molecular alterations. Int. J. Cancer 2010, 127, 1562–1569. [Google Scholar] [CrossRef]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Abdollahi, E.; Ghasemi, F.; Caraglia, M.; Sahebkar, A. The novel role of pyrvinium in cancer therapy. J. Cell Physiol. 2018, 233, 2871–2881. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Zhang, S.; Wang, W.; Hu, P. Targeting of Wnt/beta-Catenin by Anthelmintic Drug Pyrvinium Enhances Sensitivity of Ovarian Cancer Cells to Chemotherapy. Med. Sci. Monit. 2017, 23, 266–275. [Google Scholar] [CrossRef]
- Harada, Y.; Ishii, I.; Hatake, K.; Kasahara, T. Pyrvinium pamoate inhibits proliferation of myeloma/erythroleukemia cells by suppressing mitochondrial respiratory complex I and STAT3. Cancer Lett. 2012, 319, 83–88. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, L.; Zhou, Y.; Rajoria, P.; Wang, C. Pyrvinium selectively induces apoptosis of lymphoma cells through impairing mitochondrial functions and JAK2/STAT5. Biochem. Biophys. Res. Commun. 2016, 469, 716–722. [Google Scholar] [CrossRef]
- Deng, L.; Lei, Y.; Liu, R.; Li, J.; Yuan, K.; Chen, Y.; Liu, Y.; Lu, Y.; Edwards, C.K., III; Huang, C.; et al. Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis. 2013, 4, e614. [Google Scholar] [CrossRef]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.-H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef]
- Feng, J.; Jiang, W.; Liu, Y.; Huang, W.; Hu, K.; Li, K.; Chen, J.; Ma, C.; Sun, Z.; Pang, X. Blocking STAT3 by pyrvinium pamoate causes metabolic lethality in KRAS-mutant lung cancer. Biochem. Pharmacol. 2020, 177, 113960. [Google Scholar] [CrossRef]
- Corona, S.P.; Walker, F.; Weinstock, J.; Lessene, G.; Faux, M.; Burgess, A.W. Dual drug targeting to kill colon cancers. Cancer Med. 2022, 11, 2612–2626. [Google Scholar] [CrossRef] [PubMed]
- Faux, M.C.; Weinstock, J.; Gogos, S.; Prato, E.; Azimpour, A.I.; O’Keefe, R.; Cathcart-King, Y.; Garnham, A.L.; Ernst, M.; Preaudet, A.; et al. Combined Treatment with a WNT Inhibitor and the NSAID Sulindac Reduces Colon Adenoma Burden in Mice with Truncated APC. Cancer Res. Commun. 2022, 2, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Schatoff, E.M.; Goswami, S.; Zafra, M.P.; Foronda, M.; Shusterman, M.; Leach, B.I.; Katti, A.; Diaz, B.J.; Dow, L.E. Distinct Colorectal Cancer–Associated APC Mutations Dictate Response to Tankyrase Inhibition. Cancer Discov. 2019, 9, 1358–1371. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dodge, M.E.; Tang, W.-J.; Lu, J.; Ma, Z.; Fan, C.-W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef]
- Yip, H.Y.K.; Tan, C.W.; Hirokawa, Y.; Burgess, A.W. Colon organoid formation and cryptogenesis are stimulated by growth factors secreted from myofibroblasts. PLoS ONE 2018, 13, e0199412. [Google Scholar] [CrossRef]
- Hirokawa, Y.; Clarke, J.; Palmieri, M.; Tan, T.; Mouradov, D.; Li, S.; Lin, C.; Li, F.; Luo, H.; Wu, K.; et al. Low-viscosity matrix suspension culture enables scalable analysis of patient-derived organoids and tumoroids from the large intestine. Commun. Biol. 2021, 4, 1067. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Alieva, M.; Wellens, L.M.; Ariese, H.C.R.; Jamieson, P.R.; Vonk, A.M.; Amatngalim, G.D.; Hu, H.; Oost, K.C.; Snippert, H.J.G.; et al. High-resolution 3D imaging of fixed and cleared organoids. Nat. Protoc. 2019, 14, 1756–1771. [Google Scholar] [CrossRef]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [Google Scholar] [CrossRef]
