

Resistance to MAPK Pathway Inhibition in BRAF-V600E Mutant Colorectal Cancer Can Be Overcome with Insulin Receptor/Insulin-like Growth Factor-1 Receptor Inhibitors

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Organoid Culture
2.2. In Vitro Drug Screens and Cell Viability Assays
2.3. Regrowth Assays
2.4. Phospho-RTK Array
2.5. Western Blot
2.6. Xenograft Tumour Growth
2.7. Data Analysis and Statistical Analysis
3. Results
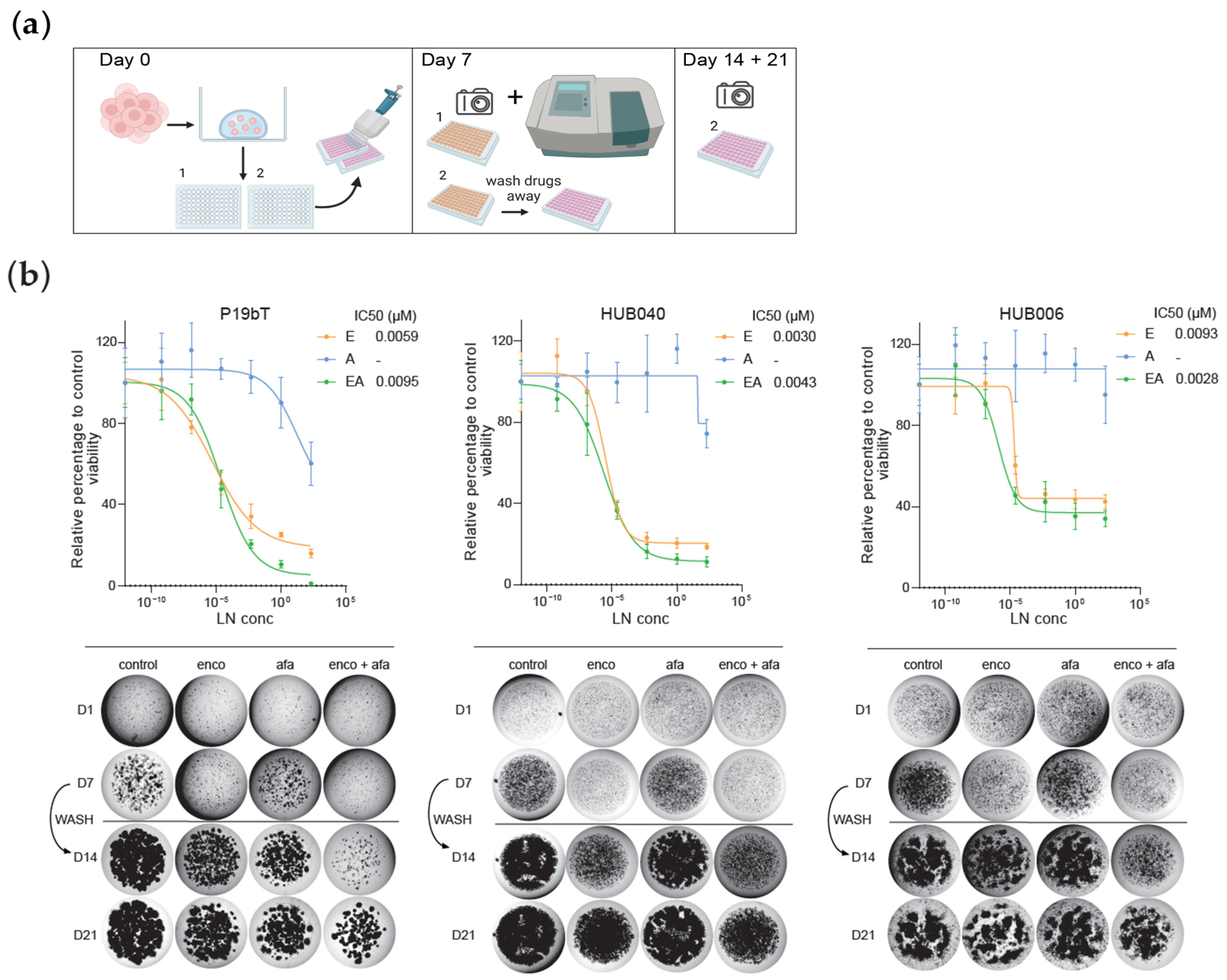
3.1. Inhibition of BRAF and EGFR in BRAF-V600E Mutant CRC PDOs Causes Potent Growth Inhibition, Followed by Rapid Regrowth After Drug Removal
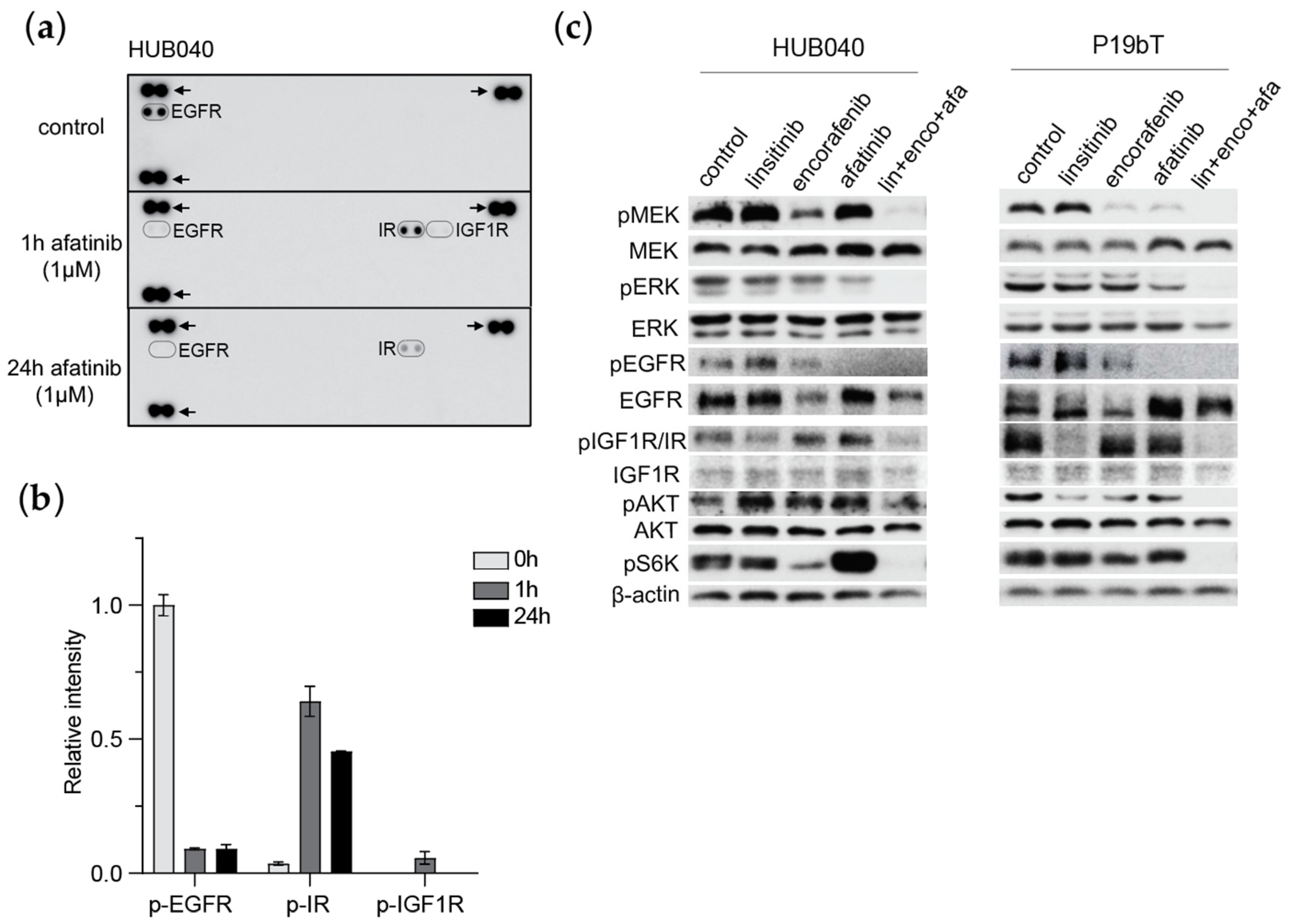
3.2. EGFR Inhibition Induces Compensatory Activation of IR and IGF1-R
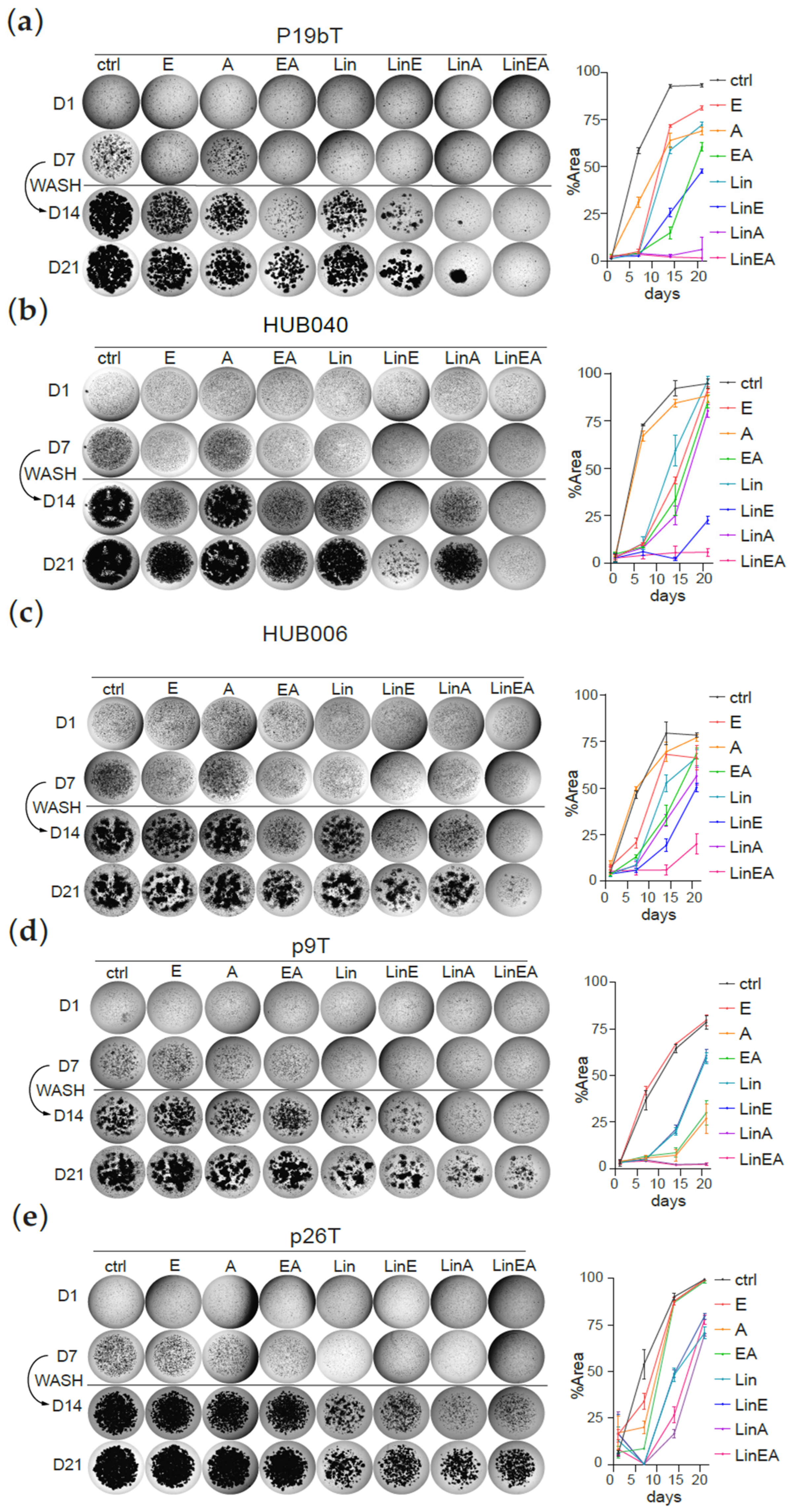
3.3. IR/IGF1R Inhibition Prevents Regrowth of CRC PDOs
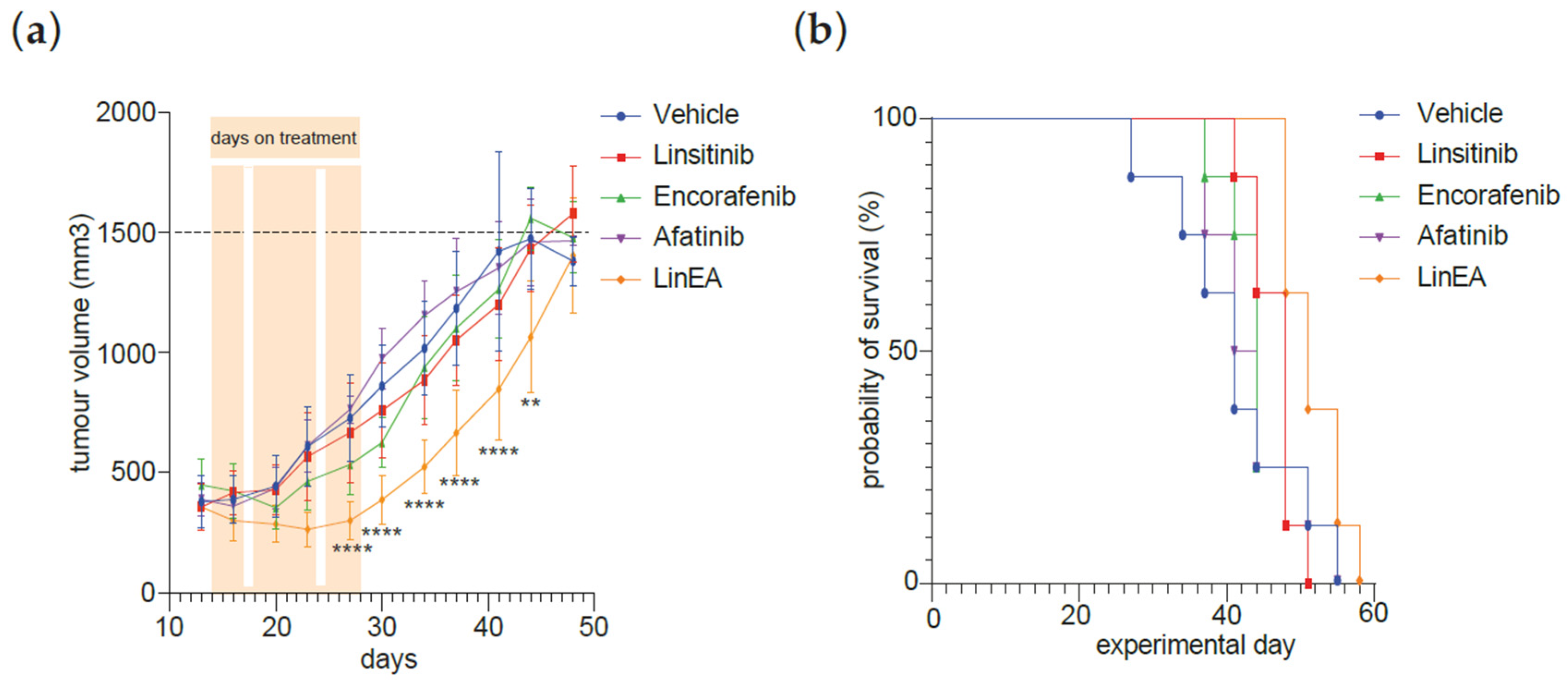
3.4. Combined Treatment MAPK Inhibitors and Linsitinib Decreases Tumour Growth in CRC PDO Xenograft
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef]
