
In Silico Evaluation of Diketopiperazine (DPK) Derivatives as Potential Inhibitors for G-Protein-Coupled Receptors (GPCRs) †


Abstract
1. Introduction
2. Methods
3. Results and Discussion
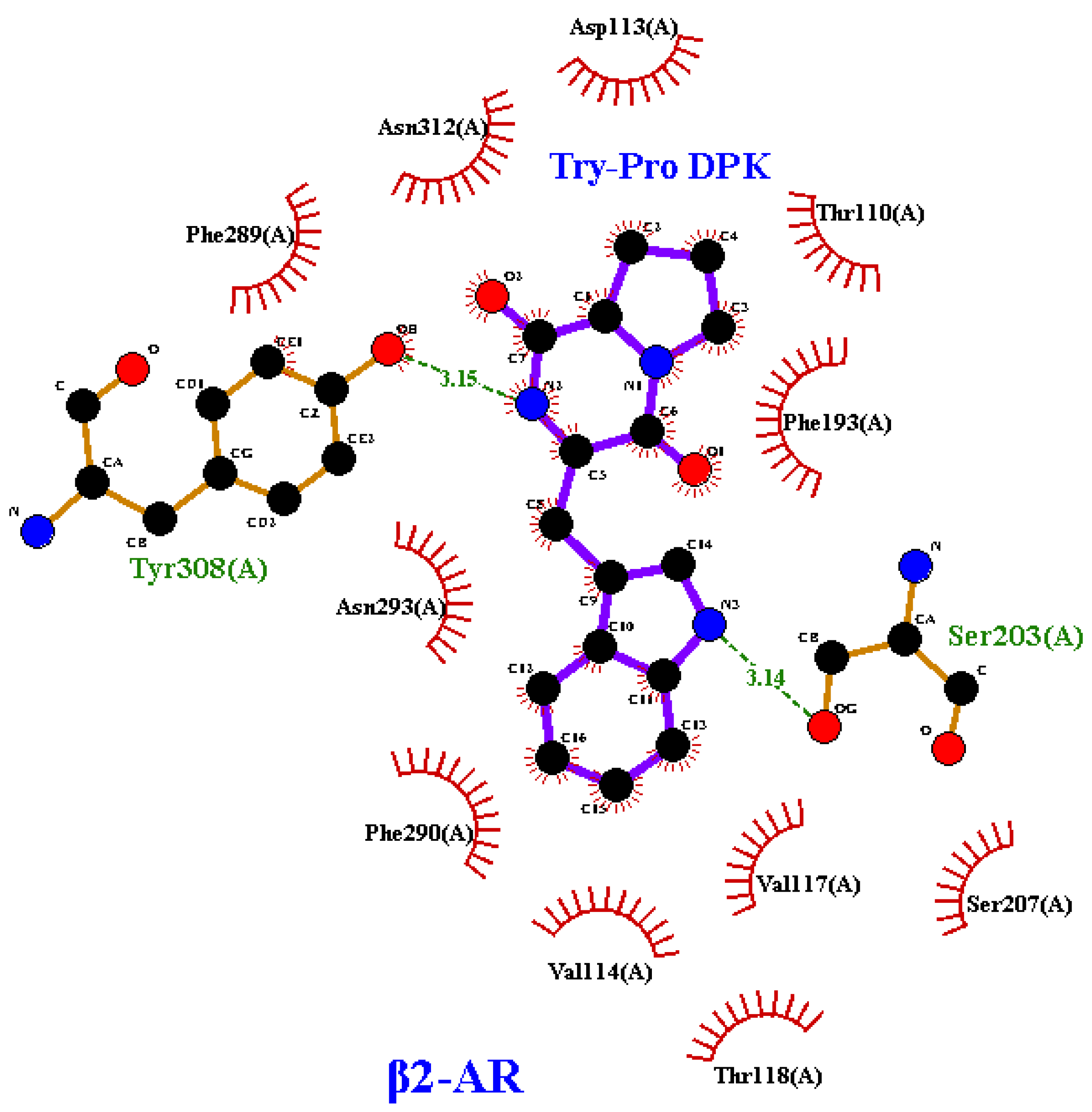
- Aromatic Interactions: The presence of tryptophan in compound 3 introduces aromaticity that may facilitate π-π stacking interactions with residues in the β2-AR binding pocket, thereby enhancing the compound’s binding strength.
- Structural Rigidity: The proline residue in the compound provides rigidity, ensuring a stable conformation within the binding pocket, which further improves the compound’s fit and binding potential.
- Hydrogen Bonding: The two hydrogen bonds formed by compound 3 contribute to the stability of the receptor–ligand complex, suggesting a strong interaction with β2-AR.