- Moré, J.J.; Garbow, B.S.; Hillstrom, K.E. User Guide for Minpack-1; CERN: Geneva, Switzerland, 1980. [Google Scholar]
- Bliss, C.I. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.W.; Nevler, A. Pyrvinium Pamoate: Past, Present, and Future as an Anti-Cancer Drug. Biomedicines 2022, 10, 3249. [Google Scholar] [CrossRef]
- Faux, M.C.; King, L.E.; Kane, S.R.; Love, C.; Sieber, O.M.; Burgess, A.W. APC regulation of ESRP1 and p120-catenin isoforms in colorectal cancer cells. Mol. Biol. Cell 2021, 32, 120–130. [Google Scholar] [CrossRef]
- Schwab, R.H.M.; Amin, N.; Flanagan, D.J.; Johanson, T.M.; Phesse, T.J.; Vincan, E. Wnt is necessary for mesenchymal to epithelial transition in colorectal cancer cells. Dev. Dyn. 2018, 247, 521–530. [Google Scholar] [CrossRef]
- Luo, M.-J.; Palmieri, M.; Riffkin, C.D.; Sakthianandeswaren, A.; Djajawi, T.M.; Hirokawa, Y.; Shuttleworth, V.; Segal, D.H.; White, C.A.; Nhu, D.; et al. Defining the susceptibility of colorectal cancers to BH3-mimetic compounds. Cell Death Dis. 2020, 11, 735. [Google Scholar] [CrossRef]
- Ramesh, P.; Lannagan, T.R.M.; Jackstadt, R.; Taboada, L.A.; Lansu, N.; Wirapati, P.; van Hooff, S.R.; Dekker, D.; Pritchard, J.; Kirov, A.B.; et al. BCL-XL is crucial for progression through the adenoma-to-carcinoma sequence of colorectal cancer. Cell Death Differ. 2021, 28, 3282–3296. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Tan, T.; Mouradov, D.; Lee, M.; Gard, G.; Hirokawa, Y.; Li, S.; Lin, C.; Li, F.; Luo, H.; Wu, K.; et al. Unified framework for patient-derived, tumor-organoid-based predictive testing of standard-of-care therapies in metastatic colorectal cancer. Cell Rep. Med. 2023, 4, 101335. [Google Scholar] [CrossRef]
- D’aldebert, E.; Quaranta, M.; Sébert, M.; Bonnet, D.; Kirzin, S.; Portier, G.; Duffas, J.-P.; Chabot, S.; Lluel, P.; Allart, S.; et al. Characterization of Human Colon Organoids from Inflammatory Bowel Disease Patients. Front. Cell Dev. Biol. 2020, 8, 363. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.M.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef] [PubMed]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; Van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; Van De Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef]
- Dattilo, R.; Mottini, C.; Camera, E.; Lamolinara, A.; Auslander, N.; Doglioni, G.; Muscolini, M.; Tang, W.; Planque, M.; Ercolani, C.; et al. Pyrvinium Pamoate Induces Death of Triple-Negative Breast Cancer Stem-Like Cells and Reduces Metastases Through Effects on Lipid Anabolism. Cancer Res. 2020, 80, 4087–4102. [Google Scholar] [CrossRef]
- Schultz, C.W.; McCarthy, G.A.; Nerwal, T.; Nevler, A.; DuHadaway, J.B.; McCoy, M.D.; Jiang, W.; Brown, S.Z.; Goetz, A.; Jain, A.; et al. The FDA-Approved Anthelmintic Pyrvinium Pamoate Inhibits Pancreatic Cancer Cells in Nutrient-Depleted Conditions by Targeting the Mitochondria. Mol. Cancer Ther. 2021, 20, 2166–2176. [Google Scholar] [CrossRef]