- Morris, V.; Overman, M.J.; Jiang, Z.Q.; Garrett, C.; Agarwal, S.; Eng, C.; Kee, B.; Fogelman, D.; Dasari, A.; Wolff, R.; et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin. Color. Cancer 2014, 13, 164–171. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E–Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Lee, R.J.; Nolop, K.B.; Saltz, L. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J. Clin. Oncol. 2010, 28, 3534. [Google Scholar] [CrossRef]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; Gerwen, B.V.; van der Ven, M.; Wetering, M.V.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. Elife 2016, 5, e18489. [Google Scholar] [CrossRef]
- Lackner, M.R.; Wilson, T.R.; Settleman, J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012, 8, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.F.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. EMBO Mol. Med. 2012, 4, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Patel, H.; Mishra, R.; Yacoub, N.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. IGF1R/IR Mediates Resistance to BRAF and MEK Inhibitors in BRAF-Mutant Melanoma. Cancers 2021, 13, 5863. [Google Scholar] [CrossRef]
- Buck, E.; Eyzaguirre, A.; Rosenfeld-Franklin, M.; Thomson, S.; Mulvihill, M.; Barr, S.; Brown, E.; O’Connor, M.; Yao, Y.; Pachter, J.; et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res. 2008, 68, 8322–8332. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Lobe, C.; Tahraoui, S.; Claperon, A.; Mergey, M.; Merabtene, F.; Wendum, D.; Coulouarn, C.; Housset, C.; Desbois-Mouthon, C.; et al. The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin. Cancer Res. 2018, 24, 4282–4296. [Google Scholar] [CrossRef]
- Puzanov, I.; Lindsay, C.R.; Goff, L.; Sosman, J.; Gilbert, J.; Berlin, J.; Poondru, S.; Simantov, R.; Gedrich, R.; Stephens, A.; et al. A phase I study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor-1 and insulin receptors, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 701–711. [Google Scholar] [CrossRef]