- Hydrophobic Contributions: The compound’s hydrophobic features likely contribute to its overall stability in the hydrophobic regions of the receptor’s binding pocket.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Heydenreich, F.M.; Flock, T.; Miljus, T.; Balaji, S.; Bouvier, M.; Veprintsev, D.B.; Tate, C.G.; et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 2016, 536, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Dimri, M. Biochemistry, G protein coupled receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Schiöth, H.B.; Fredriksson, R. The GRAFS classification system of G-protein coupled receptors in comparative perspective. Gen. Comp. Endocrinol. 2005, 142, 94–101. [Google Scholar] [CrossRef]
- Tandale, A.; Joshi, M.; Sengupta, D. Structural insights and functional implications of inter-individual variability in β2-adrenergic receptor. Sci. Rep. 2016, 6, 24379. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Biochemical basis of asthma therapy. J. Biol. Chem. 2011, 286, 32899–32905. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L. Novel β2-adrenergic receptor signaling pathways. J. Allergy Clin. Immunol. 2002, 110, S229–S235. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J. G-protein-coupled receptors: Past, present and future. Br. J. Pharmacol. 2006, 147, S27–S37. [Google Scholar] [CrossRef]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczynski, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed, E.H.; Elnagdy, S. Cyclic dipeptides: The biological and structural landscape with special focus on the anti-cancer proline-based scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H. A global review on short peptides: Frontiers and perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-drug conjugates: A new hope for cancer management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- El-Hachem, N.; Haibe-Kains, B.; Khalil, A.; Kobeissy, F.H.; Nemer, G. AutoDock and AutoDockTools for protein-ligand docking: Beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) as a case study. In Neuroproteomics: Methods and Protocols; Humana Press: New York, NY, USA, 2017; pp. 391–403. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, Inc. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, Inc.: New York, NY, USA, 2015. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
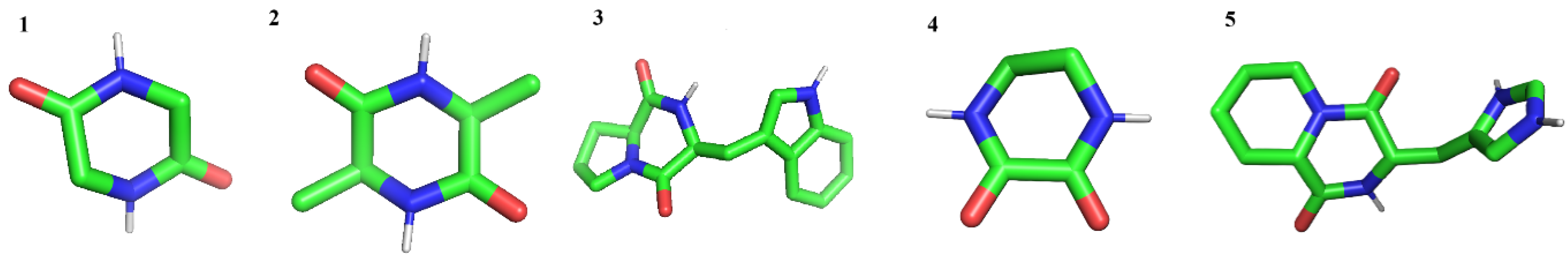
| Name of DPKs | Binding Energy (Kcal/mol) | No. H Bond | Molecular Weight (g/mol) | Hydrophobicity Log P | Net Charge at pH 7.4 | |
|---|---|---|---|---|---|---|
| 1 | 2,5 Piperazinedione | −3.00 | 2 | 114.10 | −1.1100 | 0 |
| 2 | Alanine diketopiperazine | −3.70 | 2 | 142.1559 | −0.3330 | 0 |
| 3 | Tryptophan proline diketopiperazine | −5.89 | 2 | 283.32 | 1.4665 | 0 |
| 4 | Diketopiperazine | −2.95 | 2 | 114.10 | −1.1100 | 0 |
| 5 | Histidylproline diketopiperazine | −5.13 | 2 | 248.2810 | 0.0984 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jafari, S.; Bojarska, J. In Silico Evaluation of Diketopiperazine (DPK) Derivatives as Potential Inhibitors for G-Protein-Coupled Receptors (GPCRs). Med. Sci. Forum 2025, 34, 2. https://doi.org/10.3390/msf2025034002
Jafari S, Bojarska J. In Silico Evaluation of Diketopiperazine (DPK) Derivatives as Potential Inhibitors for G-Protein-Coupled Receptors (GPCRs). Medical Sciences Forum. 2025; 34(1):2. https://doi.org/10.3390/msf2025034002
Chicago/Turabian StyleJafari, Sepideh, and Joanna Bojarska. 2025. "In Silico Evaluation of Diketopiperazine (DPK) Derivatives as Potential Inhibitors for G-Protein-Coupled Receptors (GPCRs)" Medical Sciences Forum 34, no. 1: 2. https://doi.org/10.3390/msf2025034002
APA StyleJafari, S., & Bojarska, J. (2025). In Silico Evaluation of Diketopiperazine (DPK) Derivatives as Potential Inhibitors for G-Protein-Coupled Receptors (GPCRs). Medical Sciences Forum, 34(1), 2. https://doi.org/10.3390/msf2025034002