- Shen, C.; Nayak, A.; Neitzel, L.R.; Yang, F.; Li, B.; Williams, C.H.; Hong, C.C.; Ahmed, Y.; Lee, E.; Robbins, D.J. The Casein kinase 1alpha agonist pyrvinium attenuates Wnt-mediated CK1alpha degradation via interaction with the E3 ubiquitin ligase component Cereblon. J. Biol. Chem. 2022, 298, 102227. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Wang, L.; Doherty, G.A.; Judd, A.S.; Tao, Z.-F.; Hansen, T.M.; Frey, R.R.; Song, X.; Bruncko, M.; Kunzer, A.R.; Wang, X.; et al. Discovery of A-1331852, a First-in-Class, Potent, and Orally-Bioavailable BCL-XL Inhibitor. ACS Med. Chem. Lett. 2020, 11, 1829–1836. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Puglisi, M.; van Doorn, L.; Blanco-Codesido, M.; De Jonge, M.J.; Moran, K.; Yang, J.; Busman, T.; Franklin, C.; Mabry, M.; Krivoshik, A.; et al. A phase I safety and pharmacokinetic (PK) study of navitoclax (N) in combination with docetaxel (D) in patients (pts) with solid tumors. J. Clin. Oncol. 2011, 29 (Suppl. 15), 2518. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dong, X.; Huang, D.C.S.; Xu, P.; Zhao, Q.; Chen, B. Current Advances and Future Strategies for BCL-2 Inhibitors: Potent Weapons Against Cancers. Cancers 2023, 15, 4957. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Sex | Adenoma Type | Adenoma Site | Number of Adenomas | Adenoma Size & | Differentiation | Invasive Carcinoma | Passage Organoid Cryopreserved |
|---|---|---|---|---|---|---|---|---|---|
| 009 | 74 | F | 1. tubulovillous 2. benign fibrous tissue 3. tubulovillous | 1. caecum 2. transverse colon 3. transverse colon | 1. multiple 2. 3 3. multiple | 1. 20 × 12 × 9 mm 2. 2 to 3 mm 3. 17 × 16 × 7 mm | 1. high-grade dysplasia 2. nil 3. high-grade dysplasia | No | 1. >P1 2. not cultured 3. not cultured |
| 013 | 81 | M | tubulovillous | rectum | 1 | 16 × 15 × 9 mm | low-grade dysplasia | No | P0 |
| 015 | 70 | M | 1. traditional serrated 2. tubular | 1. proximal ascending colon 2. mid ascending colon | 1. multiple 2. 1 | 1. 20 × 12 × 10 mm 2. 3 mm | 1. low-grade dysplasia 2. low-grade dysplasia | No | 1. >P1 2. not cultured |
| 016 | 79 | F | tubular | caecum | multiple | 12 × 4 × 2 mm | low-grade dysplasia | No | >P1 |
| 017 | 64 | F | tubulovillous | ascending colon | multiple | 27 × 22 × 15 mm | low- and high-grade dysplasia | No | >P1 |
| 018 | 59 | M | tubulovillous | ileocaecal valve | multiple | 16 × 11 × 5 mm | high-grade dysplasia * | No | >P1 |
| 020 | 76 | F | traditional sessile serrated | 1. Ileocaecal valve 2. caecum 3. ascending colon 4. hepatic flexure | 4 | 1. 14 × 10 × 6 mm 2. 10 mm 3. 21 × 11 × 1 mm 4. 14 × 6 × 4 mm | 1. low-grade dysplasia 2. low-grade dysplasia 3. no cytological dysplasia 4. no cytological dysplasia | Not stated | 1. >P1 2. not cultured 3. not cultured 4. not cultured |
| 021 | 65 | F | tubulovillous | hepatic flexure | multiple | 1 to 5 mm | low-grade dysplasia | Not stated | >P1 |
| 022 | 55 | M | tubulovillous | caecum | multiple | 5 to 15 mm | low-grade dysplasia | No | >P1 |
| 024 | 70 | M | 1. sessile serrated 2. tubular | 1. hepatic flexure 2. transverse colon | 1. 1 2. 2 | 1. 2 to 6 mm 2. 2 to 7 mm | low-grade dysplasia | No | 1. >P1 2. not cultured |
| 025 | 68 | F | sessile serrated | hepatic flexure | 2 | 8 and 10 mm | no cytologic dysplasia | Not stated | P0 |
| 026 | 70 | F | tubulovillous | ileocaecal valve | multiple | 12 × 2 × 7 mm | low-grade dysplasia | No | P0 |
| 027 | 50 | F | tubulovillous | transverse colon | multiple | 15 × 10 × 9 mm | low-grade dysplasia | No | P0 |
| 029 | 59 | F | tubular | caecum | not stated | Not stated | low grade and high grade | No | P0 |
| 035 | na | na | sessile tubulovillous | caecum | 1 | 50 mm | moderately differentiated adenocarcinoma | Yes | n/a |
| 039 | 74 | M | villous | rectum | not stated | Not stated | high-grade dysplasia ^ | No | >P1 |