- Fassnacht, M.; Berruti, A.; Baudin, E.; Demeure, M.J.; Gilbert, J.; Haak, H.; Kroiss, M.; Quinn, D.I.; Hesseltine, E.; Ronchi, C.L.; et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A double-blind, randomised, phase 3 study. Lancet Oncol. 2015, 16, 426–435. [Google Scholar] [CrossRef]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R., Jr.; Dayyani, F.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, I.I.; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef]
- van Geel, R.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Bouazzaoui, L.; Raats, D.A.E.; Verheem, A.; Rinkes, I.H.M.B.; Snippert, H.J.G.; Maurice, M.M.; Kranenburg, O. Resistance to MAPK Pathway Inhibition in BRAF-V600E Mutant Colorectal Cancer Can Be Overcome with Insulin Receptor/Insulin-like Growth Factor-1 Receptor Inhibitors. Organoids 2025, 4, 14. https://doi.org/10.3390/organoids4020014
El Bouazzaoui L, Raats DAE, Verheem A, Rinkes IHMB, Snippert HJG, Maurice MM, Kranenburg O. Resistance to MAPK Pathway Inhibition in BRAF-V600E Mutant Colorectal Cancer Can Be Overcome with Insulin Receptor/Insulin-like Growth Factor-1 Receptor Inhibitors. Organoids. 2025; 4(2):14. https://doi.org/10.3390/organoids4020014
Chicago/Turabian StyleEl Bouazzaoui, Layla, Daniëlle A. E. Raats, André Verheem, Inne H. M. Borel Rinkes, Hugo J. G. Snippert, Madelon M. Maurice, and Onno Kranenburg. 2025. "Resistance to MAPK Pathway Inhibition in BRAF-V600E Mutant Colorectal Cancer Can Be Overcome with Insulin Receptor/Insulin-like Growth Factor-1 Receptor Inhibitors" Organoids 4, no. 2: 14. https://doi.org/10.3390/organoids4020014
APA StyleEl Bouazzaoui, L., Raats, D. A. E., Verheem, A., Rinkes, I. H. M. B., Snippert, H. J. G., Maurice, M. M., & Kranenburg, O. (2025). Resistance to MAPK Pathway Inhibition in BRAF-V600E Mutant Colorectal Cancer Can Be Overcome with Insulin Receptor/Insulin-like Growth Factor-1 Receptor Inhibitors. Organoids, 4(2), 14. https://doi.org/10.3390/organoids4020014