| 040 | 72 | M | tubulovillous | rectum | multiple | 18 × 12 × 12 mm | low-grade dysplasia | No | >P1 |
| 041 | 75 | M | villous | caecum | 1 | 30 × 20 × 12 mm | high-grade dysplasia | No | >P1 |
| 042 | 71 | M | tubulovillous | ascending colon | 2 | 1. 50 × 50 × 15 mm 2. 22 × 14 × 13 mm | high-grade dysplasia | No | 1. >P1 2. not cultured |
| 044 | 64 | F | tubulovillous | sigmoid colon | 1 | 26 × 13 × 9 mm | low- and high-grade dysplasia | No | >P1 |
| 045 | 62 | F | tubulovillous | transverse colon | multiple | 18 × 13 × 10 mm | low- and high-grade dysplasia | No | >P1 |
| 048 | 63 | F | tubulovillous | caecum | 1 | 45 × 35 × 5 mm | low-grade dysplasia | No | >P1 |
| 049 | 62 | F | 1. sessile serrated 2. tubular | 1. transverse colon 2. rectum | 1. 1 2. 2 | 1. 15 × 10 × 2 mm 2. 12 × 8 × 2 mm | 1. no sessile dysplasia 2. low-grade dysplasia | No | >P1 |
| 050 | 59 | F | tubulovillous | caecum | 1 | 45 × 35 × 10 mm | low-grade dysplasia | No | >P1 |
| 051 | 73 | F | tubular | sigmoid colon | 1 | 18 mm | low-grade dysplasia | No | P0 |
| 052 | 88 | F | 1. tubular 2. villous | 1. ascending colon 2. rectosigmoid | 1. 1 1. 1 | 1. 2 to 4 mm 2. 60 × 30 × 5 mm | 1. low-grade dysplasia 2. low-grade dysplasia | No | 1. P0 2. not cultured |
| 053 | 80 | M | sessile serrated | caecum | multiple | 1 to 10 mm | focal high-grade dysplasia | No | >P1 |
| 036 a | 80 | M | villous | transverse colon | not stated | 40 mm | low-grade dysplasia | No | >P1 |
| 051 a | 66 | M | sessile serrated adenoma | hepatic flexure | not stated | 80 mm | no dysplasia | No | >P1 |
| 054 a | 67 | M | tubulovillous | hepatic flexure | not stated | 40 mm | low-grade dysplasia | No | >P1 |
| 056 a | 72 | M | tubulovillous | transverse colon | not stated | 50 mm | low-grade dysplasia | No | >P1 |
| 057 a | 74 | M | tubulovillous adenoma | splenic flexure | not stated | 30 mm | low-grade dysplasia | No | >P1 |
| 058 a | 53 | M | sessile serrated adenoma | caecum | not stated | 30 mm | no dysplasia | No | >P1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faux, M.C.; Ma, C.; Kane, S.R.; Samson, A.; Hirokawa, Y.; Priebe, I.; Cosgrove, L.; Singh, R.; Christie, M.; Brown, G.; et al. Pyrvinium Pamoate and BCL-XL Inhibitors Act Synergistically to Kill Patient-Derived Colorectal Adenoma Organoids. Organoids 2025, 4, 15. https://doi.org/10.3390/organoids4030015
Faux MC, Ma C, Kane SR, Samson A, Hirokawa Y, Priebe I, Cosgrove L, Singh R, Christie M, Brown G, et al. Pyrvinium Pamoate and BCL-XL Inhibitors Act Synergistically to Kill Patient-Derived Colorectal Adenoma Organoids. Organoids. 2025; 4(3):15. https://doi.org/10.3390/organoids4030015
Chicago/Turabian StyleFaux, Maree C., Chenkai Ma, Serena R. Kane, Andre Samson, Yumiko Hirokawa, Ilka Priebe, Leah Cosgrove, Rajvinder Singh, Michael Christie, Gregor Brown, and et al. 2025. "Pyrvinium Pamoate and BCL-XL Inhibitors Act Synergistically to Kill Patient-Derived Colorectal Adenoma Organoids" Organoids 4, no. 3: 15. https://doi.org/10.3390/organoids4030015
APA StyleFaux, M. C., Ma, C., Kane, S. R., Samson, A., Hirokawa, Y., Priebe, I., Cosgrove, L., Singh, R., Christie, M., Brown, G., Fung, K. Y. C., & Burgess, A. W. (2025). Pyrvinium Pamoate and BCL-XL Inhibitors Act Synergistically to Kill Patient-Derived Colorectal Adenoma Organoids. Organoids, 4(3), 15. https://doi.org/10.3390/organoids4030015